

Abstract
Background:
To identify fine specificity anti-citrullinated protein antibodies (ACPA) associated with incident rheumatoid arthritis-associated interstitial lung disease (RA-ILD).
Methods:
This nested case-control study within the Brigham RA Sequential Study matched incident RA-ILD cases to RA-noILD controls on time of blood collection, age, sex, RA duration, and rheumatoid factor status. A multiplex assay measured ACPA and anti-native protein antibodies from stored serum prior to RA-ILD onset. Logistic regression models calculated odds ratios (OR) with 95% confidence intervals (CI) for RA-ILD, adjusting for prospectively-collected covariates. We estimated optimism-corrected area under the curves (AUC) using internal validation. Model coefficients generated a risk score for RA-ILD.
Findings:
We analyzed 84 incident RA-ILD cases (mean age 67 years, 77% female, 90% White) and 233 RA-noILD controls (mean age 66 years, 80% female, 94% White). We identified six fine specificity antibodies that were associated with RA-ILD. The antibody isotypes and targeted proteins were: IgA2 to citrullinated histone 4 (OR 0.08 per log-transformed unit, 95% CI 0.03–0.22), IgA2 to citrullinated histone 2A (OR 4.03, 95% CI 2.03–8.00), IgG to cyclic citrullinated filaggrin (OR 3.47, 95% CI 1.71–7.01), IgA2 to native cyclic histone 2A (OR 5.52, 95% CI 2.38–12.78), IgA2 to native histone 2A (OR 4.60, 95% CI 2.18–9.74), and IgG to native cyclic filaggrin (OR 2.53, 95% CI 1.47–4.34). These six antibodies predicted RA-ILD risk better than all clinical factors combined (optimism-corrected AUC=0·84 versus 0·73). We developed a risk score for RA-ILD combining these antibodies with the clinical factors (smoking, disease activity, glucocorticoid use, obesity). At 50% predicted RA-ILD probability, the risk scores both without (score=2·6) and with (score=5·9) biomarkers achieved specificity ≥93% for RA-ILD.
Interpretation:
Specific ACPA and anti-native protein antibodies improve RA-ILD prediction. These findings implicate synovial protein antibodies in the pathogenesis of RA-ILD and suggest clinical utility in predicting RA-ILD once validated in external studies.
Funding:
National Institutes of Health
INTRODUCTION
Approximately 5–10% of individuals with rheumatoid arthritis (RA) have clinically significant RA-associated interstitial lung disease (RA-ILD).1 Having RA-ILD confers two to ten times the odds of mortality compared to RA patients without ILD,2 with median survival ranging between three to eight years after diagnosis in previous studies.1,2 As a result, it is one of the leading causes of premature death among RA patients.1 Improving prediction of RA-ILD is therefore critical to allow for earlier diagnosis and treatment prior to worsening lung damage and death.
Biomarkers may hold the key to predicting RA-ILD development. For example, rheumatoid factor (RF) and anti-cyclic citrullinated peptide (anti-CCP) have been long associated with RA-ILD.3 More recently, the gain-of-function MUC5B promoter variant was found to increase risk of RA-ILD by three-fold.4 Recent studies have also shown that anti-modified peptide antibodies (AMPAs)5 and fine specificity anti-citrullinated protein antibodies (ACPA) such as anti-citrullinated alpha enolase (anti-CEP1),6 anti-citrullinated heat shock protein 90 (anti-HSP90),7 and anti-fibrinogen8 are associated with RA-ILD. Identifying and testing additional fine specificity ACPAs may be important, as anti-CCP testing does not encompass all ACPAs. Furthermore, certain fine specificity ACPA could be more associated with RA-ILD than others. One cross-sectional study suggested an association between the number of fine-specificity ACPA and subclinical RA-ILD.3 However, prospective studies examining each fine specificity ACPA separately in RA-ILD are needed.
In addition to identifying novel biomarkers, another critical need is to develop an accurate RA-ILD risk prediction score using such markers. Our previous work showed that combining established clinical risk factors achieves only a modest area under the curve (AUC) in predicting RA-ILD.9,10 However, adding biomarkers such as matrix metalloproteinase 7, pulmonary and activation-regulated chemokine, and surfactant protein D significantly improved RA-ILD prediction.10 Juge et al. also improved the AUC for RA-ILD prediction to 0·82 by including the MUC5B promoter variant, though this study detected presence of subclinical RA-ILD (rather than future development of RA-ILD) and used cross sectional data.11 Another risk score recently developed for subclinical RA-ILD with cross-sectional data highlighted the urgent need for both additional biomarkers and prospective studies to improve prediction.12 Therefore, an RA-ILD risk score incorporating more biomarkers based on prospective data collection and clinical RA-ILD (for improved clinical utility) is needed.
To address these two gaps, we leveraged a prospective cohort of incident RA-ILD cases. We aimed (1) to identify fine specificity ACPA or anti-native protein antibodies associated with incident RA-ILD and (2) to develop an RA-ILD risk score for incident RA-ILD. We hypothesized that several specific ACPAs such as anti-citrullinated fibrinogen8 would be associated with increased risk for incident RA-ILD beyond the effects of the MUC5B promotor variant, and that an RA-ILD risk score incorporating such biomarkers would improve prediction of this deadly disease.
METHODS
Study population and design
This nested case-control study took place within the prospective Brigham Rheumatoid Arthritis Sequential Study (BRASS). BRASS is an ongoing, prospective registry of over 1,500 adult patients with RA containing semi-annual collection of RA characteristics, physician/patient-reported measures, blood, and DNA samples.13 BRASS recruited participants from Brigham and Women’s Hospital outpatient clinics from March 2003 to present, with an overall participation rate of approximately 18%. All participants met 1987 American College of Rheumatology (ACR) or 2010 ACR/EULAR criteria for RA as verified by a rheumatologist. Criteria for inclusion in this substudy consisted of availability of banked serum at a BRASS study visit for the ACPA assay. We defined the index date of RA-ILD onset as the initial chest CT scan showing RA-ILD, or matched date for controls. For each participant, we also chose the earliest study visit where blood was obtained prior to the index date for both antibody measurement and covariate assessment. This study received institutional review board approval (2016P000226) and complies with the Declaration of Helsinki.
RA-ILD cases
We identified RA-ILD cases occurring between March 1, 2003 and April 14, 2016. RA-ILD cases all had incident RA-ILD as outlined previously.9 We excluded patients with prevalent RA-ILD at BRASS baseline. All incident RA-ILD cases after baseline had blood banked for these studies. In short, we defined RA-ILD as interstitial changes on clinically-indicated chest CT after BRASS study enrollment, confirmed by two attending thoracic radiologists and one attending pulmonologist using a validated sequential reading method.14 Prior studies confirmed this definition to be clinically significant, with over 75% of cases having additional evaluations or follow-up for RA-ILD.15 We also assessed for RA-ILD subtypes, severity, and collected data on clinically-performed pulmonary function tests near time of RA-ILD onset.
RA-noILD controls
We matched each RA-ILD case with up to three RA-noILD controls on duration between study visit/blood date and index date (±six months), age (±five years), sex, RA duration (±five years), and RF status (positive vs. negative). The pool of eligible controls never had RA-ILD throughout the entire follow-up. We required RA-noILD controls to have banked blood available at the study visit/blood date. To ensure controls did not have ILD, we also required absence of ICD-9 codes for ILD (515, 516·30, 516·31), absence of patient-reported ILD, and absence of physician-reported history of ILD, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, bronchiectasis, bronchiolitis obliterans-organizing pneumonia, drug-induced pneumonitis, or tuberculosis.
Research multiplex ACPA assay
We determined antibody levels using a research multiplex flow cytometry assay. Previous work identified key fine specificity proteins and peptides from RA synovium and cartilage and combined them into a validated, bead-based multiplex assay.16 In brief, protein antigens were conjugated to beads using the Bio-Plex multiplex assay platform (Bio-Rad Laboratories, Hercules, CA, USA) and analyzed using the Luminex 200 (Luminex, Austin, TX, USA). Each plate used established samples with no, low, medium, or high reactivity as internal controls. he specific amino acid sequences, citrullinated residues, and reproducibility for the targeted antigens for each antibody have been previously published17. The assay has previously passed validation standards that included blinded remeasurements from the same samples18.
We performed this research multiplex assay for all RA-ILD cases and controls in a single batch at Stanford University (Palo Alto, CA). Serum from each participant was added to the bead mix, and antibody reactivity was measured in raw fluorescent intensity units. Citrullinated antigens tested for this study included beta-actin, biglycan, collagen type II, enolase-1-alpha, histone 2A, histone 2B, histone 4, fibrinogen A, fibrinogen B, fibronectin, filaggrin, and vimentin. Native (non-citrullinated) antigens tested included histone 2A, histone 2B, fibrinogen B, filaggrin, tenascin C1, tenascin C5, type II collagen, and vimentin. Some proteins had multiple epitope targets, and we also studied three immunoglobulin (Ig) isotypes for each target. Thus, we measured a total of 78 fine specificity ACPA and 39 anti-native protein antibodies. The assay did not measure antibodies to some native antigens due to previous studies showing no appreciable reactivity to the native antigens to either patients with RA or healthy controls17. A previous study using this assay found that 2% of healthy controls were ACPA-positive19. We also examined possible correlations between anti-CCP titer and each fine specificity ACPA and found modest correlations.
MUC5B promoter variant
We assessed MUC5B promoter variant status (rs35705950 G>T) from genetic testing performed through the Mass General Brigham Biobank using the Illumina genotyping array, as previously detailed.20 For participants not included or tested in the Mass General Brigham Biobank but with DNA available for genotyping, we directly measured the SNP using a custom TaqMan assay performed at Mass General Brigham HealthCare Dana-Farber/Harvard Cancer Center.
Covariates
We obtained covariates known to be associated with RA-ILD9 at the time of earliest available study visit with blood draw before index date. These included age (continuous), sex (male vs. female, self-reported), RA duration (continuous), RF positivity (present vs. absent), anti-CCP positivity (present vs. absent), race and ethnicity (White non-Hispanic vs. other), education (less than university vs. more), body mass index (BMI, continuous), smoking status (never, past, current), smoking pack-years (continuous), C-reactive protein (CRP, continuous), disease activity score with 28 joints and CRP (DAS28-CRP, continuous), multi-dimensional health assessment questionnaire (MDHAQ, continuous), bone erosions (present vs. absent), rheumatoid nodules (present vs. absent), biologic disease modifying anti-rheumatic drug (bDMARD) use (never, past, current), methotrexate use (never, past, current), and glucocorticoid use (never, past, current). We also categorized RF and anti-CCP as negative (≤ upper limit of normal [ULN], low-positive (>1 to 3x ULN), and high-positive (>3x ULN).
Of note, if covariates were not available at the date of the earliest blood draw, we took them from the next closest BRASS study visit that preceded the index date. All covariates were measured for research rather than clinical care. Third-generation anti-CCP measurements were performed using validated enzyme-linked immunosorbent assays from Inova Diagnostics (San Diego, CA) and Euro Diagnostica (Minneapolis, MN) with upper limit of normal of 19·9. RF measurements were performed by an immunoturbidimetric technique on the Cobas Integra 700 analyzer (Roche Diagnostics; Indianapolis, IN), using reagents and calibrators from Roche with upper limit of normal of 15.
Statistical analysis
For demographic characteristics, we compared normally distributed continuous variables using t-tests, non-normally distributed continuous variables using the Wilcoxon rank sum tests, and categorical variables using the chi-squared or Fisher’s exact tests (for variables with low cell sizes). When reporting medians, we also included interquartile ranges (IQR).
To identify specific ACPA and anti-native protein antibodies associated with RA-ILD, we log-transformed these biomarkers due to skewed distributions. We then performed multivariable conditional logistic regression models for each log-transformed unit antibody to obtain adjusted odds ratios (aOR) for RA-ILD. These models adjusted for all covariates statistically significant in univariate analyses including BMI, smoking pack-years, DAS28-CRP, MDHAQ, rheumatoid nodules, bDMARD use, methotrexate use, and glucocorticoid use. We also included MUC5B genotype to determine the associations of these novel biomarkers beyond MUC5B, the best known genetic risk factor for RA-ILD. Due to the number of antibodies tested, we adjusted p-values for multiple comparisons using Benjamini and Hochberg’s procedure with a false discovery rate (FDR) of 5%. To assess the added benefit of significant biomarkers (FDR p<0·05) on RA-ILD prediction, we fit unconditional logistic regression including both the predictors of interest and the matching factors to obtain receiver operator characteristic (ROC) curves and associated AUC. We also plotted ROC curves and reported AUCs for each antibody identified in the association analyses as well as two groups by citrullinated and native proteins. We performed internal validation using a bootstrapping optimism correction21 and then reported the optimism-corrected AUC. In brief, we performed 1000 bootstrap replications to obtain resamples with AUC and 95% CI metrics. Since RA-ILD subtypes may have different pathogenesis, we also investigated antibody associations for usual interstitial pneumonia (UIP)/fibrotic non-specific interstitial pneumonia (NSIP) and cellular NSIP subtypes since these were most frequent. We also performed analyses stratified by seropositive RA, seronegative RA, anti-CCP-positive RA, and anti-CCP-negative RA. To investigate potential for cross-reactivity of antibodies against citrullinated and non-citrullinated antibodies, we performed a sensitivity analysis subtracting results of non-citrullinated from citrullinated biomarkers for each specific antigen.
To develop an RA-ILD risk score, we again performed multivariable conditional logistic regression models for RA-ILD. However, this time we included only the top predictors that were independently associated with RA-ILD. To facilitate easy clinical application, we dichotomized clinical predictors and divided antibody biomarkers into tertiles based on the distribution of RA-noILD controls. We dichotomized each antibody as the highest tertile vs. the lowest two tertiles (reference group). For biomarkers inversely associated with RA-ILD, we considered the lowest tertile as the risk group, and the top two tertiles as the reference group. We also constructed a score without biomarkers for clinical encounters where biomarkers are not available. While the MUC5B promoter variant is not available clinically, it is already an established RA-ILD risk factor. Thus, we included the “Established RA-ILD risk factors” model that also included this. Unconditional logistic regression was used for AUCs since some of the model components were also matching factors. We compared the AUCs of individual models using DeLong’s statistic. Once constructed, we calculated predicted probabilities of developing RA-ILD via unconditional logistic regression with offset function, using the full BRASS cohort as the reference population (n=1,581).22 We chose RA-ILD probability cutoffs of 30% (expected high sensitivity), 50%, and 80% (expected high specificity) as different thresholds that may be implemented clinically.
There were no missing data for any of the biomarkers or covariates except for 15 RA-ILD cases and 2 RA-noILD controls missing MUC5B genotype. We imputed these values using multiple imputation with all covariates and case/control status as predictors. For the antibody biomarker analysis, FDR-corrected p-value of <0·05 was considered statistically significant. The threshold for statistical significance in the rest of the study was a two-sided p<0·05. We pre-specified all analyses in our protocol and performed them using SAS version 9·4 (SAS Institute Inc., Cary, NC).
Role of the funding source
This work was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant award K23 AR069688 to Dr. Sparks). The funders had no role in the study design; collection, analysis, or interpretation of data; writing the report; or decision to publish this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard University, its affiliated academic health care centers, or the National Institutes of Health.
RESULTS
Demographics and RA-ILD case characteristics
We identified 84 incident RA-ILD cases (mean age 67, 77% female) and matched them to 233 RA-noILD controls (Table 1). Blood was obtained at study visits median of 1·5 years (IQR 0·6,2·5) before index date of RA-ILD onset for cases and 1·9 years (IQR 1·6,5·6) before the assigned index date for RA-noILD controls (p=0·84). In univariate analyses, RA-ILD cases had higher BMI, smoking history, disease activity, rheumatoid nodules, and current glucocorticoid use, along with lower current bDMARD and methotrexate use than RA-noILD controls (Table 1). There were no differences in RF or anti-CCP status/levels between cases and controls. RA-ILD subtypes and clinical characteristics are included in Supplemental Table S1.
Table 1. Characteristics of 84 incident RA-ILD cases and 233 RA-noILD controls at the time of blood draw.
| Characteristic | Number (%) |
||
|---|---|---|---|
| RA-ILD Cases (N=84) | RA Controls (N=233) | p-value | |
| Years from Blood to Index/Matched Date, median | |||
| (IQR) | 1·5 (0·6,2·5) | 1·9 (1·6,5·6) | 0·84* |
| Age in years, mean (SD) | 67 (10) | 66 (11) | 0·41* |
| Female Sex | 65 (77) | 186 (80) | 0·64* |
| RA Duration in years, mean (SD) | 20 (12) | 20 (11) | 0·99* |
| RF Positive | 73 (87) | 195 (84) | 0·48* |
| RF level | 0·53 | ||
| Negative (≤ULN) | 11 (13%) | 38 (16%) | |
| Low Positive (>1 to 3x ULN) | 21 (25%) | 67 (29%) | |
| High Positive (>3x ULN) | 52 (62%) | 128 (55%) | |
| Anti-CCP Positive | 70 (83) | 190 (82) | 0·71 |
| Anti-CCP level | 0·86 | ||
| Negative (≤ULN) | 14 (17%) | 43 (18%) | |
| Low Positive (>1 to 3x ULN) | 8 (11%) | 21 (9%) | |
| High Positive (>3x ULN) | 61 (73%) | 169 (73%) | |
| White, non-Hispanic | 76 (90) | 219 (94) | 0·28 |
| Education Less than University | 30 (36) | 98 (42) | 0·31 |
| Body Mass Index in kg/m2, mean (SD) | 28 (6) | 27 (6) | 0·03 |
| Smoking Status | 0·03 | ||
| Never | 33 (39) | 124 (53) | |
| Past | 43 (51) | 100 (43) | |
| Current | 8 (10) | 9 (4) | |
| Smoking Pack-Years, median (IQR) | 7 (0,31) | 0 (0,10) | <0·001 |
| Disease Activity Score-28-CRP, mean (SD) | 3·6 (1·6) | 3·0 (1·3) | 0·001 |
| C-Reactive Protein in mg/L, median (IQR) | 3·6 (1·3,9·9) | 2·1 (0·8,5·1) | 0·003 |
| MDHAQ Score, mean (SD) | 0·8 (0·6) | 0·5 (0·5) | <0·001 |
| Bone Erosions | 49 (58) | 144 (62) | 0·58 |
| Rheumatoid Nodules | 42 (50) | 80 (34) | 0·01 |
| Biologic DMARD Use | 0·03 | ||
| Never | 29 (35) | 76 (33) | |
| Past | 19 (23) | 27 (12) | |
| Current | 36 (43) | 130 (56) | |
| Methotrexate Use | 0·03 | ||
| Never | 18 (21) | 24 (10) | |
| Past | 32 (38) | 93 (40) | |
| Current | 34 (41) | 116 (50) | |
| Glucocorticoid Use | 0·007 | ||
| Never | 5 (6) | 31 (13) | |
| Past | 42 (50) | 140 (60) | |
| Current | 37 (44) | 62 (27) | |
| Copies of MUC5B Promoter Variant (i.e. Genotype) | 0·16 | ||
| None (GG) | 48/69 (70) | 184/231 (80) | |
| One (GT) | 19/69 (28) | 43/231 (19) | |
| Two (TT) | 2/69 (2.9) | 4/231 (1.7) | |
BRASS = Brigham Rheumatoid Arthritis Sequential Study, CRP = C-reactive protein, DMARD = disease-modifying anti-rheumatic drug, ILD = interstitial lung disease, IQR = interquartile range, MDHAQ = multi-dimensional health assessment questionnaire, RA = rheumatoid arthritis, RF = rheumatoid factor, SD = standard deviation, ULN = upper limit of normal.
matched factor
Fine specificity ACPA and anti-native protein antibodies and RA-ILD risk
After adjusting for the above covariates plus MUC5B genotype, three fine specificity ACPA were associated with RA-ILD risk at FDR of 5% (Table 2). The top two were IgA2 antibodies to citrullinated histone antigens (histone 4 33–48 aOR 0.08 per log-transformed unit, 95% CI 0·03–0·22; histone 2A/a-2 1–20 aOR 4·03, 95% CI 2·03–8·00). The third was IgG to citrullinated cyclic filaggrin 48–65 (aOR 3·47, 95% CI 1·71–7·01), though IgA antibodies to citrullinated cyclic filaggrin also showed an association (Table 2). The association between the remaining 68 ACPAs and RA-ILD are shown in Supplementary Table S1.
Table 2. Top associations between autoantibodies to citrullinated and native antigens and incident RA-ILD in 84 BRASS RA-ILD cases and 233 RA-noILD controls.
| Targeted antigen* | Adjusted** OR (95% CI) | FDR p-value |
|---|---|---|
| Citrullinated | ||
| H4 33–48 cit 39 (IgA2) | 0·08 (0·03,0·22) | <0·001 |
| H2A/a-2 1–20 cit (IgA2) | 4·03 (2·03,8·00) | 0·003 |
| Filaggrin 48–65 cit2 cyclic (IgG) | 3·47 (1·71,7·01) | 0·014 |
| FibrinogenB 36–52 cit (IgA2) | 0·37 (0·16,0·86) | 0·397 |
| Clusterin 231–250 cit cyclic (IgG) | 1·27 (1·03,1·56) | 0·397 |
| H4 33–48 cit 39 40 (IgG) | 1·25 (1·02,1·53) | 0·397 |
| Filaggrin 48–65 cit2 cyclic (IgA2) | 5.04 (1·10,23·1) | 0·419 |
| H2A/a 1–20 cit cyclic (IgA2) | 1·60 (1·00,2·57) | 0·455 |
| Fibronectin cit 1035 1036 (IgG) | 1·22 (0·99,1·50) | 0·455 |
| Filaggrin 48–65 cit2 cyclic (IgA1) | 1·87 (0·98,3·59) | 0·455 |
| Non-Citrullinated (Native) | ||
| H2A/a 1–20 cyclic (IgA2) | 5·52 (2·38,12·78) | 0·001 |
| H2A/a-2 1–20 (IgA2) | 4·60 (2·18,9·74) | 0·001 |
| Filaggrin 48–65 cyclic (IgG) | 2·53 (1·47,4·34) | 0·010 |
| H2A/a 1–20 cyclic (IgG) | 1·91 (1·18,3·10) | 0·088 |
| H2A/a-2 1–20 (IgG) | 1·86 (1·14,3·05) | 0·105 |
| tenascin C 1 (IgG) | 0·69 (0·50,0·95) | 0·148 |
| tenascin C 1 (IgA1) | 0·73 (0·54,0·98) | 0·195 |
| Vimentin 58–77 cyclic (IgG) | 1·51 (1·02,2·24) | 0·195 |
| tenascin C 1 (IgA2) | 0·50 (0·22,1·15) | 0·455 |
| Vimentin 58–77 cyclic (IgA2) | 1·75 (0·84,3·63) | 0·530 |
BMI = body mass index, BRASS = Brigham Rheumatoid Arthritis Sequential Study, CI = confidence interval, cit = citrullinated, CRP = C-reactive protein, DAS = disease activity score, bDMARD = biologic disease-modifying anti-rheumatic drug, FDR = false discovery rate, H = histone, Ig = immunoglobulin, ILD = interstitial lung disease, MDHAQ = multi-dimensional health assessment questionnaire, OR = odds ratio, RA = rheumatoid arthritis, RF = rheumatoid factor
Additional results are shown in Supplemental Tables 2 and 3.
Presented by level of significance within each category.
Per log-transformed unit. Bold values indicate FDR p<0.05 (and nominal p<0.001).
Adjusting for significant covariates including BMI, smoking pack-years, DAS28-CRP, MDHAQ, rheumatoid nodules, bDMARD use, methotrexate use, glucocorticoid use, and MUC5B genotype.
Three anti-native (non-citrullinated) antibodies were also associated with RA-ILD (Table 2). Once again, the top two were IgA2 antibodies to histone antigens (histone 2A/a 1–20 cyclic aOR 5·52, 95% CI 2·38–12·78; histone 2A/a-2 1–20 aOR 4·60, 95% CI 2·18–9·74), with the third representing IgG to cyclic filaggrin 48–65. Antibodies to these three antigens (histone 2A/a 1–20 cyclic, histone 2A/a-2 1–20, and cyclic filaggrin 48–65) were associated with RA-ILD in both their native and citrullinated forms. The association between remaining 29 antibodies to native antigens are shown in Supplemental Table S2-S3. Unadjusted associations are shown in Supplemental Table S4-S5. Results of autoantibodies associations for the cellular NSIP (n=44) and UIP/fibrotic NSIP (n=29) RA-ILD subtypes are shown in Supplemental Tables S6-S9). Results of ROC curves for individual antibodies and those grouped by citrullinated or native proteins are shown in Supplemental Figures S1-S2. Results of analyses stratified by seropositive/seronegative RA and anti-CCP-positive/anti-CCP-negative RA are shown in Supplemental Tables S10-S17. Results of the analysis subtracting results of non-citrullinated from citrullinated biomarkers are shown in Supplemental Table S18.
Improved RA-ILD prediction using biomarkers
Figure 1 shows ROC curves for predicting incident RA-ILD risk when adding the six novel fine specificity protein antibody biomarkers (three ACPA and three anti-native protein antibodies) described above. We then calculated the AUC for these curves, including AUC corrected for optimism (Table 3). Confidence intervals between the “clinical factors” model and “all factors combined” model were non-overlapping. Indeed, combining all clinical factors including demographic, lifestyle, and RA factors (including anti-CCP) together achieved an optimism-corrected AUC of 0·73 for RA-ILD risk. After adding biomarkers including MUC5B and the six novel biomarkers from this study, optimism-corrected AUC increased to 0·84. However, nearly all the predictive capacity came from the six novel biomarkers alone (AUC=0·84) (Table 3). Comparisons between AUCs of individual models are shown in Supplemental Table S19.
Figure 1. Receiver operating characteristic curves for incident RA-ILD risk among 84 cases and 233 RA-noILD controls in BRASS, adding novel biomarkers and MUC5B to clinical factors.
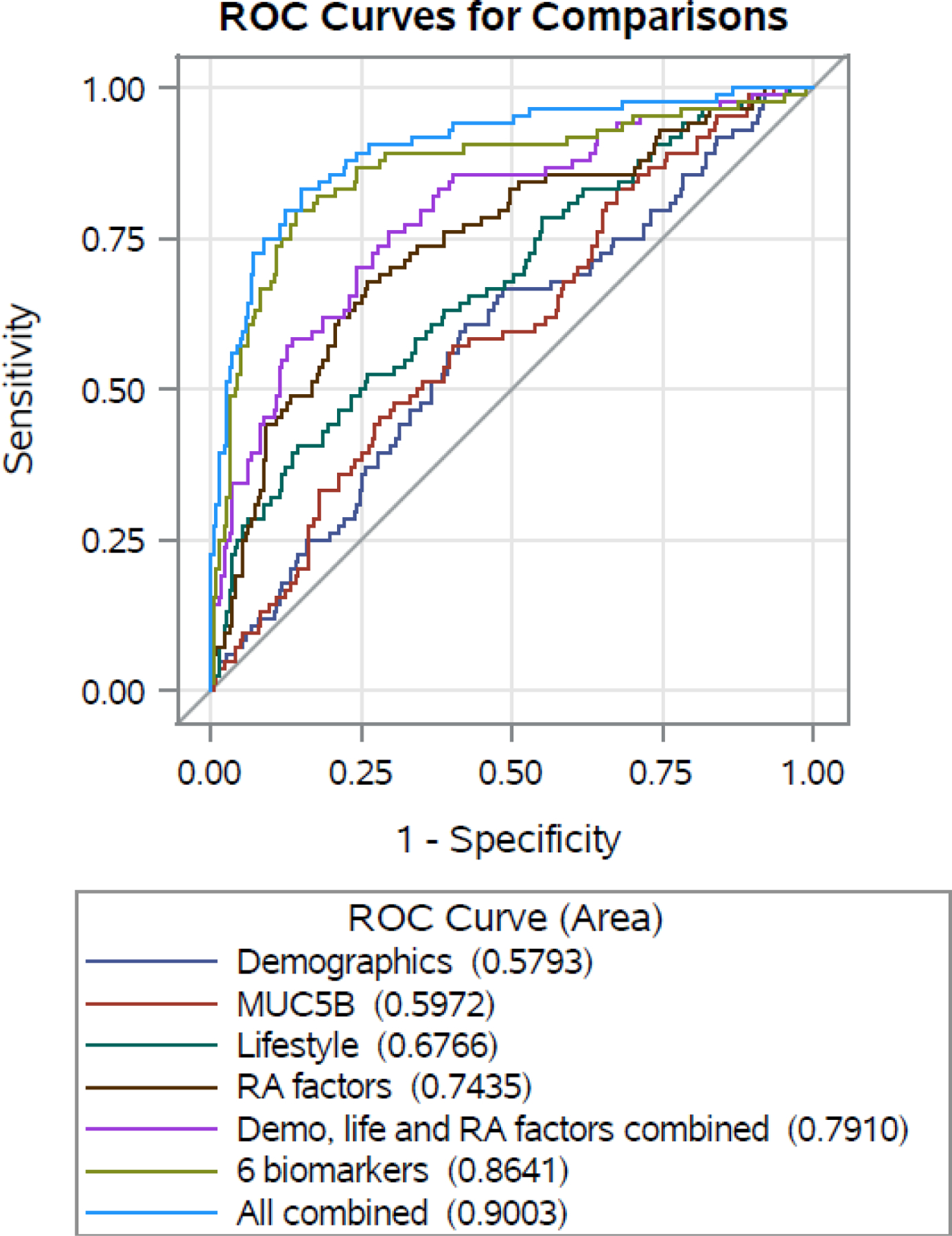
ROC = receiver operating characteristic, RA = rheumatoid arthritis. Area under curve is noted in parentheses next to each model name. Demographic = age, race and ethnicity, education; Lifestyle = BMI, smoking pack-years; RA factors = RA duration, RF, anti-CCP, bone erosion, rheumatoid nodules, methotrexate use, glucocorticoid use, biologic DMARD use, DAS28-CRP, MDHAQ; Clinical factors combined = demographic + lifestyle + RA factors; Novel biomarkers = H4 33–48 cit 39 (IgA2), H2A/a-2 1–20 cit (IgA2), Filaggrin 48–65 cit2 cyclic (IgG), H2A/a 1–20 cyclic (IgA2), H2A/a-2 1–20 (IgA2), Filaggrin 48–65 cyclic (IgG)
Table 3. Optimism-corrected area under the curve for predicting RA-ILD risk by adding novel biomarkers and the MUC5B promoter variant to clinical factors.
| Model* | Apparent AUC (95% CI) | ||
|---|---|---|---|
| Demographic | 0·58 (0·51,0·65) | 0·07 | 0·51 (0·45, 0·57) |
| MUC5B promoter variant | 0·60 (0·53,0·67) | 0·05 | 0·55 (0·48, 0·61) |
| Lifestyle | 0·68 (0·62,0·75) | 0·04 | 0·64 (0·58, 0·70) |
| RA factors | 0·74 (0·68,0·81) | 0·06 | 0·68 (0·62, 0·74) |
| Clinical factors combined | 0·79 (0·73,0·85) | 0·06 | 0·73 (0·67, 0·78) |
| Established RA-ILD factors | 0·80 (0·74, 0·85) | 0·07 | 0·73 (0·67, 0·78) |
| Novel biomarkers (N=6) | 0·86 (0·81,0·92) | 0·03 | 0·84 (0·79, 0·89) |
| All factors combined | 0·90 (0·85,0·94) | 0·06 | 0·84 (0·80, 0·88) |
AUC = area under curve, BMI = body mass index, CI = confidence interval, cit = citrullinated, CRP = C-reactive protein, DAS = disease activity score, DMARD = disease-modifying anti-rheumatic drug, H = histone, Ig = immunoglobulin, ILD = interstitial lung disease, MDHAQ = multi-dimensional health assessment questionnaire, OR = odds ratio, RA = rheumatoid arthritis, RF = rheumatoid factor
Demographic = age, race and ethnicity, education; Lifestyle = BMI, smoking pack-years; RA factors = RA duration, RF, anti-CCP, bone erosion, rheumatoid nodules, methotrexate use, glucocorticoid use, biologic DMARD use, DAS28-CRP, MDHAQ; Clinical factors combined = demographic + lifestyle + RA factors; Established RA-ILD factors = demographic + lifestyle + RA factors + MUC5B; Novel biomarkers = H4 33–48 cit 39 (IgA2), H2A/a-2 1–20 cit (IgA2), Filaggrin 48–65 cit2 cyclic (IgG), H2A/a 1–20 cyclic (IgA2), H2A/a-2 1–20 (IgA2), Filaggrin 48–65 cyclic (IgG)
RA-ILD risk score
Using the above novel biomarkers plus known clinical risk factors for RA-ILD, we developed a risk score for developing RA-ILD (Table 4). Although we showed above that biomarkers alone are predictive of RA-ILD risk, we also included an RA-ILD risk score without biomarkers to facilitate use in clinical settings where such testing is not possible (Table 4). Model coefficients and odds ratios used for risk score development are shown in Supplemental Table S20. Applying the clinical score (without biomarkers) in the BRASS dataset, RA-ILD cases scored median 1·6 (IQR 1·2,4·3) and RA-noILD controls scored 1·0 (IQR 0·0,1·2). Applying the biomarker score to the BRASS dataset, RA-ILD cases scored median 6·8 (IQR 5·7,7·7) and RA-noILD controls scored 3·0 (IQR 1·3,4·3), indicating good separation between cases and controls.
Table 4. Risk score for developing RA-ILD (with and without biomarkers).
| Score Points | ||
|---|---|---|
| Variable* | Without biomarkers | With biomarkers |
| Smoking pack-years ≥30 | 1·9 | 2·3 |
| DAS28-CRP ≥3.2 | 1·2 | 1·2 |
| Current glucocorticoid use | 1·0 | 1·2 |
| BMI ≥30 kg/m2 | 0·4 | 0·8 |
| Filaggrin 48–65 cit 2 cyclic (IgG), highest tertile | 2·0 | |
| H4 33–48 cit 39 (IgA2), lowest tertile | 1·9 | |
| H2A/a-2 1–20 (IgA2), highest tertile | 1·8 | |
| MUC5B promoter variant present | 0·1 | |
BMI = body mass index, CI = confidence interval, CRP = C-reactive protein, Ig = immunoglobulin, ILD = interstitial lung disease, OR = odds ratio, RA = rheumatoid arthritis
Intended to be recorded as of time of calculation
Finally, we calculated predicted probability of developing RA-ILD by risk score level (Table 5). The score with biomarkers showed a higher sensitivity and specificity for RA-ILD than the score without biomarkers for all predicted probabilities of RA-ILD (Table 5). However, both scores showed high specificity (≥93%) for developing RA-ILD at the threshold of 50% predicted probability of RA-ILD (2·6 for the clinical score and 5·9 for the biomarker score, Table 5).
Table 5. Performance characteristics of incident RA-ILD risk score by thresholds of predicted probability.
| Without biomarkers |
With biomarkers |
|||||
|---|---|---|---|---|---|---|
| Score | Sensitivity | Specificity | Score | Sensitivity | Specificity | |
| 30% probability of RA-ILD | 1·6 | 0·62 | 0·79 | 5·0 | 0·83 | 0·87 |
| 50% probability of RA-ILD | 2·6 | 0·25 | 0·93 | 5·9 | 0·67 | 0·94 |
| 80% probability of RA-ILD | 4·1 | 0·07 | 0·99 | 7.3 | 0.34 | 0.98 |
BRASS = Brigham Rheumatoid Arthritis Sequential Study, ILD = interstitial lung disease, RA = rheumatoid arthritis
DISCUSSSION
This prospective study of incident RA-ILD identified several fine specificity ACPA and anti-native protein antibodies associated with developing RA-ILD. For example, antibodies against cyclic filaggrin and histone 2A were associated with higher risk of RA-ILD. In contrast, antibodies against histone 4 were inversely associated with RA-ILD, suggesting ACPA profiles may identify distinct clinical trajectories for patients with RA. Many of the associations were IgA, suggesting a link between mucosal autoantibody production and RA-ILD pathogenesis. These biomarkers dramatically improved RA-ILD prediction beyond clinical factors (including anti-CCP) and MUC5B. Second, we used these novel biomarkers to develop a risk score for incident RA-ILD, which showed high sensitivity and specificity at several thresholds. These findings fill an urgent need of predicting future development of clinically-significant RA-ILD, thus allowing for earlier recognition and treatment of this common and deadly disease among patients with RA.
Several fine specificity ACPA were associated with incident RA-ILD risk. While one prior study showed that the number of fine specificity ACPA are associated with RA-ILD, it exclusively examined subclinical ILD in a cross-sectional manner and did not test each ACPA separately.3 This study showed that the strength of association varied significantly for each fine specificity ACPA, with association for antibodies to histone 4, histone 2A, and filaggrin. Anti-citrullinated histone is not part of the traditional anti-CCP assay. Nevertheless, citrullinated histones are associated with RA23 and are arthritogenic in mice.24 The protective effect seen for the antibody to citrullinated histone 4 may result from a genetic polymorphism in the peptidylarginine deaminase 4 (PADI4) gene responsible for citrullinating histones, leading to decreased anti-native and anti-citrullinated histone antibodies as well as higher risk of RA.25 Meanwhile, the association between anti-citrullinated filaggrin and RA-ILD also has biological plausibility given its sensitivity and specificity for RA diagnosis.26 Although filaggrin is a protein that binds keratin fibers in epithelial cells primarily in skin, it is also expressed in the salivary glands, explaining higher anti-citrullinated filaggrin levels in individuals with subgingival P gingivalis and thereby its connection to RA.27 Genetic variants resulting in filaggrin deficiency are also associated with asthma,28 a disease recently implicated in RA risk.29 Furthermore, filaggrin-deficient mice have spontaneous pulmonary inflammation.30 As hypothesized and suggested previously,8 anti-citrullinated fibrinogen was also associated with RA-ILD. Autophagy generates citrullinated peptides in human synoviocytes and has also been implicated in idiopathic pulmonary fibrosis31,32. Shortened telomeres are associated with higher risk of fibrotic lung diseases including RA-ILD33, perhaps through accelerated biologic aging. Histones are important components of telomeres, so this may explain some of the findings implicating antibodies against histones for RA-ILD risk. Another mechanism relates to netosis, a form of cell death occurring in inflammatory conditions where nuclear contents, including histones, are externally exposed. Netosis has been linked to cardiac and pulmonary fibrosis34. Thus, mechanistic studies should be performed to link autophagy, telomere shortening, and netosis in RA-ILD pathogenesis. The improved RA-ILD prediction using such antibody biomarkers demonstrates the importance of incorporating them into RA-ILD prediction methods.
Using the top biomarkers for RA-ILD discovered in this study, we developed a well-performing risk score for RA-ILD. To our knowledge, this is the first risk score for future development of RA-ILD using prospectively collected data, as other scores have used cross-sectional data to predict an outcome of subclinical RA-ILD.11,12 The score we developed with biomarkers had high sensitivity (>80%) at the threshold of 30% predicted probability of RA-ILD, whereas the score without biomarkers did not. However, both scores showed high specificity (≥93%) for RA-ILD at the score thresholds corresponding to predicted probability of RA-ILD 50% and higher. Of note, 50% of patients with scleroderma also have ILD,35 and guidelines already recommend baseline high-resolution chest CT screening for ILD in this population. The combination of the high mortality of RA-ILD,1,2 the risk score’s high specificity, and the recommendation for screening in comparable populations underscores the importance of validating and then implementing such risk scoring for obtaining RA-ILD chest CT screening in clinical practice. While we hypothesized the association of fine specificity ACPA with RA-ILD and their ability to improve disease prediction, we also observed a few unanticipated findings. First, IgA antibodies generally showed an association with RA-ILD than most IgG antibodies. However, previous work has also shown an association of IgA than IgG antibodies for both RA and RA-ILD,5 supporting the growing recognition of the importance of mucosal surfaces in RA pathogenesis and autoantibody production. Second, anti-citrullinated enolase was not associated with RA-ILD as they had been in previous studies,6 though those studies did not adjust for some potential confounders. Finally, antibodies to several non-citrullinated (native) antigens were also associated with RA-ILD. Although not studied as often as ACPA, antibodies to native antigens have shown an association with RA previously including anti-filaggrin with RA36 and anti-fibrinogen with RA-ILD.8 These studies support our findings and encourage future exploration of biomarkers to non-citrullinated antigens.
Strengths of this study include prospectively collected exposure data and its prediction of incident RA-ILD, an area of great need. In addition, this study verified both RA and RA-ILD cases, included a comprehensive panel of antibodies, and adjusted for many important confounders. There are also several important limitations to consider. The study population was neither population-based nor diverse, which is important as ACPA fine specificities vary in different populations.37 Most participants were female with longstanding RA, so results may not generalize to male patients with newly diagnosed RA. The number of incident RA-ILD cases was relatively low, which may have limited ability to detect true associations, for example with MUC5B genotype4 or anti-citrullinated enolase.6 Larger studies are needed to identify antibody signatures for specific RA-ILD subtypes as well as among patients with classically seropositive and seronegative RA. It is possible that some of the controls may have had subclinical RA-ILD. However, this would bias results towards null. Alternatively, we could have required controls to have had a clinically-indicated chest CT without ILD, but this may have introduced bias due to the indication of the chest CT, and these patients may not be representative of the general RA population. We chose controls without chronic lung disease from the RA general population so that findings may be relevant to the general RA population and lower the chances of confounding by indication of imaging and misclassification. In addition, this study included RA meeting classification criteria and RA-ILD cases independently verified by review of CT images by at least two experts so that misclassification was unlikely. Future studies should investigate whether antibody profiling may be helpful in identifying patients at risk for RA-ILD incorporating other control groups such as healthy individuals, RA patients who had chest CT imaging performed, RA patients with other forms of chronic lung disease, and serial chest CT imaging in a prospective research protocol. It is possible that participants in BRASS may not be reflective of the general RA population38. However, the nested case-control study design protects against possible differences in attrition since even those who only participated at baseline could be included in the study since the outcome was determined through data from routine clinical care. Due to the matched case-control design, the RA prediction curves and scores in reported this study could not fully incorporate the predictive performance of the matching factors, potentially reducing model performance. It is also possible that such performance metrics may have been artificially elevated due to overfitting, though we performed optimism-corrected analyses to account for this. We did not update covariates after the date of blood collection since this may have mediated associations between circulating antibodies and RA-ILD risk. Thus, covariates do not reflect changes that may have occurred between the blood date and RA-ILD onset. However, using the date before RA-ILD made it unlikely that RA-ILD influenced variables such as medications that may be either prescribed or avoided in RA-ILD. There were some overlapping findings of autoantibodies to citrullinated and native antibodies that may be explained by cross-reactivity of the assay or due to separate antibodies. However, analyses comparing individual and grouped by citrullinated/native targets suggested most biomarkers provided unique prediction ability, generally higher for the antibodies to citrullinated antigens. Several of our findings were of the IgA2 isotype. While other groups have also found this isotype to be important in RA39, others have been equivocal, and it may be more difficult to detect circulating levels40. The assay did not include other antibodies such as anti-CEP1 and anti-HSP90 that may be important in RA-ILD risk6,7. Future studies should also incorporate other anti-modified antibodies to other processes, such as carbamylation and secretory ACPA, to predict RA-ILD risk41,42. We performed many tests, so some findings could have been due to chance. However, all analyses were pre-specified in our protocol, and we used false discovery rate for the antibody analysis. At the same time, we did not test all antibodies to citrullinated antigens that may be important such as anti-HSP90.7 Since we had relatively low sample size, we relied on optimism-correction to perform internal validation rather than other methods such as splitting the sample into derivation and validation sets. Finally, future work should validate the findings from this study in external populations and determine duration of validity before ILD onset. Studies are also needed to implement screening strategies and to test whether early treatment may improve clinical outcomes.
In conclusion, both fine specificity ACPA and anti-native protein antibodies are associated with RA-ILD risk and improve its prediction. Given the high mortality of RA-ILD and the high specificity of the risk score developed herein, the clinical utility of screening high-risk RA patients for ILD should be urgently determined.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before this study
To ascertain a full understanding of previous published literature on RA-ILD before performing the study, we searched PubMed using the following terms: “rheumatoid arthritis[MeSH Terms] AND interstitial lung disease[MeSH Terms].” RA-ILD is a leading cause of excess mortality burden in RA. However, the predictive value of clinical factors is modest, and previous studies have focused on detecting prevalent subclinical RA-ILD rather than predicting future RA-ILD.
Added value of this study
Several fine specificity ACPA and anti-native protein antibodies, especially to histone and filaggrin, were associated with increased risk of developing incident RA-ILD. Furthermore, the risk score developed in this study had high sensitivity and specificity for RA-ILD.
Implications of all the available evidence
Rheumatology clinical practice and policy should begin implementing RA-ILD screening in high-risk RA patients using scoring systems such as outlined in this manuscript. Future research should validate this RA-ILD risk score in other populations. In addition, research should continue to discover and validate biomarkers including fine specificity protein antibodies that influence RA-ILD risk.
Funding:
This work was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant award K23 AR069688 to Dr. Sparks). Dr. Doyle is supported by the National Heart, Lung, and Blood Institute (R01 HL155522). Dr. McDermott is supported by T32 AR007530. Dr. Hatabu is supported by R01CA203636, U01 CA209414, R01 HL111024, R01 HL135142, and R01 HL130974. Dr. Sparks is also supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant numbers R01 AR077607, P30 AR070253, and P30 AR072577), the R. Bruce and Joan M. Mickey Research Scholar Fund, and the Llura Gund Award for Rheumatoid Arthritis Research and Care.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: Dr. Yoshida has received consulting fees from OM1 unrelated to this work. Dr. Davis has received research support from Pfizer, has a patent pending for assessing and treating arthritis and serve on a data safety monitoring board for a rheumatoid arthritis clinical trial sponsored by the National Institutes of Health, all unrelated to this work. Dr. Dellaripa has received consulting fees from Boehringer Ingelheim, Bristol Myers Squibb, and Genentech and receives royalties from UpToDate unrelated to this work. Dr. Hatabu has received research support from Canon Medical Systems and Konica-Minolta as well as consulting fees from Canon Medical Systems and the Mitsubishi Chemical Company unrelated to this work. Dr. Nishino has received research support from AstraZeneca, Canon Medical Systems, and Daiichi Sankyo, and consulting fees from AstraZeneca, Daiichi Sankyo unrelated to this work. Dr. Sokolove is currently an employee at GlaxoSmithKline and owns shares in GlaxoSmithKline, and his work on this project preceded this employment. Dr. Weinblatt has received research support from Amgen, Bristol Myers Squibb, Eli Lilly, Aqtual, and Janssen; consultancy fees from AbbVie, Aclaris, Amgen, Aqtual, Bayer, Bristol Myers Squibb, CorEvitas, EqRX, Genosco, GlaxoSmithKline, Gilead, Johnson & Johnson, Kyvrena, Eli Lilly, Pfizer, Rani, Revolo, Sanofi, Scipher, SciRom, SetPoint, and Tremeau; stock options from Canfite, Inmedex, and Scipher, all unrelated to this work. Dr. Shadick has received research grants from AbbVie, Amgen, Aqtual, Bristol Myers Squibb, Eli Lilly, and Mallinckrodt unrelated to this work. Dr. Doyle has received research support from Bristol Myers Squibb; consulting fees from Boehringer Ingelheim and L.E.K. consulting; speaking fees and travel support from Aura; and has been part of a clinical trial funded by Genentech, unrelated to this work. Dr. Sparks has received research support from Bristol Myers Squibb and performed consultancy for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Optum, and Pfizer unrelated to this work.
Data sharing: Data are not available without approval from the Brigham Rheumatoid Arthritis Sequential Study Scientific Advisory Board. Requests should be sent to the corresponding author.
REFERENCES
- 1.Bongartz T, Nannini C, Medina-Velasquez YF, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum 2010; 62(6): 1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyldgaard C, Hilberg O, Pedersen AB, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis 2017; 76(10): 1700–6. [DOI] [PubMed] [Google Scholar]
- 3.Giles JT, Danoff SK, Sokolove J, et al. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann Rheum Dis 2014; 73(8): 1487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juge PA, Lee JS, Ebstein E, et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N Engl J Med 2018; 379(23): 2209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Moreno C, Gómara MJ, Castellanos-Moreira R, Sanmartí R, Haro I. Peptides Bearing Multiple Post-Translational Modifications as Antigenic Targets for Severe Rheumatoid Arthritis Patients. Int J Mol Sci 2021; 22(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alunno A, Bistoni O, Pratesi F, et al. Anti-citrullinated alpha enolase antibodies, interstitial lung disease and bone erosion in rheumatoid arthritis. Rheumatology (Oxford) 2018; 57(5): 850–5. [DOI] [PubMed] [Google Scholar]
- 7.Harlow L, Rosas IO, Gochuico BR, et al. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum 2013; 65(4): 869–79. [DOI] [PubMed] [Google Scholar]
- 8.Liao KP, Sparks JA, Hejblum BP, et al. Phenome-Wide Association Study of Autoantibodies to Citrullinated and Noncitrullinated Epitopes in Rheumatoid Arthritis. Arthritis Rheumatol 2017; 69(4): 742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kronzer VL, Huang W, Dellaripa PF, et al. Lifestyle and clinical risk factors for incident rheumatoid arthritis-associated interstitial lung disease. J Rheumatol 2020. [DOI] [PMC free article] [PubMed]
- 10.Doyle TJ, Patel AS, Hatabu H, et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease Is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med 2015; 191(12): 1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juge PA, Granger B, Debray MP, et al. A Risk Score to Detect Subclinical Rheumatoid Arthritis-Associated Interstitial Lung Disease. Arthritis Rheumatol 2022. [DOI] [PMC free article] [PubMed]
- 12.Qin Y, Wang Y, Meng F, et al. Identification of biomarkers by machine learning classifiers to assist diagnose rheumatoid arthritis-associated interstitial lung disease. Arthritis Res Ther 2022; 24(1): 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iannaccone CK, Lee YC, Cui J, et al. Using genetic and clinical data to understand response to disease-modifying anti-rheumatic drug therapy: data from the Brigham and Women’s Hospital Rheumatoid Arthritis Sequential Study. Rheumatology (Oxford) 2011; 50(1): 40–6. [DOI] [PubMed] [Google Scholar]
- 14.Doyle TJ, Dellaripa PF, Batra K, et al. Functional impact of a spectrum of interstitial lung abnormalities in rheumatoid arthritis. Chest 2014; 146(1): 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sparks JA, He X, Huang J, et al. Rheumatoid arthritis disease activity predicting incident clinically-apparent RA-associated interstitial lung disease: A prospective cohort study. Arthritis Rheumatol 2019. [DOI] [PMC free article] [PubMed]
- 16.Monach PA, Hueber W, Kessler B, et al. A broad screen for targets of immune complexes decorating arthritic joints highlights deposition of nucleosomes in rheumatoid arthritis. Proc Natl Acad Sci U S A 2009; 106(37): 15867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokolove J, Bromberg R, Deane KD, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One 2012; 7(5): e35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arkema EV, Goldstein BL, Robinson W, et al. Anti-citrullinated peptide autoantibodies, human leukocyte antigen shared epitope and risk of future rheumatoid arthritis: a nested case-control study. Arthritis Res Ther 2013; 15(5): R159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner CA, Sokolove J, Lahey LJ, et al. Identification of anticitrullinated protein antibody reactivities in a subset of anti-CCP-negative rheumatoid arthritis: association with cigarette smoking and HLA-DRB1 ‘shared epitope’ alleles. Ann Rheum Dis 2015; 74(3): 579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDermott G, Gill R, Gagne S, et al. Associations of the MUC5B Promoter Variant with Timing of Interstitial Lung Disease and Rheumatoid Arthritis Onset. Rheumatology (Oxford) 2022. [DOI] [PMC free article] [PubMed]
- 21.Steyerberg EW, Harrell FE Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol 2001; 54(8): 774–81. [DOI] [PubMed] [Google Scholar]
- 22.Qian J, Payabvash S, Kemmling A, Lev MH, Schwamm LH, Betensky RA. Variable selection and prediction using a nested, matched case-control study: Application to hospital acquired pneumonia in stroke patients. Biometrics 2014; 70(1): 153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng X, Ezzati P, Smolik I, Bernstein CN, Hitchon CA, El-Gabalawy HS. Characterization of Autoantigens Targeted by Anti-Citrullinated Protein Antibodies In Vivo: Prominent Role for Epitopes Derived from Histone 4 Proteins. PLoS One 2016; 11(10): e0165501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sohn DH, Rhodes C, Onuma K, et al. Local Joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol 2015; 67(11): 2877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mergaert AM, Bawadekar M, Nguyen TQ, et al. Reduced Anti-Histone Antibodies and Increased Risk of Rheumatoid Arthritis Associated with a Single Nucleotide Polymorphism in PADI4 in North Americans. Int J Mol Sci 2019; 20(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Won P, Kim Y, Jung H, et al. Pathogenic Role of Circulating Citrullinated Antigens and Anti-Cyclic Monoclonal Citrullinated Peptide Antibodies in Rheumatoid Arthritis. Front Immunol 2021; 12: 692242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikuls TR, Payne JB, Yu F, et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol 2014; 66(5): 1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice NE, Patel BD, Lang IA, et al. Filaggrin gene mutations are associated with asthma and eczema in later life. J Allergy Clin Immunol 2008; 122(4): 834–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kronzer VL, Westerlind H, Alfredsson L, et al. Respiratory Diseases as Risk Factors for Seropositive and Seronegative Rheumatoid Arthritis and in Relation to Smoking. Arthritis Rheumatol 2021; 73(1): 61–8. [DOI] [PubMed] [Google Scholar]
- 30.Saunders SP, Moran T, Floudas A, et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J Allergy Clin Immunol 2016; 137(2): 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma P, Alizadeh J, Juarez M, et al. Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis. Cells 2021; 10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celia AI, Colafrancesco S, Barbati C, Alessandri C, Conti F. Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches. Cells 2022; 11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natalini JG, England BR, Baker JF, et al. Associations between shortened telomeres and rheumatoid arthritis-associated interstitial lung disease among U.S. Veterans. Respir Med 2022; 201: 106943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimmeler S, Zeiher AM. Netting Insights into Fibrosis. N Engl J Med 2017; 376(15): 1475–7. [DOI] [PubMed] [Google Scholar]
- 35.Bergamasco A, Hartmann N, Wallace L, Verpillat P. Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease. Clin Epidemiol 2019; 11: 257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi KH, Lee EB, Yoo CD, et al. Clinical significance of anti-filaggrin antibody recognizing uncitrullinated filaggrin in rheumatoid arthritis. Exp Mol Med 2005; 37(6): 546–52. [DOI] [PubMed] [Google Scholar]
- 37.Too CL, Murad S, Hansson M, et al. Differences in the Spectrum of Anti-Citrullinated Protein Antibody Fine Specificities Between Malaysian and Swedish Patients With Rheumatoid Arthritis: Implications for Disease Pathogenesis. Arthritis Rheumatol 2017; 69(1): 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iannaccone CK, Fossel A, Tsao H, Cui J, Weinblatt M, Shadick N. Factors associated with attrition in a longitudinal rheumatoid arthritis registry. Arthritis Care Res (Hoboken: ) 2013; 65(7): 1183–9. [DOI] [PubMed] [Google Scholar]
- 39.Sokolova MV, Hagen M, Bang H, et al. IgA anti-citrullinated protein antibodies are associated with flares during DMARD tapering in rheumatoid arthritis. Rheumatology (Oxford) 2022; 61(5): 2124–31. [DOI] [PubMed] [Google Scholar]
- 40.Derksen V, Allaart CF, Van der Helm-Van Mil AHM, Huizinga TWJ, Toes REM, van der Woude D. In rheumatoid arthritis patients, total IgA1 and IgA2 levels are elevated: Implications for the mucosal origin hypothesis. Rheumatology (Oxford) 2022. [DOI] [PMC free article] [PubMed]
- 41.Castellanos-Moreira R, Rodriguez-Garcia SC, Gomara MJ, et al. Anti-carbamylated proteins antibody repertoire in rheumatoid arthritis: evidence of a new autoantibody linked to interstitial lung disease. Ann Rheum Dis 2020; 79(5): 587–94. [DOI] [PubMed] [Google Scholar]
- 42.Roos Ljungberg K, Joshua V, Skogh T, et al. Secretory anti-citrullinated protein antibodies in serum associate with lung involvement in early rheumatoid arthritis. Rheumatology (Oxford) 2020; 59(4): 852–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.