

Abstract
A central focus of biological research is understanding the structure/function relationship of macromolecular protein complexes. Yet conventional transmission electron microscopy techniques are limited to static observations. Here we present the first direct images of purified macromolecular protein complexes using in situ liquid scanning transmission electron microscopy. Our results establish the capability of this technique for visualizing the interface between biology and nanotechnology with high fidelity while also probing the interactions of biomolecules within solution. This method represents an important advancement towards allowing future high-resolution observations of biological processes and conformational dynamics in real-time.
Keywords: In situ TEM, Liquid STEM, Nanolipoprotein, Aberration corrected STEM
1. Introduction
Transmission electron microscopy (TEM) exhibits the unique ability of visualizing all the length scales relevant to life science research at nanometer resolution or better. However, structural biology using TEM remains limited to static observations of macromolecules. Although the vitrification of protein samples helps to optimally preserve the integrity of biological material (Adrian et al., 1984), the freezing process employed during sample preparation for high-resolution cryo-electron microscopy (cryo-EM) physically immobilizes the samples and therefore prevents dynamic observation. In an effort to overcome this limitation, time-lapse cryo-EM was previously developed using either laser flash photolysis, microfluidic mixing, or directed spraying of an aerosol onto a sample coated grid to trigger a reaction just prior to plunging the grid into a volume of liquefied ethane (Berriman and Unwin, 1994; Shaikh et al., 2009; Subramaniam and Henderson, 1999; White et al., 2003). Unfortunately, time-lapse cryo-EM remains restricted to millisecond temporal resolution and solely provides “snapshots” of processes frozen in time. Thus, to permit real-time TEM observations of biological dynamics that occur on the microsecond or faster timescale, the samples need to be imaged in a fully hydrated and non-frozen state.
The concept of a windowed environmental cell for studying biological samples in a wet environment using TEM has been around for more than 75 years (Abrams and McBain, 1944; Marton, 1935). Early environmental chambers using thin film membranes provided the first electron micrographs of whole biological cells and three-dimensional protein nanocrystals in a wet and unaltered state (Abrams and McBain, 1944; Parsons, 1974). Recent applications of gas environmental chambers inside a TEM have also imaged muscle thick filaments and myosin filaments in a hydrated state by maintaining an equilibrium with saturated water vapor (Minoda et al., 2011; Sugi et al., 2008). However, these prior chamber designs did not permit flowing or exchanging liquid solutions during the course of an experiment. Additionally, the use of thick fluid path lengths and membrane thicknesses prevented resolving individual macromolecular complexes. Even applications of in situ liquid scanning TEM (STEM) for imaging fully hydrated biological materials have so far been limited to visualizing whole cells within a fluid path length of several micrometers (de Jonge et al., 2010; Peckys et al., 2009). In those experiments, the outline of human cells were detectable but the fine ultrastructural details related to the internal cellular architecture were not achieved and the identification and localization of proteins were inferred from the specific labeling by nanoparticles (de Jonge et al., 2010; Peckys et al., 2009). Recently, in situ liquid STEM of whole yeast cells has shown internal cellular structures with a stated resolution of 32 nm (Peckys et al., 2011). Yet, even higher resolution will be required to fully monitor and understand protein conformational dynamics. Here we report the first direct imaging of small isolated macromolecular protein complexes in a fully hydrated environment using in situ liquid STEM without staining or chemical fixation. This method uses very thin fluid path lengths and can be used to image biological proteins alone or at the interface with nanotechnology at better than 2 nm spatial resolution.
2. Methods
2.1. In situ environmental chamber assembly and sample loading
We utilized a continuous flow in situ liquid stage manufactured by Hummingbird Scientific (Lacey, WA) for the experiments in this paper. The sample loading and assembly of the environmental chamber were performed as previously described (Evans et al., 2011). Briefly, after cleansing the tip with an acetone wash to ensure a pristine environmental chamber, the inlet and outlet lines for the stage were primed with distilled water. Two square silicon chips containing silicon nitride membranes (50 nm thick) were then loaded into the tip of the continuous flow in situ liquid stage with membranes facing each other. Gold spacers deposited on the four corners of one chip (50 or 100 nm tall) dictated the nominal fluid path length for imaging. Prior to placing the upper window into the holder tip, 1 μl of the diluted protein sample was deposited directly on the surface of the lower window. After final assembly the tip was tested for maintaining vacuum in a Pfeiffer vacuum test chamber before loading in the electron microscope.
2.2. Ferritin and nanolipoprotein solutions
Ferritin (Sigma Aldrich, Prod. # F4503) and apoferritin (Sigma Aldrich, Prod. # A3641) were purchased from Sigma Aldrich and diluted 500 fold with distilled water. Nanolipoprotein complexes were produced using an E. coli based cell-free coupled transcription/translation reaction (Invitrogen, MembraneMax HN kit). Nanolipoproteins containing inserted bacteriorhodopsin molecules were subsequently purified using His-tag affinity purification with a Ni-NTA resin slurry followed by size exclusion chromatography (Princeton Separations, Centri-Spin 40) into a buffer containing 10 mM Tris, 100 mM NaCl, pH 7.5. The resulting nanolipoprotein sample was diluted 1000× into distilled water for all experiments.
2.3. Cryogenic and conventional transmission electron microscopy
Room temperature and cryo-negative stain images of nanolipoprotein solutions were prepared using standard procedures (Adrian et al., 1998; Ohi et al., 2004). Samples were stained with either 1% solution of uranyl formate or 2% ammonium molybdate prior to drying or plunge freezing. Images were acquired on a JEM-2100F Transmission Electron Microscope (JEOL, Japan) operating at 200 keV. Micrographs were recorded at nominal magnifications of 50,000× on a 4096 × 4096 pixel Tietz CCD camera (TVIPS, Gauting, Germany) following standard low-dose imaging procedures.
2.4. Scanning transmission electron microscopy (STEM) and image analysis
The in situ liquid stage was inserted into a JEM-2100F/Cs (JEOL, Japan) with a spherical aberration corrector for STEM imaging mode. Prior to sample insertion the microscope was aligned to 1-Å resolution using the standard Platinum/Iridium calibration sample for the CEOS corrector system. STEM images were collected simultaneously on bright field and dark field detectors with dwell times of 2 or 10 μs per pixel giving rise to single frame acquisition times of either 524 ms or 2.62 s for a 512 × 512 pixel image. The STEM probe had an average current density of 30 pA, yielding a dose of 60e−/Å2 (5 Å/pixel and 2 μs dwell time) or 150e−/Å2 (7 Å/pixel and 10 μs dwell time). Digital Micrograph (Gatan, Inc.) was utilized for all data acquisition. Line-profiles were calculated using the ImageJ “plot profile” command while image simulations of ferritin (PDB file 1AEW) were performed using the SPIDER software suite.
3. Results
3.1. Benefits of STEM
To demonstrate the capabilities of the continuous flow in situ liquid stage for imaging biological materials, we chose to utilize spherical aberration corrected STEM instead of conventional TEM. High angle annular dark field STEM imaging yields higher contrast than conventional TEM since the resulting image is mostly incoherent and the signal intensity in dark field imaging is approximately correlated to the atomic number squared (James and Browning, 1999). This increase of contrast is of critical importance for the experiments described here since biological samples typically exhibit weak scattering and the assembly of the liquid environmental chamber introduces extra material (silicon nitride membranes) through which the imaging electrons must be transmitted (Fig. 1). Using these imaging conditions, we have previously shown 2.1 Å lattice fringes for lead sulfide nanoparticles adhered to the upper membrane of the environmental chamber (Evans et al., 2011). However, the same resolution was not seen for equivalent lead sulfide nanoparticles attached to the lower window due to electron probe broadening.
Fig. 1.
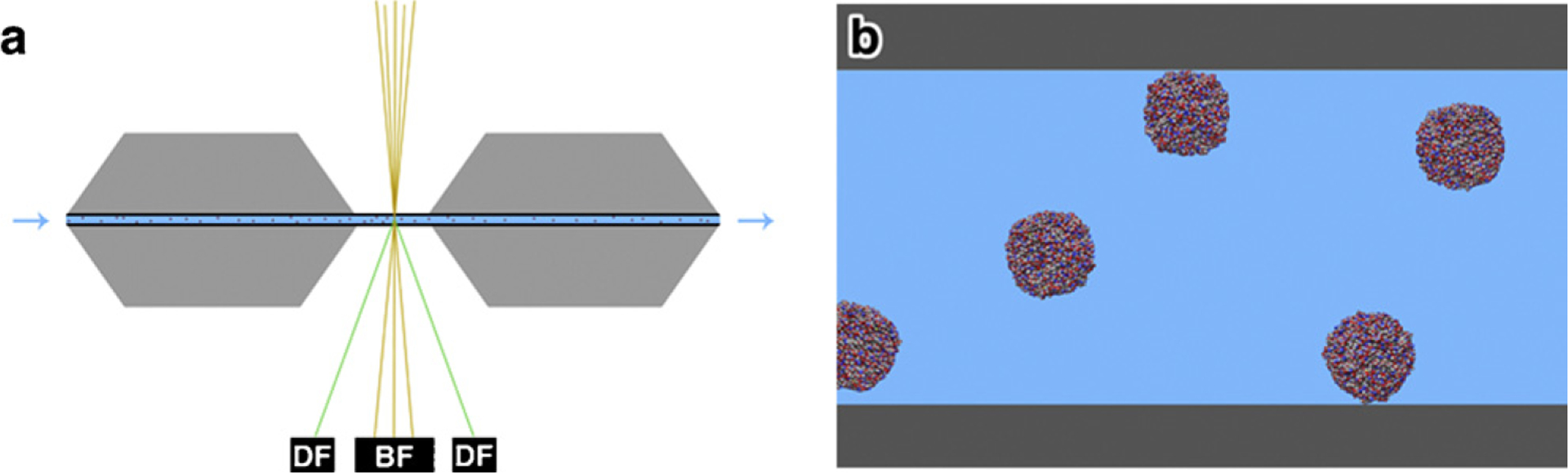
Overview of the environmental cell assembly. (a) Two silicon chips with silicon nitride membranes are oriented with membranes facing each other. A liquid suspension is contained between the electron transparent membranes while inlet and outlet fluid lines permit both static and continuous flow conditions. Downstream bright field (BF) and dark field (DF) detectors capture the signal during scan acquisition. (b) Zoomed-in region of (a) showing the two silicon nitride membranes sandwiching the liquid suspension. Ferritin molecules are depicted as freely floating or attached to the upper or lower membrane.
3.2. Imaging the interface of biology and nanotechnology
Following our previous work incorporating thin fluid path lengths to track the growth of lead sulfide nanoparticles from a precursor solution using in situ liquid STEM, we knew nanoparticles as small as 0.8 nm diameter could be successfully imaged (Evans et al., 2011). Therefore, as a positive control test sample we imaged the biomolecule ferritin, a naturally occurring interface between biology and nanotechnology. The ferritin protein complex is composed of twenty-four copies of the ferritin monomer that assemble into an octahedral protein shell of approximately 12 nm (outer diameter) surrounding an inner cavity with a diameter of 7.5 nm (Granier et al., 1997). The native role of this protein is to sequester iron molecules from the blood to prevent toxic levels of iron from adversely affecting healthy cells. Thus, in the presence of iron, ferritin acts as a biological cage housing an iron oxide nanoparticle up to ~7.5 nm diameter.
The outer surface of ferritin has regions that are partially charged or hydrophobic and can therefore interact with the silicon nitride membranes of the environmental chamber (Fig. 1b). Non-specific interactions between ferritin and the silicon nitride membranes would cause it to be partially immobilized and permit high-resolution imaging. Alternatively, any particles freely floating in solution will likely tumble or move during acquisition due to Brownian motion or fluid flow thereby causing the image to appear blurred. As can be seen in Fig. 2, purified ferritin molecules can be imaged using aberration corrected STEM within a liquid environment. Our real-time imaging of ferritin captured evidence of both static and mobile particles during acquisition periods ranging from milliseconds to seconds. Streaking artifacts can be seen in Fig. 2b where the white arrowhead indicates a ferritin molecule that was moving through solution while the STEM image was recorded. However, within the same image, other ferritin molecules are seen without such artifacts and confirm the presence of both the less dense protein shell and more dense nanoparticle core (Fig. 2c) of equivalent dimensions to the known structure. Apoferritin molecules in the absence of the nanoparticle core were also successfully imaged (Fig. 2d).
Fig. 2.

Imaging the interface of biology and nanotechnology in fluid. (a and b) In situ liquid bright field and dark field STEM images of a suspension of ferritin molecules in a buffered saline solution. The white arrowhead points to a ferritin molecule that had been moving during image acquisition. (c) Magnified view of ferritin molecule indicated by white arrow in (b) depicting both the outer protein shell and more dense (brighter) inner core with iron oxide nanoparticle. (d) In situ liquid bright field STEM image of ferritin (black arrow) and apoferritin (black arrowhead) in the same field of view. (e) Line-profiles from a ferritin macromolecule seen in (b). The line-profile shows evidence of the central iron oxide core and protein shell. The dotted and dashed lines identify the average contrast levels for the protein shell and background from which the r25–75 values were calculated (square markers). (f) Line-profiles from simulated projection images of ferritin (known structure PDB file 1AEW) with added iron oxide core and limited to spatial resolutions of 1 nm (dotted line) and 2 nm (solid line). Simulations of the projections not resolution limited and limited to 3 and 4 nm spatial resolution are not shown since they did not show similar line-profiles to the experimental data. Scale bars represent 20 nm (a and b) and 10 nm (c and d).
Since the electron probe was considerably smaller than the pixel size (0.2 nm versus 1 nm), the 25–75% rising edge width, r25–75 (Reimer, 1998), can be used as an estimate of resolution. A line-profile through one of the ferritin molecules seen in Fig. 2c depicts two r25–75 edge widths of 0.7 and 1.2 nm (Fig. 2e) followed by a contrast “plateau” corresponding to the 2 nm protein shell before rising further due to the nanoparticle core. Three other ferritin molecules yielded r25–75 values of 1.5, 1.8 and 2.0 nm (data not shown). Furthermore, comparing the line-profile from Fig. 2e to simulations of resolution limited projection images of ferritin with an iron oxide core provided a second method to verify a spatial resolution of at or better than 2 nm for these images (Fig. 2e and f). The variations in resolution for the individual ferritin molecules are attributed to differences in particle attachment or partial mobility during image acquisition. Nevertheless, these combined results prove that this integrated approach can visualize individual proteins through fluid thicknesses up to 100 nm and surrounded by two 50 nm thick silicon nitride membranes.
3.3. Observing biological macromolecules in liquid
To evaluate the versatility of imaging macromolecular biological samples within a liquid environment, we produced and purified nanolipoprotein discs (NLPs) to act as a second test specimen. NLPs have been proposed as a useful self-assembling platform for both functional and structural studies of membrane protein complexes (Bayburt and Sligar, 2003; Blanchette et al., 2008). Although the NLPs would ideally be monodisperse in solution, the direct imaging of purified NLPs by TEM depicts the presence of monomeric NLPs and discoidal stacks (Fig. 3a and b). Previous researchers have claimed that this stacking interaction is an artifact induced either by high protein concentration, salt composition (Zhang et al., 2011) or the process of drying when preparing samples for negative stain TEM (Chromy et al., 2007; Forte et al., 1971).
Fig. 3.
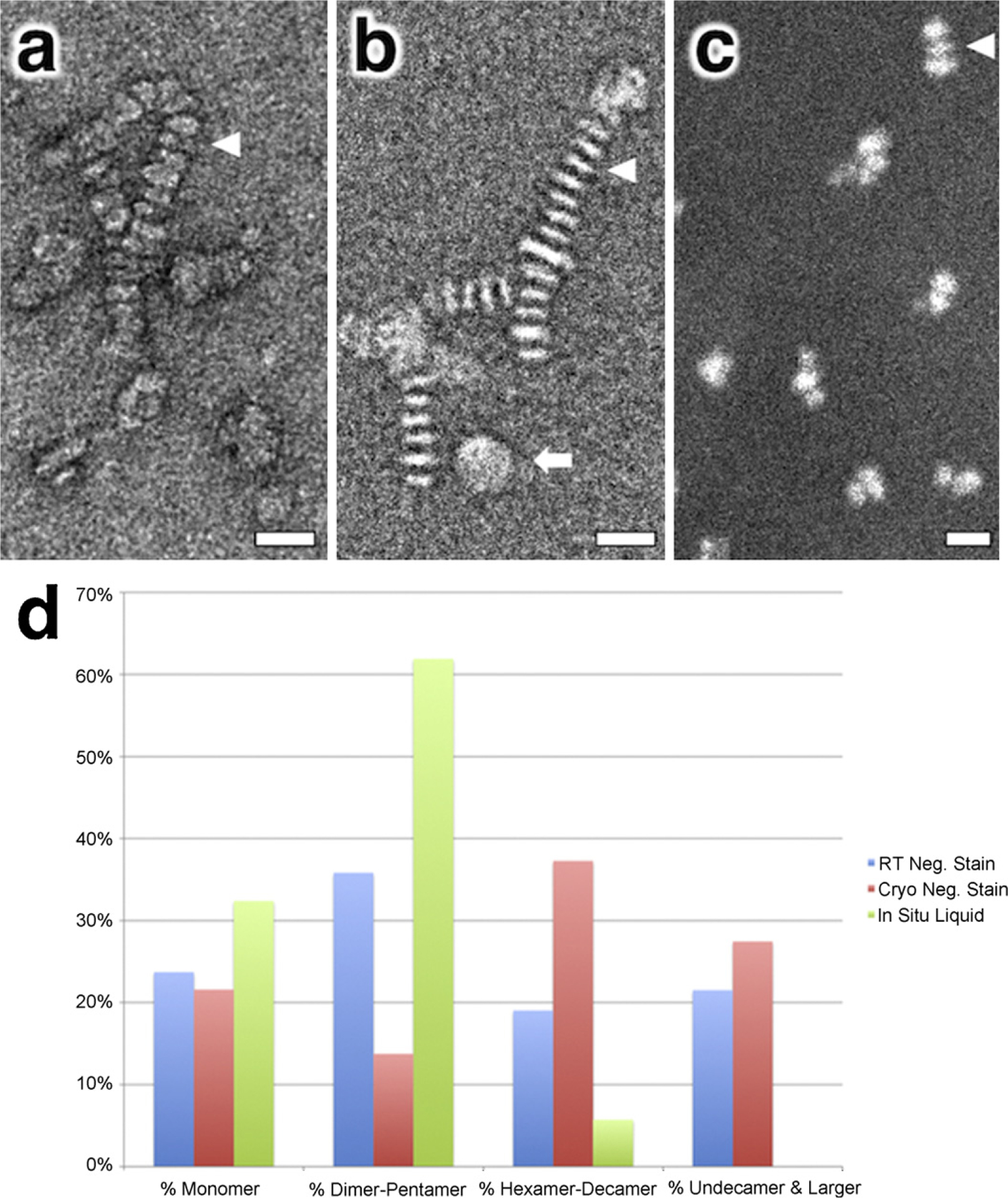
Analyzing nanolipoprotein disc (NLP) stacking with electron microscopy. (a and b) Conventional and cryogenic negative stain TEM images of nanolipoprotein discs indicating the presence of disc stacking. (c) In situ liquid dark field STEM image of NLPs fully hydrated and in suspension also depicting evidence of short stacking. (d) Histogram showing percentage of NLP population that appear as monomers or stacked oligomers of varying length. The white arrowheads in (a)–(c) point to individual NLPs in side view whereas the white arrow in (b) indicates a NLP in plan view. Note that the NLPs in (a) and (b) are arranged as long stacked chains whereas in (c) the NLPs are present in shorter stacks as dimers and trimers. Scale bars represent 20 nm.
Since the in situ liquid stage allows imaging fully hydrated samples as they appear in suspension, it provides a novel method for observing purified NLPs in a solvated state without requiring the presence of heavy metal salt stains, drying or freezing. We acquired aberration corrected STEM images of NLPs in suspension (Fig. 3c) at the same concentration used for the TEM images in Fig. 3a and b. The dark field STEM image clearly indicates the presence of short stacked NLPs even though the samples were of low protein and salt concentrations and were not subjected to drying. The population histograms from cryogenic and conventional room temperature negatively stained NLPs (Fig. 3d) both indicate that about 20% of the total population of NLPs are present as monomers while the majority are arranged in oligomeric stacks for those imaging conditions. In comparison, the histogram of the NLP population acquired using in situ liquid STEM shows a shift towards a smaller oligomeric stack (Fig. 3d) and a corresponding increase of the monomeric form to about 30%. Therefore, although the number of NLPs in a single stack may vary depending on the imaging technique, the fact that short stacks are present in the in situ image suggests that the stacking association is not an artifact caused by TEM sample preparation. Rather, these results seem to provide additional evidence that the stacking association is likely due to an electrostatic interaction as has recently been proposed (Zhang et al., 2011). Additional research probing the influence of salt concentration, buffer composition and pH are currently being explored.
4. Conclusion and discussion
Our results demonstrate that individual proteins and macromolecular complexes can be detected using in situ liquid scanning transmission electron microscopy. The images of ferritin and apoferritin prove that you can simultaneously image the interface of biology and nanotechnology, while the existence of nanolipoprotein disc stacks in suspension verifies the ability to observe complex interactions between neighboring molecules. By imaging proteins in a fully hydrated state this approach represents a necessary first step towards permitting the visualization of biological processes in real-time.
The next major hurdle that must be overcome to observe conformational dynamics at high resolution is to improve the rate at which images can be acquired. Faster read-out speeds would help limit blurring artifacts due to particle mobility during imaging. Of course the optimal temporal resolution needed for each experiment will vary depending on the type of conformational change that is expected. Intermolecular protein interactions are generally slow (>ms) and are immediately amenable to observation with the approach demonstrated here. However, by crossing the millisecond time barrier through the use of a direct electron detector (Denes et al., 2007), one could begin to image intramolecular conformational changes that occur on the ms to μs timescale such as protein domain, hinge-bending and subunit motion (McPherson and Eisenberg, 2011).
Finally, pushing the temporal resolution even further by incorporating ultrafast imaging (Reed et al., 2009) would provide access to the μs to ns timescale and facilitate examining loop or rigid-body motions that can lead to activation or inactivation of a protein’s functional state (McPherson and Eisenberg, 2011). It should be noted that it may take several years to fully evaluate the potential of integrating ultrafast TEM with in situ liquid microscopy. In the meantime, developments assimilating techniques from other fields for functionalizing silicon nitride surfaces with chemical tethers may help limit particle mobility during imaging and facilitate the study of biological processes at unprecedented resolution.
Acknowledgment
J.E.E. and N.D.B. acknowledge NIH funding support from grant number 5RC1GM091755. A portion of this work was performed at the Pacific Northwest National Laboratory which is operated by Battelle Memorial Institute for the U.S. Department of Energy under Contract No. DE-AC05-76RL01830.
Abbreviations:
- TEM
transmission electron microscopy
- STEM
scanning transmission electron microscopy
- Cryo-EM
cryogenic transmission electron microscopy
- NLP
nanolipoprotein
References
- Abrams IM, McBain JW, 1944. A closed cell for electron microscopy. Science 100, 273–274. [DOI] [PubMed] [Google Scholar]
- Adrian M, Dubochet J, Fuller SD, Harris JR, 1998. Cryo-negative staining. Micron 29, 145–160. [DOI] [PubMed] [Google Scholar]
- Adrian M, Dubochet J, Lepault J, McDowall AW, 1984. Cryo-electron microscopy of viruses. Nature 308, 32–36. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG, 2003. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 12, 2476–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman J, Unwin N, 1994. Analysis of transient structures by cryo-microscopy combined with rapid mixing of spray droplets. Ultramicroscopy 56, 241–252. [DOI] [PubMed] [Google Scholar]
- Blanchette CD, Law R, Benner WH, Pesavento JB, Cappuccio JA, Walsworth V, Kuhn EA, Corzett M, Chromy BA, Segelke BW, Coleman MA, Bench G, Hoeprich PD, Sulchek TA, 2008. Quantifying size distributions of nanolipoprotein particles with single-particle analysis and molecular dynamic simulations. J. Lipid Res 49, 1420–1430. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Arroyo E, Blanchette CD, Bench G, Benner H, Cappuccio JA, Coleman MA, Henderson PT, Hinz AK, Kuhn EA, Pesavento JB, Segelke BW, Sulchek TA, Tarasow T, Walsworth VL, Hoeprich PD, 2007. Different apolipoproteins impact nanolipoprotein particle formation. J. Am. Chem. Soc 129, 14348–14354. [DOI] [PubMed] [Google Scholar]
- de Jonge N, Poirier-Demers N, Demers H, Peckys DB, Drouin D, 2010. Nanometer-resolution electron microscopy through micrometers-thick water layers. Ultramicroscopy 110, 1114–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes P, Bussat J-M, Lee Z, Radmillovic V, 2007. Active pixel sensors for electron microscopy. Nucl. Instrum. Meth. Phys. Res. A 579, 891–894. [Google Scholar]
- Evans JE, Jungjohann KL, Browning ND, Arslan I, 2011. Controlled growth of nanoparticles from solution with in situ liquid transmission electron microscopy. Nano Lett. 11, 2809–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte T, Norum KR, Glomset JA, Nichols AV, 1971. Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency: structure of low and high density lipoproteins as revealed by electron microscopy. J. Clin. Invest 50, 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier T, Gallois B, Dautant A, Langlois d’Estaintot B, Precigoux G, 1997. Comparison of the structures of the cubic and tetragonal forms of horse-spleen apoferritin. Acta Crystallogr. D: Biol. Crystallogr 53, 580–587. [DOI] [PubMed] [Google Scholar]
- James EM, Browning ND, 1999. Practical aspects of atomic resolution imaging and analysis in STEM. Ultramicroscopy 78, 125–139. [Google Scholar]
- Marton L, 1935. La Microscopie Electronique des Objets Biologiques. Bull. Acad. Roy. Med. Belg 21, 600–617. [Google Scholar]
- McPherson A, Eisenberg D, 2011. In: Donev R (Ed.), Advances in Protein Chemistry & Structural Biology: Protein Structure and Diseases. Academic Press, pp. 189–190. [Google Scholar]
- Minoda H, Okabe T, Inayoshi Y, Miyakawa T, Miyauchi Y, Tanokura M, Katayama E, Wakabayashi T, Akimoto T, Sugi H, 2011. Electron microscopic evidence for the myosin head lever arm mechanism in hydrated myosin filaments using the gas environmental chamber. Biochem. Biophys. Res. Commun 405, 651–656. [DOI] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T, 2004. Negative staining and image classification – powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DF, 1974. Structure of wet specimens in electron microscopy. Improved environmental chambers make it possible to examine wet specimens easily. Science 186, 407–414. [DOI] [PubMed] [Google Scholar]
- Peckys DB, Mazur P, Gould KL, de Jonge N, 2011. Fully hydrated yeast cells imaged with electron microscopy. Biophys. J 100, 2522–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys DB, Veith GM, Joy DC, de Jonge N, 2009. Nanoscale imaging of whole cells using a liquid enclosure and a scanning transmission electron microscope. PLoS One 4, e8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BW, Armstrong MR, Browning ND, Campbell GH, Evans JE, LaGrange T, Masiel DJ, 2009. The evolution of ultrafast electron microscope instrumentation. Microsc. Microanal 15, 272–281. [DOI] [PubMed] [Google Scholar]
- Reimer L, 1998. Scanning Electron Microscopy: Physics of Image Formation and Microanalysis, 2nd ed. Springer-Verlag, Heidelberg. [Google Scholar]
- Shaikh TR, Barnard D, Meng X, Wagenknecht T, 2009. Implementation of a flash-photolysis system for time-resolved cryo-electron microscopy. J. Struct. Biol 165, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Henderson R, 1999. Electron crystallography of bacteriorhodopsin with millisecond time resolution. J. Struct. Biol 128, 19–25. [DOI] [PubMed] [Google Scholar]
- Sugi H, Minoda H, Inayoshi Y, Yumoto F, Miyakawa T, Miyauchi Y, Tanokura M, Akimoto T, Kobayashi T, Chaen S, Sugiura S, 2008. Direct demonstration of the cross-bridge recovery stroke in muscle thick filaments in aqueous solution by using the hydration chamber. Proc. Natl. Acad. Sci. U.S.A 105, 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HD, Thirumurugan K, Walker ML, Trinick J, 2003. A second generation apparatus for time-resolved electron cryo-microscopy using stepper motors and electrospray. J. Struct. Biol 144, 246–252. [DOI] [PubMed] [Google Scholar]
- Zhang L, Song J, Cavigiolio G, Ishida BY, Zhang S, Kane JP, Weisgraber KH, Oda MN, Rye KA, Pownall HJ, Ren G, 2011. Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J. Lipid Res 52, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]