

Key Points
-
•
The presence of a KMT2A rearrangement is highly associated with the development of a lineage switch after CD19-CAR.
-
•
Outcomes following post–CD19-CAR relapse are poor and particularly dismal in patients with KMT2A rearrangements.
Visual Abstract
Abstract
Relapse following chimeric antigen receptor (CAR) T-cell therapy directed against CD19 for relapsed/refractory B-acute lymphoblastic leukemia (r/r B-ALL) remains a significant challenge. Three main patterns of relapse predominate: CD19 positive (CD19pos) relapse, CD19 negative (CD19neg) relapse, and lineage switch (LS). Development and validation of risk factors that predict relapse phenotype could help define potential pre- or post-CAR T-cell infusion interventions aimed at decreasing relapse. Our group sought to extensively characterize preinfusion risk factors associated with the development of each relapse pattern via a multicenter, retrospective review of children and young adults with r/r B-ALL treated with a murine-based CD19-CAR construct. Of 420 patients treated with CAR, 166 (39.5%) relapsed, including 83 (50%) CD19pos, 68 (41%) CD19neg, and 12 (7.2%) LS relapses. A greater cumulative number of prior complete remissions was associated with CD19pos relapses, whereas high preinfusion disease burden, prior blinatumomab nonresponse, older age, and 4-1BB CAR construct were associated with CD19neg relapses. The presence of a KMT2A rearrangement was the only preinfusion risk factor associated with LS. The median overall survival following a post-CAR relapse was 11.9 months (95% CI, 9-17) and was particularly dismal in patients experiencing an LS, with no long-term survivors following this pattern of relapse. Given the poor outcomes for those with post-CAR relapse, study of relapse prevention strategies, such as consolidative hematopoietic stem cell transplantation, is critical and warrants further investigation on prospective clinical trials.
Introduction
Despite impressive remission induction rates for B-cell acute lymphoblastic leukemia (B-ALL) with CD19-directed chimeric antigen receptor (CD19-CAR) T cells,1,2 relapse remains a significant problem.3 Factors influencing a patient’s risk of relapse following CD19-CAR are becoming better established and include disease burden, depth of remission attained, CD19-CAR construct used and functional CD19-CAR persistence.4, 5, 6 Refining our ability to predict relapse following CD19-CAR is imperative to improve patient management and overall survival.
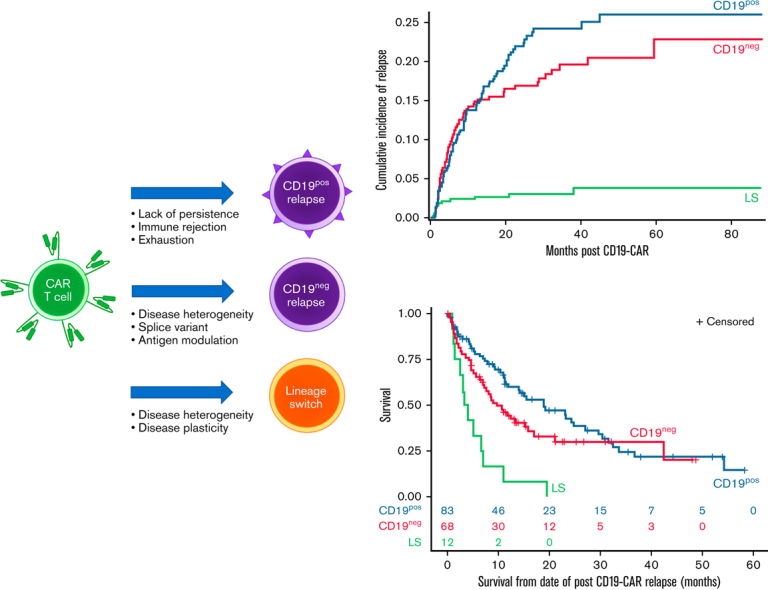
Complicating the landscape of post-CAR relapse is the immunophenotypic heterogeneity of relapse. Largely categorized into 3 predominant patterns, relapse can present with retention of CD19 (CD19-positive [CD19pos]), loss of CD19 (CD19-negative [CD19neg]), or lineage switch (LS) from a lymphoid to a myeloid phenotype (Figure 1A). Whereas CD19pos relapse generally represents loss of functional CD19-CARs, permitting disease recurrence, CD19neg relapses result from several potential mechanisms. Genomic changes, such as the acquisition of a splice variant or heterogeneous pre-CAR populations, are among the more common etiologies of CD19neg relapse.7,8 The rarest but potentially most concerning pattern of relapse is LS. Whereas CD19neg relapses may be uniquely associated with immunotherapeutic pressure, B-ALL undergoing LS following conventional therapy is seen, particularly in patients with infant B-ALL with KMT2A rearrangements (KMT2Ar).9,10 Unfortunately, the incidence of LS has increased with the higher use of CD19 targeting.11, 12, 13, 14, 15 Due to the rarity, literature surrounding LS has largely been limited to descriptions within study reports or single case reports.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23
Figure 1.

Categories of relapse phenotype and overall outcomes following relapse. (A) Categories of relapse phenotype. (B) CONSORT flow diagram. Figure made using BioRender. Unknown relapse category due to unavailable immunophenotype at time or relapse. ∗Includes 2 patients who had rapid emergence of LS at first restaging timepoint. (C) Overall survival following relapse (n = 166). Median OS was 11.9 months (95% CI, 9.0-17.0). The 6-, 12-, and 24-month OS rates were 69.8% (95% CI, 62.0% to 76.3%), 49.4% (95% CI, 41.1% to 57.2%), and 34.0% (95% CI, 25.7% to 42.5%), respectively.
The type of relapse following CD19-CAR has therapeutic and likely prognostic implications. Therefore, identifying factors predisposing to a pattern of relapse could potentially inform post-CAR relapse prevention strategies, including hematopoietic stem cell transplantation (HSCT). While we previously identified risk factors associated with any relapse of B-ALL following CD19-CAR,4 there is limited information about factors associated with each specific relapse phenotype.24 Given the critical impact of relapse phenotype on salvage options and to facilitate potential consolidative measures to extend durable remissions in patients at high risk of relapse, we sought to compare the different patterns of relapse following CD19-CAR and identify preinfusion risk factors associated with each pattern.
Methods
Study design
With a primary focus on relapse immunophenotype following CD19-CAR in children and young adults with B-ALL, we retrospectively reviewed outcomes in those receiving a first CD19-CAR product, with one of 3 unique constructs across 7 different institutions. Inclusion criteria were age ≤25 years at B-ALL diagnosis, ≥1 disease assessment evaluation after CAR infusion, and 30 days of follow-up or an event (nonresponse, disease progression, second malignancy, or treatment-related mortality) prior to 30 days. The data reported here are a sub-aim of our initial analysis of overall outcomes in 420 patients previously analyzed and reported.4 All patients received infusion between 1 January 2012, and 31 December 2019. Disease assessments were performed before and after CAR infusion at standard timepoints, as previously described.4 High disease burden was classified as ≥5% bone marrow blasts. B-cell aplasia (BCA) definitions were institutional specific and are detailed in the appendix. Relapse phenotype was defined by CD19 status and LS only in those for whom relapse phenotype was available. LS was defined by retention of cytogenetics from initial diagnosis and/or rapid emergence of myeloid disease at the first disease restaging timepoint. The study was reviewed and approved or considered exempt by each center’s Institutional Review Board. Additional methods are presented in the supplemental Appendix.
Statistical analysis
The primary objectives of this study were to evaluate the incidence and risk factors associated with relapse of CD19pos, CD19neg, and LS following CD19-CAR. Secondary objectives included evaluation of the cumulative incidence of relapse (CIR) as stratified by pattern of relapse alongside assessment of overall survival by relapse phenotype. Patient and disease demographics, along with outcomes (eg, response and relapse phenotype) were descriptively characterized.
Event-free survival (EFS) was defined as the time from CD19-CAR infusion to no response, relapse, or death from any cause. Patients who developed LS at the first restaging timepoint were categorized as relapse. Patients without any event were censored at last contact. Overall survival (OS) was defined as the time from CD19-CAR infusion to death from any cause or last contact. CIR, using D0 as the starting timepoint, was determined for each relapse outcome (CD19pos, CD19neg, and LS) with both death and an alternate relapse phenotype as competing risks, with Gray’s test comparing CIR curves. Preinfusion baseline factors (age at diagnosis, cytogenetics, disease burden) were assessed to identify their association with relapse phenotype. Follow-up was estimated using the reverse Kaplan-Meier method. Preinfusion risk factors for each form of relapse based on the cumulative incidence results were evaluated for their joint effect using multivariable Cox proportional hazards analysis, censoring for either no relapse or an alternative relapse phenotype or death. Backward selection was used to identify the final model for each relapse phenotype. Factors of interest were initially identified by univariate analysis; factors associated with relapse with P < .10 were included in the multivariable model. Subgroup analysis was performed in patients with LS, KMT2Ar, and infant ALL (defined as initial diagnosis prior to 12 months of age). Descriptive association of BCA with relapse was also performed.
Results
A total of 420 patients who received a first CD19-CAR were analyzed. Disease characteristics of the full cohort were recently reported4 and listed in supplemental Table 1. Median follow-up was 30.1 months (interquartile range [IQR]: 21.5-48.4). Among 376 patients achieving a complete remission (CR) and 2 additional patients who had rapid emergence of LS at the first restaging, 166 (43.9%) experienced a relapse (Figure 1B). Clinical characteristics associated with worse EFS included high disease burden, active extramedullary disease, circulating peripheral blood blasts, CD19/28ζ CAR construct type, and poor response to blinatumomab, as recently reported.4 The median OS following relapse was 11.9 months (95% CI, 9.0-17.0) (Figure 1C; supplemental Table 2). Patient, disease, and treatment characteristics of 163 patients for whom relapse immunophenotype was available are presented in Table 1 and constitute the analysis cohort.
Table 1.
Patient, disease, and treatment characteristics of all (n = 420) and relapsed (n = 163) patients
| All (n = 420) | Relapsed (n = 163∗) | |
|---|---|---|
| Demographics | ||
| Female, n (%) | 156 (37.1) | 71 (37.1) |
| Median age at B-ALL diagnosis, y (IQR) | 7.6 (3.4-13.8) | 7.3 (0.02-24.3) |
| Median age at CAR infusion, y (IQR) | 12.7 (7.1-17.5) | 11.8 (0.8-30.4) |
| Race (%) | ||
| White | 275 (65.5) | 108 (66.2) |
| Black | 17 (4.0) | 5 (3.1) |
| Asian | 20 (4.8) | 8 (4.9) |
| Other (mixed)/unknown | 108 (25.7) | 38 (23.3) |
| Ethnicity (%) | ||
| Non-Hispanic | 255 (60.7) | 109 (66.9) |
| Hispanic | 134 (31.9) | 42 (25.8) |
| Unknown | 31 (7.4) | 12 (7.4) |
| Prior therapy (prior to CAR T cells) | ||
| Primary refractory disease, n (%) | 92 (21.9) | 29 (17.8) |
| No. of prior CR, median (range) | 2 (0-7) | 2 (0-7) |
| Prior blinatumomab, n (%) | 33 (7.9) | 35 (21.5) |
| Prior HSCT, n (%) | 159 (37.9) | 76 (46.6) |
| Cytogenetics (%) | ||
| Normal | 41 (9.8) | 18 (11) |
| ETV6-RUNX1 | 24 (5.7) | 16 (9.8) |
| KMT2Ar | 38 (9) | 15 (9.2) |
| Ph+/Ph-like | 61 (14.5) | 17 (10.4) |
| Hypodiploid | 12 (2.9) | 7 (4.3) |
| Disease status pre-CAR (%) | ||
| M1 or MRD-negative marrow | 217 (51.7) | 64 (39.3) |
| M2/M3 marrow | 203 (48.3) | 99 (60.7) |
| CNS3 | 4 (0.9) | 1 (0.01) |
| Active EM disease | 22 (5.2) | 9 (5.5) |
| Active PB blasts | 56 (13.3) | 30 (18.4) |
| CAR T-cell construct infused (%)† | ||
| CD19/4-1BB | 277 (66.0) | 115 (70.6) |
| Tisagenlecleucel (Kymriah) | 88 (21.0) | 34 (20.9) |
| CD19/28z | 55 (13.1) | 14 (8.6) |
| Relapse phenotype (%) | ||
| CD19pos | N/A | 83 (50.9) |
| CD19neg | N/A | 68 (41.7) |
| Lineage switch | N/A | 12 (7.4) |
Blina, blinatumomab; EM, extramedullary; KMT2Ar, KMT2A-rearranged; MRD, minimal residual disease (defined as <.01% bone marrow blasts by multiparameter flow cytometry); N/A, not applicable; PB, peripheral blood.
A total of 166 patients experienced relapse, but immunophenotype at relapse was only available in 163 patients, which constituted the analysis cohort.
4-1BB CAR T-cell constructs were comprised of 1 of 2 available constructs, including the construct that eventually was FDA approved; tisagenlecleucel refers to the commercially available construct.
CD19pos relapse (n = 83)
Eighty-three of 163 (50.9%) relapses were CD19pos (Figure 1B). The 24-month CIR for a CD19pos relapse was 22% (95% CI, 17.7% to 26.5%) (Figure 2A). The median time from CAR infusion to CD19pos relapse was 244 days (range, 36-1367) (Figure 2B). Median OS for CD19pos patients following relapse was 18.9 months (95% CI, 11.2-27.0) (Figure 2C). Among the 78 patients with available data on BCA, 29 (37.2%) had ongoing BCA at a timepoint proximal to relapse (Figure 2D).
Figure 2.

Outcomes stratified by relapse immunophenotype. (A) CIR stratified by relapse immunophenotype. Corresponding data in supplemental Table 2. (B) Median time to relapse, stratified by relapse immunophenotype. (C) OS following relapse, stratified by relapse immunophenotype. Median OS for CD19pos relapse was 18.9 months (95% CI, 11.2-27.0). Median OS for CD19neg relapse was 9.7 months (95% CI, 6.9-15.9). Median OS for LS was 3.7 months (95% CI, 1.2-7.0). (D) BCA at time of relapse, stratified by relapse immunophenotype. BCA, and loss thereof, was based on local assessment and was generally defined by 1 of the following parameters: (1) >1% bone marrow CD19+ hematogones; (2) >1% increase in CD19+ cells in bone marrow or peripheral blood; or (3) >3% CD19+ B cells of total peripheral blood lymphocytes or >50 CD19+ cells/μL, verified by 2 consecutive timepoints.
In a multivariable Cox regression model, 2 or more previous remissions was the only variable independently associated with an increased risk of CD19pos relapse (Table 2). There were no significant associations with sex, age at CAR infusion, race, ethnicity, KMT2A status, prior HSCT, prior blinatumomab exposure, prior blinatumomab response, type of CAR costimulatory domain, or disease status before CAR.
Table 2.
Multivariable analysis of factors that may be associated with each relapse immunophenotype
| Parameter | Parameter estimate | Standard error | χ2 | P | Hazard ratio | 95% CI |
|---|---|---|---|---|---|---|
| CD19posrelapse | ||||||
| ≥ 2 CRs | 0.26 | 0.1 | 7.06 | .008 | 1.3 | 1.07-1.58 |
| CD19negrelapse | ||||||
| Younger age (<7 y) at CAR | 1.23 | 0.27 | 20.78 | <.001 | 3.42 | 2.02-5.81 |
| KMT2Ar presence | −2.51 | 1.02 | 6.03 | .01 | 0.08 | 0.01-0.6 |
| CAR type∗ | 1.51 | 0.72 | 4.37 | .04 | 0.22 | 0.05-0.91 |
| High disease burden | 1.64 | 0.29 | 32.28 | <.001 | 5.17 | 2.93-9.11 |
| Blina nonresponse | 1.05 | 0.37 | 8 | .005 | 2.85 | 1.38-5.89 |
| Lineage switch | ||||||
| KMT2Ar presence | 3.48 | 0.67 | 27.11 | <.0001 | 32.35 | 8.74-119.71 |
CD28z CAR associated with lower risk of CD19neg relapse.
CD19neg relapse (n = 68)
Sixty-eight of 163 (41.7%) relapses were CD19neg (Figure 1B). The 24-month CIR for a CD19neg relapse was 16.3% (95% CI, 13.2% to 21.0%) (Figure 2A). The median time from CAR infusion to CD19neg relapse was 148 days (range, 30-1159) (Figure 2B). Median OS following CD19neg relapse was 9.7 months (95% CI, 6.9-15.9) (Figure 2C). Of 63 patients with available data, 43 (68.3%) had ongoing BCA at a timepoint proximal to relapse (Figure 2D). As previously reported, among patients with dim CD19 expression before CAR (n = 29), 4 were nonresponders and 13 experienced relapses, including 9 of 13 (69.2%) with CD19neg disease.4
In a multivariable Cox regression model, age <7 years at CD19-CAR infusion, lack of KMT2Ar, 4-1BB CAR type, prior blinatumomab nonresponse, and high disease burden (≥5% blasts) before CAR were each associated with an increased risk of CD19neg relapse (Table 2). There were no significant associations with sex, age at diagnosis, race, ethnicity, prior HSCT, or cumulative number of prior CRs.
LS (n = 12)
Twelve of 163 (7.4%) relapses were LS (Figure 1B). Eleven of 12 patients (91.7%) with LS converted to acute myeloid leukemia; 1 patient converted to mixed phenotype acute leukemia.
The 24-month CIR for LS was 3% (95% CI, 1.6% to 5.2%) (Figure 2A). All but 2 LSs occurred during the first year following CD19-CAR infusion, with a median time from CAR infusion to LS of 65.5 days (range, 21-1159) (Figure 2B). Median OS following LS was 3.7 months (95% CI, 1.2-7.0), substantially shorter than OS following either a CD19pos (P < .0001) or CD19neg (P = .0018) relapse (Figure 2C). Importantly, there were no long-term survivors following LS, with all patients dying from progressive disease. Subsequent therapy ranged from various myeloid-directed intensive chemotherapies to palliative care (supplemental Table 3). All 8 patients with available data had ongoing BCA at time of LS (Figure 2D).
KMT2Ar was the predominant cytogenetic abnormality seen in patients with LS, present in 9 of 12 (75%) patients with LS compared with 20 of 408 (7.1%) patients without LS patients (P < .001) (Table 3; Figure 3A). Given the association of KMT2Ar with infant ALL, patients with LS were expectedly younger at initial diagnosis compared with the remaining cohort (median age, 1.6 years vs 7.7 years; P = .001). In a multivariable Cox regression model, KMT2Ar was the only independent predictor of LS (hazard ratio, 32.35; P < .0001; Table 2). There were no significant associations with sex, age at CAR infusion, race, ethnicity, prior HSCT, cumulative number of prior CRs, prior blinatumomab exposure, prior blinatumomab response, type of CAR, or disease status before CAR.
Table 3.
Disease characteristics based on KMT2Ar status
| KMT2A (n = 38) | Non-KMT2A (n = 382) | P | |
|---|---|---|---|
| Demographics | |||
| Median age at diagnosis, y (range) | 0.6 (0.02-11.1) | 8.5 (0.8-25.1) | <.0001 |
| Median age at infusion, y (range) | 3.0 (0.6-16.2) | 13.3 (1.7-30.4) | <.0001 |
| Female, n (%) | 16 (42.1) | 140 (36.6) | .6 |
| Male, n (%) | 22 (57.9) | 242 (63.4) | |
| Prior therapy (%) | |||
| Primary refractory | 7 (18.4) | 85 (22.2) | .68 |
| Prior HSCT | 18 (47.4) | 141 (36.9) | .22 |
| Prior blina | 12 (31.6) | 65 (17.0) | .04 |
| Prior blina nonresponse | 3 of 12 (25) | 31 of 65 (47.7) | .21 |
| Disease burden (%) | |||
| M1 | 17 (44.7) | 200 (52.4) | .4 |
| ≥M2 | 21 (55.3) | 182 (47.6) | |
| CAR type (%) | |||
| 41BB | 36 (94.7) | 329 (86.1) | .20 |
| CD28 | 2 (5.3) | 53 (13.9) | |
| CAR response (%)∗ | |||
| CR | 31 (86.1) | 345 (91.8) | .23 |
| No CR (PR/SD/PD) | 5 (13.9) | 31 (8.2) | |
| Relapse phenotype (%) | |||
| LS | 9 (23.7) | 3 (0.8) | <.0001 |
| CD19pos | 5 (13.2) | 78 (20.4) | |
| CD19neg | 1 (2.6) | 67 (17.5) | |
Number of patients evaluable for response: KMT2A (n = 36); non-KMT2A (n = 376).
Figure 3.

Outcomes for patients with KMT2Ar ALL. (A) Intersection graph showing association between KMT2Ar, infant ALL diagnosis and lineage switch. (B) Flow diagram showing overall outcomes of patients with KMT2Ar ALL following CD19 CAR. ∗2 patients died of CAR toxicity; ˆ1 died of post-HSCT TRM. CR, complete remission; NE, nonevaluable; NR, nonresponse (including partial response, stable disease, and progressive disease). (C) Relapse phenotype of non-KMT2Ar patients. (D) Relapse phenotype of KMT2Ar patients. (E) Outcomes for individual patients with KMT2Ar ALL.
KMT2Ar (n = 38)
Given the strong association between LS and KMT2Ar, as visualized in the intersection graph (Figure 3A), we further analyzed outcomes of all patients with KMT2Ar. Overall, 38 of 420 patients (9%) had a KMT2Ar, with 9 of 38 (23.7%) experiencing an LS. Outcomes for this cohort, stratified by disease burden prior to CAR infusion, are shown in Figure 3B. Patients with KMT2Ar were younger at diagnosis (median age, 0.6 years vs 8.5 years; P < .0001) and CAR infusion (median age, 3 years vs 13.3 years; P < .0001) compared with non-KMT2Ar patients. KMT2Ar patients were also more likely than non-KMT2Ar patients to have previously received blinatumomab (31.6% vs 17%%; P = .04) and had comparable CR rates to blinatumomab (75% vs 52.3%; P = .21). Otherwise, there were no substantial baseline differences between the 2 groups, including CD19-CAR response (Table 3). Relapse immunophenotype, however, was skewed toward LS in KMT2Ar patients, as discussed above (Figure 3C-D).
Individual outcomes for KMT2Ar patients are shown in Figure 3E. Thirty-one of 38 (81.6%) KMT2Ar patients achieved a CR after CAR. Seven patients received a consolidative HSCT in remission (representing 4 first and 3 second HSCTs) at a median of 100 days (range, 60-429) after CAR. Three (42.9%) patients receiving a consolidative HSCT remain alive in remission (median follow-up of 1164 days after CAR). Among the other 4 patients, there were 2 CD19pos relapses, 1 CD19neg relapse, and 1 transplant-related mortality after HSCT. No KMT2Ar patient experienced post-HSCT LS. Of the 24 KMT2Ar patients who did not receive HSCT after CAR, 3 (12.5%) experienced a CD19pos relapse, 7 (30.4%) developed LS, and 14 (60.9%) are alive with a median follow-up of 864 days after CAR. These 24 patients did not receive a consolidative HSCT in remission due to early LS (n = 5), pre-CAR HSCT (n = 11), or patient/provider preference (n = 8).
Of 7 (18.4%) CD19-CAR nonresponding patients with KMT2Ar, 2 (28.6%) had rapid emergence of LS by the first restaging timepoint, and the remaining patients died of disease (n = 3) or CAR-related toxicity (n = 2).
The EFS for KMT2Ar patients (median, 14.1 months; 95% CI, 12.2 not estimable [NE]) was similar to non-KMT2Ar patients (20.2 months; 95% CI, 13.9-28.4; P = .47) (Figure 4A). However, the median OS for KMT2Ar patients was inferior to non-KMT2Ar patients (25.3 months [95% CI, 7.9 NE] vs 51.9 months [95% CI 42.0 NE]; P = .02) (Figure 4B). Specifically, no KMT2Ar patient experiencing an event following CD19-CAR was a long-term survivor.
Figure 4.

Overall and event-free survival in patients with KMT2Ar and infant ALL. (A) EFS KMT2Ar vs non-KMT2Ar. EFS for KMT2Ar patients at 6, 12, and 24 months was 55.3% (95% CI, 38.3% to 69.3%), 52.5% (95% CI, 35.6% to 66.9%), and 43.8% (95% CI, 27.6% to 58.8%), respectively, with a median EFS of 14.1 months (95% CI, 2.2 NE). EFS for non-KMT2Ar patients at 6, 12, and 24 months was 69.5% (95% CI, 64.6% to 73.9%), 58.0% (95% CI, 52.9% to 62.7%), and 47.0% (95% CI, 41.7% to 52.2%), respectively, with a median EFS of 20.2 months (95% CI, 13.9-28.4). P = .47. (B) OS KMT2Ar vs non-KMT2Ar. OS for KMT2Ar patients at 6, 12, and 24 months was 71.1% (95% CI, 53.9% to 82.8%), 57.7 (95% CI, 40.5% to 71.5%), and 51.9% (95% CI, 34.9% to 66.5%), respectively, with a median OS of 25.3 months (95% CI, 7.9 NE). OS for non-KMT2Ar patients at 6, 12, and 24 months was 85.6% (95% CI, 81.6% to 88.7%), 76.3 (95% CI, 71.7% to 80.3%), and 65.9% (95% CI, 60.6% to 70.7%), respectively, with a median OS of 51.9 months (95% CI, 42% NE). P = .02. (C) EFS infant ALL vs noninfant ALL. EFS for infant patients at 6, 12, and 24 months was 55.2% (95% CI, 35.6% to 71.0%), 55.2% (95% CI, 35.6% to 71.0%), and 50.9% (95% CI, 31.5% to 67.5%), respectively, with a median EFS not reached. EFS for noninfant patients at 6, 12, and 24 months was 69.2% (95% CI, 64.3% to 73.5%), 57.7% (95% CI, 52.6% to 62.5%), and 46.5% (95% CI, 41.2% to 51.6%), respectively, with a median EFS of 19.5 months (95% CI, 13.9-27.0). P = .88. (D) OS Infant ALL vs noninfant ALL. OS for infant patients at 6, 12, and 24 months was 69% (95% CI, 48.8% to 82.5%), 58.2% (95% CI, 38.3% to 83.8%), and 54.1% (95% CI, 34.2% to 70.3%), respectively, with a median OS of 35.8 months (95% CI, 5.6 NE). OS for noninfant patients at 6, 12, and 24 months was 85.4% (95% CI, 81.5% to 88.5%), 75.8 (95% CI, 71.2% to 79.8%), and 65.4% (95% CI, 60.2% to 74.2%), respectively, with a median OS of 49.1 months (95% CI, 42.0 NE). P = .15.
Other predominant cytogenetic alterations in our cohort included Ph+ (n = 32), Ph-like (n = 29), ETV6-RUNX1 (n = 24), and hypodiploidy (n = 12). There was no significant association between these alterations and a specific relapse immunophenotype (supplemental Table 4).
Infant ALL (n = 29)
Overall, 29 of 420 (6.9%) patients had infant ALL (defined as initial diagnosis within the first 12 months of life). The median age at CD19-CAR infusion was 2.1 years (range, 0.6-11.2), with only 3 patients receiving CD19-CAR during their first year of life. Twenty-seven of 29 (93.1%) patients with infant ALL had a KMT2Ar, and 25 of 29 (86.2%) achieved a CR following CD19-CAR, 24 (96%) of which were minimal residual disease (MRD) negative.
Nine of 29 (31%) patients with infant ALL experienced relapse following CD19-CAR, including 2 CD19pos relapses, 1 CD19neg relapse, and 6 LSs. All patients with infant ALL experiencing an LS had a co-occurring KMT2Ar. The median EFS (not reached for infants vs 19.5 months for noninfants; P = .88; Figure 4C) and median OS (35.8 months for infants vs 49.1 months for noninfants; P = 015; Figure 4D) for patients with infant ALL were similar to patients with noninfant ALL.
Discussion
While CD19-CAR has changed the landscape for treatment of relapsed/refractory B-ALL, relapse after CAR remains a major challenge. This risk of relapse has informed next-generation CAR strategies targeting relapse prevention, including bispecific CARs,25 addition of checkpoint inhibitors,26 and episodic antigen exposure.27 However, the best approach to prevent post-CAR relapse remains unclear. To better understand post-CAR relapse and assist in the development of relapse prevention strategies, we evaluated relapse stratified by immunophenotype and identified pre-CAR risk factors specific to each pattern of relapse in a large, multicenter setting. These predictive factors are vital for treating clinicians and may inform optimal planning for peri-CAR strategies.
Among the 163 patients who relapsed after CD19-CAR, CD19pos relapses, CD19neg relapses, and LS accounted for 50.9%, 41.7%, and 7.4% of relapses, respectively. Focusing first on CD19pos and CD19neg relapse, Dourthe et al recently reported on risk factors associated with these events in a cohort of 51 patients following treatment with tisagenlecleucel.24 They found that low disease burden and loss of BCA were associated with CD19pos relapse. In contrast, they observed that high disease burden and detectable MRD at day 28 after CD19-CAR were associated with an increased risk of CD19neg relapse. With a primary focus on preinfusion factors, we similarly identified high disease burden as the most important risk factor for CD19neg relapse, followed by prior blinatumomab nonresponse, age at CAR infusion, and CAR construct.
The association of CD19neg relapses with high disease burden may reflect a heterogenous pre-CAR disease population with a CD19neg clone obscured by bulk disease, a so-called “needle in the haystack” phenomenon. Following the eradication of bulk CD19pos disease, CD19neg disease could emerge unimpeded by ongoing CD19-CAR persistence. Notably, our prior efforts incorporated extensive evaluation of CD19 expression phenotype, and there was no association between pre-CAR CD19 expression and relapse immunophenotype, potentially highlighting the challenge of describing large populations of cells (supplemental Table 1).4 This analysis is inherently limited because patients with decreased or partial expression of CD19 may have been ineligible for CD19-CAR treatment or allocated to alternative therapies.
Interestingly, several of the variables associated with CD19neg relapses have also been linked to prolonged CD19-CAR persistence. The 4-1BB CAR T-cell construct has longer persistence compared with its CD19/28ζ counterpart and is more associated with CD19neg relapse. Additionally, patients who receive a CD19/28ζ construct are typically allocated to a consolidative HSCT,28 which may potentially prevent the development of LS. The observation that younger age at infusion is associated with CD19neg relapses (and less likely to have CD19pos relapse) warrants further study, specifically to evaluate if age at the time of collection influences T-cell function. Indeed, preclinical and clinical studies have shown that older patients have decreased persistence and effectiveness of their CAR product compared with younger patients, leading to an increased risk of CD19pos relapses and, in turn, potentially decreasing the incidence of CD19neg relapses.29,30 These findings are consistent with the hypothesis that ongoing immunotherapeutic pressure associates with or potentially facilitates the emergence of CD19neg disease. Further support for this hypothesis is the observation that most patients with CD19neg relapse in our cohort had ongoing BCA (a surrogate for functional CD19-CAR persistence) and could not be used to predict CD19neg escape.
While blinatumomab exposure itself did not increase the risk of developing a CD19neg relapse, a prior lack of response to blinatumomab increased the risk for eventual CD19neg relapse, suggesting a highly refractory nature of these patients’ disease. Although the reason for blinatumomab failure in this cohort was not the development of CD19neg disease because these patients would not have been eligible for CD19-CAR therapy, a small proportion did have CD19 modulation after blinatumomab, which may have been the prelude to eventual antigen escape.4 Given that blinatumomab nonresponders have worse long-term outcomes following CD19-CAR and that the predominant relapse type for this population is CD19neg, for which there are limited salvage strategies, prioritizing these patients for next-generation CAR strategies or consolidative HSCT to decrease relapse risk could be considered.
In contrast to CD19neg relapse, the only factor in our study associated with a CD19pos relapse was the cumulative number of CRs, which serves as a proxy for additional lines of therapy needed and overall treatment burden. Specifically, higher numbers of CRs were associated with worse EFS.4 We have also previously shown that primary refractory patients have a more favorable EFS following CD19-CAR.4 We hypothesize that these variables may reflect an element of T-cell fitness and surmise that heavily pretreated patients may generate CAR T cells with shortened functional persistence, predicating to CD19pos relapse.
While LS events were rare overall, the presence of a KMT2Ar was strongly identified as a risk factor. This profound enrichment of KTM2Ar in patients developing LS following CD19-directed immunotherapy is consistent with the culmination of single case reports.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 Notably, there were no LS events following treatment with a CD19/28ζ CAR. Similar to CD19neg relapse, ongoing immunotherapeutic pressure likely facilitates LS, presumably via disease plasticity.
Patients with KMT2Ar had a similar EFS compared with non-KMT2Ar patients. However, the OS for patients with KMT2Ar was inferior, with no long-term survivors following an event after CAR. This was largely driven by the increased proportion of LS events in the KMT2Ar patients. Although there are some case reports of long-term survivors following LS,15 this entity poses a challenging clinical situation for treating clinicians. Effective immunotherapeutic targets are often lacking, their clones are likely chemotherapy resistant given the heavy pretreatment, and many of these patients have already had an HSCT. Concerningly, the KMT2Ar patients with non-LS events showed a similar lack of salvageability, which suggests that KMT2Ar relapses are challenging to salvage regardless of the relapse immunophenotype.
Not answered by our data set is the question of our ability to prevent LS, specifically as it relates to post-CAR HSCT. Some have postulated that a consolidative HSCT may be important or required to prevent LS via high-dose conditioning or a graft-versus-leukemia effect. However, avoidance of HSCT, especially for younger patients, is often desired given the associated short- and long-term toxicity. While no patients who received a post-CAR HSCT experienced an LS, our numbers are too small to indicate a protective benefit of HSCT in these patients. Additionally, several patients were unable to proceed to HSCT because their event happened very early after CD19-CAR infusion, precluding HSCT. Given the association between KMT2Ar and infant ALL, a population for whom bridging to CAR T-cell therapy and manufacturing a product is challenging and in whom there is equipoise regarding the role of HSCT,31,32 it is abundantly clear that this population remains at very high risk. Importantly, we found that neither blinatumomab exposure nor blinatumomab failure put patients with KMT2Ar at higher risk for LS. This is a key consideration given the upfront incorporation of blinatumomab in ongoing clinical trials and for treating clinicians seeking to prioritize salvage strategies.
While our study focused on pre-CAR risk factors, analysis of BCA revealed that most patients with CD19neg and LS had ongoing BCA prior to their event, supporting the notion that BCA monitoring is suboptimal for prediction of approximately 50% of relapses occurring after CD19-CAR. Even more worrisome, however, are the data that 37.2% of patients with CD19pos relapse had ongoing BCA proximal to their event, suggesting that its role as a predictive tool in monitoring for any relapse may be limited. Because of the variability in how BCA was captured in this retrospective analysis, our findings are descriptive at best. However, Pulsipher et al similarly describe in their cohort of 143 patients with more stringent definitions and well-defined monitoring of BCA that 22 of 25 (88%) patients with CD19neg relapse had ongoing BCA, as did 3 of 14 (21.2%) patients with CD19pos relapse, also raising concerns regarding the limited use of BCA monitoring in the post-CAR setting.6
An important, but unanticipated, finding was regarding the timing of relapse. Generally, most relapses have been reported within the first year after CD19-CAR.1,6 In our dataset, which is both expansive and longitudinal, the median time to relapse varied by relapse immunophenotype, but the relapse risk remained present even at later timepoints, particularly for CD19pos relapse. Indeed, the median time to CD19pos relapse was 244 days (IQR, 107-480), suggesting that late relapses are occurring well beyond the first year. This presents a potential window of opportunity in which continued monitoring with methods such as next-generation sequencing as recently reported6 may be highly informative in identifying those at high risk of relapse. Additionally, the mechanism of CD19pos relapses may differ based on timing, with late relapses being more amicable to reinfusion of CD19-CAR, but further investigation is warranted. Unfortunately, but not unexpectedly, we demonstrate, as others in adult oncology have also reported, that survival following post–CD19-CAR relapse is dismal.33, 34, 35 Relapse prevention strategies for patients in whom there is time to intervene prior to relapse will be essential.
In our analysis, we opted to use the first relapse as the primary event upon which to base our statistical analysis, particularly as it related to competing risk. We recognize, however, and particularly in the era of immunotherapy, that immunophenotypic expression is in evolution (eg, a patient whose first relapse phenotype is CD19pos may evolve to CD19neg in the future). Nevertheless, because the event of interest was first relapse, which would prompt additional therapy and represent failure following CD19-CAR, our approach of basing our analysis on the most predominant population seen at relapse remains highly informative with respect to post–CD19-CAR outcomes. We also opted to use only pre-CAR infusion characteristics to define risk but recognize the important role of post-CAR monitoring, particularly MRD and BCA in the context of a CD19-CAR response. Lastly, given the longer time to CD19pos relapse, it is important to note that we were not able to determine if a consolidative HSCT triggered by loss of BCA changed the trajectory of CD19pos relapse, a limitation of this retrospective analysis. In concert with our recent efforts that identified high disease burden and blinatumomab nonresponse as risk factors for relapse, our results identifying preinfusion risk factors stratified by relapse phenotype facilitate a more fine-tuned approach to individual patients and potential outcomes, allowing for more informed planning and decision-making for each child, adolescent, and young adult undergoing CD19-CAR.
In summary, in the context of the largest retrospective pediatric CD19-CAR dataset established to date, we provide a conclusive association of KMT2Ar with LS, identify risk factors associated with CD19neg and CD19pos relapse, further characterize the timing of relapse by immunophenotype, and comprehensively describe outcomes of patients with KMT2Ar and infant ALL who received CD19-CAR. Given the dismal outcomes for those with post-CAR relapse, relapse prevention strategies will be critical, and further development and validation of risk factors is a key next step. As next-generation CAR strategies become more widespread, the incidence of LS and relapse overall will need to be established to see if these strategies show benefit. Similarly, larger studies will need to be performed to evaluate the role of post-CAR HSCT. Our efforts serve to further refine risk factors that may help to appropriately identify patients at highest risk following CD19-CAR.
Conflict-of-interest disclosure: M.J.B. received honoraria from Amgen and Blueprint Medicines. D.W.L. has consulted for Harpoon Therapeutics, advised for Amgen and BMS, and received research funding from Kite Pharma and Gilead. S.A.G. received research funding from Novartis, Kite, Vertex, and Servier, has consulted for Novartis, Roche, GSK, Humanigen, CBMG, Eureka, Janssen/JNJ, and Jazz Pharmaceuticals, and has advised for Novartis, Adaptimmune, TCR2, Cellectis, Juno, Vertex, Allogene, Jazz pharmaceuticals, and Cabaletta. M.R.V. advised for Novartis, Equillium, Mederus, and Takeda. L.G. has consulted for Amgen, Novartis, and Roche and advised for Amgen, Novartis, and Celgene and holds equity in Amgen, Anchiano, Blueprint Medicines, Celgene, Clovis, Mirati, and Sanofi Paris. P.A.B has advised for Novartis, Takeda, Amegen, Kura, and Kite. S.R.R. received research funding from Pfizer. M.A.P has advised for Mesoblast, Novartis, Equillium, Medexus, and Vertex, received research funding from Adaptive and Miltenyi, and received honoraria from Novartis, Miltenyi, and Bellicum. R.A.G. has consulted for Novartis and received patents and royalties from BMS. T.W.L. has advised for Bayer, Cellectis, Novartis, Deciphera, Juno, and Y-mAbs Therapeutics, received honoraria from Bayer, Cellectis, Novartis, Deciphera, Juno, and Y-mAbs Therapeutics, and received research support from Pfizer and Bayer.
The current affiliation for A.T. is Janssen Research & Development, LLC, Raritan, NJ.
Acknowledgments
The authors would like to thank the treating and referring centers, care providers, supporting staff, and referring physicians who cared for the patients included in this study. The authors would also like to thank Elizabeth Holland for figure preparation using BioRender.
This work was supported in part by the Intramural Research Program, National Cancer Institute, National Institutes of Health (grant CA046934) (L.G.), the Center for Cancer Research, and the Warren Grant Magnuson Clinical Center (ZIA BC 011823) (N.N.S.).
Authorship
Contribution: A.J.L. and N.N.S. wrote the first draft of the manuscript; A.T., R.M.M., L.G., P.A.B., M.A.P., D.B., S.M.S., and N.N.S designed the study and analysis plan; R.M.M., A.T., A.J.L., A.E.K., J.S., B.Y., T.F., P.C., L.C., C.A., S.J., D.B., P.A.B., T.W.L., L.G., R.A.G., S.A.G., S.R.R., M.A.P., and N.N.S. all contributed to data collection; S.M.S. and N.N.S. performed statistical analyses; A.E.K., M.J.B., B.W., M.S-S., C.M.Y., S.AG., and V.P. performed institution-specific flow cytometry review; and all authors substantially contributed to the final version of the manuscript and approved the submission.The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Footnotes
Data from this study are available from the corresponding author: adam.lamble@seattlechildrens.org.
The full-text version of this article contains a data supplement.
Contributor Information
Adam J. Lamble, Email: adam.lamble@seattlechildrens.org.
Nirali N. Shah, Email: nirali.shah@nih.gov.
Supplementary Material
References
- 1.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers RM, Taraseviciute A, Steinberg SM, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2021;40:932–944. doi: 10.1200/JCO.21.01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz LM, Baggott C, Prabhu S, et al. Disease burden affects outcomes in pediatric and young adult B-cell lymphoblastic leukemia after commercial tisagenlecleucel: a pediatric real-world chimeric antigen receptor consortium report. J Clin Oncol. 2021;40:945–955. doi: 10.1200/JCO.20.03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pulsipher MA, Han X, Maude SL, et al. Next-generation sequencing of minimal residual disease for predicting relapse after tisagenlecleucel in children and young adults with acute lymphoblastic leukemia. Blood Cancer Discov. 2021;3:66–81. doi: 10.1158/2643-3230.BCD-21-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asnani M, Hayer KE, Naqvi AS, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia. 2020;34(4):1202–1207. doi: 10.1038/s41375-019-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pui CH, Chessells JM, Camitta B, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17(4):700–706. doi: 10.1038/sj.leu.2402883. [DOI] [PubMed] [Google Scholar]
- 10.Knez V, Liu X, Schowinsky J, et al. Clinicopathologic and genetic spectrum of infantile B-lymphoblastic leukemia: a multi-institutional study. Leuk Lymphoma. 2019;60(4):1006–1013. doi: 10.1080/10428194.2018.1508667. [DOI] [PubMed] [Google Scholar]
- 11.Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer. 2016;63(6):1113–1115. doi: 10.1002/pbc.25953. [DOI] [PubMed] [Google Scholar]
- 12.Oberley MJ, Gaynon PS, Bhojwani D, et al. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr Blood Cancer. 2018;65(9) doi: 10.1002/pbc.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wölfl M, Rasche M, Eyrich M, Schmid R, Reinhardt D, Schlegel PG. Spontaneous reversion of a lineage switch following an initial blinatumomab-induced ALL-to-AML switch in MLL-rearranged infant ALL. Blood Adv. 2018;2(12):1382–1385. doi: 10.1182/bloodadvances.2018018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659. doi: 10.1038/s41408-017-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du J, Chisholm KM, Tsuchiya K, et al. Lineage switch in an infant B-lymphoblastic leukemia with t(1;11)(p32;q23); KMT2A/EPS15, following blinatumomab therapy. Pediatr Dev Pathol. 2021;24(4):378–382. doi: 10.1177/10935266211001308. [DOI] [PubMed] [Google Scholar]
- 16.Mo G, Wang HW, Talleur AC, et al. Diagnostic approach to the evaluation of myeloid malignancies following CAR T-cell therapy in B-cell acute lymphoblastic leukemia. J Immunother Cancer. 2020;8(2) doi: 10.1136/jitc-2020-001563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aldoss I, Song JY. Extramedullary relapse of KMT2A(MLL)-rearranged acute lymphoblastic leukemia with lineage switch following blinatumomab. Blood. 2018;131(22):2507. doi: 10.1182/blood-2018-02-834911. [DOI] [PubMed] [Google Scholar]
- 18.Balducci E, Nivaggioni V, Boudjarane J, et al. Lineage switch from B acute lymphoblastic leukemia to acute monocytic leukemia with persistent t(4;11)(q21;q23) and cytogenetic evolution under CD19-targeted therapy. Ann Hematol. 2017;96(9):1579–1581. doi: 10.1007/s00277-017-3050-6. [DOI] [PubMed] [Google Scholar]
- 19.Fournier E, Inchiappa L, Delattre C, et al. Increased risk of adverse acute myeloid leukemia after anti-CD19-targeted immunotherapies in KMT2A-rearranged acute lymphoblastic leukemia: a case report and review of the literature. Leuk Lymphoma. 2019;60(7):1827–1830. doi: 10.1080/10428194.2018.1562185. [DOI] [PubMed] [Google Scholar]
- 20.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haddox CL, Mangaonkar AA, Chen D, et al. Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: pre- and post-switch phenotypic, cytogenetic and molecular analysis. Blood Cancer J. 2017;7(9):e607. doi: 10.1038/bcj.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He RR, Nayer Z, Hogan M, et al. Immunotherapy- (blinatumomab-) related lineage switch of KMT2A/AFF1 rearranged B-lymphoblastic leukemia into acute myeloid leukemia/myeloid sarcoma and subsequently into B/myeloid mixed phenotype acute leukemia. Case Rep Hematol. 2019;2019 doi: 10.1155/2019/7394619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoghbi A, Zur Stadt U, Winkler B, Müller I, Escherich G. Lineage switch under blinatumomab treatment of relapsed common acute lymphoblastic leukemia without MLL rearrangement. Pediatr Blood Cancer. 2017;64(11) doi: 10.1002/pbc.26594. [DOI] [PubMed] [Google Scholar]
- 24.Dourthe ME, Rabian F, Yakouben K, et al. Determinants of CD19-positive vs CD19-negative relapse after tisagenlecleucel for B-cell acute lymphoblastic leukemia. Leukemia. 2021;35(12):3383–3393. doi: 10.1038/s41375-021-01281-7. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel JY, Patel S, Muffly L, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27(8):1419–1431. doi: 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li AM, Hucks GE, Dinofia AM, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. 2018;132(suppl 1):556. [Google Scholar]
- 27.Annesley C, Gardner R, Wilson A, et al. Novel CD19t T-antigen presenting cells expand CD19 CAR T cells in vivo. Blood. 2019;134(suppl 1):223. [Google Scholar]
- 28.Shah NN, Lee DW, Yates B, et al. Long-term follow-up of CD19-CAR T-cell therapy in children and young adults with B-ALL. J Clin Oncol. 2021;39(15):1650–1659. doi: 10.1200/JCO.20.02262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guha P, Cunetta M, Somasundar P, Espat NJ, Junghans RP, Katz SC. Frontline science: functionally impaired geriatric CAR-T cells rescued by increased α5β1 integrin expression. J Leukoc Biol. 2017;102(2):201–208. doi: 10.1189/jlb.5HI0716-322RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takachi T, Watanabe T, Miyamura T, et al. Hematopoietic stem cell transplantation for infants with high-risk KMT2A gene-rearranged acute lymphoblastic leukemia. Blood Adv. 2021;5(19):3891–3899. doi: 10.1182/bloodadvances.2020004157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koh K, Tomizawa D, Moriya Saito A, et al. Early use of allogeneic hematopoietic stem cell transplantation for infants with MLL gene-rearrangement-positive acute lymphoblastic leukemia. Leukemia. 2015;29(2):290–296. doi: 10.1038/leu.2014.172. [DOI] [PubMed] [Google Scholar]
- 33.Wudhikarn K, Flynn JR, Rivière I, et al. Interventions and outcomes of adult patients with B-ALL progressing after CD19 chimeric antigen receptor T-cell therapy. Blood. 2021;138(7):531–543. doi: 10.1182/blood.2020009515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow VA, Gopal AK, Maloney DG, et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am J Hematol. 2019;94(8):e209–e213. doi: 10.1002/ajh.25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spiegel JY, Dahiya S, Jain MD, et al. Outcomes of patients with large B-cell lymphoma progressing after axicabtagene ciloleucel therapy. Blood. 2021;137(13):1832–1835. doi: 10.1182/blood.2020006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.