

Abstract
Aberrant transcriptional activity of androgen receptor (AR) is one of the dominant mechanisms for developing of castration-resistant prostate cancer (CRPC). Analyzing AR-transcriptional complex related to CRPC is therefore important towards understanding the mechanism of therapy-resistance. While studying its mechanism, we observed that a transmembrane protein called neuropilin-2 (NRP2) plays a contributory role in forming a novel AR-transcriptional complex containing nuclear pore proteins. Using immunogold electron microscopy, high-resolution confocal microscopy, chromatin immunoprecipitation, proteomics, and other biochemical techniques, we delineated the molecular mechanism of how a specific splice variant of NRP2 becomes sumoylated upon ligand stimulation and translocates to the inner nuclear membrane. This splice variant of NRP2 then stabilizes the complex between AR and nuclear pore proteins to promote CRPC specific gene expression. Both full-length and splice variants of AR have been identified in this specific transcriptional complex. In vitro cell line-based assays indicated that depletion of NRP2 not only destabilizes the AR-nuclear pore protein interaction but also inhibits the transcriptional activities of AR. Using an in vivo bone metastasis model, we showed that the inhibition of NRP2 led to the sensitization of CRPC cells toward established anti-AR therapies such as enzalutamide. Overall, our finding emphasize the importance of combinatorial inhibition of NRP2 and AR as an effective therapeutic strategy against treatment refractory prostate cancer.
Keywords: NRP2, Nucleoporins, Androgen Receptor, Nuclear pore complex, Transcription regulation, enzalutamide resistance
Introduction
Majority of metastatic castration-resistant prostate cancer (mCRPC) relapses due to the failure of second-generation anti-androgen therapies(1–3). In mCRPC, androgen receptor (AR) activity is still the major driving force for the progression of the disease(4, 5). Therefore, targeting AR-signaling pathways is important to achieve the treatment benefit. Present FDA-approved AR-targeting drugs are raised against its C-terminal regions(6). During the progression of prostate cancer (PCa), formation of AR splice variants with C-terminal deletions, generation of AR point mutation at the C-terminal ligand-binding site of AR, or hyper-activity of the various growth factor signaling pathways is responsible for evasion of existing AR-directed therapies(7–9). Therefore, efforts are under way to target other domains of AR. However, there is an unsuccessful clinical trial to target N-terminal trans-activating domain, thus raising the concerns of direct targeting of AR in advanced PCa(10). An alternative approach has been taken to inhibit AR-signaling in CRPC. Interestingly, one of the family members of bromodomain and extra-terminal domain (BET) protein, named BRD4, has been shown to interact with AR(11, 12). A specific BRD4 inhibitor, JQ1, has been studied in advanced PCa. However, irrespective of its early success, JQ1 failed in PCa clinical trial(13, 14). Therefore, there is an urgent need to develop a new druggable target for the AR-signaling pathway for treating mCRPC patients.
While studying the molecular signature responsible for CRPC, we observed that increased expression of Neuropilin-2 (NRP2) is associated with poor PCa survival. NRP2 is a transmembrane non-tyrosine kinase protein and is known to serve as co-receptors for other receptors such as plexins in neuronal cells and vascular endothelial growth factor receptors (VEGFRs) in endothelial cells(15) (16). Broadly, NRP2 has two major membrane bound isomers, called NRP2A and NRP2B(17). However, the functional significance of these two isomers has not been adequately studied.
In this current study, we have analyzed NRP2 expression in human PCa tissues and detected that a fraction of NRP2 is localized in the nucleus of aggressive PCa cells. Utilizing immunogold transmission electron-microscopy (ITEM), structural illumination microscopy (SIM), and other biochemical approaches we have observed the ligand dependent localization of NRP2B in the inner nuclear membrane (INM) of PCa cells, indicating its novel nuclear-specific role. Sumoylation at the C-terminal region of NRP2B has been detected as the required molecular event for its retrograde transport to INM. Our study further indicates that NRP2B stabilizes the transcriptional complex between nuclear pore proteins (nucleoporins) and AR at the nucleus and thus promotes the AR-driven gene expression in mCRPC. Interestingly, both full length and AR-splice variants have been observed to be a part of NRP2B-nuclear pore protein complex. Depletion of NRP2B thus can deregulate this specific AR-transcriptional complex and can enhance the therapeutic efficiency of the anti-androgen inhibitors, such as enzalutamide. This has been observed in both in vitro tissue culture systems and intratibial mouse models of PCa. Therefore, the results presented here have highlighted the presence of unique AR-NRP2B transcriptional complex and the benefits of combining NRP2 inhibition and anti-androgen therapies for treating mCRPC patients.
Results
NRP2 is present in the nuclear membrane of PCa cells:
Nuclear NRP2 expression is associated with a poor PCa prognosis.
NRP2 expression was analyzed within a tissue microarray consists of 396 formalin-fixed paraffin-embedded (FFPE) specimens from patients with prostate acinar adenocarcinoma who were treated at the Department of Urology, University Hospital Dresden, Germany between 1996 and 2005 and underwent radical prostatectomy. Around 80% of these PCa patients had a high risk of disease progression (pT>/=3, Gleason score >/=8 or pN1) (26). Earlier, evaluation of the above tissue microarray, we have demonstrated that nearly 33% high-risk group patients [stage ≥ pT3 33%, GS ≥8 34%, pN1 31%] are positive for membranous and cytoplasmic staining of NRP2 (Table 1). In this study, re-analysis of NRP2 staining revealed that almost one third of adenocarcinomas with Gleason grade 5 showed a distinct nuclear staining pattern for NRP2 (Fig. 1A, 1B). In contrast, only eight percent in adenocarcinomas with Gleason 4 pattern and only about two percent in adenocarcinomas with Gleason 3 pattern exhibited nuclear staining (Fig. 1A, 1B). Overall, our results suggested an association of nuclear-specific NRP2 staining with high Gleason grading and thus with aggressive PCa.
Table 1:
NRP2 expression in various prostate cancer group
| Group | n= no of people |
|---|---|
| High risk prostate cancer group | 325 |
| Intermediate risk prostate cancer | 24 |
| Low risk prostate cancer | 51 |
| NRP2 positive in overall cases | 268 |
| NRP2 positive in High risk group | 107 |
Fig. 1: NRP2 localization detected within the nucleus:
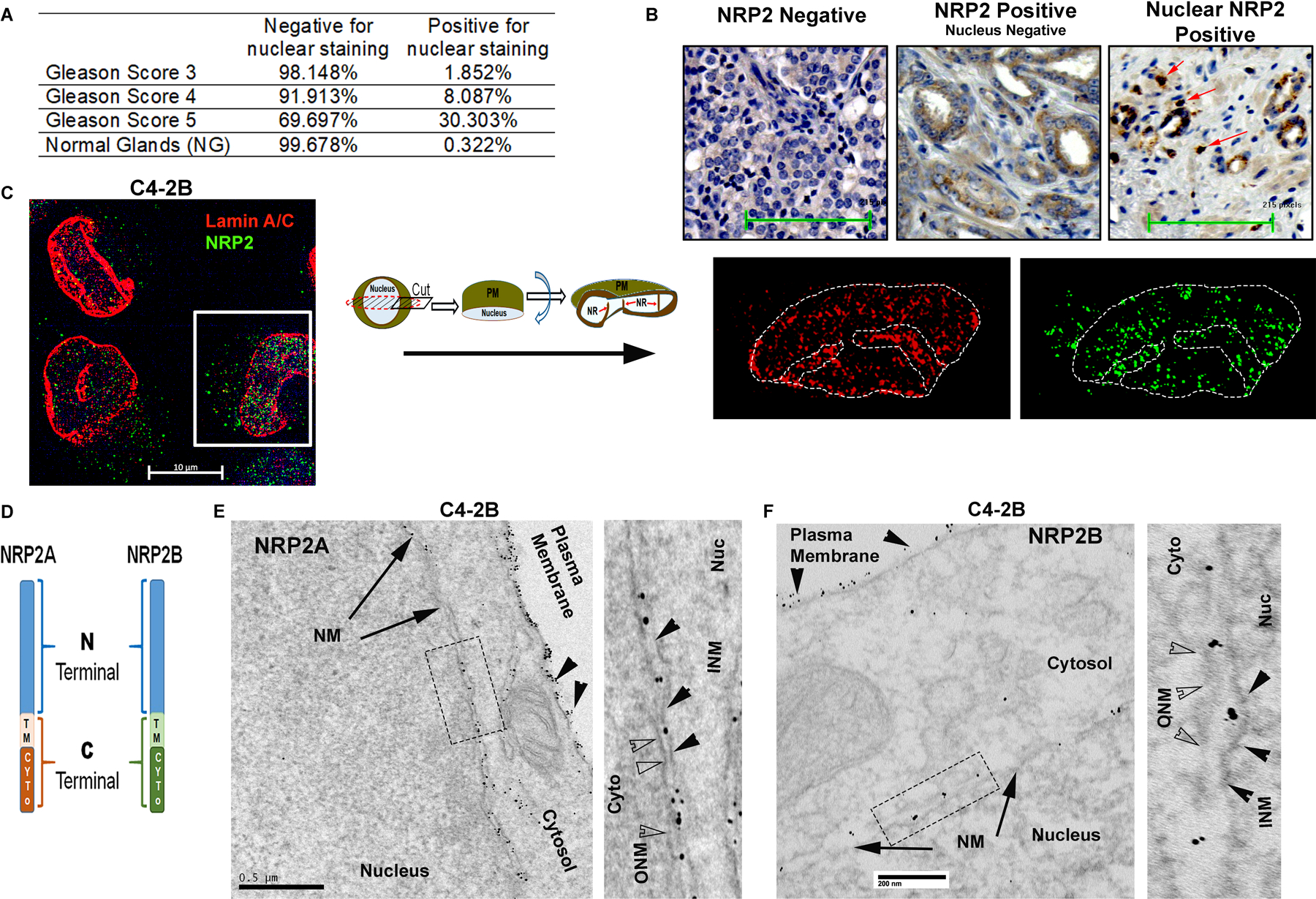
A. Percentage of nuclear NRP2-positive cells across the Gleason score. B. Representative images of immunohistochemical staining for nuclear NRP2 in human prostate cancer tissues. Arrows indicate the nuclear positive NRP2. C. Endogenous NRP2 staining (Alexa 488) in prostate cancer cell line C4–2B. INM was stained with lamin A/C (Alexa 594). DAPI indicates nuclear staining. Using Zen Blue lite software, cells were cut through the middle plane and then rotated across the x-axis. Inset is the magnified image of the nucleus of the cells. D. Schematic diagram of NRP2 isomers. E. TEM images of immunogold staining of untagged NRP2A in C4–2B cells ectopically expressing NRP2A. Cells were stained with NRP2 antibody and counterstained with gold-labeled secondary antibody. NRP2 staining appears as black punctate structures inside the cells. The nuclear and plasma membranes are indicated by the arrow and arrowhead, respectively. Inset is magnified. In the magnified image, ONM and INM are represented by opaque and black arrowheads, respectively. Scale Bar 500 nm. F. Representative TEM images of immunogold staining of HA-tagged NRP2B ectopically expressed in C4–2B. Cells were stained with HA-antibody and counterstained with gold-labeled secondary antibody. The nuclear membrane and plasma membrane are indicated by arrows and arrowheads, respectively. Insets is magnified. ONM and INM are represented by opaque and black arrowheads. Scale Bar 200 nm.
Nuclear-NRP2 localizes within the nuclear envelope in an isoform-specific manner
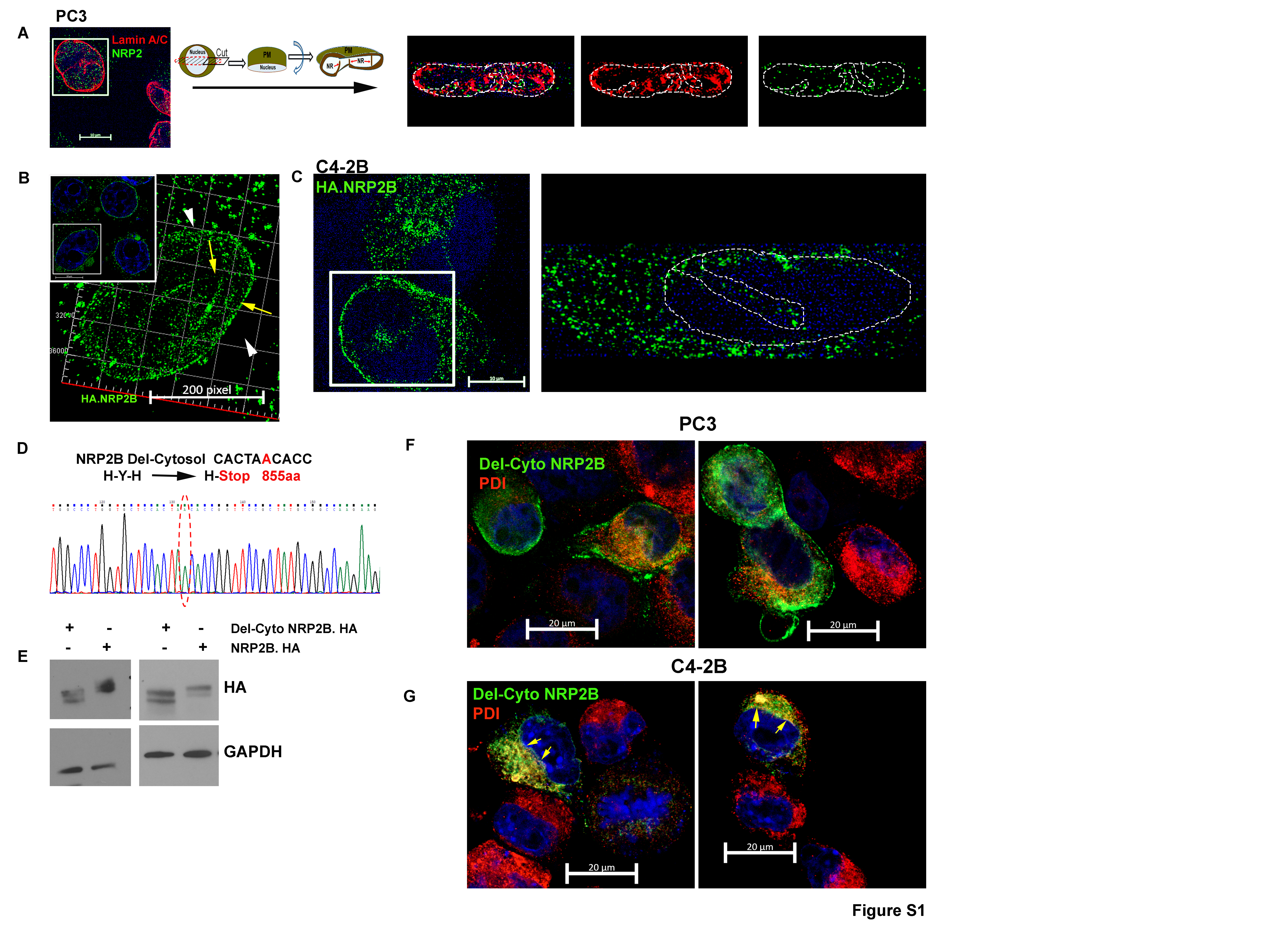
To understand the exact location of NRP2 in the nucleus, we immunostained endogenous NRP2 in PCa cell lines such as LNCaP C4–2B and PC3 (Fig. 1C, Fig. S1A). Our results with SIM indicated the presence of NRP2 at the nucleus. Because the NRP2 antibody used for structured illumination microscopy (SIM) only recognizes the membrane-bound extracellular region and not the soluble form of NRP2, we assumed that the NRP2 was anchored to the membrane/lipid droplets in the nucleus. Nuclear membrane bound NRP2 was further confirmed when we co-immunostained with Lamin A/C (Fig. 1C, Fig. S1A). Localization of NRP2 occurs around the nuclear membrane and often runs into the nuclear invaginations or the nucloplasmic reticulum (18, 19) (observed when we made virtual sections of the cells through the x-axis and rotated 90 degrees around the plane) (Fig. 1C, Fig. S1A). The ectopic expression of full length NRP2 showed the similar nuclear localization as endogenous NRP2 with characteristic nuclear membrane invaginations (Fig. S1B, S1C), while membrane-bound NRP2 devoid of the C-terminal cytosolic domain (NRP2ΔC) showed defuse non-nuclear staining of NRP2 (Fig. S1D - S1G) often residing within the ER. Our results therefore indicated the C-terminal region of NRP2 is required for its localization at the nuclear membrane.
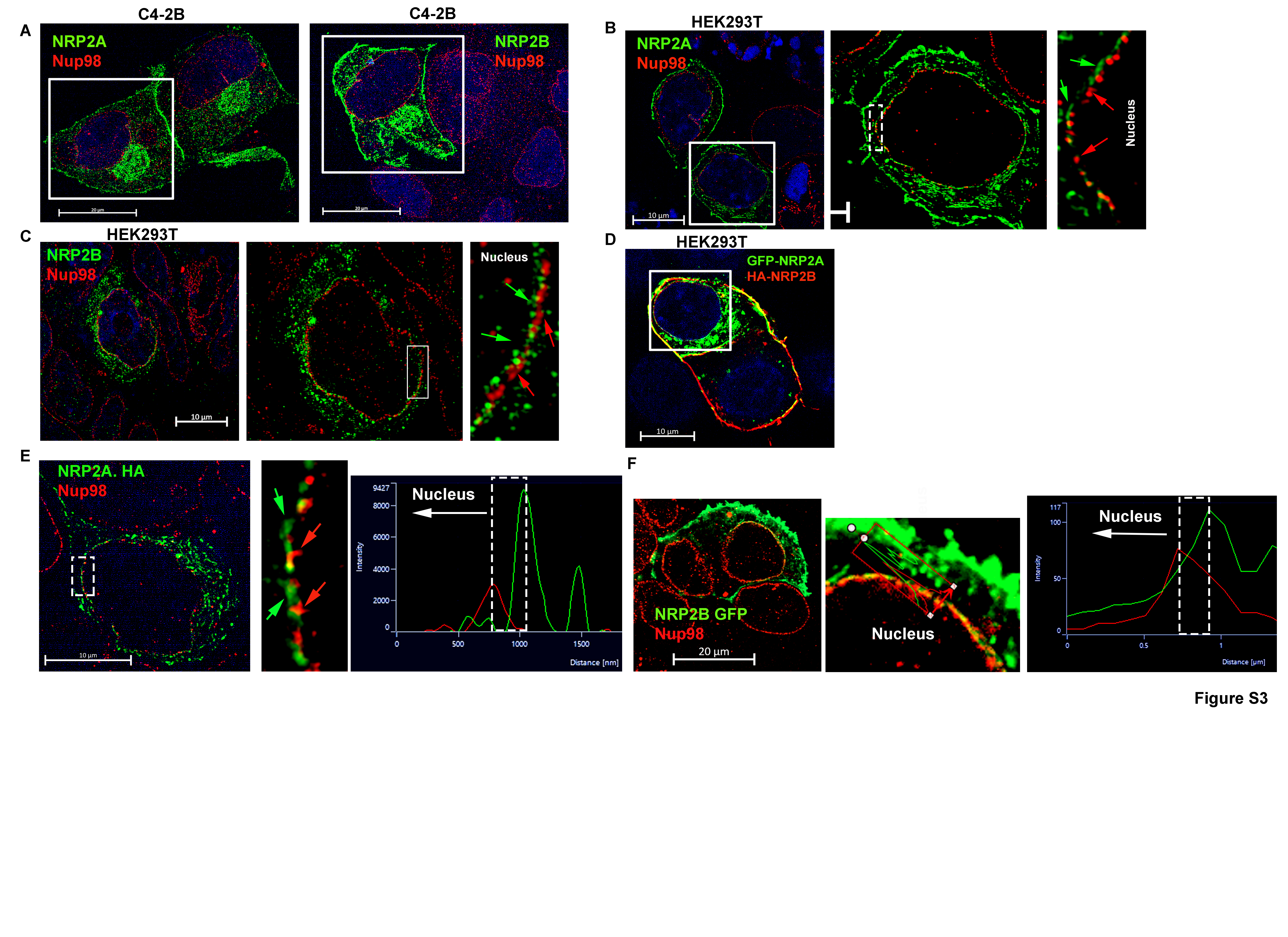
NRP2 has two major splice variants or isoforms, NRP2A and NRP2B. The isoforms are identical in structure and sequence in large parts of the N-terminal domains (a1, a2, b1 and b2), whereas they are different in the transmembrane and C-terminal cytoplasmic tail regions (Fig. 1D) (17). Because the C-terminus is important for nuclear localization, we wanted to determine whether both NRP2A and NRP2B could localize to the nuclear membrane. No isoform-specific antibody for NRP2 is commercially available; therefore, we ectopically expressed either NRP2A or NRP2B in cancer cells and analyzed their sub-cellular locations by immunogold transmission electron-microscopy (ITEM). We observed the nuclear membrane localization of both NRP2A and 2B as gold-labeled puncta in a PCa cell line C4–2B and human embryonic kidney cell line HEK293T (Fig. 1E, 1F, Fig. S2A-S2E). The electron microscopy data thus also confirmed our immunostaining data that NRP2 can be present at the nuclear membrane. Immunoblot analysis from NRP2A and 2B-expressing PCa cells also detected both variants in the nuclear extracts (Fig. S2F-S2G) further confirming the ITEM results. The antibodies used in ITEM detect the N-terminal region of NRP2 and thus stained both variants in the intranuclear membrane space. However, we observed a distinct spatial distribution of NRP2A and NRP2B in the nuclear membrane through structural illumination microscopy (SIM), when we co-immunostained with nuclear pore protein Nup98 (Fig. 2A, 2B, Fig. S3A-S3C). NRP2A at the nuclear membrane was mainly detected at the outer side of Nup98 (close to the cytoplasm) (Fig. 2A, Fig. S3A). This observation was further validated by analyzing the intensities using of fluorophore pixels of NRP2A (green line) and Nup98 (red lines), which indicated that NRP2A is further from the nucleus than Nup98 and close to outer nuclear membrane (ONM). As Nup98 is a centrally located Nup (central FG Nups), it implies that the membrane-bound NRP2A is localized within the ONM (with the N-terminal facing towards the perinuclear space). However, NRP2B was detected at a side of Nup98, which is closer to the nucleus (Fig. 2B, Fig. S3A) suggesting its localization at the INM. A similar pattern of nuclear localization of NRP2A and 2B were observed in HEK293T cells (Fig. S3B, S3C). Finally, when we expressed both GFP-tagged NRP2A (C-terminal) and HA-tagged NRP2B (N-terminal) in Hek293T cells or in C4–2B cells, we observed that they did not co-localize at the nuclear membrane. NRP2B was close to the nuclear membrane suggesting its location at the INM, whereas NRP2A was close to the cytoplasmic side indicating its presence at the ONM (Fig. 2C, 2D and Fig. S3D). Because the ONM is continuous with the endoplasmic reticulum (ER) and proteins from the ER can diffuse to the ONM; the ONM proteins often co-localize with ER markers. This is not the case for proteins located at the INM that need an active transport system to cross the nuclear pore regions for their entry into the INM. This is indeed the case for NRP2A and NRP2B. NRP2A showed significant co-localization with ER marker, protein disulfide-isomerase (PDI), near the peri-nuclear region; however, insignificant overlapping was observed between NRP2B and PDI staining (Fig. 2E-2G). To rule out the possibility that the C-terminal GFP-tag hinders the INM localization of NRP2A, we ectopically expressed N-terminal HA-tagged NRP2A. Once again, NRP2A-HA was detected at the side of Nup98 close to cytoplasm (as indicated by the line graph) (Fig. S3E) confirming its location at ONM. Since protein moves to INM from ONM by an active transport through nuclear pore, often its size exposed to the cytoplasm is the determinant factor for its passage. It has been experimentally shown that adding a bulky group to the cytoplasmic-exposed part of INM proteins hinders their movement. We observed similar phenomenon for NRP2B when we switched HA to GFP (a bulkier protein) at the C-terminus of NRP2B, as we detected no localization of GFP-tagged NRP2B in the INM. Our result thus suggested that NRP2B localizes to INM through an active transport and is regulated by its cytoplasmic tail. (Fig. S3F).
Fig. 2: NRP2 present in the nuclear membrane of the cell:
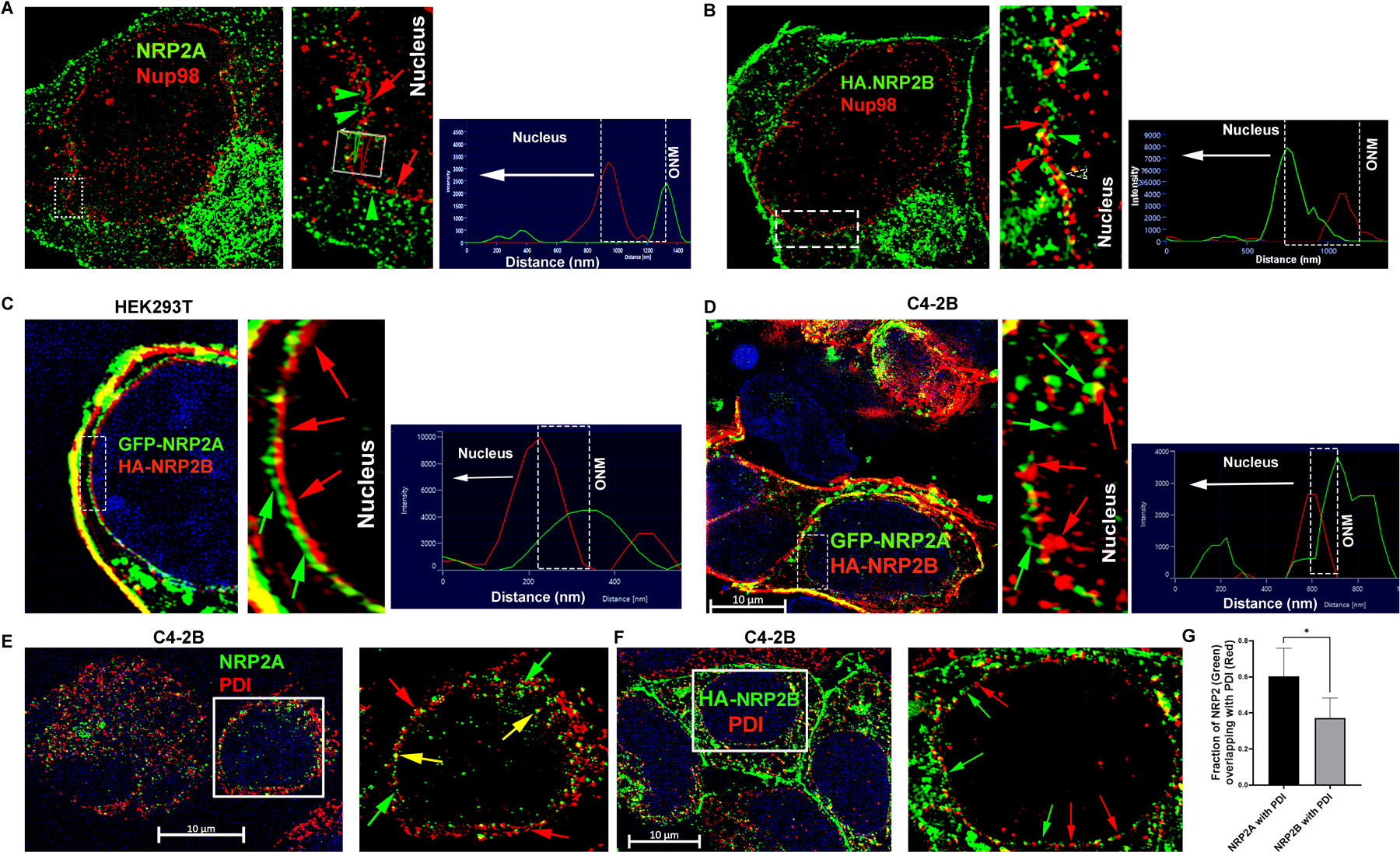
A. Immunofluorescence image of ectopically expressed untagged NRP2A (Alexa 488) in C4–2B cells. NRP2A staining was carried out with NRP2 antibody. The nuclear pore was stained with pore protein Nup98 (Alexa 594). Inset is magnified. Green arrowhead indicates NRP2A, whereas red arrows indicate Nup98. Green and red peaks in the line graph represent the relative position of NRP2A and Nup98 around the nucleus. B. Immunofluorescence image of ectopically expressed HA-tagged NRP2B (Alexa 488) in C4–2B cells. NRP2B staining was carried out with HA-antibody. The nuclear pore was stained with central pore protein Nup98 (Alexa 594). Inset is magnified. NRP2B and Nup98 are indicated by green arrowheads and red arrows, respectively. Red and green peaks in the line graph represent the relative positions of NRP2B and Nup98 around the nucleus. GFP-tagged NRP2A was ectopically co-expressed with HA-tagged NRP2B in C. Hek293T and D. C4–2B cells. Inset is magnified. NRP2B was stained with HA (Alexa 594). The INM and ONM are indicated by the red and green arrows. Scale Bar 10 µm. The nucleus was stained with DAPI. E-F. NRP2A and HA-tagged NRP2B were ectopically over-expressed in C4–2B cells and were counterstained with PDI (Alexa 594). Insets are magnified views. Red and green arrows indicate the relative localization of NRP2 and PDI around the nucleus. Yellow arrows indicate colocalization. Scale Bar 10 µm. G. Bar graph indicates NRP2 and PDI colocalization quantitation.
Inner nuclear membrane localization of NRP2B depends on ligand dependent retrograde transport
VEGF-C promotes nuclear translocation of NRP2B
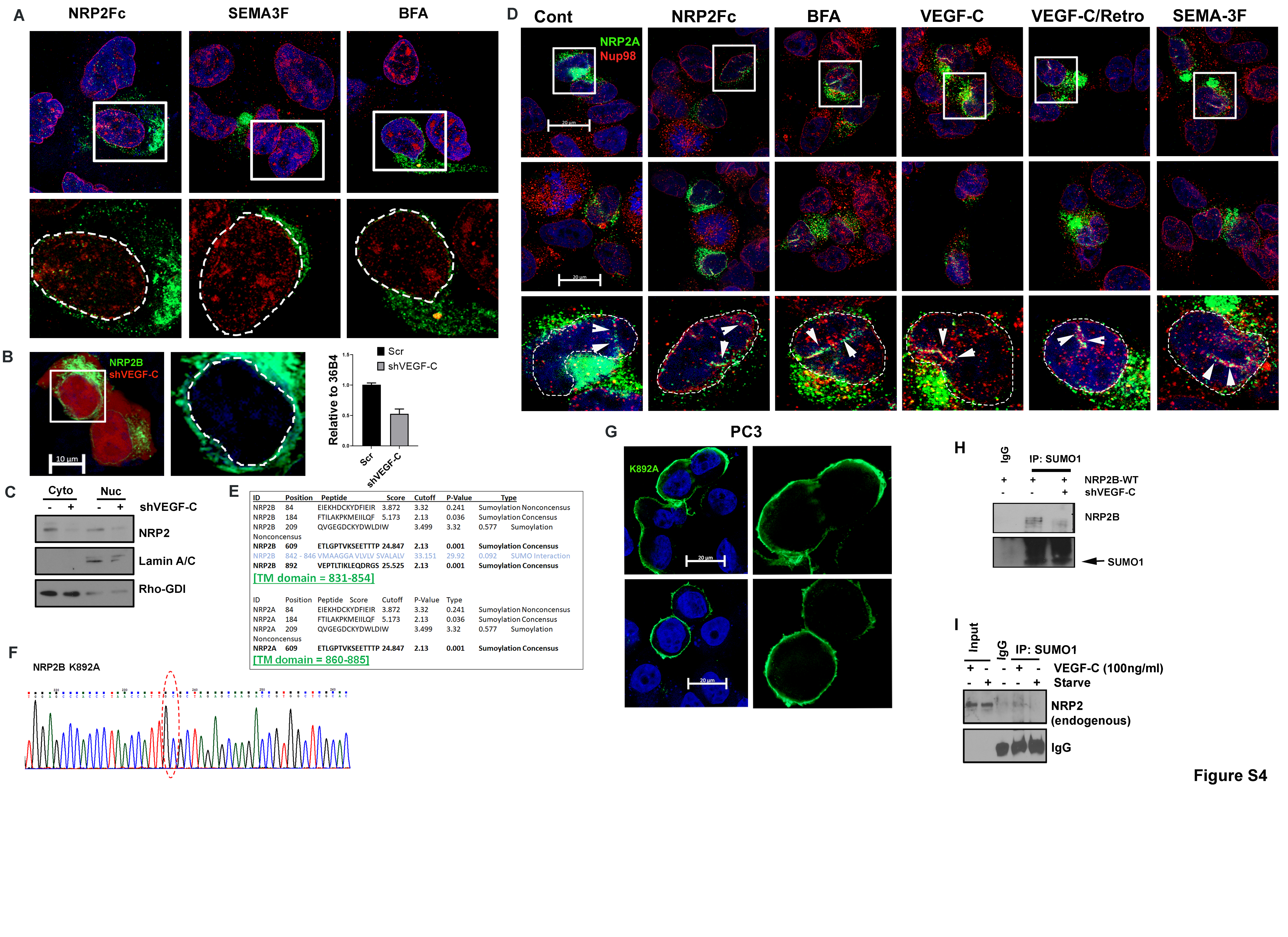
We studied whether the ligands of NRP2, such as VEGF-C and semaphorin-3F (SEMA3F), are needed for its nuclear translocation. We initially tested this with NRP2-Fc, which is a chimeric protein of the extracellular domain of NRP2 and the human IgG1 Fc fragment. Purified recombinant NRP2-Fc neutralizes the ligands of NRP2 and thus prevents their binding to cellular NRP2. NRP2-Fc treatment significantly inhibited the translocation of NRP2B into the nuclear membrane implicating the requirement of NRP2 ligands in this process (Fig. S4A, Fig. 3A, 3B). Interestingly, while VEGF-C promotes the translocation of NRP2B into the nucleus, SEMA3F has no effect (Fig. 3A, 3B, Fig. S4A). The requirement of VEGF-C in this process was further proved by genetically knocking down VEGF-C (with shRNA, tagged with RFP), which inhibited nuclear translocation of NRP2 (cells expressing RFP) shown either through immunofluorescence or by WB (Fig. S4B, S4C). Our results suggested that specific ligand binding to NRP2 has the ability in promoting nuclear translocation. We further observed that we could block the VEGF-C induced nuclear translocation of NRP2B, when we inhibited the retrograde transport of the PCa cells using a retrograde transport inhibitor, Retro-2 (Fig. 3A, 3B). Moreover, when ER to Golgi transport was inhibited using Brefeldin-A (BFA), nuclear transport of NRP2B was also arrested (Fig. S4A, Fig. 3B). Our results therefore suggested that the translocation of NRP2 occurs from plasma membrane to the nuclear membrane via retrograde transport through the Golgi-ER. This ligand-induced retrograde mode of nuclear translocation is specific for NRP2B, as we did not observe this mode of transport for NRP2A (Fig. S4D). There was no additional increase in nuclear translocation of NRP2A upon VEGF-C addition, and retro-2 or BFA was unable to block the translocation of NRP2A.
Fig. 3: VEGF-C-mediated retrograde transport and post-translational SUMOylation of NRP2B:
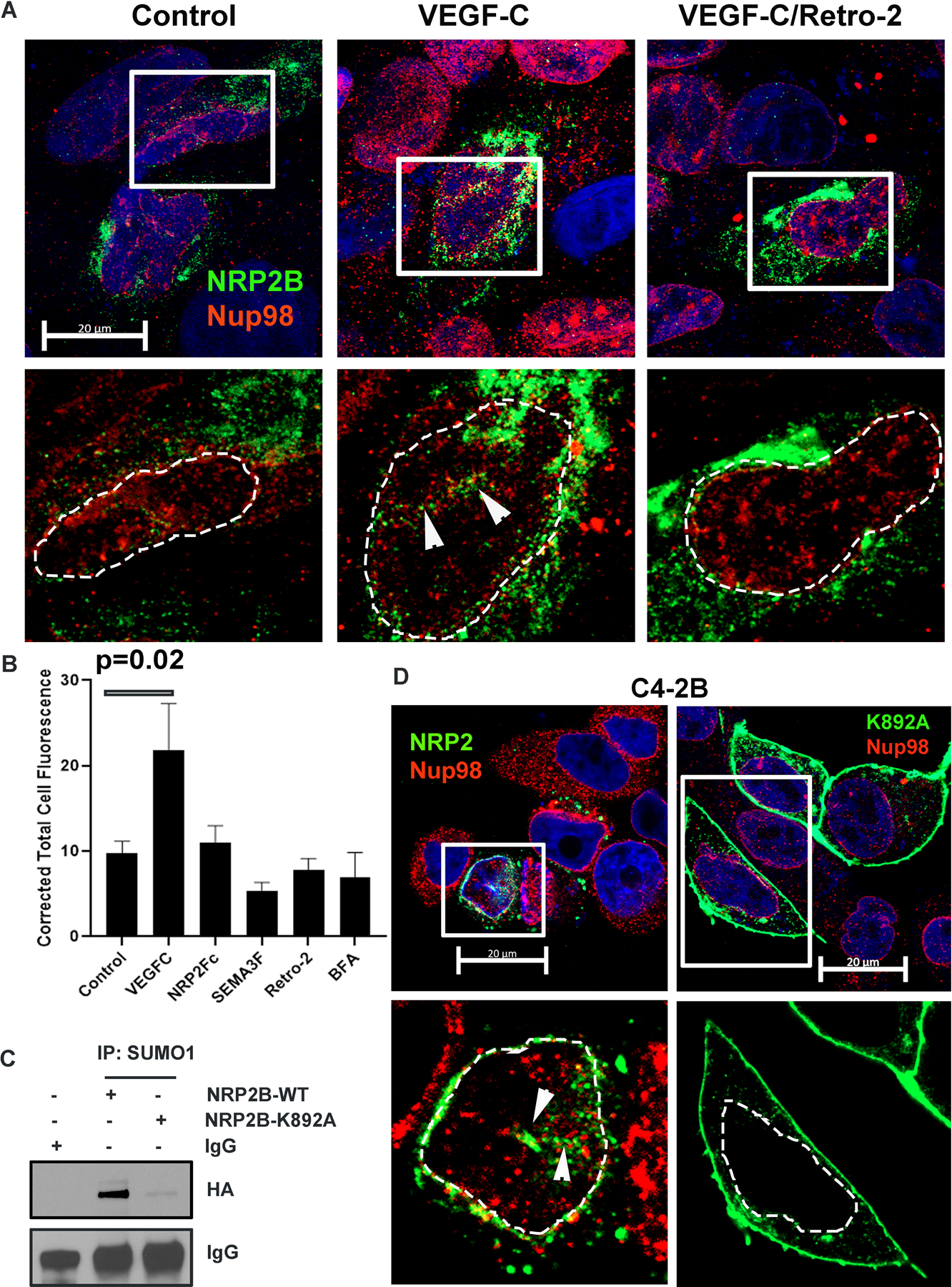
A. HA-tagged NRP2B was ectopically expressed in C4–2B cells. Immunofluorescence images representing the nuclear translocation of NRP2B under various conditions. NRP2B was detected using HA primary antibody and counterstained with Alexa 488-tagged secondary antibody. Arrowheads indicate invagination of the nuclear envelope. Nup98 was co-stained with NRP2. Inset is magnified. DAPI was used for nuclear staining. Nuclei are marked with dotted lines. Scale Bar 10 µm. B. Bar graph shows quantification of nuclear NRP2B. C. Immunoprecipitation with SUMO1 was carried out in C4–2B cells. HA-tagged wild type NRP2B and HA-tagged NRP2B K892A mutant were ectopically expressed in C4–2B cells. Immunoblots were carried out with HA-antibody. IgG bands show equal pulldown. D. Immunostaining of HA-tagged wild type and mutant K892A NRP2B in the C4–2B cell line. NRP2B was stained with HA-antibody and counterstained with secondary antibody labeled with Alexa 488. Nup98 was stained with Alexa 594. Arrowhead represents nuclear invagination in C4–2B cells expressing wild type NRP2B. Insets are magnified views. Nuclei are demarcated with dotted lines. DAPI indicates nucleus. Scale Bar 20 µm.
SUMOylation is important for nuclear translocation of NRP2B
Next, we wanted to study the molecular mechanism by which VEGF-C promotes translocation to the INM. In silico analysis of NRP2 isoforms revealed that the C-terminal part of NRP2B contains a potential SUMOylation site at lysine-892 (Fig. S4E). SUMOylation of proteins is known to regulate their nuclear translocation. Hence, we tested whether NRP2 is SUMOylated and is required for nuclear translocation. Pull-down with SUMO1 followed by western blot with NRP2 suggested that NRP2B is post-translationally modified by SUMOylation. Moreover, mutation at lysine-892 to alanine (K892A) decreased the SUMOylated form of NRP2B confirming lys-892 as the site for SUMOylation (Fig. 3C, Fig. S4F). K892A-NRP2B also failed to translocate to the nuclear membrane and predominantly accumulated at the cell surface (Fig. 3D, Fig. S4G). All these results suggested that the C-terminal tail of NRP2B is SUMOylated and this is required for nuclear translocation. Finally, NRP2 SUMOylation was inhibited when VEGF-C was depleted (Fig. S4H) suggesting SUMOylation of NRP2 is dependent on VEGF-C. In addition, treatment with recombinant VEGF-C under starved condition enhanced the SUMOylated form of NRP2 in PCa cells (Fig. S4I). All these results therefore indicated that VEGF-C mediated SUMOylation at the C-terminal of NRP2B regulates its nuclear translocation from the cell membrane.
Identification of the binding partners of nuclear NRP2B
To better understand the function of nuclear NRP2, specifically NRP2B, it is necessary to identify its interacting partners. Mass spectroscopy (MS) analysis of immuno-precipitated samples with anti-NRP2 antibody from enriched nuclear extract of NRP2B expressing LNCaP C4–2B was conducted. The protein identification threshold value was 99% (peptide threshold 95%, at-least five independent peptide identifiers for each protein in two biological replicates) (Fig. 4A). We detected 176 proteins, which are common in two independent experiments and therefore have a significant probability to interact with nuclear NRP2 (Fig. 4B, Table S1). Using the gProfile, we have classified the proteins according to their functional involvement (Fig. S5A). Our analysis revealed that these proteins are involved in RNA metabolism, splicing, replications and translations processes.
Fig. 4: NRP2 Mass-Spectrometry predicted its interactions with various nuclear-associated proteins:
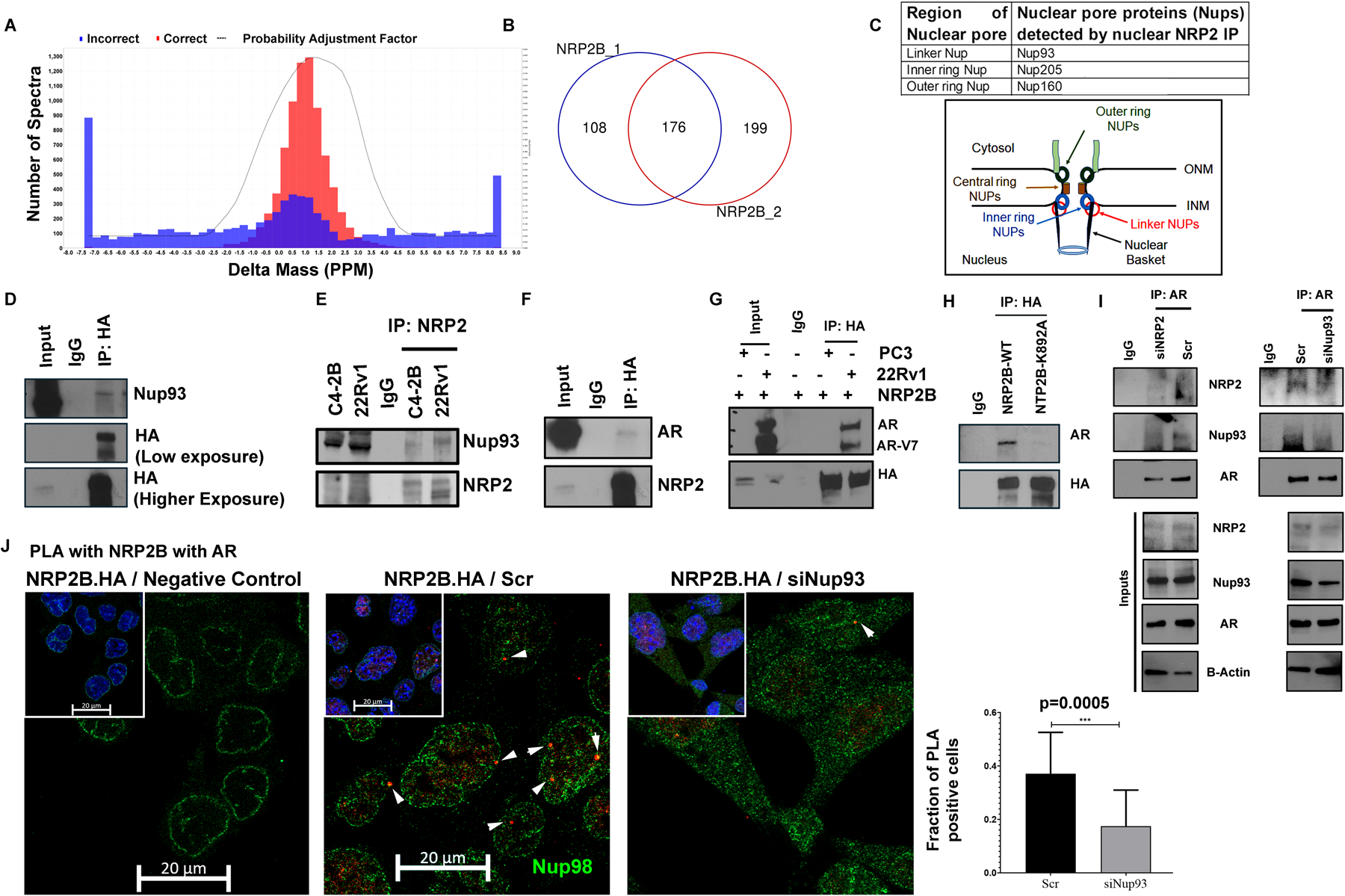
Following ectopic expression of untagged NRP2B in C4–2B cells, nuclear and post-nuclear fractions were separated, and a pull-down assay was performed on the nuclear fraction with NRP2 antibody. Mass-Spectrometry was carried out with the pull-down samples. Using the genes detected in NRP2 Mass-Spectrometry. A. Sum of all the spectra associated with NRP2B pulldown sample is determined by the spectral graph. B. Venn-diagram represent the overlapping genes identified in two independent NRP2B mass-spectrometry assay. C. Group of nuclear pore proteins (Nups) identified through NRP2B Mass-Spectrometry. Schematic diagram indicating the relative positions of Nups around the nuclear pore. D. Validation of NRP2-Nup93 interactions in C4–2B. C4–2B cells were transfected with HA-tagged NRP2B. IP was carried out with HA-antibody and immunoblots were carried out with anti-Nup93. E. Endogenously, NRP2 and Nup93 interaction was validated in C4–2B and 22Rv1 cell lines. NRP2 was pull down with NRP2-specific antibody and IB with Nup93 antibody. F. NRP2B and AR interaction was carried out in C4–2B following ectopic interaction of HA-tagged NRP2B. Pulldown was carried out with HA-antibody and IP was carried out against NRP2 and AR. G. NRP2 and AR interaction was also monitored in PC3 and 22Rv1 following ectopic expression of HA-tagged NRP2B. IB was carried out against HA and AR. H. Co-IP for AR with NRP2 was carried out with pulldown of NRP2 by HA-antibody in C4–2B cells expressing wild type and mutant K892A NRP2B isoforms. NRP2B immunoblot was carried out with HA-antibody. I. IP with AR antibody was carried out to test the interaction among AR, Nup93 and NRP2 under the presence or absence of NRP2 and Nup 93 from the nuclear fraction of LNCaP C4–2B cells. An immunoblot was performed with anti-AR, anti-Nup93 and anti-NRP2 antibody. The Co-IP was carried out in the presence of 50ng/ml VEGF-C. J. PLA was carried out to validating the NRP2-AR interaction within the nucleus. Following ectopic expression of HA-tagged NRP2B, PLA was carried out with HA and endogenous AR antibodies under the presence or absence of NUP93. As a negative control, only HA-antibody was used for PLA reaction. Arrowhead indicates the immune-reactive PLA puncta. Nucleus was counter-stained with NUP98 to demarcate the nuclear periphery. DAPI used for nuclear staining. PLA quantification was shown in Bar diagram.
We detected NRP2B in the nuclear pore regions through electron microscopy (Fig. S2E) and our mass spectrometry (MS)-based proteomics data also identified a specific group of Nups associated with NRP2B (Fig. 4C). For example, we detected 16-peptides of Nup93 through MS (Fig. S5B), indicating, NRP2B and Nup93 are in same immune-complex. We were further able to validate this complex through co-IP-IB experiment (Fig. 4D, 4E). Similar validation was performed for Nup205 (Fig. S5C). Recent studies have shown that nuclear pores in addition to its role as gatekeepers for selective import or export of proteins/RNAs across the nucleus, also serve as active sites of gene transcription. A group of Nups such as basket or linker Nups serves as scaffolds for the organization of transcriptional hubs that regulate the expression of specific genes (20, 21). These Nups (Nup 210, Nup 93, Nup 98 and Nup160) are present in the inner core of the nuclear pore and form a dynamic multi-protein complex with TFs, chromatin modulators like SAGA proteins and RNA polymerases for gene transcription (22–26). Because of our previous observation that NRP2 functions as a transcriptional regulator in advanced PCa (27), we hypothesized that NRP2, by associating with NUPs, interacts with the transcriptional machinery in PCa cells and thus regulates gene expression. Interestingly, our MS analysis revealed interaction of NRP2 with androgen receptor (AR), a transcription factor important for PCa progression (Fig. S5D). We have validated NRP2-AR interaction through coIP-IB (Fig. 4F, 4G). Using 22RV1 cell line, we found that NRP2 also interacts with AR splice variant v7 (AR-V7) (Fig. 4G). Moreover, pulling down endogenous NRP2 and immunoblot with AR-V7 specific antibody further validating NRP2-AR-V7 interaction (Fig. S5E). We further studied the association between AR and splice variant NRP2B in cancer cells. Our result indicated that wild type NRP2B can efficiently interact with AR. As expected, K892A NRP2B, which failed to translocate to INM because of its mutation at SUMOylation site, interacted less efficiently with AR (Fig. 3H).
NRP2 is required for the stabilization of the complex between Nups and transcription factors in aggressive PCa cells
We analyzed the nature of the complex formed with NRP2, NUPs and AR. Our results indicated that NRP2, AR or its splice variant AR-V7 interacted with the Nup93 (Fig. 4I, Fig. S5E, S5F). However, depletion of either NRP2 or Nup93 decreased the association between full length AR, Nup93 and NRP2 (Fig. 4I). Further, treating PCa cells either with siNRP2 (recognizes both the isoforms) or siNRP2B could decrease the nuclear AR localization suggesting the importance of NRP2-containing complex in the retention of AR in the nucleus of PCa cells (Fig. S5G). The complex between NRP2B and AR was further validated by the proximity ligation assay (PLA). The PLA puncta were formed close to nuclear periphery (shown through Nup98-positive staining) (Fig. 4J). However, depletion of Nup93 significantly decreased the association between AR and NRP2B. Our results therefore indicated that NRP2 (specifically NRP2B) recruits AR by forming a complex with Nups.
NRP2 facilitates recruitment of transcription factors to chromatin
Since, we observed NRP2 in the INM and formed a complex with Nups and AR, we wanted to investigate whether NRP2 directly interacts with nuclear chromatin or indirectly through other proteins to regulate transcription. We therefore performed a chromatin immunoprecipitation (ChIP) experiment using cell extracts of LNCaP C4–2B. NRP2-ChIP with formaldehyde as a cross-linker failed to pull down any DNA fragments. As formaldehyde forms a methylene-bridge between two macromolecules, that are ~ 2 A◦ apart, it can only crosslink molecules that are in proximity. Our result thus indicated that NRP2 is not in close enough proximity to DNA for a direct interaction. However, NRP2-ChIP following two-step crosslinking with both disuccinimidyl glutarate (DSG, with a spacer length of ~8A◦) and formaldehyde was able to reproducibly pull-down chromatin in two independent biological replicates, thus implying an indirect interaction of NRP2 with the chromatin structure (Fig. S6A, S6B). Our result therefore suggested that NRP2 indirectly interacts with DNA while being a part of the large chromatin-interacting protein complex. Further analysis revealed that NRP2 interacts with the promoter regions (2000bp upstream to 500bp downstream of transcriptional start site) of nearly 1 percent of genes- (Fig. S6C, S6D). In silico analysis using the promoter sequences following ChIP-Seq, revealed consensus transcriptional regulator binding motifs (Supplementary Fig. 6E). Using the HOCOMOCO database as an input in the TOMTOM-MEME-motif analyzer, we identified possible TFs that bind to these motifs (Fig. 5A, 5B). Interestingly, promoter consensus analysis indicated that NRP2 binding regions with chromatin overlapped with AR further validating our hypothesis that NRP2, Nup and AR forming a complex to activate AR-dependent gene expression in PCa cells (Fig. 5B). We also confirmed the interactions between NRP2 and another transcription factor, KLF4, in PCa cells by pull-down assays suggesting that NRP2 facilitates recruitment of some transcription factors to chromatin for gene transcription (Fig. S6F).
Fig. 5: Analysis of AR-NRP2 regulated gene expression:
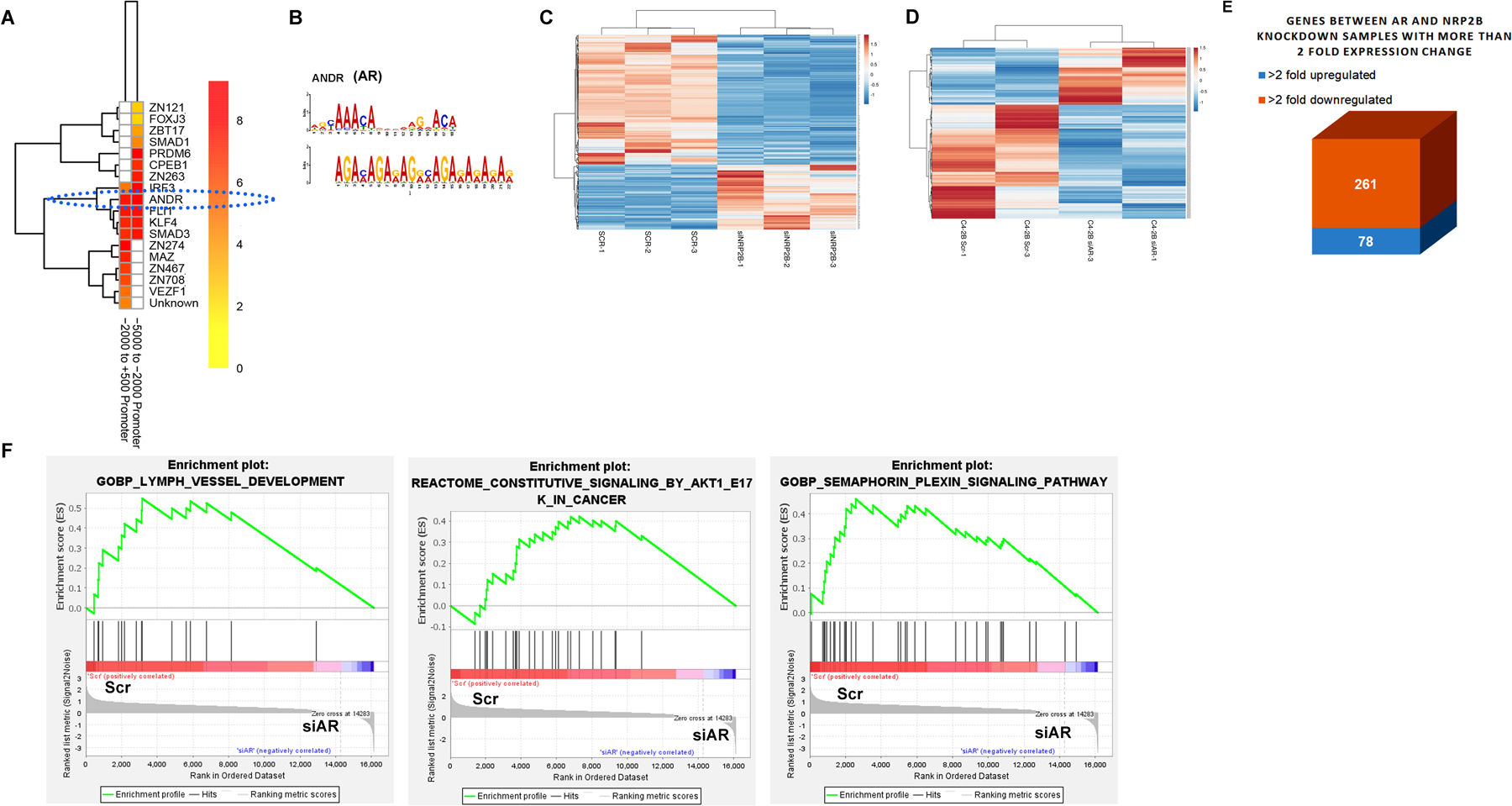
A. TFs’ binding probability within the consensus sequence derived from the NRP2-ChIP-sequencing was predicted through the heat map analysis. Consensus sequence map was generated using the indicated regions of promoter sequence from the NRP2 ChIP-Seq. B. AR motifs was analyzed using the TOMTOM-MEME-motif analyzer. C. Heat map for RNA-sequencing analysis of 200 differentially downregulated and 100 upregulated genes following depletion of NRP2B. D. Heat map of gene expression profile following the depletion of AR by siRNA. E. Box diagram representing the common up and down-regulated gene expression following AR and NRP2B depletion. F. GSEA analysis using AR-regulated genes.
NRP2 regulates AR functions
Since, AR forms complex with NRP2 and Nup93, we hypothesized that NRP2 being a part of the complex, regulates the expression of some AR-driven genes in PCa cells. Using C4–2B as a model system, we thus compared the gene expression in PCa cells by RNA sequencing following depletion of either NRP2B or AR (Fig. 5C and D) and identified the common genes that are regulated by both AR and NRP2 (Fig. 5E). Out of the 339 common genes that were jointly regulated by AR-NRP2 axis, 78 genes were upregulated, and 261 genes were downregulated following NRP2 depletion (Fig. 5E). Analysis of Gene enrichment sequence analysis (GSEA) revealed that these common gene-sets involved in lymph vessel development, AKT pathway, plexin-semaphorin signaling and others (Fig. 5F, Fig. S7A); implicating NRP2-AR complex regulates the expression of genes required for cancer progression. Interestingly, the regulation of NRP2 on the expression of AR-regulated genes seemed to be more pronounced when the PCa cells were treated with androgen (dihydroxy testosterone; DHT) (Fig. 6A). We also performed ChIP-qPCR to understand whether depletion of NRP2 can affect the binding of AR to the regulatory regions of these genes especially when the cells were treated with DHT. Our results confirmed that NRP2 depletion affected AR binding to the regulatory regions of the AR-regulated genes such as KLK3 (PSA) and KLK2 in both C4–2 and its syngeneic bone-metastatic version, C4–2B cells following androgen stimulation (Fig. 6B-6D). Efficiency of NRP2 depletion was confirmed by western blot (Fig. S7B, S7C). In presence of DHT, we also observed a significant reduction of KLK3 (PSA)-promoter activity in both C4–2 and C4–2B cells upon NRP2 depletion (Fig. 6E, 6F). Since, Nup93 is a part of the NRP2-AR complex, we tested whether depletion of Nup93 can affect the AR-transcriptional activity. Using the KLK3 promoter luciferase assay we were able to show that depletion of Nup93 affected the AR-transcriptional activity similar to the depletion of NRP2 (Fig. 6F, 6G). Our results therefore suggested the presence of NRP2-Nup93-AR complex for AR-driven gene expression in PCa cells. Interestingly, the regulation of NRP2 on AR transcription was observed in PCa cells that express high level of NRP2. LNCaP, which is an androgen dependent PCa cells, expresses low level of NRP2 (Fig. S7D). LNCaP behaves like less aggressive primary PCa cells. Deletion of NRP2 in LNCaP did not change the efficiency of AR binding to the promoters of KLK3 and KLK2 (Fig. S7E). In conclusion, these results thus suggested that NRP2 expression is increased in cancer cells during the progression of PCa, which then facilitates a specific NRP2-AR complex for the transcription of some AR-driven genes.
Fig. 6: AR-DNA interaction decreases following NRP2 depletion:
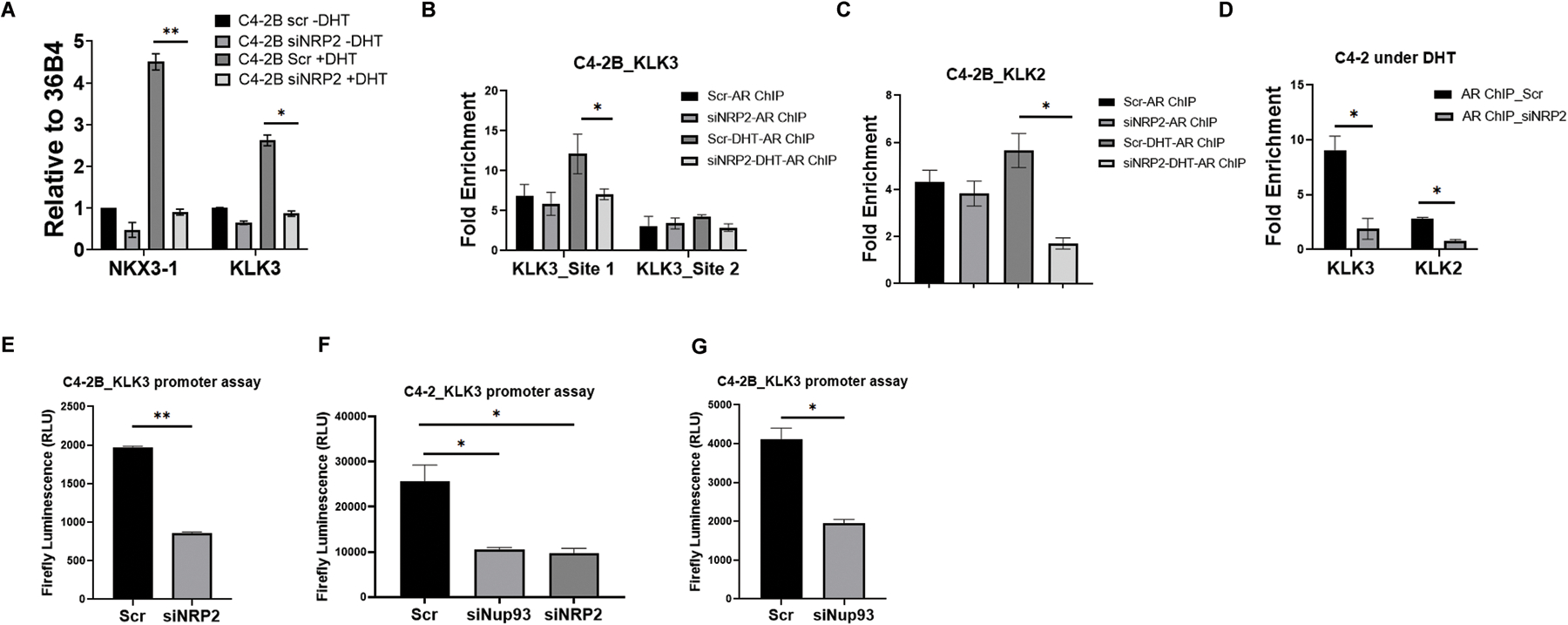
A. RT-PCR of NKX3–1 and KLK3 was carried out under steady state and DHT induced condition following the depletion of NRP2. B-D. AR ChIP was carried out following the pulldown of crosslinking DNA with AR specific antibodies under steady state and DHT induced condition under the presence and absence of NRP2. Using pulldown DNA, RT-PCR was carried out with the primer of promoter sequences of these genes. Initially, NRP2 dependence on two AR-binding sites was assayed. In B, proximal AR-binding site (Site 1) has more effect on NRP2 depletion. KLK2 binding was also analyzed using the qPCR in C4–2B as well as C4–2 as Fig C and D. E-G: KLK3 promoter luciferase assay was carried out following the depletion of NRP2 or Nup93 (in F and G). Firefly luciferase activity is represented as relative luciferase unit (RLU).
Blocking NRP2 enhances the sensitivity of enzalutamide in castration resistant metastatic prostate cancer cells:
Our results suggested that NRP2 promotes AR function in CRPC by recruiting it in a specific transcription complex that has the ability to promote several cancer promoting genes. We therefore hypothesized that blocking NRP2-mediated AR recruitment would enhance the therapeutic efficacy of enzalutamide, a second-generation anti-androgen frequently used in clinics to treat CRPC. As previously described, we blocked NRP2 function by treating LNCaP C4–2B cells with NRP2-Fc (soluble NRP2). The experiment was carried out in the presence of VEGF-C (50ng/ml) to activate nuclear translocation of NRP2 (Fig. 7A). As expected, of NRP2Fc significantly affected colony sizes compared to control. Interestingly, addition of enzalutamide and simultaneously addition of NRP2Fc significantly affected (p=0.008) the colony number as well as colony sizes compared to control VEGF-C treated condition. Thus, the colony formation assay revealed that NRP2Fc in combination with enzalutamide can enhance the anti-tumor effect of enzalutamide. Similar to the result of colony formation assay mentioned before, we observed that inactivation of NRP2 axis can affect the tumor growth in mice. We performed the experiment in subcutaneous xenograft tumor model developed by implanting inducible shNRP2 expressing C4–2B cells in the athymic nude mice (Fig. S8). For that we have implanted one million C4–2B cells carrying doxycycline inducible shNRP2 with low growth factor containing Matrigel in a 1:1 ratio. Our results indicated that depletion of NRP2 (by adding doxycycline) itself affect the tumor growth and proliferation (Fig. S8A-S8C). Moreover, depletion of NRP2 along with the addition of enzalutamide further decreased the tumor growth compared to NRP2 depletion itself (Fig S8A-S8B). Overall, our results in both colony formation assay and xenograft tumor model indicate that depletion of NRP2/inactivation of the NRP2 signaling axis can increase therapeutic efficacy of enzalutamide therapy.
Fig. 7: Inhibition of nuclear transport of NRP2 increased AR-targeted therapeutic efficacy:
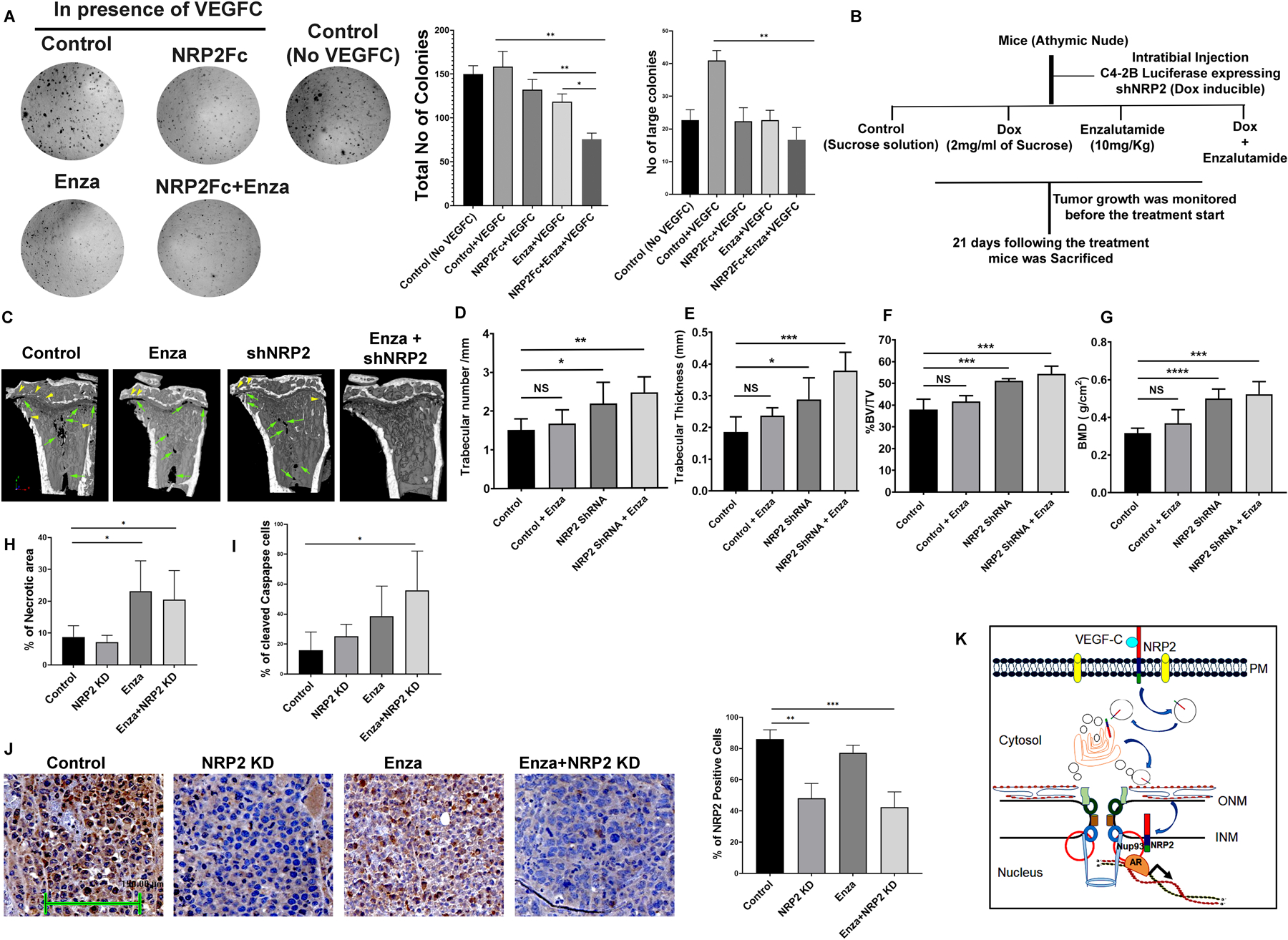
A. A colony formation assay was performed under VEGF-C treated condition to test for sensitivity to enzalutamide following the addition of NRP2Fc. The colony formation was also analyzed with VEGF-C untreated cells. Right, bar diagrams represent the number of colonies and the relative size of the colonies under the experimental conditions. B. Schematic diagram of intratibial injection. C. micro-CT images of tumor containing bone under various condition. D-G. Bone morphometric analysis in terms of trabecular number, Trabecular thickness, Bone volume (BV)/ total volume (TV) and bone mass density (BMD) of bone under different condition. H. Analysis of tumor necrosis under various condition. I. IHC analysis for Cleaved caspase staining under different experimental condition. J. IHC for NRP2 staining under different experimental condition to show the knockdown efficiency in tumor cell. K. Schematic diagram representing how NRP2 is translocated from the plasma membrane to the nucleus of the cells to regulate TF-DNA interactions.
NRP2 inhibition increases the therapeutic efficacy of enzalutamide in vivo model.
Using intratibial mice model with LNCaP C4–2B cells, we compared the tumor growth within the bone following NRP2 depletion alone or in combination with enzalutamide (Fig. 7B). Here we used the luciferase expressing stable C4–2B cell line with doxycycline inducible shNRP2 (Fig. S7F). NRP2 was depleted in tumor by adding doxycycline (2mg/ml) in the drinking water containing 2% sucrose solution. In addition, enzalutamide was injected intraperitoneally. After confirming the tumor growth by the IVIS, we randomized the mice into four groups. The control group received no enzalutamide and doxycycline. The second and the third groups were treated with only enzalutamide and doxycycline (to deplete, NRP2) respectively. The fourth group was treated with both enzalutamide and doxycycline. We performed the bone morphometric analysis using micro-CT for all the groups (Fig. 7C). Micro-CT analysis indicated that there is a significant loss of bone mineral density in control indicating tumor-induced bone degradation (a measure of tumor growth inside the bone cavity). NRP2 depletion alone and especially in combination of enzalutamide (Fig. 7C) showed less tumor-induced bone degradation compared to control or enzalutamide treated mice. Using micro-CT images, bone morphometric analysis also revealed that depletion of NRP2 in tumor cells decreased the bone loss as compared to control and enzalutamide treatment alone (Fig. 7D-7G). However, NRP2 depletion in combination with enzalutamide treatment showed an improvement of the overall bone morphology both in terms of retention of bone volume and maintenance of trabecular bone (Fig. 7D-7G) over the rest of the groups. Further, our analysis of the cancer cell cellularity by H&E staining in de-calcified bone section suggested a significant increase in necrotic zones for combination treatment compared to control (Fig. 7H, Fig. S7G). Our results also indicated that the combination therapy elicited enhanced cleaved caspase-3 expression within the cancer cells in bone (Fig. 7I, Fig. S7H). This might be due to the fact that NRP2 depletion on the tumor cells can decreased vascularity, increased stress which may result in increased necrosis and apoptosis as we seen in this experiment. Depletion of NRP2 was validated by IHC (Fig. 7J).
In summary, our results suggested that targeting NRP2 in combination with enzalutamide would be a better approach than enzalutamide alone for the treatment of PCa with bone metastasis.
Discussion
Amplification of AR signaling is one of the key features of CRPC due to increased expression of AR and generation of its constitutively active forms by alternate splicing and mutations at its ligand-binding domain (28). In addition, AR forms distinct collaborative networks with other transcriptional regulators in PCa cells to promote expression of genes necessary for cancer progression (29). Studies have reported changes in AR binding profile correlates with disease progression and is mainly caused by specific AR-transcription complex and thus divergence of AR binding profiles were detected in CRPC with respect to untreated PCa. For example, in CRPC tissue, specific sets of metabolic genes are enriched only near AR binding sites (30, 31). Targeting AR collaborating network in CRPC can therefore inhibit the cancer-specific gene expression and enhance the efficacy of second-generation AR therapy. Our current study reports that NRP2 is upregulated significantly in CRPC and aids AR-driven gene expression. We show that NRP2-AR mediated transcriptional complex is predominantly observed in CRPC and important for cancer progression and therapy resistance.
Elevated NRP2 expression correlates with poor cancer-specific survival in a PCa patient cohort (32). Our study provides a mechanistic insight into how NRP2 regulates AR driven gene transcription in PCa cells. We observed that NRP2B, a specific splice variant of NRP2, localizes in the INM (inner nuclear membrane) of aggressive PCa cells and forms a complex with nucleoporins (Nups) and AR. Nups at the nuclear pore regulate nucleocytoplasmic transport as well as are reportedly involved in DNA damage repair, chromatin silencing and transcriptional activation of genes (20, 33–35). Studies with yeast, flies, metazoan and mammalian cells indicated that transcription factors are associated with the nuclear pore complex (NPC) through Nups and are required for the enhancement of transcriptional memory as well as positioning of euchromatic regions around the pore (34, 36–40). Nups such as Nup93, Nup98, Nup153 and Nup210, regulate transcription in mammalian cells (23, 25, 41–44). For example, the interaction between Nup210 and the transcription factor, Mef2C necessitates the transcription of genes required for muscle differentiation. Importantly, downregulation of Nup210 hinders the expression of genes for myogenic differentiation without any effect on nucleocytoplasmic transport through NPCs (45). These result underscores the direct role of Nups in gene transcription. This evidence therefore indicate that certain Nups function as a scaffold to facilitate the organization of the transcription complex for gene expression. One advantage of active transcription near the nuclear pore is that the transcribed mRNA is efficiently exported to the cytoplasm for translation. This Nup-dependent transcriptional complex is highly dynamic, and its formation is dependent on various physiological or pathological contexts. In cancer, Nup-regulated gene expression is evident in acute myeloid leukemia (AML), liver cancer, and PCa (24, 46, 47). In PCa, Myc is a key driver of tumorigenesis and development of CRPC. Interestingly, the involvement of NPC in Myc-mediated transcription of specific genes was reported. Nup153 (a basket protein present in NPC) associates with Myc and facilitates its interaction with the SAGA complex for transcription of genes necessary for migration and proliferation, highlighting the importance of NPC-dependent gene transcription in PCa (41, 48). Currently, the molecular players involved in cancer cells for such a dynamic formation of transcriptional complexes and their roles in cancer progression are not well understood. In this context, our finding that NRP2B translocates to INM to stabilize the Nup -AR transcriptional complex has provided an in-depth understanding of how the dynamic transcriptional complexes with Nups can form in PCa. Since the expression of NRP2 increases with the progression of PCa, the possibility of the formation of these AR-NRP2-Nup complexes is high, which could be important for differential transcription of genes observed in aggressive PCa.
We observed a ligand dependent (VEGF-C) retrograde transport of NRP2B in the INM, whereas NRP2A moves to ONM (outer nuclear membrane) by passive diffusion. The two isoforms differ in their 93-amino acid long carboxyl-terminal region. Therefore, we concluded that the carboxyl-terminal domains of the isoforms are responsible for their specific localization and function in the nucleus. Our in-silico analysis and biochemical assays indicated that the lysine at 892 of NRP2B undergoes VEGF-C dependent SUMOylation. Mutation caused by conversion of lysine to alanine abrogated the nuclear transport of NRP2B as well as its interactions with AR. This result provided a mechanistic understanding of how NRP2B translocates differently than NRP2A and thus function distinctly. Although, VEGF-C is the most potent NRP2 ligand, we speculate other ligands such as VEGF-A or HGF which are also present in the cancer microenvironment, might activate its nuclear translocation. Further experiments are needed to validate the effects of other ligands. Recently, the non-redundant functions of individual isoforms of NRP2 were recognized. Interestingly, the expression of NRP2B in human tissue was correlated with an advanced stage of the cancer and poor progression-free survival of patients (49).
To understand the contribution of NRP2 in genome-wide AR-mediated gene transcription, RNA-sequencing following NRP2, or AR depletion was performed with PCa cells. Our results indicated that a group of genes were equally affected by both NRP2 and AR depletion. This implies that AR and NRP2 jointly regulate these genes. To validate that AR binding in these genes is affected by the presence of NRP2, we have performed the AR-ChIP following NRP2 depletion. We observed that specific AR-binding sites in chromosomes decreased when the PCa cells were induced with DHT, indicating a role of NRP2 in the binding of activated AR at specific regions of chromatin. We observed a decrease in the promoter activity (by luciferase reporter assay) and total mRNA expression (by RT-PCR) of AR-stimulated genes following NRP2B depletion and stimulation with DHT in CRPC cells. All these results thus corroborate that targeting NRP2 axis in CRPC cells affects the AR-regulated gene expression and thus improves the efficacy of AR-targeted therapy. We tested our hypothesis using in vivo intratibial model of PCa bone metastasis and show that depletion of NRP2 with enhances the sensitivity of enzalutamide and recovered the overall bone health as evidenced by increased trabecular number and volume over untreated animals. Currently, hormone sensitive PCa patients with metastatic dissemination are treated with upfront second-line AR inhibitors such as enzalutamide or abiraterone acetate. However, these patients often become resistant to these therapies. AR-splice variants are one of the causative factors for such resistance. Since NRP2 inhibition blocks both full length AR and its splice variants, simultaneous inhibition of NRP2 and AR-axis can be an effective therapeutic approach in these patients.
Overall, our results unravel a novel role of NRP2 as a transcriptional regulator of AR and indicate a stabilizing function of NRP2 in the transcriptional complexes comprised of AR and Nups in CRPC cells. NRP2 therefore either initiates or maintains sustained expression of genes important for cancer progression and therapy resistance. Targeting NRP2, especially its function at the nuclear membrane could therefore be an effective therapeutic strategy in advanced cancer. One way to achieve this goal would be by preventing its translocation to the INM. Small molecule inhibitors against NRP2 can be developed in the future to inhibit its nuclear translocation and function in enhancing AR activity.
Methods
Cell culture, plasmid constructs and transfection:
Human prostate cancer cell lines PC3 (ATCC, Manassas, VA, ATCC CRL1435), LNCaP (Manassas, VA, ATCC CRL‐1740), LNCaP C4–2, and LNCaP C4–2B (Kind gift from Prof. Allen Gao) were cultured in RPMI 1640 (Thermo Fisher Scientific, Gibco, NY, 11875093) with 10% FBS in the presence of antibiotics penicillin-streptomycin. Wild type NRP2A (Origene, RG220706 and RC220706) and NRP2B (Origene, RG210928 and RC210928) plasmids were purchased from Origene (Rockville, MD). An HA-tag was added after 21 amino acid residues from N-terminal end of NRP2 and a stop codon was also introduced at C-terminus end just after the NRP2-coding regions. Point mutation at 892 and at 856 of the plasmids were generated through site-directed mutagenesis (Agilent, CA, QuikChange II Site-Directed Mutagenesis Kit, Catalog #200523) as per the manufacturer instructions. KLK3 promoter luciferase construct was kind gift of Dr. Paramita Ghosh (UC Davis) (50). siRNA for VEGF-C (Dharmacon RNA Technologies, Chicago, IL & Qiagen, Valencia, CA, L-012071–00-0020, LQ-012071–00-0005), NRP2 (Dharmacon RNA Technologies, L-017721–00-0010, LU-017721–00-005; Qiagen, SI04995011, SI04995039, SI04995018, SI04995025), and Nup93 (Dharmacon RNA Technologies, L-020767–01-0005), AR (Dharmacon RNA Technologies, L-003400–00-0010) as well as mCherry-tagged shVEGF-C (Rockville, MD, Genecopoeia, HSH111156-mU6) were used for depletion of respective proteins. Cells were transfected using TransIT-X2 Transfection Reagent (Madison, WI, Mirus, MIR6000) (Dharmacon RNA Technologies, T-2005–02). Tissue procurements as well as tissue classification are described in Borkowetz et. al 2019.
Western blot, ELISA, Membrane protein isolation and co-immunoprecipitation (Co-IP):
Western blot and Co-IP was carried out as per our previously published materials (Dutta et. al., 2016). Nuclear and post nuclear/cytosolic fractions were separated using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78833) following the manufacturer protocol. For pulling down the SUMOylated proteins, N-ethyl maleimide was used at 10mM working conditions. Surface Biotinylation assay was performed with EZ-Link NHS-Biotin (Thermo Fisher Scientific, 20217) as per the manufacturer instruction. Proteins were analyzed by western blot. Antibodies used for this study were as followings;
NRP2 (Santa Cruz, sc-13117; R&D System AF2215; Protein Atlas, HPA054974), AR (Cell Signaling, 5153), ARv7 (Cell Signaling, 19672), HDAC1 (Cell Signaling, 34589), KLF4 (R&D System, HPA054974; Cell Signaling, 12173), PDI (Cell signaling 3501), Rho-GDI (Santa Cruz, sc-360; Cell Signaling, 2564), GAPDH (Cell Signaling, 5174), Nup98 (Cell Signaling, 2598), HA-antibody (Cell Signaling, 3724 and 2367), Lamin A/C (Cell Signaling, 4777), SMC1 (Cell Signaling, 4802), SUMO1 (Cell Signaling, 4930), Histone H3 (Cell Signaling, 5192), Cleaved Caspase-3 (Cell Signaling, 9579), NRP2 IHC specific antibody (Sigma-Aldrich HPA039980), Nup93 (Bethyl Laboratories Inc, A303–979A), HRP-conjugated secondary antibody, conformation specific (Cell Signaling, 3678), Nup205 (Bethyl Laboratories Inc, 303–935A), Nup188 (Bethyl Laboratories Inc, A302–323A).
Plasmid modification and site-directed modification:
A hemagglutinin (HA) epitope tag was introduced downstream of the predicted signal sequence cleavage site by a three-step modification involving site-directed mutagenesis. An EcoRI fragment of 181 bp covering the translation start of an expression vector containing the NRP2 coding sequence (NRP2 Human Tagged ORF Clone, OriGene Technologies, Herford, Germany) was cloned in pBluescript KSII(–). Site-directed mutagenesis following the manufacturer’s protocol (Agilent, CA, QuikChange II Site-Directed Mutagenesis Kit, 200523) and using complementary primers (forward: 5’-TGAGAGGCCAAGCGGTCCCACCGTGCGGA-3’; reverse: 5’-TCCGCACGGTGGGACCGGTTGGCCTCTCA-3’) generated an AgeI restriction site, which was used to insert a 5’-phosphorylated complementary DNA oligo hybrid (forward: 5’-CCGGAATATCCATATGATGTTCCAGATTATGCT-3’; reverse: 5’-CCGGAGCATAATCTGGAACATCATATGGATATT-3’). HA-tagged NRP2 expression vectors were generated by replacement with the modified EcoRI fragment. Coding sequence determination by the Sanger method verified the predicted amino acid modification of NRP2 of the mature protein (R21G22Q23P24EYPYDVPDYAPVP26P27C28G29…). Recovery of the NRP2 carboxyterminal sequences in myc/DDK-tagged NRP2 expression vectors (NRP2A, NRP2B, OriGene) modified with HA-tag was performed by PCR-based insertion of stop codons at the native sites. 3’-terminal fragments of NRP2 expression vectors were PCR-modified (common forward: 5’-CACCATGGAGTTCCAGTACCAGGC-3’; NRP2A reverse: 5’-CGAGCGGCCGCGTACGCTATGCCTC-3’; NRP2B reverse: 5’-CGAGCGGCCGCGTACGCTAGCAGTG-3’). Antisense primers were designed to introduce a TAG stop codon at the native site, followed by a NotI restriction site. After cloning into pCR4-TOPO (TOPO TA cloning kit, Life Technologies, Darmstadt, Germany), NotI restriction was used to generate NRP2A and NRP2B expression vectors by replacing parental expression vectors with the modified fragments. The Sanger method was used to verify NRP2 isoform coding sequences.
Using the site directed mutagenesis kit, NRP2B-Del-Cyto construct was produced by the primers specified in the primer table. HA-tagged NRP2B (NM_201267.2) mentioned above was used as template. At the 856aa position of NRP2B (NM_201267.2), a STOP codon (TAA instead of TAC) has been inserted to get the cytosolic-fragment deleted membrane-bound NRP2 fragment (NRP2∆C). Similarly, using the same HA-tagged NRP2B as template, a point mutation (Lysine to Alanine) at aa892 was introduced using the primer specified in the primer table.
Confocal and electron microscopy:
Confocal microscopy was carried out as per our published protocol (Dutta et. al., 2016). Cells were grown on poly-D/L lysine (BD Biosciences, Sparks, MD) coated cover slips before fixation for confocal analysis. All the confocal images were captured either with a Zeiss LSM 800 with Airyscan or with a Zeiss ELYRA PS.1 Super Resolution Microscope and data were analyzed and processed with Zeiss Zen Blue software.
For nuclear localization of NRP2B analysis under various experimental condition, C4–2B cells were transfected with either NRP2A-HA or NRP2B-HA plasmid. After 24hrs of transfection, the cells were serum starved for overnight. Following starvation most of the NRP2 were in the membrane. NRP2B or NRP2A localization were chased with various experimental treatment condition for 1hrs under the following reagents VEGF-C (50ng/ml, R&D System, 752-VC-025), NRP2Fc (100ng/ml, R&D System, 2215-N2–025), SEMA3F (100ng/ml, R&D System, 9878-S3–025), Brefeldin A (0.5µM, Sigma, B6542). After 1hrs of chase, cells were fixed with 4% paraformaldehyde for 20mins at room temperature. Following fixation, cells were washed and block with blocking solution (1% BSA+0.2% Saponin in PBS). Cells were stained with respective primary antibodies (NRP2 and Nup98) for overnight at 4◦C. Next, cells were washed with 1% Tween-20 containing PBS and incubated with secondary antibodies in blocking buffer for 1hr at room temperature. After thorough washing (10mins x3 times), cells were counterstained with DAPI and mount for images.
For electron microscopy, cells were cultured in 35mm dishes (MatTek, Ashland, MA, P35G-2–14-CGRD). Before staining, cells were rinse with 0.1 M phosphate buffer pre-warmed to 37°C. Then cells were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer pH7.4 containing 7.5% sucrose for 30 min at room temperature. Dishes were washed with 0.1 M phosphate buffer (3× 5min each) followed by blocking with aldehydes with 50 mM glycine in 0.1 M phosphate buffer for 15 min. Then cells were permeabilized with 0.25% saponin in 0.1 M phosphate for 30 minutes at room temperature. Cells were rinsed with 0.02% saponin in 0.1 M phosphate buffer (2× 5 min each). Blocking was carried out with 0.1 M phosphate buffer containing 0.02% saponin, 1% BSA, 0.2% fish gelatin and 5% goat serum in 0.1 M phosphate buffer (blocking buffer) for 30 minutes. Then cells were incubated with no primary antibody (control) or with primary antibodies overnight at 4°C. Primary antibodies were made up in blocking buffer (about 100–200 µl /MatTek dish). Cells were kept in the refrigerator overnight. The next day, dishes were rinsed with 0.1M phosphate buffer containing 0.02% saponin and 1% BSA (4× 5 min each) and Incubated with appropriate fluoronanogold secondary antibodies (Nanoprobes, Inc. Yaphank, NY, Cat. No: 7504 and 7502) in blocking buffer for 2 h at room temperature. After secondary antibody incubation, cells were further rinsed with buffer containing 0.02% saponin and 1% BSA (3× 5min each), then 0.02% saponin (3× 5min each) and finally just 0.1 M phosphate buffer (3× 5min each). Finally, cells were again incubated with fixative (4% paraformaldehyde with 0.05–0.1% glutaraldehyde in 0.1 M phosphate buffer pH 7.4) for 15mins and then washed with 50mM Glycine in 0.1 M phosphate buffer 3 times for a total of 5 min. Next gold enhancement was carried out and TEM images was taken in FEI Tecnai G2 Spirit Biotwin microscope.
ChIP-Seq:
ChIP was performed as per the manufacturer protocol using the ChIP-kit from MAGnify Chromatin Immunoprecipitation System (Thermo Fisher Scientific) describe elsewhere (Dutta et. al., 2016). Crosslinking was carried out with both disuccinimidyl glutarate (DSG) as well as formaldehyde. DSG was added to 107 cells to a final concentration of 2mM and incubated for 45 minutes at RT on a rotating condition. At the end of fixation, samples were centrifudge at 1500 rpm for 10 minutes. Cell pellet was washed with 20ml PBS1X/1mM PMSF and further centrifuged at 1500 rpm for 5 minutes. After twice wash, cells were re-suspended in 10ml PBS1X/1mM PMSF. Next, 270µl of second cross-linker in terms of 37% formaldehyde was added in that mixture and incubated for 15 minutes at RT. Next, we followed the kit protocol as described. Using anti-NRP2 and anti-AR antibodies, Libraries were prepared using the New England Biolabs NEB Next Ultra II DNA Library Prep Kit for Illumina and sequenced by the UNMC Sequencing Core Facility using an Illumina NextSeq 500 Genome Analyzer. Initial raw sequence files were processed as follows. Adaptor sequences and low quality (Phred score < 20) ends were trimmed from sequences using Trim Galore software package. Resulting fastq files were aligned to the human genome (GRCh38/hg38) using the sequence aligner software Bowtie2. The software package Picard routine Mark Duplicates was used to remove sequence duplications. Peak calling (FDR adjusted p-value ≤0.05) was determined using MACS2 software. The UCSC genome browser was used for visualization.
mRNA isolation and qRT-PCR:
RNA was extracted from cells using RNeasy mini kit (Qiagen, 74104) and quality was analyzed using Bio-analyzer. cDNA was prepared from 1µg of mRNA following Transcriptor First Strand Synthesis Kit protocol (Roche, Indianapolis, IN, 04379012001). Quantitative real-time PCR (RT-PCR) was carried out with Power SYBR®Green master mix (Life Technologies, Grand island, NY, 4368706) as per our previously published protocol (51) using the primer sets shown in the table below.
RNA-Seq.
RNA-seq was carried out from the control vs siNRP2 and control vs siAR in triplicate samples. RNA was extracted using the RNeasy Mini Kit (Qiagen, Germantown, MD) and RNA integrity quality was analyzed using the Agilent Bio-analyzer. A paired end read (2× 50 bp) sequencing run of RNA libraries were carried out with Illumina NextSeq 500. Sequences were aligned to human reference genome hg38. Data analysis was performed with the help of the Bioinformatics Core at UNMC. RNA abundance was estimated with feature Counts from the Sub-read package version 1.6.3. Downstream analyses were performed with the DESEQ2 R package version 1.18.1. A Principal component analysis (PCA) was performed using https://biit.cs.ut.ee/clustvis/. Differentially expressed genes (DEGs) were identified by pairwise comparisons with the DESEQ2 package (v.1.12.3). Genes were retained as differentially expressed when the fold-change (FC) was >2 or <−2.
Mass-Spectrometry analysis:
Mass-Spectrometry was carried out on LNCaP C4–2B cells over-expressing NRP2B. After nuclear and post nuclear fraction separation, IP with the NRP2 antibody was carried out while rotating at 4◦C. Pull-down was carried out with magnetic beads. Following the extractions of the samples, SDS page was run for 3 min. Bands were excised from the SDS gel followed by in gel digestion with trypsin. Mass-Spectrometry analysis was carried out through LC-MS/MS using Thermo Q-Exactive-HF mass spectrometer and a nano RSLC Ultimate 3000 from Dionex. Spectra was processed using Mascot (Matrix Science, London, UK; version 2.6.1) and were subjected to a cutoff of 1% false discovery rate. Spectra was processed by MODIRO ver.1.1 (Protagen, Germany) software (from Proteomics & Metabolomics Facility of the Nebraska Center for Biotechnology at University of Nebraska, Lincoln).
Colony formation assay:
LNCaP C4–2B cells were mixed in 0.3% Noble agar (in RPMI medium supplemented with 10% FBS) at 37◦C and immediately plated at 5000 cells/well on the top of a solidified agar layer of the 6-well plates (0.6% Noble agar in the same growth medium). Media was supplemented every fourth day along with the respective drug and inhibitors (50ng/ml VEGF-C, 100nM NRP2Fc, 10nM Enzalutamide). After 21 days, colonies were stained with crystal violet solution (0.04% crystal violet-2% ethanol in PBS) for 1 hr followed by washing with PBS and then was photographed.
Proximity Ligation Assay and Immunofluorescence:
At first, HA-tagged NRP2B was ectopically expressed in NRP2 depleted cells. Following 24 hrs ectopic expression, Nup93 was depleted from the cells for the next 48 hrs. At the end of the 72hrs, cells were fixed with 4% paraformaldehyde for 20 min followed by treatment with 10% NP40 for 2–3mins. Proximity ligation assay was performed using the Duolink kit (Sigma-Aldrich, DUO92101–1KT), according to manufacturer’s protocol, using red reagent (excitation 554 nm, emission 594 nm). After completion of the protocol, Immunofluorescence was carried out with Nup98 antibodies for 1 hr and additional secondary antibodiy for 30mins (Alexa 488). DAPI was used for counter staining. Images were captured using a Zeiss confocal microscope, with a 63X objective.
Luciferase assay:
Promoter luciferase assay for AR-binding/activity was determine in C4–2, C4–2B and LNCaP cells transfected with 2 µg of pGL3-hPSA-luc. Using firefly luciferase assay kit (Promega), luminescence was determined. After 48hrs of transfection, cells were lysed and the using the equal amount of protein from various condition, luciferase activity was analyzed according to the manufacturer protocol.
Intratibial Mouse model:
In vivo mouse model of prostate cancer bone metastasis Animal study was carried in accordance with the UNMC IACUC guidelines. To assess the importance of NRP2 inhibition in combination of AR-inhibition by Enzalutamide; GFP-Luciferase and stable shNRP2 expressing C4–2B cells (50000 cells/mice in per 20μL HBSS) was intratibially injected into male athymic nude mice (8-weeks old Crl:NU(NCr)-Foxn1nu mice from Charles River, cancer cells injected in left tibia, 5 mice per group). PBS was injected as sham control into the right tibia. Appearance of tumor was monitored by IVIS imaging after 5 days of injection and once the tumor appeared, mice were randomized into four groups containing 5 mice each (achieve 80% power to detect a mean difference in dead cells of 20% using a two sided t-test). The control and enzalutamide treated group received 2% sucrose in water. The NRP2 knockdown (NRP2 KD) was carried out by adding doxycycline in drinking water containing 2% sucrose solution (doxycycline dose 2mg/ml)(52). Enzalutamide added intraperitoneally at 10mg/kg body weight. Enzalutamide was added for 5 days a week for 3 weeks and doxycycline in drinking water was also added 5 consecutive days followed by 2 days normal water for 3 weeks. At the end of three weeks treatment, mice were sacrificed and the tumor containing bones were fixed in 10% formalin. For IHC, bones were further decalcified using 0.5M EDTA for 2 weeks with intermittent replacement of fresh solution. Paraffin embedded decalcified bone section was stained with H&E, Cleaved caspase 3 and NRP2. Tumor necrotic area was analyzed through Ventana Image Viewer and using Graphpad Prism a bar diagrams were plotted. For micro-CT, only formalin fixed bones were used to take images.
In-vivo prostate cancer subcutaneous mouse model:
Subcutaneous tumor analysis was conducted in accordance with the UNMC IACUC guidelines. To analyze whether depletion of NRP2 can inhibit the tumor growth, we have developed stable doxycycline inducible shNRP2 expressing clones of C4–2B, where NRP2 can be depleted inducibly by addition of doxycycline (Dox). C4–2B shNRP2 cells (1×106 cells) were implanted into the right flank of 8 weeks athymic nude mice. Once the tumors become palpable (~30mm3), animals were randomized into four groups (n= 5). One group acts as a control (no treatment, except sucrose water). The second group received doxycycline whereas third group receive enzalutamide as mentioned above by i.p. The last group received both doxycycline as well as enzalutamide. Tumors were regularly monitored and tumor size was measured over the period of time until sacrifice (29days from day of C4–2B injection). Tumor volume was calculated by using the formula: ½ ab2 (53).
Micro-CT analysis:
Formalin-fixed bones were scanned through the X-ray micro-tomography system (Skyscan 1172, Bruker, Kontich, Belgium, at 55 kV and 181 μA, resolution 8.89 μm, exposure time 815 ms, and aluminum filter 0.5 mm-thick). To generate composite 3D images, raw data were reconstructed using NRecon software. Bone quality-control was analyzed by using the region of interest (ROI) around the growth plate. The bone morphometric analysis was carried out by using mean bone volume (BV), bone volume/tissue volume (BV/TV), trabecular thickness and bone mineral density (BMD).
Primer sequence used for either RT-PCR or for site directed mutagenesis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer 5Ꞌ to 3Ꞌ |
|---|---|
| 36B4_F | ATGCAGCAGATCCGCATGT |
| 36B4_R | TCATGGTGTTCTTGCCCATCA |
| ChIP KLK3_1F: (Proximal) | TTGTGCCACTGGTGAGAAAC |
| ChIP KLK3_1R | TCAGAGACAAAGGCTGAGCA |
| ChIP KLK3_2F | GCAGTCTAGGTGGATGCTGT |
| ChIP KLK3_2R | GGTTTGCAAGTTGTCCCAGTA |
| ChIP KLK2_F | TCTCTGTGAGCAAAGGGATG |
| ChIP KLK2_R | TCTTAGGCCCTTTCAAGCTG |
| Hum PSA_F | TTGTCTTCCTCACCCTGTCC |
| Hum PSA_R | GGGAATGCTTCTCGCACTC |
| Hum NKX3.1_F | GCCAAGAACCTCAAGCTCAC |
| Hum NKX3.1_R | AGGAGAGCTGCTTTCGCTTA |
| Hum NRP2_F | GTGAAGAGCGAAGAGACAACCA |
| Hum NRP2_R | GCAGTTCTCCCCACACTCTG |
| Hum NRP2A_F | ATCTCGGCTTTTGCAGGTGAGA |
| Hum NRP2A_R | ATTGCTCCAGTCCACCTCGTAT |
| Hum NRP2B_F | TCGGCTTTTGCAGGTGAGAA |
| Hum NRP2B_R | CACCGTGTCCACTGTGGGCT |
| NRP2B_Del Cyto_F | CTGGTGCTCCACTAACACCGGTTCCGCTATGCGGCCAA |
| NRP2B_Del Cyto_R | TTGGCCGCATAGCGGAACCGGTGTTAGTGGAGCACCAG |
| NRP2B_K892A_F | CACCCTAACCATTGCGCTAGAGCAAGACCGTGGCT |
| NRP2B_K892A_R | AGCCACGGTCTTGCTCTAGCGCAATGGTTAGGGTG |
Acknowledgments:
The authors thank all stuff members of the Advanced Microscopy Core and Genomics Core Facility of UNMC. We also extend our sincere thanks to the UNMC Bio-informatics and Bio-statistician Core for their support to analyze the data. The raw data has been deposited in Gene Expression Omnibus with the accession number GSE205150.
Funding:
This work was supported by grants for S. Dutta (1R21CA241234-01, NE-LB506, Lageschulte Fund), K. Datta (R01CA182435, R01CA239343, DoD W81XWH2110628), M. H. Muders and L.H. Hofbauer (DFG project number 273676790), and M.H. Muders (DFG project number 416001651). Michael Muders is funded by the Rudolf-Becker-Foundation for his professorship. The construction of the prostate cancer tissue microarray was funded by the DFG Forschergruppe-1586 SKELMET to Lorenz C Hofbauer and S. Fuessel.
Footnotes
Conflicts of Interest: The authors are declaring no conflict of interest.
References:
- 1.Powers E, Karachaliou GS, Kao C, Harrison MR, Hoimes CJ, George DJ, et al. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J Hematol Oncol 2020;13(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt KT, Huitema ADR, Chau CH, Figg WD. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat Rev Urol 2021;18(4):209–26. [DOI] [PubMed] [Google Scholar]
- 3.Imamura Y, Sadar MD. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int J Urol 2016;23(8):654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tucci M, Zichi C, Buttigliero C, Vignani F, Scagliotti GV, Di Maio M. Enzalutamide-resistant castration-resistant prostate cancer: challenges and solutions. Onco Targets Ther 2018;11:7353–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giacinti S, Poti G, Roberto M, Macrini S, Bassanelli M, F DIP, et al. Molecular Basis of Drug Resistance and Insights for New Treatment Approaches in mCRPC. Anticancer Res 2018;38(11):6029–39. [DOI] [PubMed] [Google Scholar]
- 6.Roumiguie M, Paoletti X, Neuzillet Y, Mathieu R, Vincendeau S, Kleinclauss F, et al. Apalutamide, darolutamide and enzalutamide in nonmetastatic castration-resistant prostate cancer: a meta-analysis. Future Oncol 2021;17(14):1811–23. [DOI] [PubMed] [Google Scholar]
- 7.Tucci M, Leone G, Buttigliero C, Zichi C, RF DIS, Pignataro D, et al. Hormonal treatment and quality of life of prostate cancer patients: new evidence. Minerva Urol Nefrol 2018;70(2):144–51. [DOI] [PubMed] [Google Scholar]
- 8.Vander Ark A, Cao J, Li X. Mechanisms and Approaches for Overcoming Enzalutamide Resistance in Prostate Cancer. Front Oncol 2018;8:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Y, Wei T, Ye Z, Orme JJ, Lin D, Sheng H, et al. A noncanonical AR addiction drives enzalutamide resistance in prostate cancer. Nature communications 2021;12(1):1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schweizer MT, Haugk K, McKiernan JS, Gulati R, Cheng HH, Maes JL, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PloS one 2018;13(6):e0198389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shafran JS, Andrieu GP, Gyorffy B, Denis GV. BRD4 Regulates Metastatic Potential of Castration-Resistant Prostate Cancer through AHNAK. Mol Cancer Res 2019;17(8):1627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510(7504):278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coleman DJ, Gao L, Schwartzman J, Korkola JE, Sampson D, Derrick DS, et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Scientific reports 2019;9(1):3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Xu M, Kao CY, Tsai SY, Tsai MJ. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J Clin Invest 2020;130(4):1782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sulpice E, Plouet J, Berge M, Allanic D, Tobelem G, Merkulova-Rainon T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood 2008;111(4):2036–45. [DOI] [PubMed] [Google Scholar]
- 16.Parker MW, Linkugel AD, Goel HL, Wu T, Mercurio AM, Vander Kooi CW. Structural basis for VEGF-C binding to neuropilin-2 and sequestration by a soluble splice form. Structure 2015;23(4):677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossignol M, Gagnon ML, Klagsbrun M. Genomic organization of human neuropilin-1 and neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics 2000;70(2):211–22. [DOI] [PubMed] [Google Scholar]
- 18.Fricker M, Hollinshead M, White N, Vaux D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. The Journal of cell biology 1997;136(3):531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drozdz MM, Vaux DJ. Shared mechanisms in physiological and pathological nucleoplasmic reticulum formation. Nucleus 2017;8(1):34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibarra A, Hetzer MW. Nuclear pore proteins and the control of genome functions. Genes Dev 2015;29(4):337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Navarro S, Fischer T, Luo MJ, Antunez O, Brettschneider S, Lechner J, et al. Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell 2004;116(1):75–86. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Oliver E, Garcia-Molinero V, Rodriguez-Navarro S. mRNA export and gene expression: the SAGA-TREX-2 connection. Biochimica et biophysica acta 2012;1819(6):555–65. [DOI] [PubMed] [Google Scholar]
- 23.Labade AS, Karmodiya K, Sengupta K. HOXA repression is mediated by nucleoporin Nup93 assisted by its interactors Nup188 and Nup205. Epigenetics & chromatin 2016;9:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sump B, Brickner JH. Nup98 regulation of histone methylation promotes normal gene expression and may drive leukemogenesis. Genes & development 2017;31(22):2201–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franks TM, Hetzer MW. The role of Nup98 in transcription regulation in healthy and diseased cells. Trends in cell biology 2013;23(3):112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang Y, Franks TM, Marchetto MC, Gage FH, Hetzer MW. Dynamic association of NUP98 with the human genome. PLoS Genet 2013;9(2):e1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutta S, Roy S, Polavaram NS, Baretton GB, Muders MH, Batra S, et al. NRP2 transcriptionally regulates its downstream effector WDFY1. Scientific reports 2016;6:23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocrine-related cancer 2016;23(12):T179–T97. [DOI] [PubMed] [Google Scholar]
- 29.Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer cell 2013;23(1):35–47. [DOI] [PubMed] [Google Scholar]
- 30.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal 2011;30(13):2719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan KN, Avery VM, Carrasco-Pozo C. Metabolic Roles of Androgen Receptor and Tip60 in Androgen-Dependent Prostate Cancer. Int J Mol Sci 2020;21(18): 6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borkowetz A, Froehner M, Rauner M, Conrad S, Erdmann K, Mayr T, et al. Neuropilin-2 Is An Independent Prognostic Factor For Shorter Cancer-Specific Survival In Patients With Acinar Adenocarcinoma Of The Prostate. International journal of cancer 2019. [DOI] [PubMed]
- 33.Palancade B, Liu X, Garcia-Rubio M, Aguilera A, Zhao X, Doye V. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Molecular biology of the cell 2007;18(8):2912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruben GJ, Kirkland JG, MacDonough T, Chen M, Dubey RN, Gartenberg MR, et al. Nucleoporin mediated nuclear positioning and silencing of HMR. PloS one 2011;6(7):e21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radman-Livaja M, Ruben G, Weiner A, Friedman N, Kamakaka R, Rando OJ. Dynamics of Sir3 spreading in budding yeast: secondary recruitment sites and euchromatic localization. The EMBO journal 2011;30(6):1012–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn TM, Capelson M. Nuclear Pore Proteins in Regulation of Chromatin State. Cells 2019;8(11): 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhn TM, Pascual-Garcia P, Gozalo A, Little SC, Capelson M. Chromatin targeting of nuclear pore proteins induces chromatin decondensation. The Journal of cell biology 2019;218(9):2945–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raices M, D’Angelo MA. Nuclear pore complexes and regulation of gene expression. Current opinion in cell biology 2017;46:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ptak C, Aitchison JD, Wozniak RW. The multifunctional nuclear pore complex: a platform for controlling gene expression. Current opinion in cell biology 2014;28:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dieppois G, Stutz F. Connecting the transcription site to the nuclear pore: a multi-tether process that regulates gene expression. Journal of cell science 2010;123(Pt 12):1989–99. [DOI] [PubMed] [Google Scholar]
- 41.Ibarra A, Benner C, Tyagi S, Cool J, Hetzer MW. Nucleoporin-mediated regulation of cell identity genes. Genes & development 2016;30(20):2253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitazawa T, Rijli FM. Nuclear Pore Protein Meets Transcription Factor in Neural Fate. Neuron. 2017;96(2):259–61. [DOI] [PubMed] [Google Scholar]
- 43.Gomez-Cavazos JS, Hetzer MW. The nucleoporin gp210/Nup210 controls muscle differentiation by regulating nuclear envelope/ER homeostasis. The Journal of cell biology 2015;208(6):671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Angelo MA. Nuclear pore complexes as hubs for gene regulation. Nucleus 2018;9(1):142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raices M, Bukata L, Sakuma S, Borlido J, Hernandez LS, Hart DO, et al. Nuclear Pores Regulate Muscle Development and Maintenance by Assembling a Localized Mef2C Complex. Developmental cell 2017;41(5):540–54 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holzer K, Ori A, Cooke A, Dauch D, Drucker E, Riemenschneider P, et al. Nucleoporin Nup155 is part of the p53 network in liver cancer. Nature communications 2019;10(1):2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Bravo V, Pippa R, Song WM, Carceles-Cordon M, Dominguez-Andres A, Fujiwara N, et al. Nuclear Pores Promote Lethal Prostate Cancer by Increasing POM121-Driven E2F1, MYC, and AR Nuclear Import. Cell 2018;174(5):1200–15 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su Y, Pelz C, Huang T, Torkenczy K, Wang X, Cherry A, et al. Post-translational modification localizes MYC to the nuclear pore basket to regulate a subset of target genes involved in cellular responses to environmental signals. Genes & development 2018;32(21–22):1398–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gemmill RM, Nasarre P, Nair-Menon J, Cappuzzo F, Landi L, D’Incecco A, et al. The neuropilin 2 isoform NRP2b uniquely supports TGFbeta-mediated progression in lung cancer. Science signaling 2017;10(462): eaag0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savoy RM, Chen L, Siddiqui S, Melgoza FU, Durbin-Johnson B, Drake C, et al. Transcription of Nrdp1 by the androgen receptor is regulated by nuclear filamin A in prostate cancer. Endocrine-related cancer 2015;22(3):369–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stanton MJ, Dutta S, Zhang H, Polavaram NS, Leontovich AA, Honscheid P, et al. Autophagy control by the VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer research 2013;73(1):160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polavaram NS, Dutta S, Islam R, Bag AK, Roy S, Poitz D, et al. Tumor- and osteoclast-derived NRP2 in prostate cancer bone metastases. Bone Res 2021;9(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kersemans V, Cornelissen B, Allen PD, Beech JS, Smart SC. Subcutaneous tumor volume measurement in the awake, manually restrained mouse using MRI. J Magn Reson Imaging 2013;37(6):1499–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.