

Abstract
Antigen-specific retargeting of cytotoxic lymphocytes against tumor-associated antigens has thus far remained largely dependent on chimeric antigen receptors (CARs) that can be constructed by the fusion of an extracellular targeting domain (classically a single-chain variable fragment from an antibody) fused with intracellular signaling domains to trigger activation of T or natural killer (NK) cells. A major limitation of CAR-based therapies is that this technology only allows for the targeting of antigens that would be located on the surface of target cells while non-surface antigens, which affect approximately three-fourths of all human genes, remain out of reach. The targeting of non-surface antigens is only possible using inherent T cell receptor (TCR) mechanisms. However, introducing a second TCR into T cells via genetic modification is problematic due to the heterodimeric nature of the TCR ligand-binding domain, which is composed of TCR α and β chains. It has been observed that the delivery of a second TCR α/β pair may lead to the mispairing of new TCR chains with the endogenously expressed ones and create mixed TCR dimers, and this has negatively affected the advancement of TCR-based T cell therapies. Recently, NK cells have been put forward as possible effectors for TCR gene therapy. Since NK cells do not endogenously express TCR chains, this seems to be an infallible approach to circumventing the problem of mispairing. Moreover, the similarity of intracellular signaling pathways and mechanisms of cytotoxicity between NK and T cells ensures that the triggering of antigen-specific responses by the TCR/CD3 complex can be used to induce antigen-specific cytotoxicity by TCR-modified NK (TCR-NK) cells. This review provides an overview of the initial studies of TCR-NK cells, identifies open questions in the field, and defines the place of this approach within the spectrum of adoptive immunotherapy techniques that rely on cytotoxic lymphocytes.
Keywords: T-cell receptor, Natural killer cells, CAR-T cells, TCR-NK cells
Abstract
Sitotoksik lenfositlerin genetik modifikasyon yoluyla tümör antijenlerine hedeflenerek kanser immünoterapisi için kullanılması, günümüzde en yoğun olarak kimerik antijen reseptörü (CAR) teknolojisiyle uygulanmaktadır. Bu teknolojide, tümör antijenini hedefleyen antikor dizilerinden elde edilen bir tek zincirli değişken fragman, T veya doğal öldürücü (NK) hücrelerin aktivasyonunu tetiklemek için hücre içi sinyal dizileriyle birleştirilerek yeni bir reseptör tasarlanmaktadır. CAR tabanlı tedaviler, yalnızca hedefteki tümör hücrelerinin yüzeyinde yer alan antijenlerin hedeflenmesine izin verirken, tüm insan genlerinin yaklaşık dörtte üçünü oluşturan hücre içi antijenlerin bu teknoloji ile erişilemeyecek durumda olması önemli bir eksikliktir. Hücre yüzeyinde bulunmayan antijenlerin hedeflenmesi, yalnızca T-hücre reseptörü (TCR) kullanılarak mümkün olmaktadır. Bununla birlikte, genetik modifikasyon yoluyla T-hücrelerine ikinci bir TCR’nin transfer edilmesi, TCR α ve β zincirlerinden oluşan ligand bağlama bölgesinin heterodimerik yapısı nedeniyle sorunludur. Genetik modifikasyon yoluyla iletilen yeni TCR α ve β zincirlerinin, endojen olarak ifade edilen zincirlerle yanlış eşleşmesi sonucu karışık TCR dimerleri oluşabileceği gözlemlenmiş ve bu sorun TCR ile modifiye T-hücre tedavilerinin geliştirilmesini olumsuz etkilemiştir. Yakın dönemde, bu sorunun üstesinden gelmek amacıyla NK hücreleri, TCR gen terapisi için olası efektör hücre tipi olarak öne sürülmüştür. NK hücreleri, TCR zincirlerini endojen olarak ifade etmediğinden, yanlış eşleşme sorununun üstesinden gelmek için yanılmaz bir yaklaşım olarak görünmektedir. NK ve T-hücreleri arasındaki hücre içi sinyal yolaklarının ve sitotoksisite mekanizmalarının benzerliği sayesinde, TCR ile modifiye edilmiş NK (TCR-NK) hücrelerinde antijene özgül sitotoksisite indüklemenin mümkün olduğu gösterilmiştir. Bu derleme, TCR-NK hücreleriyle ilgili öncül çalışmaların genel bir özetini ileterek, alandaki açık soruları tanımlamayı ve bu yeni yaklaşımın, sitotoksik lenfositlere dayalı adoptif immünoterapi teknikleri yelpazesindeki yerini tanımlamayı amaçlamaktadır.
Adoptive Immunotherapy Based on T-Cells
Mechanisms of T-Cell Recognition
T cells are essential for the development and regulation of immunological responses, homeostasis, and memory. They originate from common lymphoid progenitors, similar to B cells and natural killer (NK) cells. T cell-mediated recognition of a target occurs with the binding of the surface T-cell receptor (TCR) to the major histocompatibility complex (MHC) molecules present on the surface of all nucleated cells [1]. MHC molecules are found on target cell surfaces in complexes with short peptides that are produced as a result of the proteasome-mediated processing of proteins inside the cell. If a foreign protein is produced in the target cell (e.g., due to an intracellular infection), peptides from that protein will be presented on MHC class I molecules and can subsequently be recognized by CD8+ cytotoxic T cells through their TCRs. If the foreign protein is picked up by antigen-presenting cells (APCs) from the extracellular environment, the presentation of processed peptides to T cells will be carried out through MHC class II molecules on the cell surfaces of APCs and this will activate CD4+ helper T cells. On the other hand, T cells check not only the identity of the peptide presented but also the origin of the MHC molecules and they are activated through alloreactivity reactions if a foreign MHC is detected on the target cell [2].
The TCR complex that is responsible for this type of recognition consists of six CD3 chains and a TCR heterodimer (Figure 1). In the case of conventional T cells, the TCR is composed of α/β chains while other subsets of T cells expressing γ/δ TCR heterodimers also play important roles in the immune system [3,4]. The CD3ζ chain dimer is located at the cytoplasmic tail of the TCR/CD3 complex and plays an important role in the initiation of the signaling cascade after the TCR binds its peptide-loaded MHC (pMHC) complex [5,6].
Figure 1.
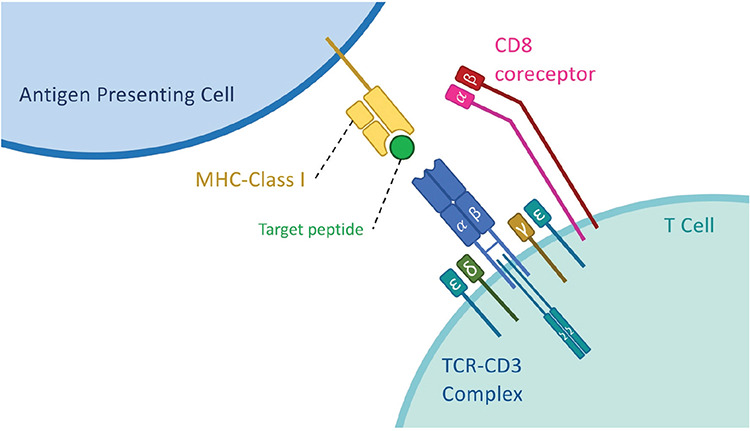
Binding of a T-cell receptor (TCR) to a major histocompatibility complex (MHC) class I molecule. The recognition of the peptide-loaded (pMHC) complex by the TCR is dependent on the binding of the TCR α/β pair to the MHC molecule presenting the processed peptide. The TCR/CD3 complex is formed with the help of four different CD3 chains binding to the TCR α/β pair. Among these, the CD3ζ chain is crucial for triggering intracellular signals upon pMHC binding. This binding and the triggering of intracellular signals can be supported by the presence of co-receptors (CD8 in this figure) that aid in stabilizing the binding and recruit intracellular signaling molecules in the vicinity of the TCR/CD3 complex.
Besides the TCR that directly binds the pMHC complex, there are also co-receptors such as CD4 and CD8 molecules that aid in the stabilization of this interaction and the triggering of the signaling cascade. These co-receptors increase the stability of the binding between the TCR and the peptide-MHC complex, which is important for signal transduction. They also have Lck on their cytoplasmic tails to start the phosphorylation cascade in TCR signaling. This signaling does not entirely depend on the co-receptors; however, they are helpful in increasing the strength of the signal [7,8] and they are also determinants of the T cell type [9,10]. During the developmental stages of T cells, thymocytes initially express both co-receptors, CD8 and CD4, and they are called double-positive thymocytes. After the selection and maturation phases, they become single positive cells and carry either CD4 or CD8 co-receptors. CD8+ cytotoxic T cells can only perform recognition through MHC class I while CD4+ T cells perform recognition through MHC class II and they have distinct roles in adaptive immunity [11].
Applications of T Cell-Based Immunotherapies
Until recently, non-targeted approaches such as chemotherapy or radiotherapy were primarily used as cancer treatments. However, these non-specific treatments are unable to distinguish between healthy cells and tumorigenic cells [12]. Alternatively, immunotherapy approaches can make use of the antigen-specific recognition properties of immune cells such as T cells and molecules such as antibodies to target and kill tumor cells.
Initial evaluations of T cell-based adoptive immunotherapy relied on tumor-infiltrating lymphocytes (TILs), which are autologous lymphocytes found inside the tumor microenvironment that could be isolated, expanded ex vivo, and reintroduced into the patient [13]. T cells from TILs have been reported to be capable of recognizing tumor-specific antigens or tumor-associated antigens (TAAs) via their TCRs [14,15]. With small sub-populations of NK and B cells, TILs are primarily made up of CD8+ cytotoxic T cells and CD4+ helper T cells [16]. T cells from TILs that are reactive against TAAs can be selectively expanded and reintroduced into the patient in order to increase the specificity and efficacy of the treatment [17,18]. However, the success of TIL-based cancer immunotherapy has been hampered by practical concerns such as problems related to the collection of TILs from patients with inoperable tumors. Additionally, it is possible that the T cells in the tumor microenvironment are already exhausted at the time of harvesting and are therefore unable to proliferate effectively [19,20].
Alternatively, the genetic modification of T cells isolated from peripheral blood to target a chosen antigen using cell surface receptors that are capable of recognizing TAAs has been a ground-breaking technique aimed at increasing the accessibility of such treatments. This can be done by delivery of either TCR genes that are targeting the tumor or a synthetic chimeric antigen receptor (CAR) to T cells [21].
Human T cells can be genetically modified with CARs, which are bioengineered tumor-targeting receptors, to redirect antigen specificity and improve functionality in adoptive immunotherapy [22]. The fundamental idea of CAR design is to connect an internal signaling module (e.g., CD3ζ) to an external ligand recognition domain, most commonly a single-chain variable fragment derived from an antibody, in order to activate T cells in response to antigen binding on the target cell surface [23]. While there was only a CD3ζ signaling domain in the first-generation CARs initially designed, second- and third-generation CAR designs include co-stimulatory domains such as CD28, 4-1BB, or OX40 to increase the proliferation, memory, and cytokine secretion of modified T cells [24]. CAR-engineered T cells can target the antigens on tumor cells in a non-MHC-dependent manner with their recognition domains. Clinical results of CAR-T cell treatments have so far been very promising and many CAR-T cell products have already been approved for clinical use [25].
While CAR-T cells have been very efficient in targeting surface antigens on tumor cells, the fact that only about one-fourth of the human genome encodes for transmembrane proteins [26] that could be expressed on cell surfaces and act as targets of CAR-T cell therapies or similar approaches such as antibody-mediated targeting is an important obstacle. This dramatically limits the possible antigenic targets that could be used for cancer immunotherapy as any intracellular or secreted proteins would remain out of reach for CAR-T cells. The only possibility for targeting intracellular antigens via immune cells seems to be the TCR-based recognition of peptides from intracellular proteins that are presented on cell surfaces via MHC molecules.
The first TAAs discovered and employed in TCR-mediated targeting through adoptive cell therapy were melanoma-associated antigens MART1, tyrosinase, and GP100 [27]. The T cells of cancer patients were genetically modified using TCR α/β chain genes that were found to be reactive against recognized TAAs [28,29,30]. However, the technology for TCR gene therapy carries the inherent burden of endogenous TCR expression in modified T cells [31]. When TCR α/β chains targeting an identified TAA are delivered to T cells, there is considerable risk of mispairing with the endogenously expressed TCR chains (Figure 2), and new TCR complexes with unknown specificity can be created when such mixing of TCR chains occurs. This may lead to autoreactivity issues and a potentially fatal graft-versus-host disease (GvHD)-like condition in vivo [32,33]. There have been various attempts to overcome this mispairing problem with different approaches, such as the introduction of extra disulfide bonds between the introduced TCR chains [34] and the use of constant regions from the TCR chains of mice [35] or small interfering RNAs downregulating endogenous TCR chain production [36]. Recent studies have also reported the use of genome editing technologies to knock out endogenous TCR genes [37,38]. Although successful to an extent, these procedures still carry a substantial risk of creating mispaired TCRs of unknown specificity, while problems such as immunogenicity related to the modification of the TCR sequence [39] or the inefficiency of the designed process impedes clinical advancement. Therefore, mispairing stands out as a major bottleneck to be addressed if safe and successful TCR gene therapy is to be achieved.
Figure 2.
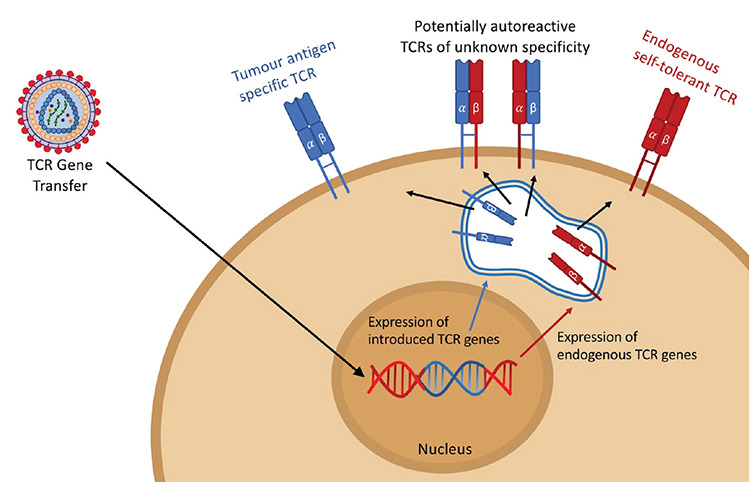
The mispairing problem in T-cell receptor (TCR) gene therapy. For the retargeting of a T cell toward a specific antigen, TCR α/β pairs that recognize the target epitopes of the major histocompatibility complex should be genetically transferred into T cells. This approach results in the expression of a new TCR α/β pair expected to form the new TCR. However, the genetically introduced TCR chains can also pair with TCR α/β chains that are already expressed in the given T cells and create novel TCRs that have not been subject to positive or negative selection. This can reduce the expression efficiency of the transferred TCRs or even cause the formation of autoreactive T cells due to the mixed TCR α/β pairs.
Adoptive Immunotherapy Based on NK Cells
Mechanisms of NK Cell Recognition
Another cytotoxic lymphocyte type in the immune system is the NK cell, which originates from common lymphoid progenitors with B and T cells. However, NK cells are classified as members of the innate immune system due to the lack of any clearly defined antigen-specific responses. With their quick cytolytic machinery, they can recognize and react against virally infected or transformed cells [40]. When NK cells were first identified in mice in the 1970s as a novel lymphocyte subset [41,42], their recognition and killing mechanisms were largely unknown. It was not until the 1980s that Kärre and Ljunggren [43] defined the missing-self hypothesis that explains the basics of NK cell-mediated cytotoxicity [44]. NK cells are now known to kill cells without antigen-specific recognition and in a non-MHC-restricted manner.
The current understanding is that the cytotoxic activity of an NK cell is regulated by the balance between various cell surface receptors that mediate its interactions with target cells [45]. Upon binding their ligands on target cell surfaces, some of these receptors work to activate NK cells and trigger effector mechanisms, while others are of an inhibitory nature and their intracellular signals counteract those delivered by activating receptors (Figure 3). It is this balance between activating and inhibitory signals that determines the outcome of an interaction of an NK cell with a target [46]. This dual receptor system ensures that NK cells can detect stressed, infected, or transformed cells through upregulated ligands of activating receptors or downregulation of MHC molecules on target cells [40,47]. Signaling pathways of activating receptors share great similarities with TCR/CD3 signaling as many NK cell activating receptors also utilize CD3ζ for signaling cascade initiation [48]. While the cell surface interactions and recognition receptors are different in NK and T cells, the intracellular pathways of signal transduction and mechanisms of target cell killing seem to be almost identical in most cases [49,50]. Once the decision to kill a target cell is made, both NK and T cells primarily act on their targets with the secretion of perforin and granzyme into the immunological synapse [51] and the triggering of apoptosis in the target cell.
Figure 3.
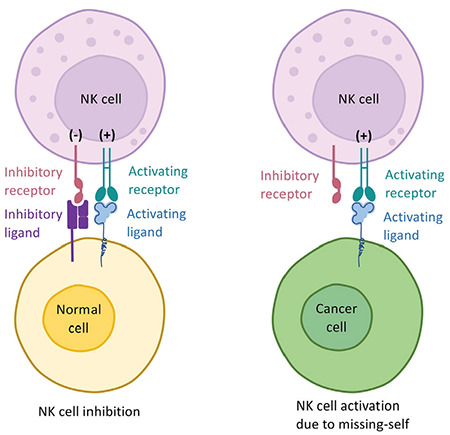
The basic mechanism of missing-self recognition in natural killer (NK) cells. The recognition mechanism in NK cells depends on two group of signals. The activating signals that trigger NK cell-mediated cytotoxicity are delivered via activating receptors and can be counteracted by negative signals delivered by inhibitory receptors such as killer cell immunoglobulin-like receptors (KIRs) that bind to major histocompatibility complex (MHC) or MHC-like molecules on the target cell’s surface. In a steady state, these signals balance each other out and the target cells are spared from NK cell attacks (left panel). In the case of MHC downregulation, which could occur due to intracellular infections or transformation, these negative signals are lost and the balance shifts toward NK cell activation in a process known as missing-self recognition.
Applications of NK Cell-Based Immunotherapies
In the 1970s, NK cells were initially recognized as tumor-killing lymphocytes [42] and their efficient cytotoxic activity against tumor cells has since been utilized with different approaches for cancer immunotherapies [52]. MHC molecules might be reduced or lost in tumor cells as an escape mechanism from the T cell-mediated immune response, but this strategy makes tumor cells prone to NK-mediated cytotoxicity, which works through a lack of MHC [53]. NK cells have been clinically tested against various cancer types including lymphoma [54], lung [55], breast [56], colon [55], and multiple myeloma [57] for adoptive immunotherapy. On the other hand, tumor cells can also develop methods to avoid NK cell cytotoxicity [58,59] and the tumor microenvironment can limit the potential of NK cells in various ways [60]. Research has been conducted to overcome this problem by increasing the release of cytokines including interleukin (IL)-2, IL-12, and IL-15 with various modifications [61,62,63]. Since 1997, a total of 420 clinical trials with NK cells have been initiated, the majority of them within the last decade [64]. Although these trials have shown promising results, the specificity of NK cells is still limited and genetic engineering is needed to better direct NK cells toward tumor cells.
CARs have been used to retarget NK cells against various tumor-specific antigens and CAR-NK cells have shown efficient killing capacity both in vivo and in vitro [65]. The source of the NK cells varies among studies from human induced pluripotent stem cell (iPSC)-derived NK cells to primary NK cells and NK cell lines [66,67]. There are currently 73 NK cell line-based and 35 primary NK cell-based clinical trials investigating CAR-NK cells [68]. Unlike CAR-T cells, which may cause cytokine release syndrome, neurotoxicity, and GvHD, CAR-NK cells have shown a milder profile regarding side effects while still delivering promising efficiency [68,69], except for one recent case of cytokine release syndrome after repeated infusions of CAR-modified NK-92 cells [70].
The distribution of clinical studies suggests that CAR-NK cells have the potential to be employed against both solid and hematological malignancies and early clinical trials have demonstrated the safety of CAR-NK cells against both. The success of CAR-T cell therapies directed at CD19 in B cell malignancies naturally makes CD19 the most often studied antigen among the target antigens of initial CAR-NK cell studies. In a recently published clinical study by Liu et al. [71], individuals with various lymphoid malignancies were treated with CAR-NK cells from allogeneic donors with anti-CD19 CARs. Seven of the 8 patients who received CAR-NK treatment achieved a full response with a lack of side effects such as cytokine release syndrome, which is frequently observed in CAR-T cell therapy.
TCR-NK Cells
While NK cells are being widely used for CAR engineering, they are also attracting attention as TCR modification of NK cells has been put forward as a solution to the TCR mispairing problem in T cells by us and others [72,73,74]. Since NK cells do not have any endogenous TCR chain expression to create problems in TCR-T cells, they are excellent candidates for TCR modification without the risk of forming mixed TCR dimers.
The close developmental link between T and NK cells, the similarity of cellular signaling and cytotoxicity mechanisms, and the expression of major TCR-related genes in NK cells provide a firm basis for the feasibility of this approach. These studies are important as they may offer a way to bypass the major setback caused by the mispairing of endogenously expressed TCRα/β chains in T cells with genetically introduced TCRs.
Initial studies on TCR-NK cells [72,73] have utilized the clinical-grade NK-92 cell line [75], which has become the most commonly used NK cell line with its ability to recognize and target various cancer types and more importantly with settled protocols for culture and genetic modification [59,60,76,77]. These previous studies were able to show that genetically modified NK-92 cells can express functional TCRs on cell surfaces when provided with the co-expression of CD3 complex proteins. As a result, TCR-NK cell lines showing antigen-specific cytotoxicity against specific antigens via the introduced TCR were successfully produced.
In the study by Parlar et al. [73], our group presented proof of the principle of functional TCR expression on NK cell surfaces using the NK-92 and YTS NK cell lines. A co-receptor-independent TCRα/β specific for the HLA-A2-restricted tyrosinase-derived “YMDGTMSQV” epitope (Tyr368-377) was delivered via lentiviral transduction to NK cell lines along with CD3 chain genes. It was demonstrated that both components of the TCR/CD3 complex (i.e., the TCR and CD3) depend on each other for stable surface expression. Since NK cells are known to have CD3ζ chain expression, the lentiviral delivery of CD3 subunits in this study included two variations, one containing the CD3ζ chain along with CD3ε, CD3γ, and CDδ (DGEZ) and another excluding the CD3ζ chain (only DGE), in order to observe whether the endogenous expression of the CD3ζ chain by NK-92 cells was enough for TCR/CD3 complex formation. It was found that both genetic modifications, with and without the CD3ζ chain, had surface expression of the TCR/CD3 complex. It was further demonstrated with in vitro degranulation and cytotoxicity assays that these cells, both NK-92 and YTS modified with DGE or DGEZ, were able to detect the target epitope of tyrosinase presented on MHC class I molecules and respond accordingly. Furthermore, a fluorescent reporter-based tumor xenograft mouse model was utilized to demonstrate the efficiency of the antigen-specific response of TCR-NK cells in vivo. Notably, the analyses of cell surface expression levels showed that the overexpression of the CD3ζ chain during genetic modification may particularly cause phenotypic changes in NK cells. Target cell recognition and the antigen-specificity of cytotoxic responses are likely to shift as a result of minor phenotypic variations and this was observed with a slight increase in non-specific cytotoxic activity against antigen-negative targets.
In another previous study, Mensali et al. [72] showed the efficient genetic modification of NK cells with TCR/CD3 complex genes and antigen-specific cytotoxic activity of TCR-NK cells. This study also utilized the NK-92 cell line but used two different TCRs, namely the DMF-5 TCR against Melan-A and the radium-1 TCR against a TGFβRII frameshift mutation peptide. These authors examined the correct folding of the TCR, TCR signaling cascade activation, phenotypic changes of TCR-NK cells, cytokine secretion, and degranulation profiles upon stimulation with target cells and confirmed the efficiency of TCR-NK cell-mediated antigen-specific cytotoxicity. They also demonstrated that TCR-NK cells show metabolic patterns similar to those of T cells upon activation. These cells effectively kill target cells in vitro and also respond to spheroids of colorectal cancer cells expressing the TGFβRII frameshift mutant in an antigen-specific manner. In vivo studies confirmed the antitumor activity of the TCR-NK cells [72]. Taken together, these studies showed the possibility of using NK cell lines, and especially NK-92, for TCR gene therapy.
Additionally, Morton et al. [74] demonstrated that it is possible to utilize primary human NK cells for TCR gene therapy. They found that primary NK cells can be successfully genetically modified with TCR α/β chains along with the addition of the CD8α/β co-receptor and the full CD3 complex including all four CD3 chains. Their study proposed a two-step retroviral transduction method for the delivery of TCR α/β chains targeting the BOB1 antigen, CD3 chains, and the CD8 co-receptor and presented positive results compared to TCR-T cells. The antigen specificity of TCR-NK cells against different targets was shown with BOB1-specific, PRAME-specific, and CMV-specific TCRs. In vivo studies further confirmed the results from in vitro experiments by showing the antigen-specific response of TCR-NK cells targeting BOB1. It was found that the cytolytic activity of TCR-NK cells is higher than that of TCR-T cells whose endogenous TCR expression was knocked down with CRISPR/Cas9, indicating that the combination of the antitumor effects of NK and T cells offers an enhanced therapeutic strategy. Degranulation and cytokine secretion assays further confirmed the antigen-specific activation of TCR-NK cells. Finally, the full extent of the antitumor activity of TCR-NK cells was demonstrated both in vitro and in vivo using cells with knocked-down MHC-I molecules to prove that the TCR-NK cells were still able to kill their targets and slow the tumor growth in a non-TCR-dependent matter, which indicated that these cells still possessed the NK cell characteristics that regulate the response against MHC loss on target cells.
Taken together, these studies offer new ways to utilize primary NK cells for TCR-based immunotherapies and enable the combination of NK cell-mediated and T cell-mediated antitumor effects [74].
Conclusion
TCR-NK technology stands out as a potentially fool-proof solution to the mispairing problem observed with TCR-T cells. However, the clinical development of TCR-NK cells is not exempt from the major hurdles faced by NK cell-based immunotherapies. For the successful translation of TCRs or otherwise genetically modified NK cells into clinical practice, efforts to optimize ex vivo NK cell expansion [64,78] and genetic modification protocols [79,80] have achieved considerable improvements in recent decades. The NK-92 cell line has been widely used in preclinical and clinical studies to develop novel immunotherapy applications [81,82], but the cost of having to irradiate the cells before infusions may be reflected in decreased activity and a lack of long-term persistence [83]. Alternatively, a wide array of autologous or allogeneic sources of NK cells, ranging from short-term [84] or long-term [85] activated peripheral blood NK cells to NK cells derived from iPSC [86], embryonic stem cells [87], or cord blood hematopoietic progenitor cells [88], are under clinical investigation as potential NK cell sources. Recent clinical experience with CAR-modified allogeneic NK cells has shown that they can expand and persist at low levels in vivo for at least 12 months [71]. Further research into the combination of NK cell infusions with immune checkpoint inhibitors is expected to further the likelihood of the long-term persistence and elevated activity of adoptively transferred NK cells [89].
While the clinical experience with CAR-NK therapies is surely paving the way to the advancement of the clinical translation of TCR-NK cells, many open questions remain to be answered about the exact mechanisms of TCR-NK cell-mediated cytotoxicity and possible approaches to optimize the production of TCR-NK cells for clinical use.
Although NK cells lack most of the TCR/CD3 complex components, they have endogenous expression of the CD3ζ chain intracellularly [90]. It has been observed that the delivery of CD3δ, CD3γ, and CD3ε is enough for the surface expression of the TCR in NK-92 cells. Additionally, it was demonstrated that overexpression of the CD3ζ chain increased the cytotoxic response of TCR-NK cells against the A375 and K562 cell lines in a non-specific manner [73]. These results suggest that the inclusion of CD3ζ in vector design does not significantly increase antigen-specific triggering. On the contrary, it may even increase the background activity against non-specific targets. Thus, the inclusion of CD3ζ in the vector may be deleterious for the function of the TCR in NK cells as a result of non-specific activation due to CD3ζ-dependent changes in NK cell-activating receptors. Whether the presence of CD3ζ shows similar effects in primary NK cells still remains a mystery since a comparison of vectors with and without CD3ζ was not undertaken in the study by Morton et al. [74]. For future applications of TCR modification in primary NK cells, the importance of all four CD3 chains needs to be carefully and separately analyzed to pinpoint the minimal requirements for functional TCR expression while sustaining NK activity and antigen specificity.
It is known that the majority of TCRs require CD4 or CD8 as a co-receptor, which are not expressed on the cell surfaces of NK cells. The importance and necessity of co-receptors for TCR-NK cells is probably related to the specific TCR α/β pairs used and is yet to be fully understood. The recruitment of Lck to the immunoreceptor tyrosine-based activation motifs (ITAMs) of the CD3 signaling complex is increased by co-receptors, but signaling is not totally dependent on the Lck contributed by the co-receptors as it has been reported that cytosolic Lck can also phosphorylate ITAMs even in the absence of co-receptors [7,8]. It is also known that co-receptors make TCR and pMHC binding more stable [10], which may contribute to better signaling. Therefore, the modulation of TCR signaling with the presence of a co-receptor may affect TCR signaling in NK cells depending on the specific characteristics of each different TCR. Our initial observations with the TCR directed against tyrosinase indicated that the CD8aa homodimer could have some suppressive effects for TCR-NK cells while CD8αβ and CD8ββ might be correlated with increased cytotoxic activity [91]. However, for a definitive answer to this question, more in-depth investigations using multiple different co-receptor-dependent and -independent TCRs are required in order to fully understand the function of co-receptors in TCR-NK signaling.
Killer cell immunoglobulin-like receptors (KIRs), which have the ability to interact with the MHC class I molecules on their targets, are the most common class of inhibitory receptors in NK cells [92]. This interaction is distinct from the way in which TCRs bind to MHC molecules and it is not dependent on the identity of the peptide presented. KIRs bind to MHC molecules from the side in a way that circumvents interaction with the presented peptide. As a result, KIRs only identify the presence of the MHC rather than the nature of the peptide [93]. Since the NK-92 cell line does not express any KIRs [75], it could be used to study the effects of specific KIR ligands in a controlled manner by overexpressing one KIR at a time to see its effect on TCR-NK function. In this regard, our initial results showed that the antigen-specific cytotoxic action of NK-92 cells is unaffected by the presence of any particular inhibitory receptor [94]. The study of TCR modification of primary NK cells conducted by Morton et al. [74] also provided evidence that KIR expression does not impede the functions of TCRs on NK cells. However, it is still unclear how the diverse range of KIRs in primary human NK cell populations may impact TCR-NK cell functions and whether certain KIR-ligand combinations would have a deleterious effect.
While many questions about the exact nature of TCR-NK cells and their direct comparison to TCR-T cells remain, initial studies have successfully put NK cells forward as a novel resource for TCR-based adoptive immunotherapy. Future studies focusing on the comparison of different approaches to producing TCR-NK cells and the interactions of TCR signals with co-receptor or inhibitory receptor signaling in NK cells are warranted to support the clinical translation of this approach for the efficient targeting of intracellular antigens, a realm that has so far remained out of reach for cancer immunotherapies such as CAR-T or CAR-NK cells.
Footnotes
Ethics
Authorship Contributions
Literature Search: Z.S.K., M.A., T.S.; Writing: Z.S.K., M.A.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Williams WV, Weiner DB, Wadsworth S, Greene MI. The antigen-major histocompatibility complex-T cell receptor interaction. A structural analysis. Immunol Res. 1988;7:339–350. doi: 10.1007/BF02935538. [DOI] [PubMed] [Google Scholar]
- 2.Felix NJ, Allen PM. Specificity of T-cell alloreactivity. Nat Rev Immunol. 2007;7:942–953. doi: 10.1038/nri2200. [DOI] [PubMed] [Google Scholar]
- 3.Lalor SJ, McLoughlin RM. Memory γδ T cells–Newly appreciated protagonists in infection and immunity. Trends Immunol. 2016;37:690–702. doi: 10.1016/j.it.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Holtmeier W, Kabelitz D. γδ T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151–183. doi: 10.1159/000086659. [DOI] [PubMed] [Google Scholar]
- 5.Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18:485–497. doi: 10.1038/s41577-018-0020-8. [DOI] [PubMed] [Google Scholar]
- 6.Samelson LE, Klausner RD, Baniyash M, Garcia-Morales P. The T-cell antigen receptor structure and mechanism of activation. Ann N Y Acad Sci. 1988;540:1–3. doi: 10.1111/j.1749-6632.1988.tb27045.x. [DOI] [PubMed] [Google Scholar]
- 7.Mørch AM, Bálint Š, Santos AM, Davis SJ, Dustin ML. Coreceptors and TCR signaling – the strong and the weak of it. Front Cell Dev Biol. 2020;8:597627. doi: 10.3389/fcell.2020.597627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schilham MW, Fung-Leung WP, Rahemtulla A, Kuendig T, Zhang L, Potter J, Miller RG, Hengartner H, Mak TW. Alloreactive cytotoxic T cells can develop and function in mice lacking both CD4 and CD8. Eur J Immunol. 1993;23:1299–1304. doi: 10.1002/eji.1830230617. [DOI] [PubMed] [Google Scholar]
- 9.Cheroutre H, Lambolez F. Doubting the TCR coreceptor function of CD8αα. Immunity. 2008;28:149–159. doi: 10.1016/j.immuni.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Kern P, Hussey RE, Spoerl R, Reinherz EL, Chang HC. Expression, purification, and functional analysis of murine ectodomain fragments of CD8αα and CD8αβ dimers. J Biol Chem. 1999;274:27237–27243. doi: 10.1074/jbc.274.38.27237. [DOI] [PubMed] [Google Scholar]
- 11.Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol. 2008;8:788–801. doi: 10.1038/nri2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arruebo M, Vilaboa N, Sáez-Gutierrez B, Lambea J, Tres A, Valladares M, González-Fernández A. Assessment of the evolution of cancer treatment therapies. Cancers (Basel) 2011;3:3279–3330. doi: 10.3390/cancers3033279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, Simpson C, Carter C, Bock S, Schwartzentruber D, Wei JP, White DE. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 14.Wölfel T, Klehmann E, Müller C, Schütt KH, Meyer zum Büschenfelde KH, Knuth A. Lysis of human melanoma cells by autologous cytolytic T cell clones. Identification of human histocompatibility leukocyte antigen A2 as a restriction element for three different antigens. J Exp Med. 1989;170:797–810. doi: 10.1084/jem.170.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van den Eynde B, Hainaut P, Hérin M, Knuth A, Lemoine C, Weynants P, van der Bruggen P, Fauchet R, Boon T. Presence on a human melanoma of multiple antigens recognized by autologous CTL. Int J Cancer. 1989;44:634–640. doi: 10.1002/ijc.2910440413. [DOI] [PubMed] [Google Scholar]
- 16.Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13:228–241. doi: 10.1038/nrclinonc.2015.215. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 18.Fisher B, Packard BS, Read EJ, Carrasquillo JA, Carter CS, Topalian SL, Yang JC, Yolles P, Larson SM, Rosenberg SA. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. 1989;7:250–261. doi: 10.1200/JCO.1989.7.2.250. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther. 2016;16:1265–1275. doi: 10.1080/14712598.2016.1214266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vacchelli E, Eggermont A, Fridman WH, Galon J, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Adoptive cell transfer for anticancer immunotherapy. Oncoimmunology. 2013;2:e24238. doi: 10.4161/onci.24238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 23.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960–968. doi: 10.1016/0006-291x(87)90502-x. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36:494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivica NA, Young CM. Tracking the CAR-T revolution: analysis of clinical trials of CAR-T and TCR-T therapies for the treatment of cancer (1997-2020) Healthcare (Basel) 2021;9:1062. doi: 10.3390/healthcare9081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10:1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 27.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, Morgan RA. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson LA, Heemskerk B, Powell DJ Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JH, Heemskerk MH. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A. 2010;107:10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525–541. doi: 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- 33.Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41:49–58. doi: 10.1007/s00281-018-0703-z. [DOI] [PubMed] [Google Scholar]
- 34.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okamoto S, Amaishi Y, Goto Y, Ikeda H, Fujiwara H, Kuzushima K, Yasukawa M, Shiku H, Mineno J. A promising vector for TCR gene therapy: differential effect of siRNA, 2A peptide, and disulfide bond on the introduced TCR expression. Mol Ther Nucleic Acids. 2012;1:e63. doi: 10.1038/mtna.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruggiero E, Carnevale E, Prodeus A, Magnani ZI, Camisa B, Merelli I, Politano C, Stasi L, Potenza A, Cianciotti BC, Manfredi F, Di Bono M, Vago L, Tassara M, Mastaglio S, Ponzoni M, Sanvito F, Liu D, Balwani I, Galli R, Genua M, Ostuni R, Doglio M, O’Connell D, Dutta I, Yazinski SA, McKee M, Arredouani MS, Schultes B, Ciceri F, Bonini C. CRISPR-based gene disruption and integration of high-avidity, WT1-specific T cell receptors improve antitumor T cell function. Sci Transl Med. 2022;14:eabg8027. doi: 10.1126/scitranslmed.abg8027. [DOI] [PubMed] [Google Scholar]
- 38.Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. doi: 10.1182/blood-2017-05-787598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis JL, Theoret MR, Zheng Z, Lamers CHJ, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res. 2010;16:5852–5861. doi: 10.1158/1078-0432.CCR-10-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krzewski K, Strominger JL. The killer’s kiss: the many functions of NK cell immunological synapses. Curr Opin Cell Biol. 2008;20:597–605. doi: 10.1016/j.ceb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herberman RB, Nunn ME, Lavkin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. 1975;16:216–229. doi: 10.1002/ijc.2910160204. [DOI] [PubMed] [Google Scholar]
- 42.Kiessling R, Petranyi G, Kärre K, Jondal M, Tracey D, Wigzell H. Killer cells: a functional comparison between natural, immune T-cell and antibody-dependent in vitro systems. J Exp Med. 1976;143:772–780. doi: 10.1084/jem.143.4.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 44.Sivori S, Vacca P, del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16:430–441. doi: 10.1038/s41423-019-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology. 2018;154:383–393. doi: 10.1111/imm.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meza Guzman LG, Keating N, Nicholson SE. Natural killer cells: tumor surveillance and signaling. Cancers (Basel) 2020;12:952. doi: 10.3390/cancers12040952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vivier E, Nunè JA, Vély F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 48.Altin JG, Pagler EB, Kinnear BF, Warren HS. Molecular associations involving CD16, CD45 and zeta and gamma chains on human natural killer cells. Immunol Cell Biol. 1994;72:87–96. doi: 10.1038/icb.1994.13. [DOI] [PubMed] [Google Scholar]
- 49.Narni-Mancinelli E, Vivier E, Kerdiles YM. The “T-cell-ness” of NK cells: unexpected similarities between NK cells and T cells. Int Immunol. 2011;23:427–431. doi: 10.1093/intimm/dxr035. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg J, Huang J. CD8+ T cells and NK cells: parallel and complementary soldiers of immunotherapy. Curr Opin Chem Eng. 2018;19:9–20. doi: 10.1016/j.coche.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapani JA, Davis J, Vivien RS, Smyth MJ. Proapoptotic functions of cytotoxic lymphocyte granule constituents in vitro and in vivo. Curr Opin Immunol. 2000;12:323–329. doi: 10.1016/s0952-7915(00)00094-7. [DOI] [PubMed] [Google Scholar]
- 52.Sutlu T, Alici E. Natural killer cell-based immunotherapy in cancer: current insights and future prospects. J Intern Med. 2009;266:154–181. doi: 10.1111/j.1365-2796.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- 53.Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10:230–252. doi: 10.1038/cmi.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller JS, Tessmer-Tuck J, Pierson BA, Weisdorf D, McGlave P, Blazar BR, Katsanis E, Verfaillie C, Lebkowski J, Radford J Jr, Burns LJ. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol Blood Marrow Transplant. 1997;3:34–44. [PubMed] [Google Scholar]
- 55.Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, Pfister K, Multhoff G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase I trial. Clin Cancer Res. 2004;10:3699–3707. doi: 10.1158/1078-0432.CCR-03-0683. [DOI] [PubMed] [Google Scholar]
- 56.Liang S, Xu K, Niu L, Wang X, Liang Y, Zhang M, Chen J, Lin M. Comparison of autogeneic and allogeneic natural killer cells immunotherapy on the clinical outcome of recurrent breast cancer. Onco Targets Ther. 2017;10:4273–4281. doi: 10.2147/OTT.S139986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nahi H, Chrobok M, Meinke S, Gran C, Marquardt N, Afram G, Sutlu T, Gilljam M, Stellan B, Wagner AK, Blomberg P, Holmqvist PH, Walther-Jallow L, Mellström K, Liwing J, Gustafsson C, Månsson R, Klimkowska M, Gahrton G, Lund J, Ljungman P, Ljunggren HG, Alici E. Autologous NK cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep Med. 2022;3:100508. doi: 10.1016/j.xcrm.2022.100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pegram HJ, Kershaw MH, Darcy PK. Genetic modification of natural killer cells for adoptive cellular immunotherapy. Immunotherapy. 2009;1:623–630. doi: 10.2217/imt.09.36. [DOI] [PubMed] [Google Scholar]
- 59.Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res. 2003;90:127–156. doi: 10.1016/s0065-230x(03)90004-2. [DOI] [PubMed] [Google Scholar]
- 60.Dahlberg CIM, Sarhan D, Chrobok M, Duru AD, Alici E. Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front Immunol. 2015;6:605. doi: 10.3389/fimmu.2015.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goding S, Yang Q, Mi Z, Robbins PD, Basse PH. Targeting of products of genes to tumor sites using adoptively transferred A-NK and T-LAK cells. Cancer Gene Ther. 2007;14:441–450. doi: 10.1038/sj.cgt.7701019. [DOI] [PubMed] [Google Scholar]
- 62.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10:1359–1373. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Tian Z, Sun R, Wei H, Zhang J. Characterization of interleukin-15 gene-modified human natural killer cells: implications for adoptive cellular immunotherapy. Haematologica. 2004;89:338–347. [PubMed] [Google Scholar]
- 64.Lamers-Kok N, Panella D, Georgoudaki AM, Liu H, Özkazanc D, Kučerová L, Duru AD, Spanholtz J, Raimo M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. 2022;15:164. doi: 10.1186/s13045-022-01382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunology. 2021;10:e1274. doi: 10.1002/cti2.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, Juan M, Eguizabal C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019;9:18729. doi: 10.1038/s41598-019-55239-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192.e5. doi: 10.1016/j.stem.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14:73. doi: 10.1186/s13045-021-01083-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, Vemuri MC. Current perspectives on “off-the-shelf” allogeneic NK and CAR-NK cell therapies. Front Immunol. 2021;12:732135. doi: 10.3389/fimmu.2021.732135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang X, Guo Y, Ji Y, Gao Y, Zhang M, Liu Y, Zhu W, Lu P. Cytokine release syndrome after modified CAR-NK therapy in an advanced non-small cell lung cancer patient: a case report. Cell Transplant. 2022;31:9636897221094244. doi: 10.1177/09636897221094244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Myhre MR, Fåne A, Gaudernack G, Kvalheim G, Myklebust JH, Inderberg EM, Wälchli S. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine. 2019;40:106–117. doi: 10.1016/j.ebiom.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parlar A, Sayitoglu EC, Ozkazanc D, Georgoudaki AM, Pamukcu C, Aras M, Josey BJ, Chrobok M, Branecki S, Zahedimaram P, Ikromzoda L, Alici E, Erman B, Duru AD, Sutlu T. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur J Immunol. 2019;49:1278–1290. doi: 10.1002/eji.201948140. [DOI] [PubMed] [Google Scholar]
- 74.Morton LT, Wachsmann TLA, Meeuwsen MH, Wouters AK, Remst DFG, van Loenen MM, Falkenburg JHF, Heemskerk MHM. T cell receptor engineering of primary NK cells to therapeutically target tumors and tumor immune evasion. J Immunother Cancer. 2022;10:e003715. doi: 10.1136/jitc-2021-003715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- 76.Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, Koehl U, Schambach A, Wels WS, Modlich U, Ullrich E. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2020;10:3123. doi: 10.3389/fimmu.2019.03123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chrobok M, Dahlberg CIM, Sayitoglu EC, Beljanski V, Nahi H, Gilljam M, Stellan B, Sutlu T, Duru AD, Alici E. Functional assessment for clinical use of serum-free adapted NK-92 cells. Cancers (Basel) 2019;11:69. doi: 10.3390/cancers11010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fang F, Xie S, Chen M, Li Y, Yue J, Ma J, Shu X, He Y, Xiao W, Tian Z. Advances in NK cell production. Cell Mol Immunol. 2022;19:460–481. doi: 10.1038/s41423-021-00808-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. 2018;2018:4054815. doi: 10.1155/2018/4054815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sutlu T, Nyström S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23:1090–1100. doi: 10.1089/hum.2012.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klingemann H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy (in press) [Internet] [DOI] [PubMed]
- 82.Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 83.Navarrete-Galvan L, Guglielmo M, Cruz Amaya J, Smith-Gagen J, Lombardi VC, Merica R, Hudig D. Optimizing NK-92 serial killers: gamma irradiation, CD95/Fas-ligation, and NK or LAK attack limit cytotoxic efficacy. J Transl Med. 2022;20:151. doi: 10.1186/s12967-022-03350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, Orchard PJ, Blazar BR, Wagner JE, Slungaard A, Weisdorf DJ, Okazaki IJ, McGlave PB. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 85.Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, Alici E. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy. 2010;12:1044–1055. doi: 10.3109/14653249.2010.504770. [DOI] [PubMed] [Google Scholar]
- 86.Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, Lee DA, Kaufman DS. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2:274–283. doi: 10.5966/sctm.2012-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175:5095–5103. doi: 10.4049/jimmunol.175.8.5095. [DOI] [PubMed] [Google Scholar]
- 88.Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, Schaap N, Dolstra H. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One. 2011;6:e20740. doi: 10.1371/journal.pone.0020740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kennedy PR, Felices M, Miller JS. Challenges to the broad application of allogeneic natural killer cell immunotherapy of cancer. Stem Cell Res Ther. 2022;13:165. doi: 10.1186/s13287-022-02769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beecher MS, Baiocchi RA, Linett ML, Krajewski CA, Caligiuri MA. Expression of the ζ protein subunit in CD3-NK effectors derived from human thymus. Cell Immunol. 1994;155:508–516. doi: 10.1006/cimm.1994.1143. [DOI] [PubMed] [Google Scholar]
- 91.Karahan ZS. Co-receptor mediated regulation of T cell receptor signaling in natural killer cells. MSc thesis, Boğaziçi University, İstanbul. 2022. [Google Scholar]
- 92.Moretta L, Moretta A. Killer immunoglobulin-like receptors. Curr Opin Immunol. 2004;16:626–633. doi: 10.1016/j.coi.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 93.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 94.Ergün EZ. Dissecting the crosstalk between inhibitory receptor signaling and the T cell receptor in natural killer cells. MSc thesis, Boğaziçi University, İstanbul. 2022. [Google Scholar]