

Abstract
Many tumor cells are impaired in adhesion-regulated apoptosis, which contributes to their metastatic potential. However, suppression of this apoptotic pathway in untransformed cells is not mediated only by adhesion to the extracellular matrix but also through the resulting ability to spread and adopt a distinct morphology. Since cell spreading is dependent on the integrity of the actin microfilament cytoskeleton, we sought to determine if actin depolymerization was sufficient to induce apoptosis, even in the presence of continuous attachment. For this study, we used a human mammary epithelial cell line (MCF10A), which is immortalized but remains adhesion dependent for survival. Treatment of MCF10A cells with latrunculin-A (LA), an inhibitor of actin polymerization, rapidly led to disruption of the actin cytoskeleton and caused cell rounding but preserved attachment. Initiation of apoptosis in LA-treated MCF10A cells was detected by mitochondrial localization of the Bax apoptotic protein, which was prevented by overexpression of Bcl-2. DNA fragmentation and poly(ADP-ribose) polymerase (PARP) cleavage in LA-treated MCF10A cells indicated progression to the execution phase of apoptosis. The MDA-MB-453 cell line, which was derived from a metastatic human mammary tumor, was resistant to PARP cleavage and loss of viability in response to actin depolymerization. Stable overexpression of Bcl-2 in the untransformed MCF10A cells was able to recapitulate the resistance to apoptosis found in the tumor cell line. We demonstrate that inhibition of actin polymerization is sufficient to stimulate apoptosis in attached MCF10A cells, and we present a novel role for Bcl-2 in cell death induced by direct disruption of the actin cytoskeleton.
Epithelial cells depend on their interaction with the extracellular matrix for survival (10). Detachment from extracellular contacts typically results in a specific form of apoptotic cell death that is termed anoikis (10). Like many other forms of apoptosis, anoikis is characterized by loss of mitochondrial membrane potential through the action of the Bcl-2 family of proteins, caspase activation, and DNA fragmentation (12). The ability of epithelial cells to survive through suppression of anoikis depends on their engagement of the extracellular matrix through a family of heterodimeric transmembrane receptors known as integrins (35). Attachment to different extracellular matrix proteins depends on specific integrin heterodimers (14). Mammary epithelial cells bind collagen and laminin through the α2β1-integrin receptor (30), and anoikis can be initiated through direct inhibition of β1-integrin in this cell type (5, 25). Many tumor cells are impaired in anoikis, and their resulting ability to grow in the absence of matrix attachment contributes to their metastatic potential (27). Loss of α2β1-integrin expression is commonly observed in mammary tumor cells, indicating that these cells have lost their normal dependence on this integrin interaction for survival (1).
Binding of integrins to extracellular matrix proteins results in spreading of epithelial cells that depends on the integrity of their internal actin cytoskeleton (19, 21). Organization of actin filaments within the cell is necessary to cluster integrin receptors and proteins associated with their cytoplasmic domain into focal adhesion complexes that provide a direct physical link between the extracellular matrix and the actin cytoskeleton (6, 20, 24). Release of integrin contacts leads to depolymerization of actin filaments and subsequent apoptosis (26). Metastatic tumor cells often display gross abnormalities in actin cytoskeletal organization (4). Comparison of normal and malignant endometrial cells demonstrates a significantly reduced level of polymerized actin in the malignant cells (34). Disruption of actin organization also correlates with progression of tumor cells to adhesion-independent growth and increased invasiveness (2, 9).
Experiments with endothelial cells demonstrate that suppression of anoikis following cellular attachment is mediated not solely by integrin engagement but, rather, through the resulting ability of the cells to spread and adopt a distinct cell shape (8, 26). Malignant epithelial cell tumors often display morphological abnormalities that correlate with independence from anoikis (4), implying that epithelial cells may employ a similar morphological requirement for survival. Since epithelial cell spreading depends on organization of the actin cytoskeleton (11), we decided to directly address the role of actin polymerization in the suppression of anoikis, using the immortalized human mammary epithelial cell line MCF10A (32). These cells do not form colonies when cultured in soft agar or form tumors when xenotransplanted into nude mice (32), indicating that they undergo adhesion-dependent growth. We have used the marine macrolide toxin latrunculin-A (LA), a specific inhibitor of actin polymerization (33), to directly disrupt actin organization in MCF10A cells without affecting their attachment state. LA treatment leads to rapid induction of apoptosis in MCF10A cells, and we find that transient expression of the cell survival protein Bcl-2 can prevent apoptotic initiation. Furthermore, we find that a mammary tumor cell line, which was isolated from a metastatic carcinoma, is resistant to apoptotic execution induced by LA. We can recapitulate this resistant phenotype in MCF10A by stably expressing Bcl-2 and demonstrate a rescue from LA-induced caspase activation and loss of viability. This proves that adherent MCF10A cells remain dependent on cytoskeletal organization for survival and ascribes a novel role for Bcl-2 in mediating apoptotic cell death induced by actin depolymerization.
MATERIALS AND METHODS
Cell culture and LA treatment.
MCF10A cells were obtained from Robert Pauley of the Barbara Ann Karmanos Cancer Institute (Detroit, Mich.) and are a high-passage clone designated MCF10A1. Cells were cultured on tissue culture plastic (Corning) in a 1:1 mixture of Dulbecco's modified Eagle's medium and F12 medium (DMEM-F12) supplemented with 5% horse serum, hydrocortisone (0.5 μg/ml), insulin (10 μg/ml), epidermal growth factor (20 ng/ml), and penicillin-streptomycin (100 μg/ml each). LA (Biomol) was reconstituted as a 5 mM solution in ethanol and stored at −20°C. All treatments of MCF10A cells with LA were performed with serum-free DMEM.
Actin staining.
MCF10A cells, grown on four-chambered plastic slides (Nunc), were treated for 30 min at 37°C with DMEM containing vehicle (0.1% ethanol) or LA (5 μM). The cells were then fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 10 min, permeabilized with 0.5% Triton-PBS for 3 min, washed with PBS, and stained with 2U of Alexa594-phalloidin (Molecular Probes) per ml and 3 μg of Hoescht 33342 (Sigma) per ml for 30 min at room temperature. The slides were washed successively with PBS and H2O for 5 min each and mounted with glass coverslips in PBS containing 15% Gelvatol (polyvinyl alcohol), 33% glycerol, and 2.5% diazobicyclo-octane (Sigma).
Fluorescence-activated cell sorter (FACS) analysis.
MCF10A cells (106) were maintained in complete medium or treated for 16 h with serum-free DMEM containing either vehicle (0.1% ethanol) or LA (5 μM). Cells were harvested by trypsinization and centrifugation at 300 × g and fixed in cold 70% ethanol overnight. They were washed twice with 5 ml of PBS, resuspended in PBS containing 0.1% (vol/vol) Triton X-100 (Sigma) and 0.2 mg of DNase-free RNase A (Sigma) per ml and incubated for 30 min at 37°C. Propidium iodide (Molecular Probes) was added to a final concentration of 50 μg/ml, and after a 15-min incubation in the dark at room temperature, at least 105 cells for each sample were analyzed for DNA content using a FACScalibur cell sorter (Becton Dickinson).
Bax immunofluorescence.
MCF10A cells, grown on plastic slides and treated with LA as indicated, were fixed in 3.7% formaldehyde–PBS and permeabilized in PBS containing 0.5% NP-40 and 50 mg of bovine serum albumin (BSA) per ml (BSA–NP-40–PBS). After being washed with PBS, the cells were stained with polyclonal anti-Bax antibody (Pharmingen) diluted 1:1,000 in BSA–NP-40–PBS and incubated for 1 h at room temperature. Secondary staining was performed with tetramethylrhodamine-5-isothiocyanate (TRITC)-conjugated goat anti-rabbit antibody (1:500; Jackson Immunoresearch) for 1 h in BSA–NP-40–PBS containing 3 μg of Hoescht 33342 per ml to stain DNA. For Mitotracker costaining, 500 nM Mitotracker CM-H2XRos was added to the medium for 30 min and then washed out for 30 min with DMEM prior to fixation and staining.
Transfections.
MCF10A cells grown in complete medium on plastic slides were transfected with 10 μl of Fugene-6 (Roche) preincubated for 20 min at room temperature with either 5 μg of pcDNA3 (Invitrogen) or 5 μg of pcDNA3-Bcl2, containing the coding region for the full-length human Bcl-2 protein subcloned into the EcoRI site. Each sample was supplemented with 1 μg of GFP-C1 (Clontech) as a transfection marker. After 24 h of expression, the cells were changed into serum-free DMEM containing either vehicle or LA as indicated, incubated for 16 h at 37°C, and then fixed and stained for Bax localization or nuclear condensation as noted above. In three independent experiments, at least 100 cells from each sample were first counted blindly for green fluorescent protein (GFP) expression and then scored independently for either Bax localization or nuclear condensation.
PARP cleavage immunoblot.
MCF10A cells grown in 10-cm dishes were treated with DMEM containing 0.1% ethanol or LA as indicated. A concentration of 1 μM LA was determined to have a maximum effect for Western blot assays and was used to conserve reagent. The cells were harvested by washing and gently scraping in ice-cold PBS. To ensure total recovery of cells, all media, washes, and scraped cells for an individual plate were added to a single centrifuge tube and pelleted by centrifugation for 5 min at 300 × g. The pellets were lysed in 200 μl of sample buffer, and 20 μl was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12% polyarylamide gel. Western blotting was then performed using 1 μg of polyclonal anti-poly(ADP-ribose) polymerase (PARP) antibody (H-250; Santa Cruz) per ml of Tris-buffered saline plus 0.1% Tween 20. (TBST). Samples were also blotted with either monoclonal anti-Bcl-2 (Transduction Labs) or monoclonal anti-β-actin (AC-15; Sigma) antibody as specified by the manufacturer. For caspase inhibition, cells were treated with 25 μM zVAD-fmk (Enzyme Systems Products) for 30 min before being treated with LA.
Trypan blue exclusion.
MCF10A cells grown in six-well tissue culture plates were treated for 48 h with DMEM containing 0.1% ethanol or 5 μM LA. The cells were then harvested by trypsinization and centrifugation at 300 × g, with any suspended cells also being collected in the same tube. Pellets were resuspended in 0.4% trypan blue solution (Sigma), and the percentage of cells staining blue was counted using a hemacytometer.
Fluorescence microscopy and photography.
All fluorescence experiments were observed using a Zeiss Axiophot upright fluorescence microscope, and images were captured using a SPOT-RT cooled digital camera (Diagnostic Instruments). Individual fluorophores were imaged in black and white without color-separating filters for maximum sensitivity and pseudocolored using Adobe Photoshop. In the experiment in Fig. 3, Mitotracker staining and Bax staining were pseudocolored green and red, respectively, to maintain consistency of Bax coloration with Fig. 4. In the Mitotracker experiment, Bax localization was detected using a fluorescein isothiocyanate-conjugated secondary antibody (Jackson Immunoresearch). Images were overlaid in Adobe Photoshop.
FIG. 3.
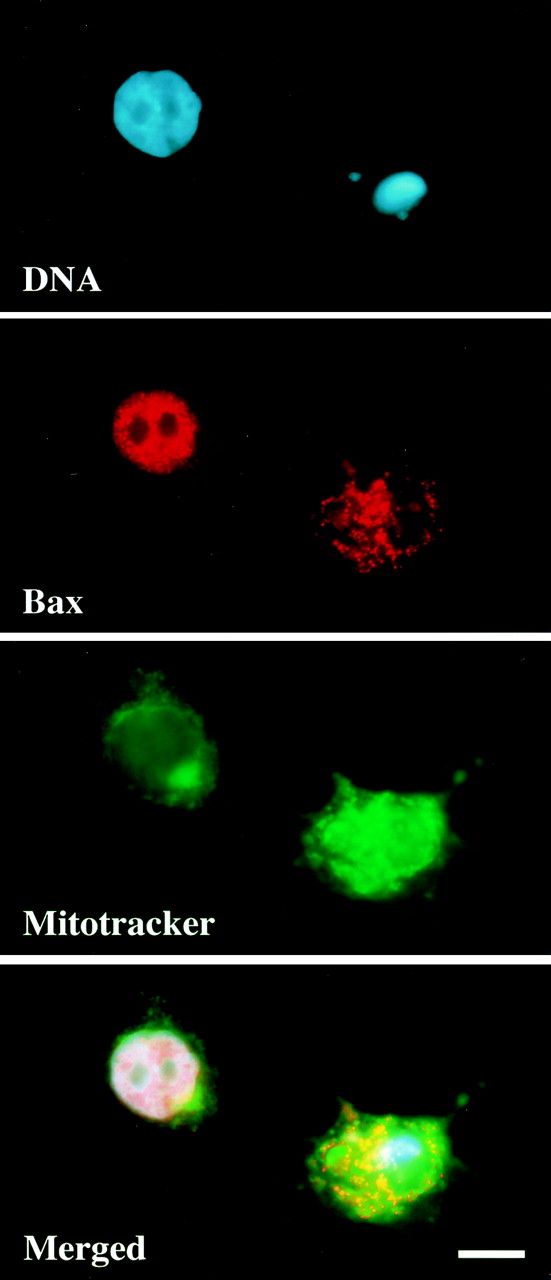
LA stimulates the movement of Bax to mitochondria. MCF10A cells were treated with LA (5 μM) for 16 h, fixed in formaldehyde, and immunostained for Bax localization. DNA was stained with Hoescht 33342, and Mitotracker CMX-H2-ROS was added 30 min prior to fixation to label mitochondria. The panels represent three independent images of the same field along with a merged overlay. Bar, 10 μm.
FIG. 4.

Bcl-2 prevents LA-induced Bax mitochondrial localization. MCF10A cells were transfected with either GFP alone or GFP plus Bcl-2 as indicated and allowed to express protein for 24 h. The cells were then treated with vehicle (0.1% ethanol) or LA (5 μM) for 16 h and costained for Bax and DNA localization. Each vertical column represents three individual images of identical fields of cells. Solid arrowheads indicate the location of the transfected cells in Bax and DNA panels, while open arrowheads mark a representative apoptotic cell in each field. Bar, 10 μm.
RESULTS
To address the direct role of the actin cytoskeleton in mediating mammary cell survival, we used the immortalized MCF10A human mammary epithelial cell line (32). When grown attached to uncoated tissue culture plates, these cells display a typical epithelial morphology and organize actin bundles along cell borders (Fig. 1). Given the hypothesis that survival of adhesion-dependent cells relies on actin-mediated cell morphology rather than simple adhesion, we sought to directly inhibit the actin cytoskeleton without affecting cellular attachment. Treatment of MCF10A cells with LA, an inhibitor of actin polymerization (33), led to significant disruption of actin microfilaments within 30 min, although the cells began to detach only after 24 h (Fig. 1). These experiments were conducted in the absence of serum and exogenous growth factors to sensitize the cells to survival signals mediated by the cytoskeleton.
FIG. 1.

LA disrupts the actin cytoskeleton of human MCF10A mammary epithelial cells. MCF10A cells were treated with vehicle (0.1% ethanol) or LA (5 μM) for 30 min. They were fixed with formaldehyde and stained for actin with Alexa594-phalloidin and for DNA with Hoescht 33342. The panels represent two horizontal rows of identical fields of cells viewed independently for phase-contrast, actin, and DNA. Bar, 10 μm.
FACS analysis of MCF10A cells treated with LA showed a significant increase in sub-G1 DNA content compared to that in cells grown in either serum-free medium alone or complete medium (Fig. 2). To ensure that this apparent DNA fragmentation was not simply the result of disrupted cytokinesis, we monitored movement of the Bax protein. Earlier studies using a Bax-GFP fusion protein and Bax immunostaining have shown that Bax moves to a mitochondrial localization following apoptotic stimuli (12, 38). Treatment with LA stimulated the movement of the Bax protein from a predominantly nuclear localization to a punctate cytoplasmic localization (Fig. 3). Costaining with Mitotracker CM-H2Xros to label mitochondria (green) demonstrated that this punctate cytoplasmic Bax staining (red) colocalizes with mitochondria of MCF10A cells, yielding a yellow composite signal when the Bax and Mitotracker images are overlaid (Fig. 3). Populations of MCF10A cells that were marked by GFP transfection and scored for LA-induced Bax localization showed that disruption of the actin cytoskeleton led to an approximately 10-fold increase in the number of cells displaying mitochondrial Bax localization (Fig. 4 and 5). These cells also displayed nuclear condensation characteristic of apoptosis. Cotransfection of an expression vector coding for the cell survival protein Bcl-2 led to significant inhibition of Bax localization to mitochondria, although nuclear condensation was actually increased in the Bcl-2-expressing population (Fig. 4 and 5). These data demonstrate that LA treatment initiates an apoptotic cascade that involves the translocation of Bax to mitochondria. However, given the apparent discrepancy between the ability of Bcl-2 to suppress LA-induced Bax translocation and nuclear condensation, it was important to determine if LA treatment committed MCF10A cells to apoptotic execution and exclude general cytotoxicity of the compound as a cause of this cell death.
FIG. 2.

Induction of DNA fragmentation in MCF10A cells treated with LA. Cells were grown for 16 h in complete medium, serum-free medium, or serum-free medium containing LA (5 μM). They were fixed in ethanol and subjected to FACS analysis following staining with propidium iodide to assess DNA content.
FIG. 5.

Bcl-2 rescues Bax mitochondrial localization but not nuclear condensation. Populations of MCF10A cells, transfected and stained as in the experiment in Fig. 4, were scored for apoptosis by both Bax localization (black bars) and nuclear condensation (grey bars). Each bar represents the mean and standard deviation of three independent experiments in which at least 100 cells were counted.
To measure apoptotic commitment, we assessed the cleavage of PARP, a DNA repair protein that is cleaved by caspases during the execution phase of apoptotic cell death (37). As seen in Fig. 6, treatment of MCF10A cells with LA led to approximately 50% cleavage of PARP by 8 h and relatively complete cleavage by 24 h. This process was largely inhibited by pretreatment of the cells with the broad-spectrum caspase inhibitor zVAD-fmk (37), indicating that LA-induced PARP cleavage occurs by a caspase-dependent mechanism. PARP cleavage in serum-starved cells was minimal following 24 h of treatment (Fig. 6). To address any possible general cytotoxicity of LA, we employed the MDA-MB-453 human mammary epithelial cell line which was isolated from a metastatic tumor (7). If the hypothesis holds true that the normal actin-dependent apoptosis pathway is inactivated in tumor cells, then MDA-MB-453 cells might be capable of surviving despite actin depolymerization. As seen in Fig. 7, MDA-MB-453 cells are highly resistant to PARP cleavage induced by LA treatment, showing virtually no cleavage even after 48 h. While this result probably excludes a general cytotoxic effect of LA, we were interested to see if solitary expression of Bcl-2 was capable of recapitulating the resistant phenotype of the metastatic cell line in the MCF10A normal human mammary epithelial cells. We generated MCF10A cell lines stably overexpressing Bcl-2 (Fig. 8A) and assessed LA-induced PARP cleavage in these cells (Fig. 8B). Compared to either parental MCF10A cells or those carrying vector alone (pcDNA3), three independent clones overexpressing Bcl-2 gained significant resistance to LA-induced PARP cleavage (Fig. 8B). These Bcl-2-expressing stable cell lines were still subject to LA-induced cell rounding, but unlike MCF10A parentals or vector controls, the Bcl-2 expressing stable cell lines did not detach from the plate even after 48 h of treatment (Fig. 9A). This phenotype of displaying rounding without detachment was also observed in the MDA-MB-453 cells (data not shown). Treatment of MCF10A cells with LA also led to eventual loss of cell viability as gauged by trypan blue exclusion (Fig. 9B). Cell death approached 90% by 48 h in both MCF10A parental cells and stable cell lines carrying the pcDNA3 vector alone. However, Bcl-2-expressing stables cell lines were also resistant to loss of cell viability after disruption of the actin cytoskeleton with LA (Fig. 9B). After 48 h of treatment with 5 μM LA, only approximately 10% of the Bcl-2 expressing stable cell lines had lost viability (Fig. 9B). In fact, transfer of fresh medium to the cells at this point was capable of relieving the LA-induced rounding and cytokinetic arrest, and the Bcl-2-expressing stable cell lines continued to grow and apparently divided normally (data not shown). Therefore, with respect to apoptotic sensitivity that is induced by disruption of the actin cytoskeleton, the untransformed MCF10A human mammary epithelial cells behave like their metastatic MDA-MB-453 counterpart when stably expressing Bcl-2.
FIG. 6.

Time course of PARP cleavage following LA treatment. MCF10A cells grown in either complete or serum-free medium were treated with vehicle (0.1% ethanol) or LA (1 μM) for the indicated times. Cell lysates were subjected to SDS-PAGE and Western blotting using a polyclonal anti-PARP antibody which recognizes both full-length and cleaved PARP as indicated. The last two samples were treated with zVAD-fmk (25 μM) 30 min prior to the addition of LA to inhibit general caspase activation.
FIG. 7.

. Metastatic mammary tumor cells are resistant to apoptosis induced by LA. MCF10A cells or MDA-MB-453 cells were grown in complete or serum-free medium and treated with LA (1 μM) for either 24 or 48 h as indicated. Lysates from each sample were subjected to Western blot analysis for PARP cleavage as in the experiment in Fig. 6.
FIG. 8.

Bcl-2 overexpression prevents LA-induced PARP cleavage in MCF10A cells. (A) Protein levels of Bcl-2 in MCF10A cells or three independent stable lines expressing either empty vector (pcDNA3, lanes a to c) or pcDNA3-Bcl2 (pcDNA3Bcl2, lanes a to c) were measured by Western blotting with a monoclonal antibody against Bcl-2. The blot was stripped and reprobed with a monoclonal antibody against β-actin to confirm equivalent loading. (B) Parental MCF10A cells or stable lines (described in panel A) were subjected to 1 μM LA treatment for 16 h in serum-free medium. PARP cleavage was assessed by Western blotting as in the experiment in Fig. 6.
FIG. 9.

Bcl-2 rescues detachment and loss of viability following LA treatment. (A) MCF10A cell lines stably expressing either vector (pcDNA3) or Bcl-2 (pcDNA3-Bcl2) were treated with either vehicle (0.1% ethanol) or 5 μM LA. Representative fields of each were photographed after 48 h. Bar, 10 μm. (B) MCF10A parental cell lines or those stably transfected with either pcDNA3 or pcDNA3-Bcl2 were grown in six-well dishes and treated with LA or left untreated, as in panel A, for 48 h. Loss of cell viability was measured by staining for trypan blue exclusion. Bars represent the mean and standard deviations of three independent experiments in which at least 200 cells were scored for cell death.
DISCUSSION
Mammary epithelial cells are dependent on attachment to the extracellular matrix for survival (35). Experiments using primary mouse mammary epithelial cells demonstrated that this attachment-mediated survival requirement relies specifically on binding to laminin (25). For our experiments we have used a spontaneously immortalized human mammary epithelial cell line, MCF10A (32). These cells grow well on tissue culture plastic but remain adhesion dependent for growth, as indicated by their inability to form colonies in soft agar or tumors in nude mice (32). MCF10A cells also secrete laminin onto tissue culture plates following attachment (13) and are therefore capable of providing themselves with a suitable extracellular matrix for survival. We were able to confirm laminin secretion in these cells by immunofluorescence (data not shown). However, since previous studies have shown that attachment-mediated survival in endothelial cells does not depend solely on integrin engagement but, rather, involves cell spreading (8, 26), we were interested in investigating the role of morphology in survival rather than direct attachment.
Since epithelial cell spreading on extracellular matrix depends on actin filaments (11), we inhibited actin polymerization in MCF10A cells that were already attached and spread to directly address whether actin organization performs a persisting role in the survival of adherent cells. For these experiments, we employed LA, a specific inhibitor of actin polymerization (33). LA is capable of directly inhibiting the polymerization of purified actin in vitro (33), and its direct binding site on the actin protein has recently been identified (22). Furthermore, earlier experiments with yeast have shown that the development of specific actin mutations confers resistance to LA, excluding a general cytotoxic role for the compound (3). MCF10A cells became rapidly rounded following treatment with LA, although they maintained attachment to the plate. Approximately half the cells had entered the execution phase of cell death by 8 h, as gauged by caspase-mediated cleavage of PARP (Fig. 6), but did not detach until after 24 h. Cell death was characterized by movement of Bax to a mitochondrial localization, which has been observed in mouse mammary epithelial cells grown without extracellular matrix attachment (12). Overexpression of Bcl-2 was capable of preventing a mitochondrial localization of Bax in response to LA (Fig. 4 and 5). However, nuclear condensation was increased in this population (Fig. 5), providing an apparent dilemma. Stable expression of Bcl-2 demonstrated rescue from both LA-mediated PARP cleavage and loss of viability (Fig. 8 and 9), indicating that apoptosis was indeed rescued. We therefore conclude that assessment of apoptosis by nuclear condensation, a common practice in the literature, may be misleading in cases of cytoskeletal disruption or suspension growth, due to the alteration of cellular morphology. The apparent increase of nuclear condensation in Bcl-2-overexpressing cells was probably due to enhanced attachment as a result of inhibition of caspase activation. Direct inhibition of caspases with zVAD-fmk can prevent cell detachment during apoptosis (36), so that Bcl-2-overexpressing cells probably tolerated fixation and washing better during immunofluorescence experiments. In this same regard, it is notable that Bcl-2-overexpressing cells remained attached even after long treatments with LA, despite rounding significantly. This indicates that eventual detachment occurs as a consequence of the apoptotic signaling cascade rather than a direct result of LA treatment. Human MCF10A mammary epithelial cells therefore maintain a persistent requirement for an organized actin cytoskeleton, even after attachment and spreading have been completed.
Apoptotic signaling following actin depolymerization was also greatly reduced in MDA-MB-453 cells. This human mammary epithelial cell line was isolated from a patient with metastatic mammary carcinoma (7). We were able to recapitulate this resistant phenotype in the untransformed MCF10A human mammary epithelial cell line by stably expressing Bcl-2. Like the Bcl-2-expressing stable cell lines, MDA-MB-453 cells also showed continual attachment after LA treatment, reinforcing the notion that apoptosis is the cause of the detachment in response to LA, rather than the reverse. Overexpression of Bcl-2 therefore confers a similar survival advantage to MCF10A cells following cytoskeletal disruption to that found in metastatic carcinoma cells. Although our cells showed a blockade of Bax localization to mitochondria by Bcl-2 overexpression, it is unlikely that this is the solitary mechanism by which Bcl-2 confers a survival advantage on these cells. Bcl-2 provides much more effective protection than does loss of Bax in the mouse mammary gland during involution (29), a process which is characterized by disruption of the extracellular matrix and epithelial cell morphology. Furthermore, Bcl-2 overexpression is not sufficient to induce tumorigenesis in mammary epithelial cells in vivo but has been shown to enhance the ability of c-myc to form tumors (16). Taken together, our results and those of others suggest that dysregulation of Bcl-2 expression confers resistance to apoptosis induced by epithelial cell shape change. Although this does not directly induce tumor formation, it may contribute to the ability of tumor cells to tolerate the gross actin cytoskeletal abnormalities found in metastatic tumor cells (2, 4, 9, 34).
Loss of the tumor suppressor protein p53 is known to suppress cell death induced by cytoskeletal depolymerization in mouse fibroblast cells (28). Studies with fibroblasts and endothelial cells (15) show that attachment to extracellular matrix can suppress p53-mediated apoptosis through the signaling of focal adhesion kinase (FAK). Tyrosine phosphorylation of FAK and its binding to numerous signaling proteins can be inhibited during attachment in serum-containing media by inhibition of actin polymerization (18, 31). Focal adhesions represent a direct mechanical connection between the extracellular matrix and the internal cytoskeleton (20). Therefore, signaling through FAK might mediate the actin-dependent survival of attached mammary epithelial cells. However, expression of a dominant negative form of FAK (FAK-CD) completely blocks localization and tyrosine phosphorylation of endogenous FAK without affecting the viability or attachment of MCF10A cells (40). It is possible that the additional organization of the epithelial actin cytoskeleton through cell-cell contacts serves as an added survival signal not found in fibroblasts.
Another possibility is that the actin-dependent survival signals transduced from FAK are not mediated by the same signaling proteins or phosphorylation events that function during stimulation with growth factors. Notably, a recent report suggests that a region in FAK that is N terminal to its focal adhesion targeting sequence may be responsible for its ability to specifically suppress p53-mediated apoptosis following detachment from the extracellular matrix (15). These authors performed their studies in the absence of serum growth factors to isolate signaling mechanisms involved in cell adhesion, and we have used the same technique. They found that survival signaling from FAK in this system was distinct from that induced in the presence of growth factors, particularly since it did not require activation of phosphatidylinositol 3-kinase (15). The difference in the dominant negative version of FAK used to induce apoptosis in fibroblasts and endothelial cells (15) and that failing to induce apoptosis in human mammary epithelial cells (40) comprises only amino acids 693 to 839 of the FAK protein. That this sequence may be responsible for survival signaling specifically in the presence of extracellular attachment or actin cytoskeletal organization definitely deserves further investigation. Interestingly, the apoptotic signal mediated by disruption of FAK signaling in serum-starved cells was also inhibitable by overexpression of Bcl-2 (15). It is therefore distinct from that induced in the presence of serum (15, 17, 39) and consistent with the mode of cell death induced by the actin cytoskeletal depolymerization described here. FAK is overexpressed in a majority of malignant breast tumors (23), and this may contribute to their ability to tolerate the disruption of actin organization thought necessary for metastatic progression.
In conclusion, we have shown that direct depolymerization of the actin cytoskeleton is sufficient to induce apoptosis in human MCF10A mammary epithelial cells despite continuous attachment. This pathway is inhibited in metastatic mammary carcinoma cells and can be blocked in untransformed MCF10A cells by overexpression of Bcl-2, providing novel evidence that Bcl-2 is involved in cell death induced by actin depolymerization.
ACKNOWLEDGMENTS
This work was supported in part by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation Fellowship, DRG-1496 (to S. S. Martin) and by the Howard Hughes Medical Institute.
We thank Juanita Campos-Torres for her help with the FACS analysis and Michel Lebel for his assistance with the Western blot assay for PARP cleavage. We are grateful to Steve Santer, Robert Pauley, and Fred Miller for providing the MCF10A cells and for instructions on their growth.
REFERENCES
- 1.Alford D, Pitha-Rowe P, Taylor-Papadimitriou J. Adhesion molecules in breast cancer: role of alpha 2 beta 1 integrin. Biochem Soc Symp. 1998;63:245–259. [PubMed] [Google Scholar]
- 2.Asch H L, Head K, Dong Y, Natoli F, Winston J S, Connolly J L, Asch B B. Widespread loss of gelsolin in breast cancers of humans, mice, and rats. Cancer Res. 1996;56:4841–4845. [PubMed] [Google Scholar]
- 3.Ayscough K R, Stryker J, Pokala N, Sanders M, Crews P, Drubin D G. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Ze'ev A. Cytoskeletal and adhesion proteins as tumor suppressors. Curr Opin Cell Biol. 1997;9:99–108. doi: 10.1016/s0955-0674(97)80158-5. [DOI] [PubMed] [Google Scholar]
- 5.Boudreau N, Sympson C J, Werb Z, Bissell M J. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 7.Cailleau R, Olive M, Cruciger Q V. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 8.Chen C S, Mrksich M, Huang S, Whitesides G M, Ingber D E. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 9.Curschellas E, Matter A, Regenass U. Immunolocalization of cytoskeletal elements in human mammary epithelial cells. Eur J Cancer Clin Oncol. 1987;23:1517–1527. doi: 10.1016/0277-5379(87)90095-2. [DOI] [PubMed] [Google Scholar]
- 10.Frisch S M, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuda M, Nishida T, Otori T. Role of actin filaments and microtubules in the spreading of rabbit corneal epithelial cells on the fibronectin matrix. Cornea. 1990;9:28–35. [PubMed] [Google Scholar]
- 12.Gilmore A P, Metcalfe A D, Romer L H, Streuli C H. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J Cell Biol. 2000;149:431–446. doi: 10.1083/jcb.149.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldfinger L E, Stack M S, Jones J C. Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J Cell Biol. 1998;141:255–265. doi: 10.1083/jcb.141.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynes R O. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 15.Ilic D, Almeida E A, Schlaepfer D D, Dazin P, Aizawa S, Damsky C H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jager R, Herzer U, Schenkel J, Weiher H. Overexpression of Bcl-2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene. 1997;15:1787–1795. doi: 10.1038/sj.onc.1201353. [DOI] [PubMed] [Google Scholar]
- 17.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne P H, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipfert L, Haimovich B, Schaller M D, Cobb B S, Parsons J T, Brugge J S. Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J Cell Biol. 1992;119:905–912. doi: 10.1083/jcb.119.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotz M M, Burdsal C A, Erickson H P, McClay D R. Cell adhesion to fibronectin and tenascin: quantitative measurements of initial binding and subsequent strengthening response. J Cell Biol. 1989;109:1795–1805. doi: 10.1083/jcb.109.4.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maniotis A J, Chen C S, Ingber D E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meredith J E, Fazeli B, Schwartz M A. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morton W M, Ayscough K R, McLaughlin P J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- 23.Owens L V, Xu L, Craven R J, Dent G A, Weiner T M, Kornberg L, Liu E T, Cance W G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55:2752–2755. [PubMed] [Google Scholar]
- 24.Parsons J T, Martin K H, Slack J K, Taylor J M, Weed S A. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 25.Pullan S, Wilson J, Metcalfe A, Edwards G M, Goberdhan N, Tilly J, Hickman J A, Dive C, Streuli C H. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J Cell Sci. 1996;109:631–642. doi: 10.1242/jcs.109.3.631. [DOI] [PubMed] [Google Scholar]
- 26.Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, Colotta F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol. 1994;127:537–546. doi: 10.1083/jcb.127.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed J C. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 28.Rubtsova S N, Kondratov R V, Kopnin P B, Chumakov P M, Kopnin B P, Vasiliev J M. Disruption of actin microfilaments by cytochalasin D leads to activation of p53. FEBS Lett. 1998;430:353–357. doi: 10.1016/s0014-5793(98)00692-9. [DOI] [PubMed] [Google Scholar]
- 29.Schorr K, Li M, Bar-Peled U, Lewis A, Heredia A, Lewis B, Knudson C M, Korsmeyer S J, Jager R, Weiher H, Furth P A. Gain of Bcl-2 is more potent than bax loss in regulating mammary epithelial cell survival in vivo. Cancer Res. 1999;59:2541–2545. [PubMed] [Google Scholar]
- 30.Shaw L M. Integrin function in breast carcinoma progression. J Mammary Gland Biol Neoplasia. 1999;4:367–376. doi: 10.1023/a:1018766317055. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y, Schaller M D. Focal adhesion targeting: the critical determinant of FAK regulation and substrate phosphorylation. Mol Biol Cell. 1999;10:2507–2518. doi: 10.1091/mbc.10.8.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soule H D, Maloney T M, Wolman S R, Peterson W D, Brenz R, McGrath C M, Russo J, Pauley R J, Jones R F, Brooks S C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 33.Spector I, Shochet N R, Blasberger D, Kashman Y. Latrunculins—novel marine macrolides that disrupt microfilament organization and affect cell growth. I. Comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- 34.Stournaras C, Stiakaki E, Koukouritaki S B, Theodoropoulos P A, Kalmanti M, Fostinis Y, Gravanis A. Altered actin polymerization dynamics in various malignant cell types: evidence for differential sensitivity to cytochalasin B. Biochem Pharmacol. 1996;52:1339–1346. doi: 10.1016/s0006-2952(96)00389-9. [DOI] [PubMed] [Google Scholar]
- 35.Streuli C H, Gilmore A P. Adhesion-mediated signaling in the regulation of mammary epithelial cell survival. J Mammary Gland Biol Neoplasia. 1999;4:183–191. doi: 10.1023/a:1018729308878. [DOI] [PubMed] [Google Scholar]
- 36.van de Water B, Nagelkerke J F, Stevens J L. Dephosphorylation of focal adhesion kinase (FAK) and loss of focal contacts precede caspase-mediated cleavage of FAK during apoptosis in renal epithelial cells. J Biol Chem. 1999;274:13328–13337. doi: 10.1074/jbc.274.19.13328. [DOI] [PubMed] [Google Scholar]
- 37.Wolf C M, Reynolds J E, Morana S J, Eastman A. The temporal relationship between protein phosphatase, ICE/CED-3 proteases, intracellular acidification, and DNA fragmentation in apoptosis. Exp Cell Res. 1997;230:22–27. doi: 10.1006/excr.1996.3401. [DOI] [PubMed] [Google Scholar]
- 38.Wolter K G, Hsu Y T, Smith C L, Nechushtan A, Xi X G, Youle R J. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiong W, Parsons J T. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol. 1997;139:529–539. doi: 10.1083/jcb.139.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L H, Yang X, Bradham C A, Brenner D A, Baldwin A S, Craven R J, Cance W G. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275:30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]