

INTRODUCTION
Transthyretin amyloid cardiomyopathy (ATTR-CM) is an elusive, underdiagnosed condition, as its clinical presentation may be similar to many other cardiac and non-cardiac conditions, such as signs and symptoms of congestive heart failure or arrhythmias. A high degree of clinical suspicion of this condition with the appropriate diagnostic tools will allow early diagnosis and treatment, which can improve outcomes for patients diagnosed with ATTR-CM. Our case study below illustrates this condition.
CASE PRESENTATION
A 56-year-old Chinese man with a past history of hyperlipidaemia and bilateral carpal tunnel syndrome presented with a 2-week history of decreased effort tolerance, breathlessness and bilateral lower limb swelling. He did not have orthopnoea, paroxysmal nocturnal dyspnoea or chest pain.
Vital signs on admission were heart rate 66 beats per minute, blood pressure 119/78 mmHg and oxygen saturation 98% on room air. His jugular venous pulse was elevated and there were no added heart sounds or murmurs. Auscultation of the lungs revealed bilateral basal crackles. He had pitting oedema up to his knees.
Initial blood tests showed a haemoglobin level of 12.7 g/L (normal range [NR] 13.1–16.6 g/L) and creatinine level of 76 μ g/L (NR 60–107 μ g/L). Serial troponin-I levels were 67.9 ng/L, 88.8 ng/L and 91.3 ng/L (NR 0.0–17.4 ng/L). Electrocardiogram (ECG) on admission demonstrated sinus rhythm, PR interval 182 ms, normal QRS complex voltages and QRS duration 112 ms [Figure 1]. Serial ECGs showed an absence of dynamic ST segment-T wave changes. Chest radiograph revealed pulmonary congestion.
Figure 1.

12-lead electrocardiogram on admission.
Transthoracic echocardiography (TTE) demonstrated a marked symmetrical increase in left ventricular wall thickness on the parasternal long axis [Figure 2a] and apical 4-chamber view [Figure 2b], with a small pericardial effusion. The left ventricular wall thickness was measured to be maximally 22 mm during diastole (normal <10 mm). TTE also demonstrated a preserved left ventricular ejection fraction of 58% and mild valvular regurgitations. Strain imaging on TTE showed impaired global longitudinal strain (GLS) of −11.5% predominantly affecting the basal and mid-ventricular myocardial segments, with sparing of the apical segments (’cherry on top’ pattern) [Figure 2c]. Given the TTE findings and the clinical presentation of heart failure, the working diagnosis was noted to be cardiac amyloidosis.
Figure 2.

(a) Transthoracic echocardiogram, parasternal long axis view. (b) Transthoracic echocardiogram, apical 4 chamber view, focused view of the left ventricle. (c) Transthoracic echocardiogram with speckle tracking, longitudinal strain map.
Coronary angiogram demonstrated the absence of significant coronary artery disease. Screening for serum light chains was negative for monoclonal bands and free kappa/lambda ratio was within normal limits.
The patient underwent endomyocardial biopsy (EMB), which confirmed the presence of Congo red-positive amyloid deposits. Histology images showed staining of the myocardium with haematoxylin and eosin (H&E) [Figure 3a] and immunoperoxidase [Figure 3b]. Liquid chromatography tandem mass spectrometry was performed and it detected a peptide profile consistent with transthyretin (TTR) amyloid deposition.
Figure 3.

Histology specimen from endomyocardial biopsy. (a) Photomicrograph shows myocardium with extracellular accumulation of hyalinised material (arrow) in between cardiomyocytes (H&E stain, x200). (b) Photomicrograph shows extracellular material that is immunoreactive for amyloid P protein (arrow) (immunoperoxidase, x200).
He was reviewed by the neurology team and diagnosed with autonomic dysfunction, sensorimotor peripheral neuropathy and carpal tunnel syndrome, which were likely associated with systemic amyloidosis. There was no evidence of ocular manifestations of amyloidosis.
He was counselled and subsequently underwent genetic testing. He was found to be heterozygous for a pathogenic variant in the TTR gene c.349G>T (p.Ala117Ser) associated with hereditary ATTR-CM (hATTR-CM).[1] Thus, a diagnosis of hATTR-CM was made.
The patient was initially started on frusemide and spironolactone and achieved a good diuretic response. When EMB confirmed the diagnosis of cardiac amyloidosis, he was commenced on treatment with doxycycline and tauroursodeoxycholic acid.
He remained well during clinic follow-up and was in New York Heart Association (NYHA) Class I. Given his diagnosis of hATTR-CM, his two children, a 22-year-old daughter and a 21-year-old son, were counselled for genetic testing. The same pathogenic variant in the TTR gene c.349G>T (p.Ala117Ser) was detected in both his children. To date, they are both pre-symptomatic and are planned for serial screening ECGs and TTEs with speckle tracking.
DISCUSSION
Our patient presented with heart failure along with neurological features of sensorimotor peripheral neuropathies and carpal tunnel syndrome. These clinical clues should prompt investigation of systemic amyloidosis, a family of disorders characterised by misfolded precursor proteins that form β -sheet-rich amyloid fibrils that are deposited extracellularly in several tissues, including cardiomyocytes.[2]
About 95% of cardiac amyloidosis is the result of two culprit proteins — monoclonal immunoglobulin light chains secreted by clonal plasma cells, or TTR, a protein produced primarily by the liver in transthyretin amyloidosis (ATTR).[2] Light chain (AL) amyloid cardiomyopathy (CM) typically presents with rapidly progressive heart failure with a median survival of 6 months from the onset of heart failure if untreated. AL-CM is treated predominantly with chemotherapy and immunotherapeutics.[3] ATTR-CM, however, tends to present as a slow progressive disease, and patients without treatment can survive from years to decades.[3] Distinguishing ATTR-CM from AL-CM is hence important for management and prognostication.
ATTR is classified into wild-type (wtATTR) or hereditary (hATTR), in which the mutant TTR gene is transmitted in an autosomal-dominant manner with variable penetrance.[2] The TTR gene is found on chromosome 18.[2] In hATTR, there are single amino acid mutations that destabilise the hetero-tetramer, leading to misfolding and aggregation in tissues.[2] TTR amyloid protein can infiltrate organs, primarily the heart and autonomic and peripheral nervous systems.[4] Cardiac infiltration of these rigid amyloid fibrils leads to stiffness and dysfunction.[2] The presence and extent of cardiac involvement is a major determinant of prognosis for patients with ATTR.
ATTR-CM remains an elusive, underdiagnosed condition, as its clinical presentation may be similar to many other cardiac and non-cardiac conditions. Patients with ATTR-CM may experience symptoms and signs of heart failure, including exertional dyspnoea and decreased functional capacity, lung crackles, hepatomegaly, ascites and lower extremity oedema.[5,6] They may subsequently develop arrhythmias and conduction system disease.[5] Patients may also present with primary peripheral and autonomic neuropathy, spinal canal stenosis, biceps tendon rupture and vitreous opacities.[5]
Laboratory findings include elevated NT-proBNP and troponin levels.[5] ECGs may demonstrate low voltages that are classically described in cardiac amyloidosis; however, this finding is less commonly seen in ATTR-CM compared to AL-CM (refer to our patient's ECG in Figure 1 which did not show low voltages).[4]
Echocardiography is a cost-effect imaging modality that is often used in the initial evaluation of suspected cardiac amyloidosis.[5] TTE classically demonstrates global left ventricular wall thickening because of increased extracellular deposition of amyloid protein [Figures 2a and b].[4] The thick and dense myocardium is also often described as having a sparkling or speckled appearance.[4] There may be bi-atrial enlargement, thickened valves and pericardial and pleural effusions.[4,6,7] In the later stages of the disease, there may be diastolic dysfunction with restrictive left ventricular filling pattern.[4] There is reduced longitudinal systolic strain and a distinctive pattern of apical sparing in which the left ventricular apical region shows preserved strain compared with the mid and basal regions [Figure 2c].[4,6] A ratio of mean apical to mean basal plus mid-longitudinal strain of >1.0 has been found to be 93% sensitive and 82% specific for cardiac amyloidosis.[7,8] GLS has been shown to be an effective marker of cardiovascular mortality and morbidity — advanced ATTR with GLS <17% has a 100% 5-year mortality rate.[9]
Cardiac MR imaging (CMR) provides detailed information about systolic function and cardiac function, and enables tissue characterisation.[5] As compared to echocardiography, CMR imaging has superior spatial resolution and provides reproducible measure of radial and longitudinal systolic function. Cine images may suggest a diagnosis of cardiac amyloidosis through MR imaging parameters such as global left ventricular and right ventricular thickening, as well as alterations in systolic and diastolic function measures, including strain pattern, mitral and tricuspid annular plain excursion and pericardial effusions.[10,11]
Late gadolinium enhancement (LGE) imaging in CMR imaging can distinguish cardiac amyloidosis from other causes of left ventricular hypertrophy such as hypertension and hypertrophic cardiomyopathy.[12] LGE imaging is challenging in amyloidosis, as amyloid infiltration within the interstitium of the heart reduces the differences in contrast signal between blood and myocardium; this may cause the two compartments to null together or reverse, due to the high uptake of gadolinium in the expanded interstitial of amyloidotic hearts.[12] The global subendocardial pattern of LGE has traditionally been considered virtually diagnostic of the disease [Figure 4]. However, the pattern of LGE can be atypical and patchy, especially in early disease.[12] LGE may be inadequate for detecting cardiac amyloidosis in cases of diffuse and symmetrical disease without normal myocardium as a point of visual reference.[12] Quantitative parameters such as extracellular volume (ECV) and T1 mapping can also be employed for evaluation of cardiac amyloidosis in patients with diffuse disease and to identify the presence of early disease.[12] Extensive infiltration of the interstitium by the amyloid matrix would lead to increased ECV and native myocardial T1.[13] Extracellular expansion in cardiac amyloidosis is significant with an average ECV of 54% in ATTR and 51% in AL amyloidosis; in contrast, ECV in healthy subjects is about 20%–26%, and this is elevated to 27%–31% in diffuse fibrotic conditions and rarely goes beyond 40%.[14]
Figure 4.

MR images of a patient with transthyretin amyloid cardiomyopathy show a diffuse subendocardial late gadolinium enhancement pattern in the mid to basal regions of the left ventricle (arrows).
Although useful for differentiating amyloidosis from other non-amyloid diseases, neither echocardiography nor cardiac MR imaging alone is considered sufficient in the diagnosis of cardiac amyloidosis.[4] Furthermore, both modalities are unable to reliably differentiate ATTR-CM from AL-CM.[4]
Histopathological diagnosis with EMB is nearly 100% sensitive and specific for cardiac amyloidosis if the biopsy specimens are collected from multiple sites and tested for amyloid deposits with Congo red staining.[2] Identification of misfolded precursor proteins with immunohistochemistry or tandem mass spectrometry analysis can determine the amyloid subtype.[2] Extracardiac biopsy has varied sensitivity and unclear diagnostic reliability.[4] EMB, however, is invasive and is not routinely available at all centres.
With the advent of nuclear scintigraphy using bone-avid radiotracers, diagnosis of ATTR-CM is now increasingly made through non-invasive imaging without the need for invasive cardiac biopsy and its attendant risks.[2] Myocardial scintigraphy with bone-avid tracers — Tc-99m-PYP (pyrophosphate), Tc-99m-DPD (diphosphono-1,2-propanodicarboxylic acid) and Tc-99m-HMDP (hydroxymethylene diphosphonate) — have high sensitivity and specificity for ATTR-CM, and may be useful for early diagnosis of ATTR-CM, even before the onset of increased wall thickness on TTE.[5] Studies using Tc-99m-PYP scan demonstrated that AL-CM and ATTR-CM can often be differentiated using quantitative or semiquantitative approaches; TTR amyloidosis has been shown to have high avidity for these radiotracers, while AL amyloidosis has low avidity.[9] However, it is prudent to concurrently screen for monoclonal proteins in the serum and urine using protein electrophoresis and immunofixation, as AL cardiac amyloidosis can uncommonly have grade 1 or higher grades of cardiac uptake on bone scintigraphy or there may be co-existence of an unrelated monoclonal gammopathy in ATTR-CM.[2] Semiquantitative grading using the Perugini visual scoring system in which the heart-to-rib uptake is compared can confer 97% specificity for ATTR-CM when grade 2 or 3 uptake is seen [Figure 5a].[9] A quantitative approach with heart-to-contralateral-chest ratio uptake of >1.5 is diagnostic of ATTR-CM, with 97% sensitivity and 100% specificity [Figure 5b], in the absence of monoclonal proteins.[9]
Figure 5.
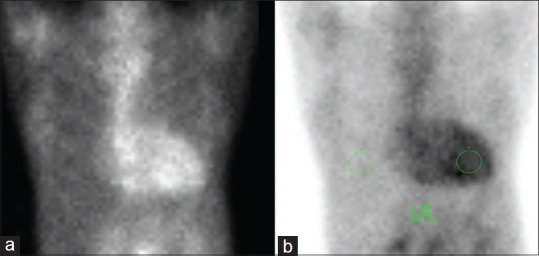
Tc-99m-PYP scintigraphy images of a patient with transthyretin amyloid cardiomyopathy. Delayed planar and single photon emission chest CT images up to 3 hours were obtained following intravenous administration of Tc-99m-PYP. (a) There was diffuse, moderately intense radiotracer uptake of the myocardium, in particular the left ventricular wall. Overall, the uptake intensity was slightly higher than that of the adjacent ribs (indicating grade 3 myocardial PYP-uptake). (b) The calculated heart uptake: contralateral chest uptake ratio at 1-hour post-radiotracer administration was approximately 2.34 (>1.5), which was highly suggestive of transthyretin cardiac amyloidosis.
Treatment of ATTR-CM is broadly divided into management of heart failure, treatment of arrhythmias and commencement of disease-modifying therapies. ATTR-CM exhibits a similar clinical phenotype as heart failure with preserved ejection fraction.[7] Diuretics are the first-line treatment for congestion, along with mineralocorticoid receptor antagonists.[7] Treatment of atrial fibrillation, which is common in ATTR-CM, is often challenging; rate control agents using beta-blockers and calcium-channel blockers may have negative inotropic effect and reduce cardiac output.[7]
Disease-modifying targets include TTR silencing (target TTR hepatic synthesis), TTR stabilisation (prevent TTR tetramer misfolding) and TTR disruption (target clearance of amyloid fibrils from tissues).[15] Tafamidis, a TTR stabiliser, in its landmark trial ATTR-ACT in 2018, has demonstrated lower all-cause mortality and cardiovascular-related hospitalisation after 30 months.[16] It is now indicated in patients with ATTR-CM that having NYHA Class I-III symptoms and early initiation may slow disease progression.[6] Doxycycline and tauroursodeoxycholic acid are TTR disruptors; there are ongoing studies to elucidate their efficacy in ATTR-CM.[7] Patisiran, a small interfering RNA that blocks expression of TTR in the liver, is a TTR silencer that has demonstrated improvement in neurological status in patients with ATTR and polyneuropathy in the phase 3 trial APOLLO; studies are ongoing to evaluate its efficacy in ATTR-CM.[15]
Genetic testing should be advised in all patients with confirmed ATTR-CM to look for pathologic TTR gene variants, regardless of patient age, because of the potential significant impact on their family members.[2,17] Hereditary ATTR is inherited in an autosomal-dominant manner, in which each child of the affected person has a 50% chance of carrying the pathogenic TTR variant. The advent of effective ATTR therapies in the past decade to treat the disease, especially in its early stages, has contributed to a shift in the perception of genetic testing. Genetic testing of asymptomatic family members of patients with ATTR is recommended, which allows early identification of individuals with the pathologic TTR variant.[18] By identifying at-risk individuals early, physicians can offer them close monitoring and prompt treatment once early signs of disease are detected.[17] There are founder mutations for TTR among families in Japan, Taiwan and elsewhere, with differing dominance between neurological and cardiac signs, but the TTR mutation spectrum has not yet been analysed in detail in Singapore.[19,20] Genetic counselling should be conducted to provide applicants with information about the disease, allowing them to understand the medical, psychological and familial implications of the genetic test results.[18] Penetrance of the disease varies among variants, with some individuals developing symptoms early in adulthood, while some remain asymptomatic throughout their lives.[18]
Recommendations on the frequency of interval follow-up, appropriate modalities for surveillance and treatment options for pre-symptomatic carriers are not yet clearly delineated in current guidelines.[18] Several groups have suggested regular monitoring 10 years before the predicted age of onset of symptomatic disease, estimated through the involved mutation, typical age of onset and age of onset in affected family members.[18] Regular cardiac and neurological assessments are recommended and ECG, TTE, CMR and scintigraphy with bone tracers may be employed.[17,18]
The diagnosis of ATTR-CM can be challenging and relies on the integration of information derived from a range of imaging modalities. Management of these patients requires a multidisciplinary approach and longitudinal follow-up.
Financial support and sponsorship
Nil.
Conflicts of interest
Foo SY is a member of the SMJ Editorial Board.
Acknowledgements
We would like to thank Professor Tan Kong Bing for reviewing and providing the histology slides for our patient.
SMC CATEGORY 3B CME PROGRAMME
Online Quiz: https://www.sma.org.sg/cme-programme
Deadline for submission: 6 pm, 28 February 2023
| Question | True | False |
|---|---|---|
| 1. Regarding transthyretin amyloidosis (ATTR): | ||
|
| ||
| (a) Point mutations destabilise the transthyretin tetramer, leading to misfolding and tissue aggregation. | ||
|
| ||
| (b) Transthyretin amyloid protein typically infiltrates cardiac tissue and the autonomic and peripheral nervous system. | ||
|
| ||
| (c) ATTR has similar outcomes and treatment modalities as light chain (AL) amyloidosis. | ||
|
| ||
| (d) Extracardiac biopsy for ATTR is very sensitive and reliable. | ||
|
| ||
| 2. Typical presenting features of ATTR include: | ||
|
| ||
| (a) Orthopnoea | ||
|
| ||
| (b) Autonomic neuropathy | ||
|
| ||
| (c) Arrhythmias | ||
|
| ||
| (d) Headache | ||
|
| ||
| 3. The following are classic diagnostic findings for cardiac amyloidosis on transthoracic echocardiography: | ||
|
| ||
| (a) Asymmetrical left ventricular hypertrophy | ||
|
| ||
| (b) Speckled appearance of the left ventricular wall | ||
|
| ||
| (c) Sparing of the left ventricular apex on strain imaging | ||
|
| ||
| (d) Diastolic dysfunction | ||
|
| ||
| 4. Regarding the role of various imaging modalities in the diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM): | ||
|
| ||
| (a) Cardiac magnetic resonance (CMR) imaging typically shows a diffuse subendocardial or transmural enhancement pattern. | ||
|
| ||
| (b) CMR imaging can help to distinguish ATTR-CM from AL amyloidosis reliably. | ||
|
| ||
| (c) Nuclear scintigraphy using bone-avid radiotracers can definitively exclude AL amyloidosis from ATTR-CM. | ||
|
| ||
| (d) Regarding Tc-99m-PYP scan, heart-to-contralateral-chest ratio uptake of >1.5 is diagnostic of ATTR-CM, in the absence of monoclonal proteins seen in the serum and urine. | ||
|
| ||
| 5. Regarding genetic testing of ATTR: | ||
|
| ||
| (a) Genetic testing should be advised to all patients with confirmed ATTR, regardless of patient’s age. | ||
|
| ||
| (b) Early identification of carriers allows close monitoring and prompt treatment if early signs are picked up. | ||
|
| ||
| (c) The TTR gene mutation that a carrier has can help to guide the timing and frequency of follow-up based on a predicted age of disease onset and recognition of likely clinical manifestations. | ||
|
| ||
| (d) There are no clear guidelines currently on the treatment of asymptomatic ATTR mutation carriers. | ||
REFERENCES
- 1.National Center for Biotechnology Information. ClinVar; [VCV000013468.5] [Last accessed on 2021 Jul 20]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000013468.5.
- 2.Ruberg FL, Grogan M, Hanna M, Kelly J, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. JACC. 2019;73:2872–91. doi: 10.1016/j.jacc.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grogan M, Dispenzieri A, Gertz MA. Light-chain cardiac amyloidosis: Strategies to promote early diagnosis and cardiac response. Heart. 2017;103:1065–72. doi: 10.1136/heartjnl-2016-310704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hafeez AS, Bavry AA. Diagnosis of transthyretin amyloid cardiomyopathy. Cardiol Ther. 2020;9:85–95. doi: 10.1007/s40119-020-00169-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12:e006075. doi: 10.1161/CIRCHEARTFAILURE.119.006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac amyloidosis: Evolving diagnosis and management: A scientific statement from the American Heart Association. Circulation. 2020;142:e7–22. doi: 10.1161/CIR.0000000000000792. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto H, Yokochi T. Transthyretin cardiac amyloidosis: An update on diagnosis and treatment. ESC Heart Fail. 2019;6:1128–39. doi: 10.1002/ehf2.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98:1442–8. doi: 10.1136/heartjnl-2012-302353. [DOI] [PubMed] [Google Scholar]
- 9.Khanna S, Wen I, Bhat A, Chen HHL, Gan GCH, Pathan F, et al. The role of multi-modality imaging in the diagnosis of cardiac amyloidosis: A focused update. Front Cardiovasc Med. 2020;7:590557. doi: 10.3389/fcvm.2020.590557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, et al. Magnetic resonance intransthyretin cardiac amyloidosis. J Am Coll Cardiol. 2017;70:466–77. doi: 10.1016/j.jacc.2017.05.053. [DOI] [PubMed] [Google Scholar]
- 11.Knight DS, Zumbo G, Barcella W, Steeden JA, Muthurangu V, Martinez-Naharro A, et al. Cardiac structural and functional consequences of amyloid deposition by cardiac magnetic resonance and echocardiography and their prognostic roles. JACC Cardiovasc Imaging. 2019;12:823–33. doi: 10.1016/j.jcmg.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Banypersad SM. The evolving role of cardiovascular magnetic resonance imaging in the evaluation of systemic amyloidosis. Magn Reson Insights. 2019;12:1178623X19843519. doi: 10.1177/1178623X19843519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, et al. Native T1 and extracellular volume in transthyretin amyloidosis [published online ahead of print 14 March 2018] JACC Cardiovasc Imaging. 2019;12:810–9. doi: 10.1016/j.jcmg.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Pan JA, Kerwin MJ, Salerno M. Native T1 mapping, extracellular volume mapping and later gadolinium enhancement in cardiac amyloidosis: A meta-analysis. JACC: Cardiovascular imaging. 2020;13:1299–310. doi: 10.1016/j.jcmg.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emdin M, Aimo A, Rapezzi C, Fontana M, Perfetto F, Seferović PM, et al. Treatment of cardiac transthyretin amyloidosis: An update. ESC. 2019;40:3699–706. doi: 10.1093/eurheartj/ehz298. [DOI] [PubMed] [Google Scholar]
- 16.Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. NEJM. 2018;379:1007–16. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 17.Conceição I, Damy T, Romero M, Galán L, Attarian S, Luigetti M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26:3–9. doi: 10.1080/13506129.2018.1556156. [DOI] [PubMed] [Google Scholar]
- 18.Grandis M, Obici L, Luigetti M, Briani C, Benedicenti F, Bisogni G, et al. Recommendations for pre-symptomatic genetic testing for hereditary transthyretin amyloidosis in the era of effective therapy: A multicenter Italian consensus. Orphanet J Rare Dis. 2020;15:348. doi: 10.1186/s13023-020-01633-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohmori H, ANdo Y, Makita Y, Onouchi Y, Nakajima T, Saraiva MJ, et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J Med Genet. 2004;41:e51. doi: 10.1136/jmg.2003.014803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao HC, Liao YC, Liu YT, Guo YC, Chang FP, Lee YC, et al. Clinical and genetic profiles of hereditary transthyretin amyloidosis in Taiwan. Ann Clin Transl Neurol. 2019;6:913–22. doi: 10.1002/acn3.778. [DOI] [PMC free article] [PubMed] [Google Scholar]