

Abstract
Introduction
Continuous measures of amyloid burden as measured by positron emission tomography (PET) are being used increasingly to stage Alzheimer's disease (AD). This study examined whether cerebrospinal fluid (CSF) and plasma amyloid beta (Aβ)42/Aβ40 could predict continuous values for amyloid PET.
Methods
CSF Aβ42 and Aβ40 were measured with automated immunoassays. Plasma Aβ42 and Aβ40 were measured with an immunoprecipitation–mass spectrometry assay. Amyloid PET was performed with Pittsburgh compound B (PiB). The continuous relationships of CSF and plasma Aβ42/Aβ40 with amyloid PET burden were modeled.
Results
Most participants were cognitively normal (427 of 491 [87%]) and the mean age was 69.0 ± 8.8 years. CSF Aβ42/Aβ40 predicted amyloid PET burden until a relatively high level of amyloid accumulation (69.8 Centiloids), whereas plasma Aβ42/Aβ40 predicted amyloid PET burden until a lower level (33.4 Centiloids).
Discussion
CSF Aβ42/Aβ40 predicts the continuous level of amyloid plaque burden over a wider range than plasma Aβ42/Aβ40 and may be useful in AD staging.
Highlights
Cerebrospinal fluid (CSF) amyloid beta (Aβ)42/Aβ40 predicts continuous amyloid positron emission tomography (PET) values up to a relatively high burden.
Plasma Aβ42/Aβ40 is a comparatively dichotomous measure of brain amyloidosis.
Models can predict regional amyloid PET burden based on CSF Aβ42/Aβ40.
CSF Aβ42/Aβ40 may be useful in staging AD.
Keywords: biomarker concordance, CSF Aβ42/Aβ40, machine learning, PET, plasma Aβ42/Aβ40
1. INTRODUCTION
Alzheimer's disease (AD) is a slowly progressive neurodegenerative disorder characterized by the presence of amyloid plaques and neurofibrillary tangles. 1 , 2 , 3 The burden of amyloid plaques in the brain can be quantified in vivo with positron emission tomography (PET) tracers such as Pittsburgh compound B (PiB). 4 Amyloid PET is well correlated with AD neuropathology, 5 , 6 , 7 and is often used as the reference standard for brain amyloidosis. 8 , 9 Amyloid PET positivity is also used as an enrollment criterion for clinical trials, including trials in cognitively normal individuals that are designed to prevent or slow AD symptom onset. 10 , 11 Multiple studies have demonstrated that amyloid plaques, as measured by PET, accumulate consistently across individuals during preclinical AD (when individuals are amyloid positive but cognitively normal), enabling staging of preclinical AD with continuous measures of amyloid PET. 12 , 13 , 14 , 15 , 16 , 17
Cerebrospinal fluid (CSF) and plasma amyloid beta (Aβ)42/Aβ40 have also been found to be accurate biomarkers of brain amyloidosis. 8 , 18 However, although amyloid PET quantifies the lifetime accumulation of amyloid plaques, CSF and plasma concentrations of Aβ42 and Aβ40 represent the overall production and clearance of Aβ peptides at the time of collection. 19 , 20 Sequestration of Aβ42 into amyloid plaques may be reflected by lower levels of Aβ42 in the CSF and plasma, 19 , 20 , 21 whereas Aβ40 remains relatively stable in the presence of amyloid plaques and, therefore, can be used to normalize for overall protein production. 20 , 22 Although the Aβ42 to Aβ40 ratio as measured in either CSF or plasma strongly predicts dichotomous amyloid PET status, 8 , 9 , 18 , 23 , 24 , 25 , 26 the relationships of CSF and plasma Aβ42/Aβ40 with continuous values for amyloid PET burden are non‐linear, and it is unclear whether CSF or plasma Aβ42/Aβ40 can be used as continuous measures of brain amyloidosis.
Because CSF and plasma measures of amyloid change monotonically over time 27 , 28 , 29 , 30 and amyloid burden accumulates in a generally consistent pattern, 12 , 13 , 14 , 15 , 16 , 17 it is possible that CSF and plasma Aβ42/Aβ40 may predict amyloid burden as measured by amyloid PET. Here we evaluated the relationships between CSF and plasma Aβ42/Aβ40 and continuous values for amyloid PET burden. We used high‐performance measures of amyloid: CSF Aβ42/Aβ40 by automated immunoassays, plasma Aβ42/Aβ40 by a high‐precision immunoprecipitation–mass spectrometry assay, and amyloid PET with PiB. We generated three types of models (linear models, generalized additive models, and artificial neural networks) to predict mean cortical and regional amyloid burden based on CSF and/or plasma Aβ42/Aβ40. In addition, we implemented a recently published approach to staging AD based on regional amyloid PET values and compared disease stage based on actual amyloid PET with predicted regional amyloid PET values.
2. METHODS
2.1. Participants
Community‐dwelling older adults enrolled in studies at the Knight Alzheimer's Disease Research Center (Knight ADRC) at Washington University in St. Louis were considered for inclusion based on the following criteria: (1) plasma and CSF collected on the same day with available Aβ42/Aβ40 data, and (2) amyloid PET with PiB obtained within 2 years of the plasma and CSF collection. Written informed consent was obtained from all participants and their study partners. All procedures were approved by Washington University's Human Research Protection Office.
Participants underwent clinical assessments using the Uniform Data Set (UDS) 31 that included the Clinical Dementia Rating (CDR). 32 A CDR of 0 indicates no cognitive impairment, a CDR of 0.5 indicates very mild impairment, and a CDR of 1 indicates mild cognitive impairment. Race and gender were self‐identified. Apolipoprotein E (APOE) genotyping was performed using either an Illumina 610 or OmniExpress chip as described previously. 33
RESEARCH IN CONTEXT
Systematic Review: Continuous measures of mean cortical and regional amyloid burden as measured by positron emission tomography (PET) are being used increasingly to stage Alzheimer's disease (AD). Cerebrospinal fluid (CSF) and plasma amyloid beta (Aβ)42/Aβ40 have a non‐linear relationship with amyloid PET measures. To facilitate comparisons of fluid biomarkers and amyloid PET, these measures are often dichotomized, which reduces the information represented by these measures.
Interpretation: CSF Aβ42/Aβ40 predicts continuous values for mean cortical amyloid PET burden over a wider range than plasma Aβ42/Aβ40, potentially because of biological differences in these measures. CSF Aβ42/Aβ40 also predicts regional amyloid PET burden. These characteristics suggest that CSF Aβ42/Aβ40 may be useful in staging AD.
Future Directions: Validation of the utility of CSF Aβ42/Aβ40 for disease staging using additional cohorts and study designs is needed. Further studies of CSF and plasma Aβ metabolism are needed to understand the differences between these measures of amyloidosis.
2.2. Cerebrospinal fluid (CSF) and plasma biomarkers
CSF and blood samples from each participant were collected at a single session at ≈8 a.m. following overnight fasting as described previously. 8 , 21 Concentrations of CSF Aβ40 and Aβ42 were measured by chemiluminescent enzyme immunoassay using a fully automated platform (LUMIPULSE G1200, Fujirebio, Malvern, PA, USA). Plasma Aβ42 and Aβ40 were measured by the C2N Diagnostics commercial laboratory using an immunoprecipitation‐mass spectrometry assay (St. Louis, MO, USA). 24 All assays were performed by personnel who were blinded to participant information.
2.3. Structural magnetic resonance imaging (MRI)
Magnetic resonance imaging (MRI) images were obtained on 3T Siemens scanners. T1‐weighted scans were segmented with FreeSurfer 5.3 (Martinos Center for Biomedical Imaging, Charlestown, MA, USA), and the Desikan–Killiany atlas was applied. Partial volume correction of amyloid PET images used volumes obtained from structural MRI.
2.4. Positron emission tomography (PET) imaging
Amyloid PET scans with 11C‐Pittsburgh Compound B (or PiB) were obtained via previously described methods, and images were processed using the PET unified pipeline (PUP, https://github.com/ysu001/PUP). 34 , 35 Briefly, dynamically acquired PET data were reconstructed into frames that underwent affine registration to correct for inter‐frame motion. 36 , 37 Standardized uptake value ratios (SUVRs) from the 30–60 min post‐injection window were calculated using the cerebellar gray matter as the reference region. 34 , 38 Images were smoothed using a gaussian kernel to achieve a spatial resolution of 8 mm. Data were then summarized in regions of interest defined by the Desikan–Killiany atlas derived from the MRI. Partial volume correction was implemented via a geometric transfer matrix approach. 35 , 39 An amyloid PET summary value was calculated from the arithmetic mean of SUVRs for the following bilateral regions (average of right‐ and left‐sided structures): precuneus, superior frontal and rostral middle frontal regions, lateral orbitofrontal and medial orbitofrontal regions, and superior temporal and middle temporal regions. 34 Individuals were classified as amyloid positive if the mean cortical SUVR was greater than 1.42. 34 Centiloid values were calculated using Equation 1. 40
| (1) |
2.5. Statistical analysis
Differences in participant characteristics by amyloid PET status were calculated using Student's t‐tests for continuous variables and chi‐square tests for categorical variables. Spearman correlations were used to evaluate the non‐parametric relationships between biomarkers.
2.6. Model development and architecture
We constructed linear, generalized additive models (GAMs) and artificial neural networks (ANNs) to predict both mean cortical and regional cortical SUVRs based on CSF and plasma Aβ42/Aβ40. For all models, we used an 80–20 train‐test split after stratifying by PET amyloid status. In addition to models based only on fluid biomarkers, we considered models with age, number of APOE ε4 alleles (using target encoding), and their interaction as potential covariates. For GAMs we fit cubic regression splines with a maximum allowed k of 4. For GAM model confidence intervals, we performed a 1000‐iteration bootstrap and used a sliding window (N = 50) to evaluate the mean average percent error (MAPE) associated with the model fit relative to amyloid burden. We next constructed feedforward ANNs that used CSF or plasma Aβ42/Aβ40 to predict 37 bilateral regional PET‐PiB SUVR values. ANNs were designed in R Studio v4.0 (R Core Development Team, 2013) using the Keras package. Feedforward ANNs map an input to an output using a directed acyclic graph composed of sets of smaller functions. 41 The ANNs consisted of an input layer, three hidden layers with Relu activation functions, and an output layer with a linear activation function (Figure 1). Dropout was used between hidden layers in the model. 42 Input features were scaled and centered, and the ANNs were trained with adaptive moment estimation (Adam) optimization with training terminated after 100 epochs. 43 The parameters for each ANN were identified with hyperparameter optimization via a coarse grid search (Table S1). Confidence intervals (Cis) were estimated from a 500‐iteration bootstrap procedure.
FIGURE 1.
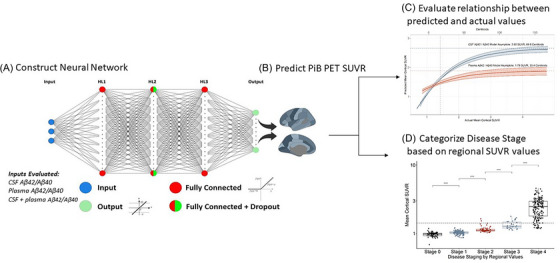
Overview of study. The objectives of this study were to examine the continuous relationships of amyloid positron emission tomography (PET) with cerebrospinal fluid (CSF) and plasma amyloid beta (Aβ)42/Aβ40, and to determine whether these fluid biomarkers could be used to stage amyloidosis. We built a three‐layer neural network with dropout to predict amyloid PET standardized uptake value ratio (SUVR) using CSF and plasma Aβ42/Aβ40 (A). We evaluated the accuracy of these predictions (B), including across the range of amyloid PET SUVR values (C). We categorized individuals according to regional amyloid PET as described by Collij et al. 13 (D) and compared the stages to those predicted by CSF Aβ42/Aβ40.
2.7. Inference from developed models
After training and optimizing the neural network models, we generated regional SUVR predictions. The range of fluid biomarkers that predicted mean cortical SUVRs was evaluated by asymptotic regression as described by Equation 2, where A and B were determined using a non‐linear least‐squares model fitted via nls(), a function in the R package nlme.
| (2) |
2.8. Staging by regional amyloid PET values
We next examined the relationship between fluid biomarkers and AD stage based on regional amyloid PET values. We categorized individuals into five disease stages based on regional amyloid PET. 13 The same individuals were then categorized based on predicted values for regional SUVRs. Using accuracy and Kendall's tau, we compared the concordance of the disease stage based on actual and predicted regional amyloid PET values. Finally, we constructed a multinomial model to directly predict disease stage with CSF Aβ42/Aβ40 as the sole input and the disease stages 13 as a categorical output. We compared the performance of this direct multinomial model to the disease stages derived from the predicted regional PiB values on the basis of both Kendall's tau and confusion matrices.
3. RESULTS
3.1. Participants
Most participants were cognitively normal (427 of 491 [87%] were rated CDR 0) and the mean age was 69.0 with a standard deviation (SD) of 8.8 years (Table 1). The cohort included 157 amyloid PET‐positive and 334 amyloid PET‐negative participants. The amyloid PET‐positive group was older (73.2 ± 6.8 years vs 67.1 ± 8.9 years, p < 0.001), was more likely to have cognitive impairment as defined by a CDR > 0 (31% vs 6%, p < 0.001), and was more likely to carry an APOE ε4 allele (64% vs 25%, p < 0.001) compared to the amyloid PET‐negative group.
TABLE 1.
Participant characteristics.
| Entire cohort | PET Amyloid Negative | PET Amyloid Positive | p | |
|---|---|---|---|---|
| N | 491 | 334 | 157 | |
| Age in years (mean ± SD) | 69.0 ± 8.8 | 67.1 ± 8.9 | 73.2 ± 6.8 | <0.001 |
| Gender, female n (%) | 270 (55%) | 194 (58%) | 76 (48%) | 0.06 |
| CDR | <0.001 | |||
| 0 | 427 (87%) | 316 (95%) | 111 (71%) | |
| 0.5 | 54 (11%) | 16 (5%) | 38 (26%) | |
| 1 | 10 (2%) | 2 (0.6%) | 8 (5%) | |
| APOE genotype, n (%) | <0.001 | |||
| ε2/ε2 | 1 (0.2%) | 1 (0.3%) | 0 (0%) | |
| ε2/ε3 | 51 (11%) | 48 (15%) | 3 (2%) | |
| ε2/ε4 | 15 (3%) | 9 (3%) | 6 (4%) | |
| ε3/ε3 | 248 (52%) | 197 (60%) | 54 (35%) | |
| ε3/ε4 | 138 (29%) | 67 (20%) | 71 (46%) | |
| ε4/ε4 | 28 (6%) | 7 (2%) | 21 (14%) | |
| Race, n (%) | 0.02 | |||
| White | 438 (89%) | 289 (87%) | 149 (95%) | |
| Black | 48 (10%) | 42 (13%) | 6 (4%) | |
| Other | 5 (1%) | 3 (0.9%) | 2 (1.2%) | |
| Years of education (mean ± SD) | 16.0 ± 2.6 | 16.2 ± 2.4 | 15.7 ± 2.8 | 0.09 |
| Years between amyloid PET and fluid biomarker collection a (mean ± SD) | −0.03 (0.57) | −0.04 (0.55) | −0.03 (0.61) | 0.835 |
| Mean cortical SUVR (mean ± SD) | 1.52 ± 0.79 | 1.06 ± 0.11 | 2.50 ± 0.73 | <0.001 |
| Mean cortical Centiloids (mean ± SD) | 20.94 ± 35.66 | 0.19 ± 4.84 | 64.97 ± 32.68 | <0.001 |
| CSF Aβ42/Aβ40 (mean ± SD) | 0.073 ± 0.023 | 0.086 ± 0.014 | 0.046 ± 0.011 | <0.001 |
| Plasma Aβ42/Aβ40 (mean ± SD) | 0.101 ± 0.009 | 0.104 ± 0.008 | 0.094 ± 0.007 | <0.001 |
Negative sign indicates that amyloid positron emission tomography (PET) imaging occurred after fluid biomarker collection.
Amyloid PET scans were typically performed within 2 weeks of fluid biomarker collection (median 8.1 days, range 0 days to 2 years); previous studies have demonstrated similar correlations between amyloid PET and CSF biomarkers when CSF was collected up to 6 years prior to or 2 years after amyloid PET. 44 There was no systematic bias whereby PET was more frequently collected either before or after the fluid biomarkers (Figure S1). CSF and plasma were collected on the same day.
3.2. Correlations of CSF and plasma Aβ42/Aβ40 with amyloid PET
Within the entire cohort, CSF Aβ42/Aβ40 was well correlated with amyloid PET as measured by mean cortical SUVR (Spearman ρ = −0.73 (95% CI –0.78 to –0.70)), and plasma Aβ42/Aβ40 was moderately correlated with mean cortical SUVR (Spearman ρ = −0.56, 95% CI –0.64 to –0.50) (Figure 2). Within the amyloid PET‐positive group (mean cortical SUVR > 1.42, N = 157), the correlation between CSF Aβ42/Aβ40 and mean cortical SUVR was reduced, but there was still a moderate correlation (Spearman ρ = −0.43, 95% CI –0.55 to –0.30). Within the group with high amyloid PET burden (mean cortical SUVR > 2.0, N = 109), there was only a trend toward a correlation between CSF Aβ42/Aβ40 and mean cortical SUVR (Spearman ρ = −0.18, 95% CI –0.36 to 0.00). There was no significant correlation between plasma Aβ42/Aβ40 and mean cortical SUVR in the amyloid PET‐positive group (Spearman ρ = −0.09, 95% CI –0.20 to 0.01) or in the group with high amyloid PET burden (Spearman ρ = 0.10, 95% CI –0.09 to 0.28). Correlations of CSF and plasma Aβ42/Aβ40 with regional mean cortical SUVR are shown in Figure S2).
FIGURE 2.
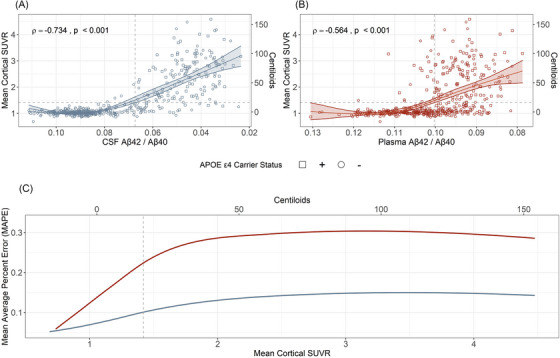
Correlations of amyloid positron emission tomography (PET) with cerebrospinal fluid (CSF) and plasma amyloid beta (Aβ)42/Aβ40. The relationship of amyloid PET with CSF Aβ42/Aβ40 (A) or plasma Aβ42/Aβ40 (B) is shown with Spearman correlations. Horizontal dashed lines denote the established cutoff for amyloid PET (standardized uptake value ratio [SUVR] 1.42). The line of best fit for (A) and (B) was determined using a generalized additive model (GAM) with bootstrapped confidence intervals (Cis). The mean average percent error (MAPE) associated with the GAM model fit for the previous 50 values is shown (C). For low amyloid PET values, the MAPE is low (good prediction) for both CSF Aβ42/Aβ40 (blue) and plasma Aβ42/Aβ40 (red). As amyloid burden increases, the sliding window MAPE increases more rapidly for the plasma Aβ42/Aβ40 GAM than the CSF Aβ42/Aβ40 GAM. A vertical line shows the established cutoff for amyloid PET positivity.
Within the entire cohort, CSF Aβ42/Aβ40 was well correlated with plasma Aβ42/Aβ40 (Spearman ρ = 0.64, 95% CI 0.58 to 0.69). The correlation between CSF and plasma Aβ42/Aβ40 was stronger in the amyloid PET‐negative group (ρ = 0.404, p < 0.001) than the amyloid PET‐positive group (ρ = 0.184, p = 0.022) (Figure S3). There was no significant correlation between CSF and plasma Aβ42/Aβ40 in the group with high amyloid PET burden (Spearman ρ = 0.019, 95% CI –0.17 to 0.21).
A GAM for mean cortical SUVR as a function of CSF Aβ42/Aβ40 had a good fit (R2 adj = 0.662, Figure 2A ); a GAM for mean cortical SUVR as a function of plasma Aβ42/Aβ40 had a moderate fit (R2 adj = 0.275, Figure 2B ). A comparison of the MAPE for the GAM models based on CSF or plasma Aβ42/Aβ40 demonstrated that both analytes were highly predictive of very low mean cortical amyloid PET burden, that is, both analytes identified individuals who did not have amyloid plaques (Figure 2C ). However, as amyloid plaque burden increased in the brain, CSF Aβ42/Aβ40 remained predictive of continuous mean cortical SUVRs even after individuals passed the threshold for amyloid PET positivity, whereas plasma Aβ42/Aβ40 had a greater error in prediction starting with the relatively low amyloid burden.
3.3. Artificial neural network models
ANN models were constructed to predict amyloid PET SUVR based on CSF and/or plasma Aβ42/Aβ40 (Figure S4). Models that predicted mean cortical SUVR using either a combination of CSF and plasma Aβ42/Aβ40 or CSF Aβ42/Aβ40 alone performed similarly (MAPE 14.8%, 95% CI 13.1% to 17.4% vs 14.2%, 95% CI 13.3% to 15.8%, respectively) (Figure 3). In contrast, the ANN predicting mean cortical SUVR using plasma Aβ42/Aβ40 had a MAPE of 29.3% (95% CI 22.6% to 35.9%). Note that a lower MAPE indicates better prediction. Because of their nearly equivalent performance, we focused on the CSF‐based model rather than the combination of CSF and plasma. The addition of age and number of APOE ε4 alleles did not significantly improve the performance of the CSF‐based model; however, the number of APOE ε4 alleles did improve the plasma‐based model to a MAPE of 23.2% (Figure S5). The addition of age did not improve the performance of any models. ANN models were compared to statistical models of amyloid PET as a function of CSF and/or plasma Aβ42/Aβ40. The ANN models substantially outperformed the linear models and outperformed the generalized additive models (Figure S6). The linear model and GAM predicting mean cortical SUVR based on CSF Aβ42/Aβ40 had a MAPE of 23.8% (95% CI 22.5% to 24.3%) and 15.1% (95% CI 14.2% to 16.1%), respectively.
FIGURE 3.
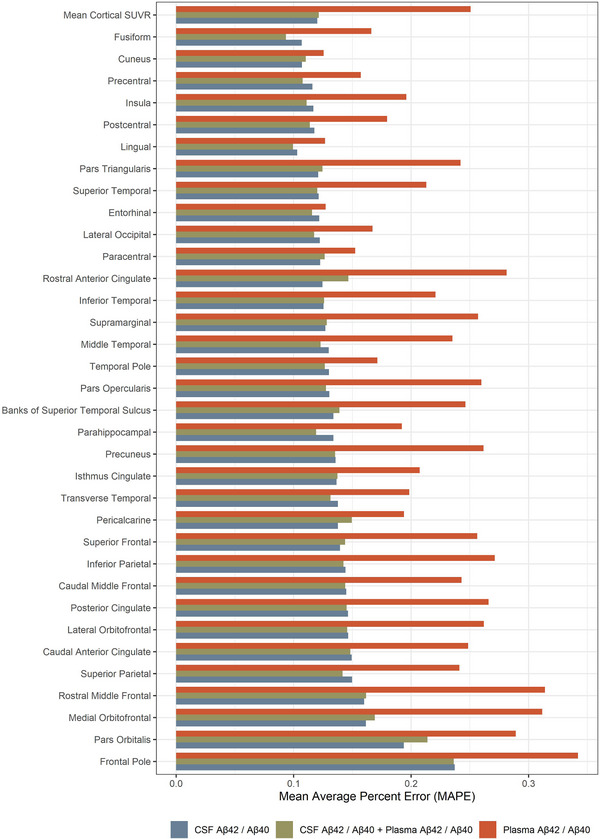
Prediction of regional amyloid positron emission tomography (PET) standardized uptake value ratio (SUVR) by neural network models. The mean average percent error (MAPE) for prediction of mean cortical SUVR and regional amyloid PET (average of right‐ and left‐sided structures) is shown for cerebrospinal fluid (CSF) amyloid beta (Aβ)42/Aβ40, CSF and plasma Aβ42/Aβ40, and plasma Aβ42/Aβ40. The most accurate prediction, by a small margin, was the combined CSF and plasma Aβ42/Aβ40 model (average MAPE over all regions, MAPEavg = 13.3%). The CSF Aβ42/Aβ40 model performed almost as well (MAPEavg = 14.1%). The plasma Aβ42/Aβ40 model did not perform as well as models that included CSF Aβ42/Aβ40 (MAPEavg = 23.6%), but performance improved with the inclusion of APOE genotype in the model (Figure S4).
After developing and tuning these models, the predicted and actual mean cortical SUVRs were compared (Figure 4). ANNs using CSF or plasma Aβ42/Aβ40 under‐predicted mean cortical SUVRs for individuals with higher amyloid PET values, suggesting that the CSF or plasma Aβ42/Aβ40 only predicted continuous SUVRs until a certain threshold value. We performed asymptotic regression analyses to find the maximum SUVR that could be predicted by CSF or plasma Aβ42/Aβ40. This approach found that the models predicted mean cortical SUVR until the following levels: CSF and plasma Aβ42/Aβ40, 2.49 SUVR (64.6 Centiloids); CSF Aβ42/Aβ40, 2.60 SUVR (69.5 Centiloids); and plasma Aβ42/Aβ40, 1.79 SUVR (33.1 Centiloids). Notably, the amyloid positivity threshold for amyloid PET with PiB is SUVR 1.42 (16.4 Centiloids).
FIGURE 4.
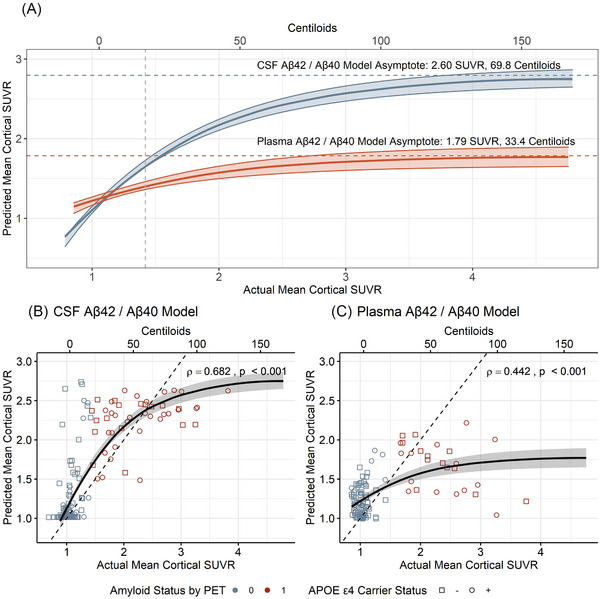
Prediction of mean cortical standardized uptake value ratio (SUVR) by neural network models. The relationship between predicted and actual mean cortical SUVR for models using cerebrospinal fluid (CSF) (A, B) or plasma amyloid beta (Aβ)42/Aβ40 (A, C) are shown. Asymptotic regression was used to identify the maximum level of amyloid burden at which CSF and plasma Aβ42/Aβ40 accurately predicted mean cortical SUVR (A).
3.4. Disease stage classification
Although mean cortical amyloid burden has been used as a measure of AD stage, another recent approach to staging emphasizes regional spread as the primary marker of disease severity and uses the number of brain regions where amyloid SUVR is higher than a relatively low threshold. 13 Individuals with virtually no amyloid accumulation are considered Disease Stage 0, whereas those with amyloid burden in most regions of the brain are considered Disease Stage 4. Participants were classified into each of the five disease stages based on actual regional amyloid PET SUVR values (Figure S7) and based on predicted regional SUVRs. The concordance between disease stage based on actual regional amyloid PET SUVRs and values predicted by the model based on CSF Aβ42/Aβ40 had high concordance (τKendall = 0.655, p < 0.001); they were the same for 61% of individuals and within one stage for 90% of individuals (Figure S8). The CSF Aβ42/Aβ40 model resulted in a more bimodal distribution than amyloid PET, classifying nearly all individuals as either Stage 1 or Stage 4, but it did identify some individuals as Stages 2 or 3 (Table S2). The model based on plasma Aβ42/Aβ40 did not categorize individuals reliably by disease stage (Table S3). We then constructed a multinomial model to directly predict disease stage using CSF Aβ42/Aβ40 as the model input. Direct prediction of disease stage from CSF Aβ42/Aβ40 resulted in a similar overall performance (τKendall = 0.627); however, this approach failed to categorize any individuals into the intermediate stages (Stage 2 or Stage 3) (Table S4).
4. DISCUSSION
Multiple studies using amyloid PET have shown that amyloid plaque burden accumulates in a generally consistent manner across individuals. 12 , 13 , 14 , 15 , 16 , 17 As amyloid burden increases, CSF Aβ42 levels and plasma Aβ42/Aβ40 decrease and then appear to plateau. 21 , 28 , 29 , 45 , 46 In this study, we sought to clarify the continuous relationships of CSF and plasma Aβ42/Aβ40 with amyloid plaque burden as measured by amyloid PET. Most studies have performed Spearman correlations to quantify these non‐linear relationships, 8 , 9 , 18 , 23 , 24 , 25 , 26 although some have performed more sophisticated modeling. 28 , 30 , 47 We found that CSF Aβ42/Aβ40 predicted mean cortical SUVR until 69.8 Centiloids, which includes most of the range of amyloid PET values that are associated with preclinical and early symptomatic AD, 16 whereas plasma Aβ42/Aβ40 predicted mean cortical SUVR until 33.4 Centiloids, which is above the threshold for amyloid positivity (16.4 Centiloids), but did not distinguish between moderate to high amyloid burden. In addition, we found that a single CSF Aβ42/Aβ40 measurement could be used to categorize individuals into a disease stage representing the regional spread of amyloidosis. Using CSF Aβ42/Aβ40 to first predict regional amyloid PET values via a neural network model and then predicting disease stage better identified individuals in the intermediate disease stages (Stage 2 and Stage 3) compared to directly predicting disease stage from CSF Aβ42/Aβ40. Although the accuracy of these predictions was limited, this finding demonstrates that the regional distribution of amyloid plaques is strongly related to the global level of brain amyloidosis as reflected by CSF Aβ42/Aβ40.
Overall, these analyses demonstrate key differences in amyloid PET, CSF Aβ42/Aβ40, and plasma Aβ42/Aβ40 as measures of amyloidosis. The dynamic range over which CSF and plasma Aβ42/Aβ40 reflect amyloid burden is smaller for plasma Aβ42/Aβ40, consistent with a previous report. 30 The different relationships of CSF and plasma Aβ42/Aβ40 with amyloid PET are likely related to differences in Aβ metabolism in the CSF and plasma. Aβ peptides that are generated by the brain enter the interstitial fluid, where they can interact directly with extracellular amyloid plaques and enter the CSF. The lower CSF Aβ42/Aβ40 levels found in individuals with brain amyloidosis likely represent the preferential sequestration of Aβ42 compared to Aβ40 into amyloid plaques. 20 In contrast, plasma Aβ has multiple sources: Aβ peptides in the brain interstitial fluid are transported across the blood‐brain barrier into the blood; Aβ peptides in the CSF are resorbed into venous blood; and Aβ peptides are produced in the periphery, including by platelets. 48 The more complex origin of plasma Aβ42/Aβ40 may explain why brain amyloidosis is associated with an ≈40% lower CSF Aβ42/Aβ40, but only 10% lower plasma Aβ42/Aβ40. 8 Furthermore, studies of Aβ kinetics suggest that differences in the balance between the peripheral and central nervous system (CNS) contributions may vary by amyloid status. 19 The reduced correlation between CSF and plasma Aβ42/Aβ40 in amyloid PET‐positive individuals further supports the idea that relationships between these biomarkers change with amyloid plaque deposition. Changes in plasma Aβ metabolism that occur with amyloidosis could underlie our finding that plasma Aβ behaves as a relatively dichotomous marker of amyloidosis, rather than a continuous measure that steadily changes with increasing amyloid plaque burden.
Consideration of the number of APOE ε4 alleles improved prediction of mean cortical SUVR in a model based on plasma Aβ42/Aβ40 but not CSF Aβ42/Aβ40, consistent with previous work demonstrating that APOE ε4 status affects the likelihood of amyloid PET positivity by plasma Aβ42/Aβ40. 8 , 24 , 26 , 49 In contrast, including age did not improve the prediction of mean cortical SUVR in models based on either plasma or CSF Aβ42/Aβ40. Moreover, inclusion of age worsened the prediction of mean cortical SUVR, suggesting that age was simply adding noise to the prediction. Previous studies have varied in finding significant effects of age on the relationship between plasma Aβ42/Aβ40 and amyloid PET 8 , 23 , 25 , 26 , 49 ; this association may be affected by the mean age and size of the cohort, as well as the PET tracer.
Limitations of this study include the relatively low frequency of individuals with high amyloid burden. Extension of this work into a cohort that includes more individuals with high amyloid burden would provide greater confidence in the range of CSF and plasma Aβ42/Aβ40 that predicts continuous values for amyloid PET. Notably, few cohorts currently have large data sets on plasma Aβ42/Aβ40 measured with a high‐precision assay, and more widely available assays may have significantly lower performance that would make interpretation less clear. 23 We used the PiB radiotracer in this study, which may be more sensitive than other amyloid PET tracers 50 and has demonstrated very consistent longitudinal trajectories in our cohort that make it ideal for staging. 16 Furthermore, although the cross‐sectional design enabled modeling of the relationships between biomarkers performed at a single time, longitudinal studies are needed to examine how these relationships change within individuals over time.
In conclusion, we observed a non‐linear but continuous relationship between CSF Aβ42/Aβ40 and amyloid PET that persisted throughout much of the range of amyloid PET values that are associated with preclinical AD. In contrast, plasma Aβ42/Aβ40 behaved as a more dichotomous measure of amyloid plaque burden. Although CSF and plasma Aβ42/Aβ40 and amyloid PET have high agreement as dichotomous measures of amyloid, and amyloid status (positive or negative) is often used in clinical practice and clinical studies, continuous levels of these measures are not interchangeable, especially at high levels of amyloid burden.
CONFLICT OF INTEREST STATEMENT
Julie K. Wisch, Anna H. Boerwinkle, Patrick H. Luckett, Yan Li, Rachel L. Henson, and Beau M. Ances report no disclosures. Brian A. Gordon receives research support from Eli Lilly and Avid Radiopharmaceuticals. James G. Bollinger and Vitaliy Ovod have submitted the US provisional patent application “Plasma Based Methods for Detecting CNS Amyloid Deposition” as co‐inventors and may receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. Tim West, Mathew R. Meyer, and Kristopher M. Kirmess are employees of C2N Diagnostics, which offers the plasma Aβ42/Aβ40 assay used in this article. Tammie L.S. Benzinger has consulted on clinical trials with Biogen, Roche, Janssen, and Eli Lilly. She receives research support from Eli Lilly and Avid Radiopharmaceuticals. Neither John C. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. Randall J. Bateman co‐founded C2N Diagnostics, which offers the PrecivityAD blood test. Washington University and Randall J. Bateman have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. Randall J. Bateman has received research funding from Avid Radiopharmaceuticals, Janssen, Roche/Genentech, Eli Lilly, Eisai, Biogen, AbbVie, Bristol Myers Squibb, and Novartis. Randall J. Bateman serves on the Roche Gantenerumab Steering Committee as an unpaid member. Suzanne E. Schindler has analyzed data provided by C2N Diagnostics to Washington University at no cost. She has not received any research funding or personal compensation from C2N Diagnostics or any other for‐profit organization. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to express our gratitude to the research volunteers who participate in AD clinical trials and their supportive families. This work was funded by the National Institutes of Health (NIH) grants R01AG070941 (S.E.S.), R01NR012907 (B.A.), R01NR012657 (B.A.), R01NR014449 (B.A.), K01 AG053474 (B.G.), P30 AG066444 (J.C.M.), P01AG003991 (J.C.M.), P01AG026276 (J.C.M.), U19 AG032438 (J.C.M.), and U19 AG024904 (J.C.M.). This work was also supported by the generous support of the Barnes‐Jewish Hospital Foundation; the Washington University Institute of Clinical and Translational Sciences Foundation (UL1 TR000448); the Hope Center for Neurological Disorders; the Paula and Rodger O. Riney Fund; the Daniel J Brennan MD Fund; and Fred Simmons Olga Mohan Fund and the Chan Zuckerberg Initiative (C.Z.I.). C2N Diagnostics provided measurements of plasma amyloid to Washington University at no cost. This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the Neurogenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/), and the Departments of Neurology and Psychiatry at Washington University School of Medicine.
Wisch JK, Gordon BA, Boerwinkle AH, et al. Predicting continuous amyloid PET values with CSF and plasma Aβ42/Aβ40. Alzheimer's Dement. 2023;15:e12405. 10.1002/dad2.12405
REFERENCES
- 1. Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. 2016;87(5):539‐47. doi: 10.1212/WNL.0000000000002923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791‐1800. [DOI] [PubMed] [Google Scholar]
- 3. Thal DR, Capetillo‐Zarate E, del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Review Sci Aging Knowledge Environ. 2006;2006(6):re1. http://sageke.sciencemag.org/cgi/content/full/2006/6/re1 [DOI] [PubMed] [Google Scholar]
- 4. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound‐B. Ann Neurol. 2004;55(3):306‐319. [DOI] [PubMed] [Google Scholar]
- 5. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐β plaques: a prospective cohort study. Lancet Neurol. 2012;11(8):669‐678. [DOI] [PubMed] [Google Scholar]
- 6. Ikonomovic MD, Buckley CJ, Abrahamson EE, et al. Post‐mortem analyses of PiB and flutemetamol in diffuse and cored amyloid‐β plaques in Alzheimer's disease. Acta Neuropathol. 2020;140(4):463‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen CD, Joseph‐Mathurin N, Sinha N, et al. Comparing amyloid‐β plaque burden with antemortem PiB PET in autosomal dominant and late‐onset Alzheimer disease. Acta Neuropathol. 2021;142(4):689‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma β‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):E1647‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimer's Dement. 2018;14(11):1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rafii MS, Sperling RA, Donohue MC, et al. The AHEAD 3‐45 study: design of a prevention trial for Alzheimer's disease. Alzheimer's Dement. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ. In vivo staging of regional amyloid deposition. Neurology. 2017;89(20):2031‐2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collij LE, Heeman F, Salvadó G, et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology. 2020;95(11):e1538‐e1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O. Staging β ‐amyloid pathology with amyloid positron emission tomography. JAMA Neurol. 2019;76(11):1319‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koscik RL, Betthauser TJ, Jonaitis EM, et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimers Dement. 2020;12(1):e12007. doi: 10.1002/dad2.12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schindler SE, Li Y, Buckles VD, et al. Predicting symptom onset in sporadic Alzheimer disease with amyloid PET. Neurology. 2021;97(18):e1823‐e1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Betthauser TJ, Bilgel M, Koscik RL, et al. Multi‐method investigation of factors influencing amyloid onset and impairment in three cohorts. Brain. 2022;45(11):4065‐4079. doi: 10.1093/brain/awac213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Aβ42/40 corresponds better than Aβ42 to amyloid PET in Alzheimer's disease. J Alzheimer's Dis. 2016;55(2):813‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer's Dement. 2017;13(8):841‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patterson BW, Elbert DL, Mawuenyega KG, et al. Age and amyloid effects on human central nervous system amyloid‐beta kinetics. Ann Neurol. 2015;78(3):439‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta;42 in humans. Ann Neurol. 2006;59(3):512‐519. [DOI] [PubMed] [Google Scholar]
- 22. Graff‐Radford J, Jones DT, Wiste HJ, et al. Cerebrospinal fluid dynamics and discordant amyloid biomarkers. Neurobiol Aging. 2022;110:27‐36. https://linkinghub.elsevier.com/retrieve/pii/S0197458021003298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐β 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78(11):1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu Y, Kirmess KM, Meyer MR, et al. Assessment of a plasma amyloid probability score to estimate amyloid positron emission tomography findings among adults with cognitive impairment. JAMA Netw Open. 2022;5(4):e228392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zicha S, Bateman RJ, Shaw LM, et al. Comparative analytical performance of multiple plasma Aβ42 and Aβ40 assays and their ability to predict positron emission tomography amyloid positivity. Alzheimer's Dement. 2022. doi: 10.1002/alz.12697. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y, Schindler SE, Bollinger JG, et al. Validation of plasma amyloid‐β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology. 2022;98(7):e688‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 2015;72(9):1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toledo JB, Bjerke M, Da X, et al. Nonlinear association between cerebrospinal fluid and florbetapir F‐18 β‐amyloid measures across the spectrum of Alzheimer disease. JAMA Neurol. 2015;72(5):571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Wolf F, Ghanbari M, Licher S, et al. Plasma tau, neurofilament light chain and amyloid‐β levels and risk of dementia; a population‐based cohort study. Brain. 2020;143(4):1220‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11(12):e11170. doi: 10.15252/emmm.201911170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dodge HH, Goldstein FC, Wakim NI, et al. Differentiating among stages of cognitive impairment in aging: version 3 of the uniform data set (UDS) neuropsychological test battery and MoCA index scores. Alzheimers Dement. 2020;6(1):e12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morris JC. Clinical Dementia rating: a reliable and valid diagnostic and staging measure for Dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(1):173‐176. doi: 10.1017/S1041610297004870 [DOI] [PubMed] [Google Scholar]
- 33. Cruchaga C, Kauwe J, Harari O, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron. 2013;78:256‐268. https://www.sciencedirect.com/science/article/pii/S0896627313001840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su Y, D'Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB‐PET with FreeSurfer ROIs. PLoS One. 2013;8(11):e73377. doi: 10.1371/journal.pone.0073377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55‐64. https://www.sciencedirect.com/science/article/pii/S1053811914009884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eisenstein SA, Koller JM, Piccirillo M, et al. Characterization of extrastriatal D2 in vivo specific binding of [18F](N‐methyl)benperidol using PET. Synapse. 2012;66(9):770‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hajnal JV, Saeed N, Soar EJ, Oatridge A, Young IR, Bydder GM. A registration and interpolation procedure for subvoxel matching of serially acquired MR images. J Comput Assist Tomogr. 1995;19(2):289‐296. [DOI] [PubMed] [Google Scholar]
- 38. Mintun MA, LaRossa GN, Sheline YI, et al. [11C]PIB in a nondemented population. Neurology. 2006;67(3):446–452. doi: 10.1212/01.wnl.0000228230.26044.a. http://www.neurology.org/cgi/content/full/67/3/446 [DOI] [PubMed] [Google Scholar]
- 39. Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39(5):904‐911. [PubMed] [Google Scholar]
- 40. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer's Dement. 2015;11(1):1‐15.e4. https://www.sciencedirect.com/science/article/pii/S155252601402500X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luckett PH, McCullough A, Gordon BA, et al. Modeling autosomal dominant Alzheimer's disease with machine learning. Alzheimer's Dement. 2021;17(6):1005‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Srivastava N, Hinton G, Krizhevsky A, Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15(1):1929‐1958. [Google Scholar]
- 43. Kingma DP, Ba J. Adam: A Method for Stochastic Optimization. arXiv. 2014; http://arxiv.org/abs/1412.6980
- 44. Boerwinkle AH, Wisch JK, Chen CD, et al. Temporal correlation of CSF and neuroimaging in the amyloid‐Tau‐neurodegeneration model of Alzheimer disease. Neurology. 2021;97(1):e76‐e87. doi: 10.1212/WNL.0000000000012123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lopez OL, Klunk WE, Mathis CA, et al. Relationship of amyloid‐β1‐42 in blood and brain amyloid: ginkgo evaluation of memory study. Brain Commun. 2020;2(1):fcz038. doi: 10.1093/braincomms/fcz038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winder Z, Sudduth TL, Fardo D, et al. Hierarchical clustering analyses of plasma proteins in subjects with cardiovascular risk factors identify informative subsets based on differential levels of angiogenic and inflammatory biomarkers. Front Neurosci. 2020;14:84. doi: 10.3389/fnins.2020.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milà‐Alomà M, Ashton NJ, Shekari M, et al. Plasma p‐tau231 and p‐tau217 as state markers of amyloid‐β pathology in preclinical Alzheimer's disease. Nat Med. 2022;28(9):1797‐1801. doi: 10.1038/s41591-022-01925-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roberts KF, Elbert DL, Kasten TP, et al. Amyloid‐β efflux from the central nervous system into the plasma. Ann Neurol. 2014;76(6):837‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. West T, Kirmess KM, Meyer MR, et al. A blood‐based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener. 2021;16(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Su Y, Flores S, Wang G, et al. Comparison of Pittsburgh compound B and florbetapir in cross‐sectional and longitudinal studies. Alzheimer's Dement. 2019;11(1):180‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information