

Key Points
-
•
The SUMO pathway is activated in MM and its magnitude associated with progression and treatment resistance.
-
•
SUMO inhibition overcomes proteasome inhibitor resistance by blocking myeloma stress resilience, irrespective of p53 state.
Visual Abstract
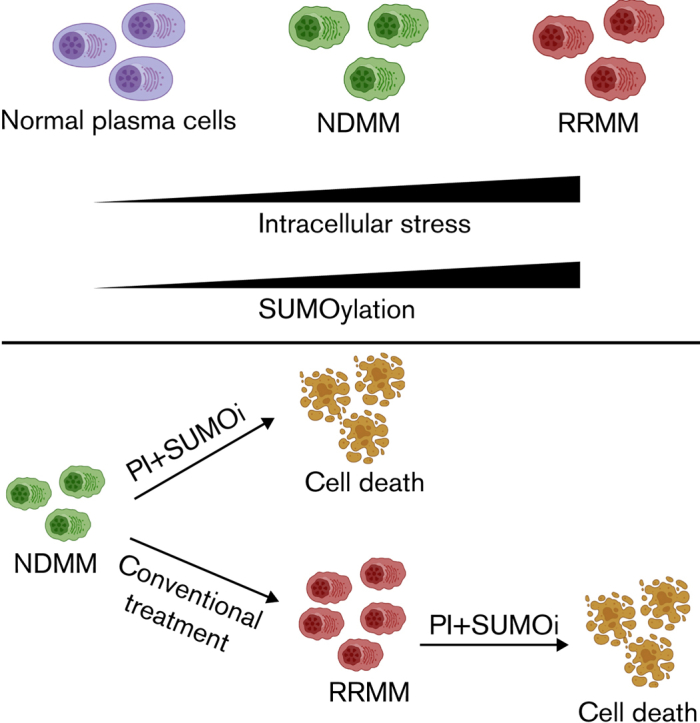
Abstract
Proteasome inhibition is a highly effective treatment for multiple myeloma (MM). However, virtually all patients develop proteasome inhibitor resistance, which is associated with a poor prognosis. Hyperactive small ubiquitin-like modifier (SUMO) signaling is involved in both cancer pathogenesis and cancer progression. A state of increased SUMOylation has been associated with aggressive cancer biology. We found that relapsed/refractory MM is characterized by a SUMO-high state, and high expression of the SUMO E1-activating enzyme (SAE1/UBA2) is associated with poor overall survival. Consistently, continuous treatment of MM cell lines with carfilzomib (CFZ) enhanced SUMO pathway activity. Treatment of MM cell lines with the SUMO E1-activating enzyme inhibitor subasumstat (TAK-981) showed synergy with CFZ in both CFZ-sensitive and CFZ-resistant MM cell lines, irrespective of the TP53 state. Combination therapy was effective in primary MM cells and in 2 murine MM xenograft models. Mechanistically, combination treatment with subasumstat and CFZ enhanced genotoxic and proteotoxic stress, and induced apoptosis was associated with activity of the prolyl isomerase PIN1. In summary, our findings reveal activated SUMOylation as a therapeutic target in MM and point to combined SUMO/proteasome inhibition as a novel and potent strategy for the treatment of proteasome inhibitor–resistant MM.
Introduction
Multiple myeloma (MM) is a genetically and clinically heterogeneous plasma cell malignancy.1,2 Genetic lesions that are associated with MM include loss-of-function of tumor suppressors and cell cycle regulators (p53, CDKN2C, and RB), as well as activation of oncogenic signaling pathways (MYC, RAS, and NF-κB), leading to aberrant cell cycle checkpoint regulation and increased proliferation.3 Although the introduction of new drugs improved outcome, MM remains incurable, and most patients die of their disease.4 Currently, the common treatment regimens for patients with MM include proteasome inhibitors, which are routinely combined with dexamethasone, chemotherapy, immunomodulatory drugs, or CD38-specific antibodies.5,6 Although such regimens can induce remissions for many years, development of drug resistance remains a major clinical problem.7,8 Therefore, new therapeutic strategies to overcome drug resistance are urgently needed.
Posttranslational modification (PTM) of proteins by small ubiquitin-like modifiers (SUMOs), termed SUMOylation, are involved in maintenance of genome integrity and in regulation of gene expression and intracellular and extracellular signaling.9 Similar to ubiquitination, protein SUMOylation occurs via a tightly controlled enzymatic pathway controlled by a SUMO-specific E1-activating enzyme (SAE1/UBA2), an E2-conjugating enzyme (UBE2I), and a subset of E3 SUMO ligases.10 Importantly, protein SUMOylation is fully reversible and executed by the SENP (sentrin-specific proteases) family of isopeptidases, making SUMOylation a finely tuned molecular switch.11,12 To date, only one heterodimeric E1 enzyme and one E2 enzyme have been identified in the SUMOylation pathway, suggesting that disruption of either will substantially inhibit global SUMO conjugation. Members of the p53 family are essential regulators mediating a plethora of tumor-suppressive functions; these members can be subdivided in p53, p63, and p73.13 They can all bind p53 consensus sequences and share some common targets, but they also have diverse roles in tumorigenesis.13, 14, 15 SUMO E3-like ligases of the PIAS family have been described as transcriptionally repressing the activity of the p53 tumor suppressor.16 Similar to p53, the other family proteins p63 and p73 are also SUMOylated. SUMOylation of p73 does not affect its transcriptional activity but rather changes its subcellular localization17; p63 SUMOylation, however, leads to an impaired transactivation activity.18,19
To date, PIN1 is the only known peptidyl-prolyl cis–trans isomerase that can specifically recognize and isomerize phosphorylated-serine/phosphorylated-threonine-proline motifs and thus change the stability of its target proteins.20 Dependent on the molecular context, PIN1 acts either as an oncogene or a tumor suppressor. PIN1 modulates many target proteins and also controls a variety of biological processes, including apoptosis.21 For example, PIN1 has been shown to modulate p53, which was required for activation of the proapoptotic protein BAX.22 PIN1 itself is also subject to various posttranslational modifications, and SUMOylation of PIN1 has been shown to impair its activity.23
Due to the wide array of effects elicited by SUMOylation, it is not surprising that deregulation of SUMOylation has been associated with tumorigenesis, tumor progression, and adverse patient outcomes in MM; SUMO inhibition may thus potentially represent a novel approach in cancer treatment.10,24, 25, 26 These and further data have led to the development of SUMO inhibitors such as subasumstat (TAK-981).27 Subasumstat blocks the enzymatic cascade of SUMOylation by forming an irreversible SUMO-subasumstat adduct, which prevents the transfer of SUMO from the E1 ligase complex to UBC9. Subasumstat has shown preclinical activity in multiple malignancies, including colorectal carcinoma and lymphoma,27 and is currently being evaluated in clinical trials for hematologic diseases (#NCT03648372 and #NCT04776018).
The current study identified synergistic effects between subasumstat and carfilzomib (CFZ) in MM, including in CFZ-resistant MM. Combination treatment resulted in proteotoxic and genotoxic stress and induced apoptosis in MM model cell lines independent of the cellular TP53 state but dependent on PIN1. A combinatorial treatment of subasumstat and CFZ therefore constitutes a potential novel therapeutic strategy for patients with MM.
Material and methods
Cell lines and culturing
AMO1, OPM2, JJN3, and NCI-H929 cells were obtained from the DSMZ (Braunschweig, Germany). MM1S cells were purchased from ATCC (Manassas, VA). Cells were cultured in RPMI 1640 (21875034; Thermo Fischer Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (A4766801; Thermo Fischer Scientific) and 1% penicillin/streptomycin (15140122; Thermo Fischer Scientific). All cell lines were cultured at 37°C and 5% carbon dioxide in a humidified incubator. All cell lines tested negative for mycoplasma contamination, as described elsewhere,28 and underwent repeated short tandem repeat profiling for authentication. Generation of CFZ-resistant cell lines is described in the supplemental Material and Methods.
Fluorescence-activated cell sorting analysis
Flow cytometry analysis was performed following standard protocols. Cell suspensions, treated as indicated, were directly labeled with fluorescently labeled antibodies against the following surface proteins: CD38 (allophycocyanin), CD138 (phycoerythrin), and Annexin V (fluorescein isothiocyanate). For primary MM patient samples, red blood cells were lysed for 20 minutes in ACK buffer (A1049201; Thermo Fischer Scientific) before antibody labeling.
Data were acquired by using a Beckman Coulter CytoFLEX flow cytometer and analyzed by using FlowJo software version 10.1 (Becton, Dickinson and Company, Ashland, OR).
Compounds
Subasumstat (TAK-981) was provided by Takeda Pharmaceutical Company Limited (Cambridge, MA). CFZ (S2853), bortezomib (S1013), pomalidomide (S1567), doxorubicin (S1208), and dexamethasone (S5956) were purchased from Selleck Chemicals (Houston, TX). Sulfopin was purchased from MedChemExpress (Monmouth Junction, NJ).
In vivo xenograft experiment
Xenograft experiments were performed as previously described.29 In summary, 1.0 × 107 MM cells were resuspended in serum-free medium, mixed with Matrigel Basement Membrane Matrix (Corning, Corning, NY) at a 1:1 ratio, and injected subcutaneously into the flanks of female NOD.CB17/AlhnRj-PrkdcSCID/Rj mice 8 to 10 weeks of age (Janvier Labs, Le Genest-Saint-Isle, France). After tumor engraftment, mice were randomly assigned to be administered CFZ (2 mg/kg) IV, subasumstat (25 mg/kg) IV, a combination of both, or vehicle control twice per week. Treatment was performed for a total of 7 days. Tumor growth was monitored by caliper measurements. Mice were housed under specific pathogen-free conditions, and animal experiments were conducted in accordance with the local ethical guidelines and approved by the responsible regional authorities (District Government of Upper Bavaria; application no. ROB-55.2-2532.Vet_02-17-230).
Primary MM patient material
Primary human MM bone marrow samples were obtained from the Department of Hematology, Oncology and Cancer Immunology of Charité University Medicine. All patients gave written informed consent, and protocols were approved by the local ethics committee of Charité (vote #EA2/142/20). Bone marrow cells were used directly to perform short-term ex vivo treatments with CFZ and subasumstat as indicated.
Proteomics, transcriptomics, cell viability, and immunoblotting
Detailed procedures are provided in the supplemental Material and Methods.
Statistical analysis
Statistical analyses were performed by using GraphPad Prism (GraphPad Software, San Diego, CA). Error bars shown in the figures represent the standard deviation. For each experiment, the statistical test that was used is indicated in the figure legend.
Results
SUMO pathway is activated in proteasome inhibitor–resistant MM cells
Dysregulation of the SUMO pathway has been associated with aggressive cancer biology and poor prognosis.10,24 Using publicly available transcriptome data of MM patient samples and healthy donors,30 we found increased expression of the SUMO core components SAE1, UBA2, UBE2I, SUMO1, SUMO2, and SUMO3 in patients with MM compared with CD138+ cells from healthy donors (supplemental Figure 1A). Hierarchical clustering was performed based on the SUMO core components on 2 different data sets,31,32 which revealed SUMOhigh and SUMOlow subgroups (Figure 1A; supplemental Figure 1B). Here, SUMOhigh status was associated with significantly lower probability of overall survival (Figure 1B; supplemental Figure 1C). In addition, gene expression of the 2 components of the heterodimeric SUMO-activating E1 complex alone, SAE1 and UBA2, was also associated with significantly lower probability of overall survival (supplemental Figure 1D). Furthermore, immunoblotting analysis revealed that primary MM patient cells showed increased protein SUMOylation status compared with CD138+ cells of healthy donors (Figure 1C; supplemental Figure 1E). To further evaluate a potential dysregulation of the SUMO pathway during progression of MM, we interrogated transcriptome data from patients sequentially biopsied at first diagnosis and after relapse33 (supplemental Table 1). Reactome database34 signatures containing components of the SUMO machinery were significantly upregulated upon relapse, indicating a possible role for SUMOylation in MM progression and drug resistance (Figure 1D).
Figure 1.

SUMO pathway is activated in MM and associated with poor prognosis. (A) Heatmap and hierarchical clustering of the SUMO core components SAE1, UBA2, UBE2I, SUMO1, SUMO2, and SUMO3 derived from transcriptome data from n = 768 patients with MM of MMRF-CoMMpass data; the data were clustered as indicated into SUMOhigh and SUMOlow groups. (B) Kaplan-Meier curves for probability of survival of SUMOhigh and SUMOlow groups as described in panel A. Curve comparison by log-rank test with indicated P value. (C) Immunoblot depicting expression of SUMO2/3 and SUMO1 in healthy CD138+ cells and CD138+ MM cells, which have been isolated from human specimen by magnetic-activated cell sorting. β-Actin served as loading control. (D) Top: a cohort of n = 13 patients with MM, biopsied at diagnosis, subsequently treated, and biopsied again after disease relapse. Biopsied material was subsequently used for transcriptome profiling. Bottom: Gene set enrichment analysis using the fgsea package reveals enrichment of indicated Reactome SUMOylation signatures of relapsed vs newly diagnosed patients with MM. (E) Top: schematic depiction of the CFZ resistance RNA interference (RNAi) resistance screen performed by Acosta-Alvear et al.35 Bottom: Enrichment indicated SUMOylation signatures upregulated in CFZ-resistant compared with CFZ-sensitive U266 cells, determined by fgsea package using the Reactome knowledgebase. (F) Top: schematic depiction of the applied strategy to cultivate CFZ-resistance MM cells. CFZ-sensitive cells were cultured in CFZ-containing medium, slowly increasing the concentration (1-2 weeks) of CFZ until cells became resistant (total duration >12 weeks). Bottom: immunoblot showing expression of SUMO2/3 and SUMO1 in AMO1 and JJN3 CFZ-resistant cells (R) compared with AMO1 and JJN3 parental (P) cells. P cells were treated with 1 μM subasumstat and/or 12 nM CFZ for 4 hours; dimethyl sulfoxide served as vehicle control (-). Resistant cells were cultured in the presence of 12 nM CFZ and cotreated with 1 μM subasumstat as indicated. Cotreatment with subasumstat depletes SUMOylation. Adjusted P values (false discovery rate), ∗P < .05, ∗∗P < .01, ∗∗∗P < .001 (panels D-E). mRNA, messenger RNA; NES, normalized enrichment score; P-adj, adjusted P value; Suba, subasumstat.
To inform whether SUMO pathway activity was associated with resistance to proteasome inhibitor treatment, we next analyzed data from an RNA interference screen that was designed to identify genes or proteins associated with resistance or sensitivity to CFZ.35 Gene set enrichment analysis showed that interference with SUMOylation-associated genes increased sensitivity to CFZ treatment (Figure 1E). AMO-1 and JJN3 MM cells that were rendered proteasome inhibitor–resistant through exposure to increasing CFZ concentrations showed enhanced levels of SUMOylated proteins (Figure 1F; supplemental Figure 2A).
In summary, these results point to a prominent role of SUMOylation in MM and link activity of the SUMO pathway to proteasome inhibitor resistance.
Synergy of SUMO and proteasome inhibition in MM cell lines
Because we identified high expression of components of the SUMO machinery and a hyperSUMO state in MM patient samples, we next tested the efficacy of the small molecule SUMO E1 inhibitor subasumstat in MM cell lines. Subasumstat potently inhibited SUMOylation, evidenced by the decrease of SUMO2/3 protein modification upon treatment of MM cells, and, accordingly, the pool of free SUMO2/3 increased (Figure 2A). To test the growth inhibitory effect of subasumstat as a single agent, we treated a panel of MM cell lines with different expression levels of the SUMO core machinery (Figure 2B-C). Strikingly, JJN3 cells, which exhibited very high SUMOylation, were most sensitive to subasumstat treatment. We next investigated which proteins are differentially expressed and which signaling pathways cause the subasumstat-mediated loss of viability in MM cells. We therefore generated a proteome of the p53 mutant cell line OPM2 after 16 hours of subasumstat treatment and observed an induction of a DNA repair signature (Reactome)34 and an apoptosis signature (C2, Molecular Signatures Database)36 (Figure 2D).
Figure 2.

Subasumstat induces cell death in MM cell lines. (A) Treatment of five MM cell lines with 250 nM subasumstat inhibits 2/3 SUMOylation and increases the pool of free SUMO2/3 compared with dimethyl sulfoxide (DMSO)-treated control cells. (B) Expression of indicated core SUMOylation machinery genes in a panel of MM cell lines (data derived from depmap.org). TP53 status is indicated for each cell line. (C) Subasumstat monotreatment on a panel of five MM cell lines. Cells were treated for 3 days with different concentrations of subasumstat; subsequently, viability was measured. Results of 3 independent measurements are shown. (D) Top: OPM2 cells were treated for 16 hours with DMSO or subasumstat and analyzed by quantitative proteomics. Bottom: fgsea plots of quantitative proteomics data. DNA repair and apoptosis proteins are significantly upregulated in subasumstat-treated OPM2 cells over DMSO-treated control cells. P-adj, adjusted P value; Suba, subasumstat.
Because increased SUMOylation was associated with proteasome inhibitor resistance (Figure 1F), we chose to explore potential synergistic action between pharmacologic inhibition of SUMOylation and the established proteasome inhibitors CFZ and bortezomib. Applying sensitivity data to SynergyFinder37 and CompuSyn (Chou-Talalay method),38 we identified dose-dependent synergistic effects in all tested MM cell lines after subasumstat and proteasome inhibitor combination treatment (Figure 3A-B; supplemental Figure 2B-C). Importantly, this effect was observed in TP53 wild-type, TP53 mutant, and TP53 null cells (Figures 2B and 3C; supplemental Figure 2D). Irrespective of efficacy of single CFZ or subasumstat treatment, all MM cell lines tested exhibited a significant response to the combination treatment (Figure 3C; supplemental Figure 2E).Cell lines of other entities also displayed a synergistic effect, except for the CML cell line K562 (supplemental Figure 2F).
Figure 3.

Combined SUMO and proteasome inhibition acts synergistically in MM cell lines. (A) Combination of subasumstat with the proteasome inhibitors bortezomib (BTZ) and CFZ has a synergistic effect on the viability of THE indicated MM cells. Synergy score has been determined by SynergyFinder using the Zero Interaction Potency method (ZIP). The presented ZIP synergy scores are the average of n = 3 independent biological experiments with n = 3 technical replicates. Cells were treated with single and combination treatments using a 4 × 6 matrix. (B) Landscape plots depicting the synergistic area of concentrations for subasumstat and CFZ combination treatment in JJN3, OPM2, and AMO1 cells. Cells were treated for 72 hours with the indicated concentrations (4 × 6 matrix) of subasumstat and CFZ, and cell viability was measured by CellTiterGlo. Subsequently, cell viability data were used to generate landscape plots using SynergyFinder. (C) Bar diagrams showing the effect on cell viability after 72 hours of treatment with CFZ, subasumstat, and the combination thereof in JJN3 (2 nM CFZ, 200 nM subasumstat), OPM2 (2 nM CFZ, 200 nM subasumstat), and AMO1 (4 nM CFZ, 200 nM subasumstat) cells. Statistical testing was determined by one-way analysis of variance. (D) Bar diagram showing the effect on cell viability after 72 hours of treatment with 6 nM (JJN3) or 12 nM (AMO1) CFZ, 200 nM (JJN3) or 1 μM (AMO1) subasumstat, and the combination thereof in parental (P) and CFZ-resistant (R) cells. Statistical testing was determined by one-way analysis of variance. ∗P ≤ .05, ∗∗P ≤ .01, ∗∗∗∗P ≤ .0001. Suba, subasumstat.
Resistance to proteasome inhibitors is a major clinical challenge in the treatment of patients with MM.39 To test if resistance to CFZ can be overcome by simultaneous subasumstat treatment, we combined subasumstat and CFZ in CFZ-resistant JJN3 and AMO1 cells (supplemental Figure 2G). Here, we observed a significant drop in viability upon combination treatment (Figure 3D). To investigate whether the uniform synergy observed upon subasumstat and proteasome inhibitor treatment was a class effect, we also tested combinations of subasumstat with other drugs used to treat MM, specifically dexamethasone and doxorubicin, and the immunomodulatory drug pomalidomide. Here, results were more ambiguous; that is, in some cell lines a subasumstat-based combination worked antagonistically and in others synergistically (supplemental Figure 2H-I).
In summary, subasumstat single-agent treatment reduced the viability of MM cell lines and was highly synergistic with proteasome inhibitors independent of the cellular TP53 status.
Subasumstat and CFZ combination treatment increases cellular stress response and apoptosis in MM cells
Having identified a synergistic effect between subasumstat and CFZ, we next aimed to investigate the underlying mechanism. We selected 3 cell lines with different expression of the SUMO core machinery and p53 status (Figure 2B; supplemental Figure 2D): JJN3 cells are characterized by a high expression of SUMO core machinery genes and a deletion of the p53 gene (p53null); OPM2 cells exhibit a low expression of SUMO core machinery genes and a dominant-negative p53R175H mutation (p53mut); and AMO1 display an intermediate expression of the SUMO core machinery and express wild-type p53 (p53wt). To globally analyze subasumstat- and CFZ-mediated changes in RNA and protein expression levels, we performed RNA-sequencing as well as quantitative proteomics after CFZ and subasumstat single and combination treatment (Figure 4A-B). Transcriptome analysis indicated enriched apoptosis signatures in all 3 cell lines (Figure 4A). In addition, activation of the unfolded protein response (UPR) and cellular stress response pathways were detected (supplemental Figure 3A-B). In the combination treatments, significantly more genes were regulated compared with the single treatments, supporting an increased cellular stress response (supplemental Figure 3C). Quantitative proteomics analysis indicated CFZ-triggered endoplasmic reticulum stress and UPR response (Figure 4B). A significant upregulation was observed of proteins involved in the UPR pathway in the combination treatment (Figure 4C; supplemental Figure 3D). Other proteins involved in UPR (CREB3L2, DNAJB1, and GADD45A) were also significantly upregulated upon combination treatment compared with CFZ single treatment.
Figure 4.

Subasumstat and CFZ combination increases cellular stress response and apoptosis. (A) Top: OPM2, JJN3, and AMO1 cells were treated for 4 hours with dimethyl sulfoxide (DMSO), 250 nM subasumstat, 5 nM CFZ, or the combination thereof and subsequently analyzed by RNA-sequencing (n = 3 biological replicates). Bottom: Gene set enrichment analysis using the fgsea package of the combination treatment (4 hours) vs DMSO control shows enriched apoptosis signatures of the Hallmark set from the molecular signature database. fgsea P values and adjusted P values (P-adj; false discovery rate [FDR]) are indicated. (B) Top: OPM2 cells were treated for 4 hours with DMSO, 250 nM subasumstat, 5 nM CFZ, or the combination thereof and analyzed by quantitative proteomics. Bottom: Enriched Gene Ontology signatures in CFZ vs DMSO-treated OPM2 cells are displayed. (C) Graphical representation of quantitative proteomics data of OPM2 cells that are treated for 4 hours with a combination of subasumstat and CFZ over DMSO-treated control (Ctrl) cells. Proteins are ranked in a volcano plot according to their statistical P value (y-axis) and their relative abundance ratio (log2 fold change [log2FC], x-axis). (D) Top: Significantly synergistically induced genes from transcriptomic data (log2FC > 0, panel A) and proteins significantly induced in the combination treatment of subasumstat and CFZ (log2FC > 0.5; indicated in panel C) were extracted, and matching genes and proteins (n = 19) were subsequently analyzed in all 3 indicated cell lines. Bottom: Heatmap of the fold change of treatment vs control of the identified indicated messenger RNA expression in the cell lines JJN3, OPM2, and AMO1. (E) Immunoblots on JJN3, OPM2, and AMO1 cell lysates treated for 4 hours with 250 nM subasumstat, 5 nM CFZ, or the combination thereof. Protein expression of gH2AX (S139), p-CHK1 (S345), and pan-CHK1 to determine DDR, XBP1 (unfolded protein response), and the apoptosis markers cleaved poly(ADP-ribose) polymerase (Cl. PARP) and cleaved caspase-3 (Cl. Casp-3) have been analyzed. β-Actin served as loading control. (F) Fluorescence-activated cell sorting analysis of JJN3, OPM2, and AMO1 cells stained with Annexin V and 4′,6-diamidino-2-phenylindole to measure apoptosis after treatment for 48 hours with 250 nM subasumstat, 5 nM CFZ, or the combination thereof. P values were determined by one-way analysis of variance. ∗P ≤ .05, ∗∗P ≤ .01, ∗∗∗P ≤ .001, ∗∗∗∗P ≤ .0001. ER, endoplasmic reticulum. Suba, subasumstat.
To compare the obtained proteome and transcriptome data, proteins that are induced in all 3 cell lines (log2 fold change >0.5) were filtered out. In addition, we added genes that can be synergistically activated from the transcriptome data and compared which candidates can be induced at both the transcriptional and protein levels isolated. This comparison resulted in a total of 19 genes/proteins (Figure 4D). These 19 candidates were then examined in all 3 indicated cell lines for their significant expression in the combination treatment. Ten overlapping candidates were identified, all of which are part of the UPR pathway and can be significantly induced synergistically.
Because we found that proteins of the DNA damage response (DDR) pathway as well as apoptosis-related proteins were upregulated in subasumstat-treated cells (Figure 2D), and we found a massive increase of the UPR (Figure 4B-D), western blot analysis was performed on selected markers for the DDR, the UPR, and apoptosis in JJN3, OPM2, and AMO1 cells (Figure 4E). Indeed, combination treatment with CFZ and subasumstat clearly enhanced the effect on these processes compared with the single treatments, evidenced by induction of γH2AX (DDR), phosphorylation of CHK1 (DDR), XBP1 (UPR), cleaved poly(ADP-ribose) polymerase (apoptosis), and cleaved caspase-3 (apoptosis). Annexin V staining revealed that dual subasumstat and CFZ treatment significantly increased the percentage of apoptotic cells compared with the single treatments (Figure 4F).
In summary, combination treatment with subasumstat and CFZ enhanced cellular stress responses and resulted in subsequent induction of cell death.
Sulfopin antagonizes subasumstat and CFZ treatment
The initiation of apoptosis occurs through various signaling pathways and is often regulated by members of the p53 protein family.13,40 Because the OPM2 and JJN3 cell lines lack intact p53, but significant p53 hallmark signatures were observed in transcriptome data of the combination treatments (Figure 5A; supplemental Figure 4A), we assumed a possible association of SUMOylation of p53 family regulators. One important regulator of the p53 family is the prolyl isomerase PIN1, which has been shown to be responsible for efficient promoter loading of p53 target genes.41 In breast cancer cells, it has been shown that in the presence of mutant p53, the transcriptional function of p63 is regulated by PIN1.42 PIN1 SUMOylation is induced by proteotoxic stress (Figure 5B)43 and is associated with decreased activity.44 Because we observed activation of p53 target genes and this activation can be PIN1 dependently regulated, we investigated whether p53 target gene–associated apoptosis could be abrogated by inhibition of PIN1. We therefore combined the specific PIN1 inhibitor sulfopin45 with subasumstat. Indeed, antagonistic effects were observed in the combination treatments in OPM2 cells (Figure 5C-D). In addition, combination of subasumstat, CFZ, and sulfopin could abrogate caspase-3 cleavage and diminish CHK1 phosphorylation (Figure 5E; supplemental Figure 4B).
Figure 5.

Induction of cell death upon combined SUMO/proteasome inhibition is associated with PIN1. (A) OPM2 cells were treated for 4 hours with dimethyl sulfoxide (DMSO), 250 nM subasumstat, 5 nM CFZ, or the combination thereof and subsequently analyzed by RNA-sequencing. GSEA analysis by the fgsea package of the combination treatment (4 hours) vs DMSO control shows enriched p53 signatures of the Hallmark set from the molecular signature database. fgsea P values and adjusted P values (P-adj; false discovery rate) are indicated. (B) PIN1 SUMOylation upon 8 hours of 10 μM MG132 treatment or 1 hour of heat shock vs control with indicated P values. Data were retrieved from the qPTM database (http://qptm.omicsbio.info/) from the study. (C) Determination of viability of OPM2 cells, treated with 2.5 μM subasumstat or 1 μM sulfopin or combination of both for 72 hours. Cell viability was measured by CellTiterGlo. (D) Landscape plots depicting the antagonistic/additive or synergistic area of concentrations for subasumstat in combination with sulfopin treatment in OPM2 cells. Cells were treated for 72 hours with the indicated concentrations (4 × 6 matrix) of subasumstat and sulfopin and cell viability was measured by CellTiterGlo. Subsequently, cell viability data were used to generate landscape plots using SynergyFinder. (E) Immunoblots on OPM2 cell lysates treated for 4 hours with 250 nM subasumstat, 5 nM CFZ, 2 μM sulfopin, or the combination thereof. Protein expression of indicated proteins was detected using specific antibodies as indicated in the Material and methods section. Actin served as loading control. CL. Casp.3, cleaved caspase-3; Suba, subasumstat; Sulf, sulfopin; ZIP, Zero Interaction Potency method.
Taken together, we show on the one hand that combination treatment of subasumstat and CFZ leads to a p53-associated response in p53-deficient cells. On the other hand, we show that PIN1 inhibition in combination with SUMO inhibitor treatment has an antagonistic effect and points to a previously unknown potential resistance mechanism in MM.
Combined subasumstat and CFZ treatment inhibits MM tumor growth in vivo and induces apoptosis in primary MM cells
We next investigate the effects of dual proteasome and SUMO inhibition in JJN3 and OPM2 in vivo xenograft models. Although sublethal doses of single CFZ and single subasumstat treatment showed only a limited effect on tumor growth, significantly reduced tumor volumes were observed in the combination treatment group (Figure 6A). Of note, subasumstat treatment did not show measurable side effects upon short-term treatment, as judged by body weight assessment of the mice. Treatment with CFZ led to weight loss in a number of mice, which was seemingly not enhanced by the combination with subasumstat (Figure 6B).
Figure 6.

Efficacy of combined SUMO and proteasome inhibition in vivo and in primary MM cells. (A) Average tumor volume over time in nude mice injected with 1 × 107 JJN3 or OPM2 cells. After tumor engraftment, mice were treated with either vehicle, subasumstat (25 mg/kg), CFZ (2 mg/kg), or the combination thereof for 7 days. P values were determined by unpaired t test. (B) Histogram showing the number of mice that lost >10% body weight (but <20%, which was the exclusion criterion) for each treatment group during the in vivo xenograft experiment. (C) Bar diagram of Annexin V staining of 5 primary MM patient samples treated with dimethyl sulfoxide, 250 nM subasumstat, 5 nM CFZ, or the combination thereof. ∗P ≤ .05, ∗∗P ≤ .01. d0, day 0; Suba, subasumstat.
We then treated primary MM patient cells (supplemental Table 2) ex vivo and determined the fraction of apoptotic cells by Annexin V/fluorescein isothiocyanate staining after 24 hours in CD38+/CD138+ MM cells (supplemental Figure 5). In 5 of 7 patient samples (relapsed and newly diagnosed) measured, cells treated with both CFZ and subasumstat exhibited increased apoptosis compared with the single treatments (Figure 6C).
In summary, in vivo treatment of MM xenografts confirmed the efficacy of the subasumstat and CFZ combination, and we nominate this combination for further evaluation in prospective clinical trials.
Discussion
SUMOylation is a posttranslational modification that affects oncogenic pathways and acts as a safeguard to maintain cellular functions in cancer cells.11 This has led to the development of specific SUMO inhibitors such as subasumstat, which has entered clinical trials.27 Here, we show that SUMOylation is hyperactivated in proteasome inhibitor–resistant patient MM samples and that high expression of core components of the SUMO machinery correlate with inferior prognosis. We show that combining the SUMO inhibitor subasumstat with CFZ efficiently kills MM cells in vitro and blocks tumor growth in vivo.
Proteasome inhibition is an effective and universally applied therapy in MM, and it has become a backbone of MM treatment. Inhibition of the proteasome results in proteotoxic and genotoxic stress, driving apoptosis of MM cells.39 We show here that the combination of subasumstat and CFZ induced elevated UPR and DDR, which ultimately triggered apoptosis in MM cells. Because loss of p53 function is linked to drug resistance and disease progression,46 we investigated cell lines with genetic alterations in p53 and found that the combination of CFZ and subasumstat consistently induced apoptosis irrespective of the p53 status. SUMO conjugation is typically triggered in response to genotoxic or proteotoxic stress, indicating that the pathway contributes to cellular stress resilience.47, 48, 49 We therefore postulate that inhibition of SUMOylation exacerbates the apoptotic effects of proteasome inhibition in MM cells, culminating in synergistic lethality.
Molecular homeostasis from apoptosis-associated proteins and its PTM is critical for tumor cell survival and therapy resistance. Despite a lack of p53 function, p53 signatures were observed in all cell models tested. All p53 family members (p53, p63, and p73) are able to bind p53 consensus sequences to induce p53 target gene expression.40 TP53 mutations are seen in only 13% of patients with MM and are associated with worse overall survival.50 Activity of p53 family proteins is regulated by different factors affecting PTM. The prolyl isomerase PIN1 has become a focus of cancer research in recent years, and a first specific PIN1 inhibitor (sulfopin) with antitumor activity in preclinical in vivo models could be developed.45 PIN1 causes stabilization of phosphorylated target proteins (including p53 family proteins) through proline isomerization.51 Depending on the cellular p53 status, PIN1 seems to regulate p63 activity in variable ways.42 PIN1 is required for efficient binding of p53 on target genes.41 In addition, PIN1 enhances TAp63a-mediated apoptosis52 and is required for p73-dependent apoptosis.53 In the p53mut cell line OPM2, we found that PIN1 inhibition abrogates the apoptotic effects of subasumstat. This points to a previously unknown resistance mechanism that may be relevant for future treatment strategies in MM, but it needs to be further investigated in future studies.
Resistance to proteasome inhibition arises almost invariably after prolonged treatment and is a major limitation in the management of patients with MM. Thus, there is an urgent clinical need of novel therapies restoring proteasome inhibitor sensitivity in relapsed patients.5 For instance, the associated “BRCAness” induced by proteasome inhibition may be exploited by combination with additional DDR-inhibiting agents such as poly(ADP-ribose) polymerase inhibitors.54 We detected increased levels of SUMOylation in MM cells that were resistant to proteasome inhibition. This is in line with the finding that inhibition of SENP2, a negative modulator of SUMOylation, promotes bortezomib resistance and increases SUMOylation.55 Fine-tuning of intracellular SUMOylation by the deSUMOylase SENP6 was recently shown to determine chromatin organization and DDR,56 and alterations in SENP6 contributed to increased genomic instability and lymphomagenesis in B-cell lymphoma.57 Because SUMOylation of proteins regulates the DDR and other cellular stress response proteins,47,48 we hypothesize that MM cells activate the SUMO machinery to cope with proteasome inhibitor–induced proteotoxic and genotoxic stress, resulting in proteasome inhibitor tolerance. As a consequence, concurrent inhibition of SUMOylation could effectively counter proteasome inhibitor resistance, suggesting that SUMO inhibition could be effective as a combination therapy in patients who have developed resistance to proteasome inhibition.
Dexamethasone-resistant MM cells can be resensitized when combining dexamethasone with subasumstat,58 supporting the notion of SUMOylation as a safeguard to buffer the cellular stress response. Although we observed inconsistent results in terms of synergy when combining dexamethasone with subasumstat, this shows that subasumstat may have beneficial effects beyond the combination with proteasome inhibitors, supporting its potential for the management of MM. SUMO inhibition counteracts not only the tumor cell–intrinsic induction of cell death59 but also immune evasion (eg, by activating cytotoxic T cells).60 This leads to massive tumor cell reduction in pancreatic ductal adenocarcinoma,61 which is known as an immune desert, making this therapy extremely promising. Further studies will show whether such a mode of action could also be transferred to MM. In addition, by cellular indexing of transcriptomes and epitopes by sequencing, we recently showed that subasumstat had only moderate effects on physiological cell populations and displays favorable tolerability.62
In summary, we found that combination of the SUMO inhibitor subasumstat and proteasome inhibitors acts synergistically in MM models by interfering with the cellular stress response, which ultimately triggers intrinsic tumor cell death. A combination of classical proteasome inhibitor–containing regimens with inhibition of the SUMO pathway could be a promising approach to address the frequently emerging therapy resistance in MM.
Conflict-of-interest disclosure: U.K. received speaker honorary/advisory fees from Roche, Janssen-Cilag, Takeda, BMS, Gilead, Hexal, Pfizer, AstraZeneca, PentixaPharm, Amgen, Novartis, and MSD; and clinical study support from Celgene, Takeda, BMS, Roche, AstraZeneca, Novartis, MSD, Janssen-Cilag, and Pfizer. F. Bassermann received honoraria and research funding from BMS/Celgene. J. Krönke received speaker honorary and advisory fees from BMS/Celgene and Takeda. The remaining authors declare no competing financial interests.
Acknowledgments
Subasumstat (TAK-981) was provided by Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The authors thank Sina Müller and Chuanbing Zang for help with preparation of primary MM patient material. They also thank the quantitative proteomics facility at IBC2 for supporting the proteomics experiments. Graphical abstract was created with BioRender.com.
This work was supported by Deutsche Forschungsgemeinschaft (grant SFB1335/P3, U.K.; SFB1335/P6, F. Bassermann; grant KE 222/10-1, U.K.; MU 1764/7-1 [# 494535244], S.M.; BA 2851/6-1, F. Bassermann; DFG Kr3886/2-2, J. Krönke), Deutsche Krebshilfe (grants 70114425 and 70114724, U.K.; 70114823, S.M.), the European Research Commission, project BCM-UPS (grant #682473, F. Bassermann), Stiftung Charité (U.K.), Wilhelm-Sander Foundation (2017.048.2, U.K.), a German Cancer Consortium (DKTK) grant (U.K., P.M., and J. Krönke), and Berliner Krebsgesellschaft (WIFF201922 MM, M.W., S.L., and U.K.; KRFF202018MM, J. Krönke and P.M.).
Authorship
Contribution: G.J.J.E.H., F. Baumgartner, U.K., and M.W. conceived and designed the study; G.J.J.E.H., F. Baumgartner, M. Heider, U.P., M. Holz, J.B., M.K., I.S., S.A.B., E.R., A.M., Y.L.D.N., U.M.D., D.L., S.L., J. Krüger, M.J., A.N., M.S., P.M., S.M, F. Bassermann, J. Krönke, U.K., and M.W. acquired the data and/or analyzed and interpreted the data; G.J.J.E.H., F. Baumgartner, U.K., and M.W. drafted the manuscript; and all authors revised the manuscript for important intellectual content and approved the final version submitted for publication.
Footnotes
Data can be accessed via the European Nucleotide Archive (accession identifier PRJEB51059) and the Proteomics Identifications database (project accession number PXD032316). Reagents and cell lines can be obtained from the corresponding author and are also available upon reasonable request (e-mail: matthias.wirth@charite.de).
The full-text version of this article contains a data supplement.
Contributor Information
Ulrich Keller, Email: ulrich.keller@charite.de.
Matthias Wirth, Email: matthias.wirth@charite.de.
Supplementary Material
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1) doi: 10.1038/nrdp.2017.46. [DOI] [PubMed] [Google Scholar]
- 3.Heider M, Nickel K, Högner M, Bassermann F. Multiple myeloma: molecular pathogenesis and disease evolution. Oncol Res Treat. 2021;44(12):672–681. doi: 10.1159/000520312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajkumar SV, Kumar S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020;10(9):94. doi: 10.1038/s41408-020-00359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan D, Richter J. Where we stand with precision therapeutics in myeloma: prosperity, promises, and pipedreams. Front Oncol. 2022;11 doi: 10.3389/fonc.2021.819127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robak P, Drozdz I, Szemraj J, Robak T. Drug resistance in multiple myeloma. Cancer Treat Rev. 2018;70:199–208. doi: 10.1016/j.ctrv.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Nass J, Efferth T. Drug targets and resistance mechanisms in multiple myeloma. Cancer Drug Resistance. 2018;1:87–117. [Google Scholar]
- 9.Eifler K, Vertegaal AC. Mapping the SUMOylated landscape. FEBS J. 2015;282(19):3669–3680. doi: 10.1111/febs.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroonen JS, Vertegaal ACO. Targeting SUMO signaling to wrestle cancer. Trends Cancer. 2021;7(6):496–510. doi: 10.1016/j.trecan.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Wirth M, Schick M, Keller U, Krönke J. Ubiquitination and ubiquitin-like modifications in multiple myeloma: biology and therapy. Cancers (Basel) 2020;12(12):E3764. doi: 10.3390/cancers12123764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunz K, Piller T, Müller S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J Cell Sci. 2018;131(6) doi: 10.1242/jcs.211904. [DOI] [PubMed] [Google Scholar]
- 13.Belyi VA, Ak P, Markert E, et al. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol. 2010;2(6):a001198. doi: 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt T, Petrovich M, Joerger AC, Veprintsev DB. Conservation of DNA-binding specificity and oligomerisation properties within the p53 family. BMC Genomics. 2009;10(1):628. doi: 10.1186/1471-2164-10-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson IR, Irwin MS. Ubiquitin and ubiquitin-like modifications of the p53 family. Neoplasia. 2006;8(8):655–666. doi: 10.1593/neo.06439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci USA. 2002;99(5):2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275(46):36316–36323. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 18.Huang YP, Wu G, Guo Z, et al. Altered sumoylation of p63alpha contributes to the split-hand/foot malformation phenotype. Cell Cycle. 2004;3(12):1587–1596. doi: 10.4161/cc.3.12.1290. [DOI] [PubMed] [Google Scholar]
- 19.Ghioni P, D’Alessandra Y, Mansueto G, et al. The protein stability and transcriptional activity of p63alpha are regulated by SUMO-1 conjugation. Cell Cycle. 2005;4(1):183–190. doi: 10.4161/cc.4.1.1359. [DOI] [PubMed] [Google Scholar]
- 20.Yu JH, Im CY, Min SH. Function of PIN1 in cancer development and its inhibitors as cancer therapeutics. Front Cell Dev Biol. 2020;8:120. doi: 10.3389/fcell.2020.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinton P, Rimessi A, Marchi S, et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315(5812):659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- 22.Follis AV, Llambi F, Merritt P, Chipuk JE, Green DR, Kriwacki RW. Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol Cell. 2015;59(4):677–684. doi: 10.1016/j.molcel.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Correction: SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2017;77(8):2173. doi: 10.1158/0008-5472.CAN-17-0403. [DOI] [PubMed] [Google Scholar]
- 24.Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184–197. doi: 10.1038/nrc.2016.143. [DOI] [PubMed] [Google Scholar]
- 25.Kukkula A, Ojala VK, Mendez LM, Sistonen L, Elenius K, Sundvall M. Therapeutic potential of targeting the SUMO pathway in cancer. Cancers (Basel) 2021;13(17):4402. doi: 10.3390/cancers13174402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Driscoll JJ, Pelluru D, Lefkimmiatis K, et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115(14):2827–2834. doi: 10.1182/blood-2009-03-211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langston SP, Grossman S, England D, et al. Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J Med Chem. 2021;64(5):2501–2520. doi: 10.1021/acs.jmedchem.0c01491. [DOI] [PubMed] [Google Scholar]
- 28.Ossewaarde JM, de Vries A, Bestebroer T, Angulo AF. Application of a Mycoplasma group-specific PCR for monitoring decontamination of Mycoplasma-infected Chlamydia sp. strains. Appl Environ Microbiol. 1996;62(2):328–331. doi: 10.1128/aem.62.2.328-331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heider M, Eichner R, Stroh J, et al. The IMiD target CRBN determines HSP90 activity toward transmembrane proteins essential in multiple myeloma. Mol Cell. 2021;81(6):1170–1186.e10. doi: 10.1016/j.molcel.2020.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chauhan D, Tian Z, Nicholson B, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22(3):345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanamura I, Huang Y, Zhan F, Barlogie B, Shaughnessy J. Prognostic value of cyclin D2 mRNA expression in newly diagnosed multiple myeloma treated with high-dose chemotherapy and tandem autologous stem cell transplantations. Leukemia. 2006;20(7):1288–1290. doi: 10.1038/sj.leu.2404253. [DOI] [PubMed] [Google Scholar]
- 32.Study TMC Multiple Myeloma Research Foundation (MMRF) CoMMpass study. https://themmrf.org/finding-a-cure/our-work/the-mmrf-commpass-study/ Available at:
- 33.Ng YLD, Ramberger E, Bohl SR, et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat Commun. 2022;13(1):1009. doi: 10.1038/s41467-022-28515-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillespie M, Jassal B, Stephan R, et al. The Reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50(D1):D687–D692. doi: 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acosta-Alvear D, Cho MY, Wild T, et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife. 2015;4 doi: 10.7554/eLife.08153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48(W1):W488–W493. doi: 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 39.Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017;36(4):561–584. doi: 10.1007/s10555-017-9707-8. [DOI] [PubMed] [Google Scholar]
- 40.Bénard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members and human cancers. Hum Mutat. 2003;21(3):182–191. doi: 10.1002/humu.10172. [DOI] [PubMed] [Google Scholar]
- 41.Mantovani F, Tocco F, Girardini J, et al. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat Struct Mol Biol. 2007;14(10):912–920. doi: 10.1038/nsmb1306. [DOI] [PubMed] [Google Scholar]
- 42.Girardini JE, Napoli M, Piazza S, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell. 2011;20(1):79–91. doi: 10.1016/j.ccr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal AC, Nielsen ML. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol. 2017;24(3):325–336. doi: 10.1038/nsmb.3366. [DOI] [PubMed] [Google Scholar]
- 44.Chen CH, Chang CC, Lee TH, et al. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013;73(13):3951–3962. doi: 10.1158/0008-5472.CAN-12-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubiella C, Pinch BJ, Koikawa K, et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat Chem Biol. 2021;17(9):954–963. doi: 10.1038/s41589-021-00786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jovanović KK, Escure G, Demonchy J, et al. Deregulation and targeting of TP53 pathway in multiple myeloma. Front Oncol. 2019;8:665. doi: 10.3389/fonc.2018.00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49(5):795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 48.Dou H, Huang C, Van Nguyen T, Lu LS, Yeh ET. SUMOylation and de-SUMOylation in response to DNA damage. FEBS Lett. 2011;585(18):2891–2896. doi: 10.1016/j.febslet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Müller S, Ledl A, Schmidt D. SUMO: a regulator of gene expression and genome integrity. Oncogene. 2004;23(11):1998–2008. doi: 10.1038/sj.onc.1207415. [DOI] [PubMed] [Google Scholar]
- 50.Chng WJ, Price-Troska T, Gonzalez-Paz N, et al. Clinical significance of TP53 mutation in myeloma. Leukemia. 2007;21(3):582–584. doi: 10.1038/sj.leu.2404524. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Wu YR, Yang HY, et al. Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death Dis. 2018;9(9):883. doi: 10.1038/s41419-018-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li C, Chang DL, Yang Z, et al. Pin1 modulates p63α protein stability in regulation of cell survival, proliferation and tumor formation. Cell Death Dis. 2013;4(12):e943. doi: 10.1038/cddis.2013.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mantovani F, Piazza S, Gostissa M, et al. Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol Cell. 2004;14(5):625–636. doi: 10.1016/j.molcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 54.Neri P, Ren L, Gratton K, et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood. 2011;118(24):6368–6379. doi: 10.1182/blood-2011-06-363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie H, Gu Y, Wang W, et al. Silencing of SENP2 in multiple myeloma induces bortezomib resistance by activating NF-κB through the modulation of IκBα sumoylation. Sci Rep. 2020;10(1):766. doi: 10.1038/s41598-020-57698-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagner K, Kunz K, Piller T, et al. The SUMO isopeptidase SENP6 functions as a rheostat of chromatin residency in genome maintenance and chromosome dynamics. Cell Rep. 2019;29(2):480–494. doi: 10.1016/j.celrep.2019.08.106. e5. [DOI] [PubMed] [Google Scholar]
- 57.Schick M, Zhang L, Maurer S, et al. Genetic alterations of the SUMO isopeptidase SENP6 drive lymphomagenesis and genetic instability in diffuse large B-cell lymphoma. Nat Commun. 2022;13(1):281. doi: 10.1038/s41467-021-27704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du L, Liu W, Aldana-Masangkay G, et al. SUMOylation inhibition enhances dexamethasone sensitivity in multiple myeloma. J Exp Clin Cancer Res. 2022;41(1):8. doi: 10.1186/s13046-021-02226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biederstädt A, Hassan Z, Schneeweis C, et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut. 2020;69(8):1472–1482. doi: 10.1136/gutjnl-2018-317856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lightcap ES, Yu P, Grossman S, et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med. 2021;13(611) doi: 10.1126/scitranslmed.aba7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Schoonderwoerd MJA, Kroonen JS, et al. Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut. 2022 doi: 10.1136/gutjnl-2021-324834. gutjnl-2021-324834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demel UM, Böger M, Yousefian S, et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J Clin Invest. 2022;132(9) doi: 10.1172/JCI152383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.