

Abstract
Spinocerebellar ataxias (SCAs) are autosomal dominant diseases characterized by cerebellar atrophy and ataxia. The SCA subtype SCA34 is caused by specific mutations in the gene ELOVL4, which encodes a fatty acid (FA) elongase that synthesizes ultra-long-chain (ULC; ≥C26) FAs. However, the pathogenesis and molecular mechanism that confers dominant inheritance remains unknown. Here, a cell-based assay demonstrated that each of the five known SCA34 mutants produced shorter ULC polyunsaturated FA-containing phosphatidylcholines (ULC-PCs) than wild-type protein, in the following order of severity: Q180P and T233M > W246G > I171T and L168F. Next, we generated knock-in mouse embryonic stem cells that contained heterozygous Q180P, heterozygous W246G, or homozygous W246G mutations. Neuronal differentiation-dependent production of ULC-PCs was reduced in heterozygous Q180P and homozygous W246G cells relative to control cells, and we observed shortening of the FA moiety in all mutant cells. This FA shortening was consistent with our prediction that amino acid residues substituted by SCA34 mutations are located in the transmembrane helices that interact with the ω-end region of the FA moiety of the substrate acyl-CoA. Hence, reduced levels and shortening of ULC-PCs in neurons may cause SCA34, and incomplete elongation of ULC polyunsaturated acyl-CoAs by mutated ELOVL4 may induce dominant inheritance.
Keywords: ELOVL4, spinocerebellar ataxia, fatty acid, neuron, polyunsaturated fatty acid
INTRODUCTION
The spinocerebellar ataxias (SCAs) are a group of cerebellar ataxias that are inherited in an autosomal dominant manner and characterized by cerebellar atrophy and ataxia.1,2 Currently, there exist 49 SCA subtypes; for most of them, causative genes have been identified.1,3 Each causative gene exhibits a particular type of mutation, such as expansion of CAG repeats encoding the polyglutamine tract, expansion of noncoding repeats, missense mutations, or frameshift mutations.1 It is thought that these mutations have gain-of-function, dominant negative, or loss-of-function (leading to haploinsufficiency) effects. They cause various cellular abnormalities, including transcriptional dysregulation, RNA toxicity, ion channel dysfunction, and mitochondrial dysfunction. One type of SCA, namely SCA34, is caused by mutations in the fatty acid (FA) elongase ELOVL4.
ELOVL4 is one of the seven FA elongases (ELOVL1–7) that exist in mammals.4,5 FAs are classified according to their chain-length; they can be categorized as long-chain FAs (C11–C20) or very-long-chain (VLC) FAs (≥ C21). VLCFAs with C26 or more are termed ultra-long-chain (ULC) FAs.4,5 FAs are also classified according to their degree of unsaturation and are categorized as saturated FAs (SFAs), monounsaturated FAs (MUFAs), or polyunsaturated FAs (PUFAs). The long-chain FAs synthesized by FA synthase in the cytosol or incorporated from dietary sources are subject to elongation via the FA elongation cycle, which consists of four sequential reactions and takes place in the endoplasmic reticulum (ER). ELOVLs catalyze the rate-limiting reaction: condensation of an acyl-CoA–the activated form of FA–with a malonyl-CoA.6 Each ELOVL isozyme exhibits characteristic substrate specificity toward acyl-CoAs with different chain-lengths and/or degrees of unsaturation.7 Of the ELOVLs, only ELOVL4 is responsible for generating ULCFAs, regardless of the saturation/unsaturation type (SFAs, MUFAs, and PUFAs).4,5,7 Thus far, five missense mutations—L168F [NM_022726.4(ELOVL4):c.504G > C (p.Leu168Phe), rs587777598], I171T [NM_022726.4(ELOVL4):c.512T > C (p.Ile171Thr), rs1554162301], Q180P [NM_022726.4(ELOVL4):c.539A > C (p.Gln180Pro)], T233M [NM_022726.4(ELOVL4):c.698C > T (p.Thr233Met), rs1554162016], and W246G [NM_022726.4(ELOVL4):c.736T > G (p.Trp246Gly), rs1131692036]—have been identified in ELOVL4 as mutations that cause SCA34. Gait ataxia, nystagmus, and dysarthria have been reported as common neurological abnormalities in patients with these mutations.8–17 Skin erythrokeratodermia was also observed in patients carrying one of the mutations L168F, Q180P, and T233M,11,12,16 while retinitis pigmentosa was observed in a subset of patients harboring the I171T mutation.17 The age of onset differs among the mutations: 10–20 years for Q180P; 10–50 years for T233M and W246G; 30–50 years for I171T; and 30–70 years for L168F. Recently, knock-in (KI) rats harboring the W246G mutation have been generated.18 Assessment using the rotarod performance test revealed that heterozygous and homozygous W246G KI rats exhibited impaired motor function without apparent histological abnormalities, while homozygous W246G KI rats exhibited impaired synaptic plasticity in parallel and climbing fibers in the cerebellum.18 However, the effects of the SCA34 mutations on ELOVL4 enzymatic activity have not been examined, and the mechanism underlying the pathogenesis and autosomal dominant inheritance mode of SCA34 remains unclear.
Distinct sets of mutations in the ELOVL4 gene are associated with two other hereditary diseases: Stargardt disease 3 (STGD3) and ISQMR (ichthyosis, spastic quadriplegia, and mental retardation).19 STGD3 is a type of juvenile macular dystrophy without other neurological or skin symptoms that is inherited in an autosomal dominant manner. Three ELOVL4 mutations (frameshift mutations N264LfsX10 and N264TfsX9 and the nonsense mutation Y270X) have been identified in STGD3.20–22 It has been predicted that these mutations produce mutant ELOVL4 proteins lacking the C-terminal of approximately 50 residues, including the ER retention motif. Several studies have demonstrated that these mutant proteins exert dominant negative effects on wild-type (WT) ELVOL4 protein in the retinal photoreceptor cells.23–26 Alternatively, the mutant proteins can be abnormally transported to the outer segment of photoreceptors and phagocytosed by the retinal pigment epithelium, where they cause noncell-autonomous toxic effects.27,28 ISQMR is an autosomal recessive neurodevelopmental and cutaneous disorder characterized by skin ichthyosis (scaly and dry skin), spastic quadriplegia, seizures, and intellectual disability. Six ELOVL4 mutations have been identified in ISQMR (nonsense mutations Y26X, R70X, and R216X; frameshift mutations I146YfsX29 and I230MfsX22; and the missense mutation C163R).29–31 R70X and C163R were found together in one compound heterozygous patient, while the other mutations have been found in homozygous patients. Except for C163R, all mutations produce a truncated ELOVL4 protein lacking more C-terminal residues than STGD3 mutant proteins. The C163R mutation causes substitution of the amino acid located near the catalytically essential histidine-motif (residues 158–162). These findings, together with the recessive mode of inheritance, suggest that the ISQMR mutations have loss-of-function effects. Since heterozygous carriers of ISQMR do not exhibit symptoms,29–31 SCA34 is not caused by the simple haploinsufficiency of ELOVL4. Other mechanisms may be responsible for rendering the disease dominant.
ELOVL4 is abundantly expressed in the retina, skin, testes, meibomian glands, and brain (mainly in neurons).32 In the retina, phosphatidylcholines (PCs) containing ULC PUFAs (ULC-PCs) are present and important for visual function.33,34 In the skin, ULC SFAs/MUFAs are essential for the formation of the epidermal permeability barrier, as they are components of ω-O-acylceramides, which are specialized barrier lipids.4,35,36 In the testes, ULC PUFAs are used to create glycosphingolipids and sphingomyelins and are essential for spermatogenesis.37 In the meibomian glands, ULC SFAs/MUFAs are used to create cholesteryl esters, wax monoesters/diesters, and (O-acyl)-ω-hydroxy FAs, which are secreted as the constituents of tear film lipids and play an important role in preventing dry eye disease.38–42 In the brain, ULC PUFAs are predominantly present in PCs, in which ULC PUFAs with four to six double bonds occupy the sn-1 position.43,44 However, the functions of ULC-PCs in the nervous system and their relationships with SCA34 and ISQMR remain unexplored.
The aim of this study was to elucidate the pathogenesis of SCA34 and the molecular mechanism that causes its dominant heritability. For this purpose, we deduced the positions of substituted amino acid residues in ELOVL4 protein based on the recently resolved crystal structure of ELOVL7.45 We then examined the intracellular localization of mutant proteins and measured the FA elongation activity of the mutant proteins in a cell-based overexpression system. Furthermore, we generated mouse embryonic stem (ES) cells carrying the Q180P or W246G mutation and measured the levels and FA composition of ULC-PCs in them. Through these analyses, we obtained clues about the pathogenesis of SCA34 and the molecular mechanism causing its dominant mode of inheritance.
RESULTS
SCA34 mutations cause amino acid substitutions in the regions involved in the binding of the FA moiety of acyl-CoA in ELOVL4 protein
It has been established that five ELOVL4 missense mutations cause SCA34 (Fig. 1). However, the position of these amino acid residues in the 3D structure of the ELOVL4 protein remains unclear. In this study, we mapped the amino acid residues of ELOVL4 substituted in SCA34 onto the recently revealed crystal structure of the human ELOVL7 protein (PDB: 6Y7F),45 which is one of the seven isozymes of the ELOVL family. The amino acid residues of ELOVL4 mutated in SCA34 were L168F, I171T, Q180P, T233M, and W246G, and correspond to residues T157, F160, L169, I222, and F238 from ELOVL7, respectively (Fig. 1). These five residues are not conserved but rather diverged among ELOVL isozymes. In the crystal structure of ELOVL7, these amino acid residues were mapped in the transmembrane helix (TMH) 4 (L168F and I171T), TMH5 (Q180P), TMH6 (T233M), and TMH7 (W246G) (Fig. 2). These residues were not located close to the catalytic site consisting of a histidine box, but rather located at the ω-end region of the FA moiety of the bound acyl-CoA analog (Fig. 2). Thus, the amino acid residues of ELOVL4 substituted in SCA34 constitute the substrate binding site and may participate in the acquisition of substrate specificity in terms of the FA chain length of acyl-CoAs.
FIG 1.

Multiple alignment of ELOVL1–7. Amino acid sequences of human ELOVL1–7 proteins were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). The numbers in the right-hand margin refer to the adjacent amino acid residues. Amino acid residues conserved in all seven isozymes are indicated by black boxes, while residues conserved in five or six isozymes are indicated by gray boxes. SCA34-substituted residues in ELOVL4 and corresponding residues in ELOVL7 are indicated by cyan and orange boxes, respectively. The catalytically important histidine box is marked by a red rectangle.
FIG 2.
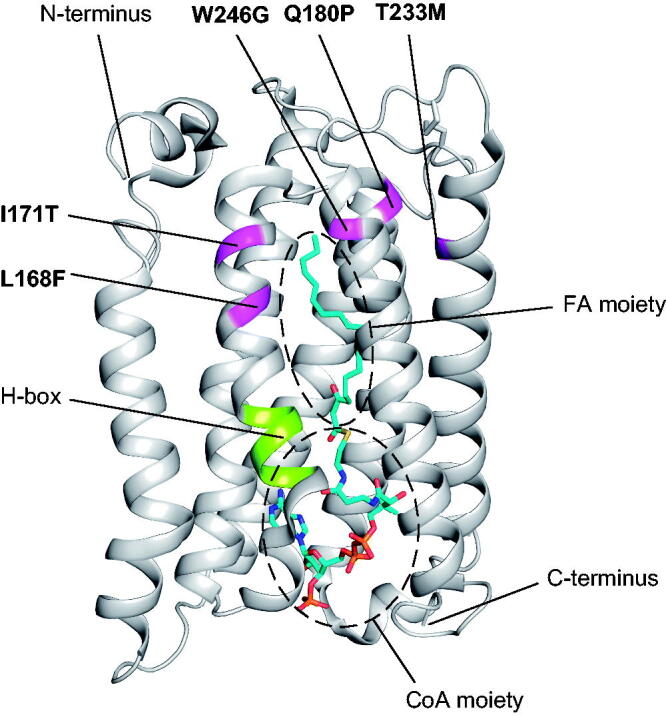
Positions of the amino acid residues of ELOVL4 that are substituted in SCA34 mapped to the crystal structure of ELOVL7. Five amino acid residues of ELOVL4 that are substituted in SCA34 (magenta) mapped on the ribbon diagram of the human ELOVL7 structure with the bound acyl-CoA analog containing the C20:0 FA moiety (PDB: 6Y7F).45 The catalytically important histidine box (H-box) is shown in lime green.
SCA34 mutant proteins are localized in the ER
Although WT ELOVL4 is localized in the ER, it has been reported that STGD3 mutations affect its subcellular localization.23,27 Conversely, the effect of SCA34 mutations on subcellular localization has not been investigated. To address this, five SCA34 mutants and WT protein as a control were transiently expressed in HeLa cells as 3 × FLAG-tagged proteins. Subsequently, their subcellular localization was examined using indirect immunofluorescence microscopy. Consistent with a previous report,7 WT ELOVL4 protein was localized in the ER, as demonstrated by its colocalization with the ER marker calnexin (Fig. 3). All five SCA34 mutant proteins were also localized in the ER, indicating that the SCA34 mutations do not affect intracellular localization.
FIG 3.

Localization of ELOVL4 SCA34 mutant proteins in the ER. HeLa cells were transfected with a vector or each of the 3 × FLAG-ELOVL4-encoding plasmids: WT and five SCA34 mutants (L168F, I171T, Q180P, T233M, and W246G). Twenty-four hours after transfection, the cells were fixed and subjected to indirect immunofluorescence microscopy using anti-FLAG antibody (red) and anti-calnexin antibody (green), which mark the ER. Nuclei were stained with DAPI (blue). Scale bar, 10 µm.
SCA34 mutants produce shorter ULC lipids than WT ELOVL4
To investigate the effect of SCA34 mutations on the ULCFA-producing activity of ELOVL4, we overproduced 3 × FLAG-tagged WT ELOVL4 or each of the SCA34 mutants in HEK 293 T cells, extracted the lipids from these cells, and measured the quantities of ceramides and PCs via liquid chromatography (LC) – tandem mass spectrometry (MS/MS). As a control, we also overproduced R216X ISQMR mutant, which lacks a C-terminus (one-third of the protein) and therefore likely has no enzymatic activity. Since ceramides can contain saturated, monounsaturated, or polyunsaturated ULCFA, measurement of ceramides is useful for monitoring the FA elongation activity of ELOVL4 toward each type of acyl-CoA. To produce ULC polyunsaturated ceramides (Fig. 4A), HA-tagged ELOVL2 [synthesizes C24 PUFAs,7 the substrates for the synthesis of ULC PUFAs (Fig. 4B)] and 3 × FLAG-tagged CERS3 [synthesizes ULC ceramides using ULC acyl-CoAs as substrates]46,47 were co-overproduced with 3 × FLAG-ELOVL4. In the immunoblot analysis, WT ELOVL4 protein was detected as two bands, an upper glycosylated form and a lower nonglycosylated form (Fig. 4C).7 All five SCA34 mutant proteins were also detected as two bands, with expression levels comparable to that of WT proteins. Two bands of the R216X ISQMR mutant protein were observed (24 and 27 kDa), consistent with the large C-terminus truncation. The levels of ceramides produced by each ELOVL4 mutant were shown as a ratio to those produced by WT ELOVL4 and displayed as a heat map (Fig. 4D). In cells expressing the R216X ISQMR mutant, the ceramide composition was similar to that noted in cells transfected with an empty vector. This composition included reduced levels of ≥ C28 saturated/monounsaturated ceramides and ≥ C32 polyunsaturated ceramides, with concomitant increases in the shorter species compared with WT ELOVL4-expressing cells (Fig. 4D and E; E indicates C32:6 and C38:6 ceramide levels as representatives). These results demonstrated that as expected, the R216X mutant does not have activity. In cells expressing the SCA34 mutants, the levels of numerous ceramide species containing ≥ C34 ULC PUFA were decreased, accompanied by increases in the levels of ≤ C32 polyunsaturated ceramides (Fig. 4D). An exception to this finding was the L168F mutant; its expression resulted in decreases in most ≥ C30 species, except C38:4 and C38:5. The chain-length-shortening effect of the SCA mutations was strongest for the Q180P and T233M mutants, followed by W246G, I171T, and L168F. Shortening was also observed for saturated and monounsaturated ceramides by many of the SCA mutants. Nevertheless, shortening was not observed for saturated ceramides by the Q180P mutant and for monounsaturated ceramides by the L168F and I171T mutants.
FIG 4.

Incomplete elongation of ULC PUFAs by ELOVL4 SCA34 mutants. (A) Structures of a representative ULC PUFA-containing ceramide (upper) and ULC-PC (lower): a ceramide that contains C32:6 FA (upper) and a PC that contains FAs (lower) making a total of C50:7 (C32:6 FA in the sn-1 position plus C18:1 in the sn-2 position). (B) Schematic representations of PUFA elongation pathways (n–6 and n–3 series). Linoleic acid (C18:2n–6) and α-linolenic acid (C18:3n–3) are subjected to a series of reactions, including desaturation, elongation by ELOVL isozymes, and β-oxidation, and some of the products are elongated to ULC PUFAs by ELOVL4. The letter E denotes ELOVL isozymes and font size reflects the relative strength of their activities. (C to G) HEK 293 T cells were transfected with a vector or each of the 3 × FLAG-ELOVL4-encoding plasmids (WT, L168F, I171T, Q180P, T233M, W246G, and R216X), together with 3 × FLAG-CERS3- and HA-ELOVL2-encoding plasmids (C to E) or HA-ELOVL2-encoding plasmid (F and G) for 48 h. (C and F) Total lysates prepared from the transfected cells were separated by SDS-PAGE, followed by immunoblotting with anti-FLAG, anti-HA, and anti-α-tubulin (for loading control) antibodies. (D, E, and G) Lipids were extracted from the transfected cells, and ceramides (D and E) or PCs (G) containing the indicated FA moiety were analyzed by LC–MS/MS. PC species are expressed as the sum of the chain-length and the number of double bonds of two constituent FAs. (D and G) Heatmaps presenting the levels of ceramide (D) or PC (G) species in each kind of ELOVL4 mutant-expressing cell relative to those in ELOVL4 WT-expressing cells (n = 3 independent cell cultures). (E) Percentages of the C32:6 and C38:6 ceramides among the total ceramides quantified in (D). Values represent means + SD (n = 3 independent cell cultures). Significant differences in comparison to ELOVL4 WT-expressing cells (*P < 0.05; **P < 0.01; Dunnett’s test) are indicated.
Next, to measure ULC-PCs (Fig. 4A), which are present in neurons, 3 × FLAG-ELOVL4 WT or each of the SCA34 mutants were overexpressed together with HA-ELOVL2 in HEK 293 T cells. The expression levels of these proteins were confirmed by immunoblot analysis (Fig. 4F). Lipids were extracted, and ULC-PCs were measured via LC–MS/MS. Each PC species was expressed according to the sum of its chain length and the number of double bonds in its two constituent FAs. Again, PC composition in cells expressing the R216X ISQMR mutant was similar to that recorded in vector-transfected cells (Fig. 4G). Numerous ≥ C50 ULC-PC species were reduced and ≤ C48 species were increased compared with those measured in WT ELOVL4-expressing cells. In the cells expressing the SCA34 mutants, the levels of PCs with ≥ C52 decreased, whereas levels of PCs with ≤ C50 increased in most cases. The heatmap pattern of ULC-PCs for each SCA34 mutant was similar to that of polyunsaturated ceramides (Fig. 4D and G), confirming the above observed activity of SCA34 mutants towards ULC polyunsaturated acyl-CoAs. Comparison of the two heatmaps suggested, for example, that the C32:6 ceramide species corresponds to the C50:7 PC species. Therefore, the C50:7 PC species mainly consists of C32:6 ULC PUFA and C18:1 FA (oleic acid) in the sn-1 and sn-2 positions, respectively (Fig. 4A). Such PC species have been reported in human and rat brains.43,44 These results indicate that the SCA34 mutations cause incomplete elongation of ULC polyunsaturated acyl-CoAs and shortening of the ULC PUFA-containing lipids.
Neuronal differentiation increases ULC-PC levels
In the brain, ELOVL4 is predominantly expressed in neurons,32 and ULC PUFAs synthesized by ELOVL4 mainly exist in PCs.43,44 In the present study, we utilized neurons differentiated from ES cells as model neurons. Mouse ES cells were subjected to differentiation into neurons via the formation of embryoid bodies, treatment with retinoic acids, and growth in neuronal differentiation medium.48 This protocol efficiently induced the differentiation of ES cells into neurons, as demonstrated by their cell morphology and the expression of the neuronal marker class III β-tubulin (Fig. 5A). To confirm Elovl4 mRNA expression and ULC-PC production in these cells, total RNA and lipids were extracted, and the levels of Elovl4 mRNAs and ULC-PCs were examined by quantitative RT-PCR and LC–MS/MS, respectively. Both Elovl4 mRNA and ULC-PC levels increased as the cells differentiated (Fig. 5B and C). Thus, neurons differentiated from ES cells are useful for the evaluation of ELOVL4 activity.
FIG 5.

Differentiation of ES cells into neurons induces Elovl4 expression and ULC-PC production. ES cells were differentiated into neurons in neuronal differentiation medium for 5 days. The cells were harvested for RNA and lipid extractions prior to the initiation of differentiation (day 0), on day 1, and on day 5. (A) On days 0, 1, and 5, bright-field images of live cells were captured, then the cells were fixed and subjected to indirect immunofluorescence microscopy using anticlass III β-tubulin antibody (green). Nuclei were stained with DAPI (blue). Scale bars, 100 µm (day 0) and 25 µm (days 1 and 5). (B) Total RNAs were prepared from the cells on days 0, 1, and 5, and were subjected to real-time quantitative RT-PCR using specific primers for Elovl4 or Actb. Values presented are the mean (± SD) levels of Elovl4 mRNA relative to those of Actb (3 independent cell cultures). (C) Lipids were extracted from the cells on days 0, 1, and 5, and ULC-PCs were analyzed by LC–MS/MS. Values presented are mean (± SD) levels of ULC-PCs (three independent cell cultures).
Neurons with SCA34 mutations produce shortened ULC-PCs
To clarify the effects of SCA34 mutations on the levels and composition of ULC-PCs, the Q180P and W246G mutations were each introduced into the ELOVL4 gene in mouse ES cells using single-stranded oligodeoxynucleotide-mediated KI with a CRISPR/Cas9 system.49 These mutations were selected as severe (Q180P) and moderate (W246G) examples in terms of the age of disease onset12,50 and their shortening effect on ULC-lipids (Fig. 4). A heterozygous KI clone, which mimics the dominant form of inheritance in SCA34, was obtained for each mutation. A homozygous KI clone, which likely exhibits more changes than the corresponding heterozygous KI clone, was obtained for the W246G mutation (Fig. 6A). These KI and control ES cells were differentiated into neurons, and the lipids prepared after 1 and 5 days of differentiation were subjected to quantification of ULC-PCs by LC–MS/MS. After 1 day of differentiation, the total levels of ULC-PCs in W246G homozygous KI cells were reduced to 42% of those noted in control cells (Fig. 6B). In contrast, heterozygous Q180P and W246G cells exhibited reduction tendencies, which were not statistically significant. The proportion of ≥ C52 ULC-PCs, which mainly consist of ≥ C34 ULC PUFAs, among the total ULC-PCs was lower in Q180P heterozygous and W246G homozygous KI cells than in control cells. These findings were suggestive of ULC PUFA shortening. The proportion of ≥ C52 ULC-PCs in W246G heterozygous KI cells was slightly higher than that recorded in control cells at this early stage of differentiation. After 5 days of differentiation, total ULC-PC levels had reduced significantly in W246G homozygous KI cells (reduction to 43%) and Q180P heterozygous KI cells (reduction to 44%). Whereas, W246G heterozygous KI cells exhibited a reduction tendency, which was not statistically significant. The proportions of ≥ C52 ULC-PCs in all three KI cells were lower than that measured in control cells. The observed reduction and shortening of ULC-PCs in KI cells were not due to the reduced expression of Elovl genes involved in the synthesis of ULC PUFA or defective differentiation into neurons. This conclusion is based on a lack of reduced expression levels of Elovl4, Elovl2, Elovl5, and the neuronal differentiation marker Tubb3 (encodes class III β-tubulin) in the KI cells after 5 days of differentiation (Fig. 6C). Rather, increases in the levels of Elovl4 and Tubb3 in Q180P heterozygous KI cells and in those of Elovl2 in W246G heterozygous KI cells were observed. These results indicate that polyunsaturated acyl-CoA elongation is incomplete in neurons possessing SCA34 mutations and suggest that decreases and/or shortening of ULC-PCs are associated with the pathogenesis of SCA34.
FIG 6.

Incomplete elongation of ULC PUFAs in Elovl4 Q180P and W246G KI neurons. (A) The gene structure of Elovl4 and sequence chromatograms of the Q180P and W246G mutations in control, heterozygous Q180P, heterozygous W246G, and homozygous W246G KI ES cells. (B and C) Control and KI ES cells were differentiated into neurons for 1 (B) and 5 days (B and C). (B) Lipids were extracted and the PC species C44:5–C56:7, which are expressed according to the sum of their chain length and the number of double bonds in their two constituent FAs, were analyzed by LC-MS/MS. Values in the left panels on days 1 and 5 represent mean (+ SD) levels of PCs (3 independent cell cultures), with each PC species being color-coded. Values in the right panels on days 1 and 5 represent the percentages of ≥ C52 ULC-PCs among the sum of C44:5–C56:7 PCs (3 independent cell cultures). Significant differences from control cells (*P < 0.05; **P < 0.01; Dunnett’s test) are indicated. (C) Total RNAs were prepared from the cells and subjected to real-time quantitative RT-PCR using specific primers for Elovl2, Elovl4, Elovl5, Tubb3, or Actb. Values presented are mean (+ SD) levels of each mRNA relative to those of Actb and expressed as a ratio to the control cells (three independent cell cultures). Significant differences from control cells (*P < 0.05; **P < 0.01; Dunnett’s test) are indicated. Het, heterozygous; Homo, homozygous.
DISCUSSION
Five ELOVL4 missense mutations have been identified in patients with SCA34.12,15,50 In the present study, all five amino acid residues substituted in SCA34 were mapped to TMHs that interact with the ω-end region of the FA moiety of the substrate acyl-CoA based on the 3D structure of ELOVL7 (Fig. 2).45 WT ELOVL4 protein exhibits activity for the elongation of C24 or C26 acyl-CoA to become C36 or C38 (Fig. 4).4,51 This indicates that its substrate-binding pocket has sufficient space to accommodate a C36 or C38 ULC acyl-CoA. Each of the SCA34 mutations may alter the pocket structure so that it can only accommodate shorter ULC acyl-CoAs, leading to incomplete elongation of acyl-CoAs (in the case of polyunsaturated acyl-CoAs, up to C32; Fig 4 and Fig 6). The effects of the Q180P, T233M, and W246G mutations on the elongation of polyunsaturated ULC acyl-CoA were greater than those of L168F and I171T (Fig. 4). There was thus a correlation between the degree of ULC-PC shortening and the age of disease onset (see Introduction).12,50 The Q180P mutation affected the elongation of polyunsaturated and monounsaturated ULC acyl-CoAs, but did not affect saturated ULC acyl-CoAs (Fig. 4). The Gln180 residue is present in TMH5, so its α-helix structure is likely disrupted by the Gln-to-Pro substitution, leading to a structural change in the substrate-binding pocket that alters the substrate specificity depending on the saturation/unsaturation status.
In this study, we found that the levels of ≥ C52 PCs were lower in Q180P and W246G heterozygous KI neurons compared with control neurons (Fig. 6B). In addition, Q180P heterozygous KI neurons showed a decrease in total ULC-PC levels. These results suggest that the shortening of ULC PUFAs and reduction in ULC-PCs in neurons are involved in the pathogenesis of SCA34. The shortening of ULC PUFAs is particularly important, since it was observed in all five SCA34 mutations (Fig. 4). The ≥ C52 ULC-PCs may play an important role in neuronal functions; hence, they cannot be replaced by shorter ULC-PCs. Although the presence of ULC-PCs has long been recognized,33,34 their function in neurons remains unknown. Neurons like the Purkinje cells in the cerebellum have very complex structures, including highly branched neurites and many synapses. Given the special structure of ULC-PCs, with a ULC PUFA in the sn-1 position, they may be enriched in membranes that make up complex structures like neurites and synapses, and the shortening of ULC PUFAs may impair their function in these structures.
In this study, we also analyzed the R216X mutation, one of the causative mutations in recessively inherited ISQMR. In a cell-based assay, the R216X mutant did not exhibit activity for the production of ULC ceramides or ULC-PCs (Fig. 4). Thus, the R216X mutation has a loss-of-function effect, which is consistent with the recessive inheritance mode of ISQMR. Since the R216X mutant lacks TMH6 and subsequently the C-terminal region, it cannot form the substrate-binding pocket for acyl-CoAs.
Although the R216X ISQMR mutant did not exhibit activity, SCA34 mutants had residual activity. The neurological symptoms of ISQMR are severe and become evident within the first few months to 1 year after birth.29 In contrast, the symptoms of SCA34 are milder than those of ISQMR, with the onset of disease mostly observed in middle age.12,50 Thus, SCA34 pathology may develop due to long-term decreases and/or changes in the quality (shortening) of ULC-PCs in neurons in the cerebellum, which may be more susceptible than other brain regions.
We speculate that the cause of the dominant inheritance mode of SCA34 is the substitution of highly functional ≥ C52 ULC-PCs (ULCFA moiety, mainly ≥ C34) for weakly functional ≤ C50 ULC-PCs (ULCFA moiety, mainly ≤ C32) (Fig. 7). This substitution may be related to the continuity of acyl-CoA elongation in the FA elongation cycle involving ELOVL4. The FA elongation cycle consists of four sequential reactions starting from condensation by ELOVLs, then reduction by 3-ketoacyl-CoA reductase, dehydration by 3-hydroxyacyl-CoA reductases, and reduction by trans-2-enoyl-CoA reductase.4,5 These are all ER-resident proteins and may form complexes,4,25,52 which enables efficient and sequential progress of reactions. Since ELOVL4 is the only enzyme involved in the elongation of ULC acyl-CoAs from C24 or C26 to C34–C38, it is reasonable to assume that the acyl-CoA synthesized in the previous cycle enters the next cycle catalyzed by the same complex, rather than dissociate from the previous complex and find and associate with another complex. Thus, one C24/C26 acyl-CoA may be elongated repetitively to become C34–C38 by the same ELOVL4-containing complex (Fig. 7). However, it is likely that C24/C26 acyl-CoA may be elongated only up to C32 by FA-elongation complexes that contain an SCA34 mutant; they would then be released from the complex and used for PC synthesis. Although carriers that are heterozygous for the ISQMR mutation are predicted to have half the ELOVL4 activity of normal individuals, they do not develop symptoms.29–31 This may be because ULC acyl-CoAs are fully elongated by the remaining WT ELOVL4 (Fig. 7). In other words, SCA34 is induced by shortening of ULC PUFAs due to incomplete elongation reactions, rather than decreased activity of ELOVL4.
FIG 7.

Model for the dominant mode of inheritance in SCA34. One C24/C26 acyl-CoA synthesized by ELOVL5 and ELOVL2 is elongated by the same WT or SCA34 mutant ELOVL4 protein (black arrows). In normal cases, in which both ELOVL4 alleles are WT, C24/C26 acyl-CoAs are elongated to become C34–C38; this process produces highly functional ULC-PCs. In patients with ISQMR, in whom both ELOVL4 alleles are ISQMR mutants, C24/C26 acyl-CoAs cannot be elongated by the ISQMR mutant protein (gray arrows with crosses), and there is no production of ≥ C28 ULC-PCs. In ISQMR carriers, in whom one allele is WT and the other allele is an ISQMR mutant, all C24/C26 acyl-CoAs are elongated by the remaining WT protein; thus, normal levels of highly functional ULC-PCs may be produced. In patients with SCA34 in whom one allele is WT and the other allele is an SCA34 mutant, a proportion of C24/C26 acyl-CoAs are elongated normally to C34–C38 by the WT protein. The remaining proportion is elongated by the SCA34 protein only up to C32, thereby producing weakly functional ULC-PCs. Gray arrows indicate reduced elongation.
In summary, this is the first biochemical analysis of SCA34 mutants, and our findings suggest that the molecular mechanism underlying the pathogenesis of SCA34 is a decrease in the levels and/or shortening of ULC-PCs. Furthermore, the dominant inheritance mode of SCA34 is caused by the substitution of ≥ C52 ULC-PCs with ≤ C50 ULC-PCs. Another SCA subtype, namely SCA38, is caused by mutations in ELOVL5,53–55 which is responsible for the elongation of polyunsaturated acyl-CoAs from C18 to C22 (Fig. 4B).4,5,7 This suggests a link between SCA34 and SCA38 and raises the possibility that the neural symptoms in these disorders are caused by the same or a similar molecular mechanism (that is to say, the shortening of ULC-PCs). Future research is needed to investigate whether SCA38 and other SCA subtypes are also accompanied by changes in the quantity or quality of ULC-PCs.
MATERIALS AND METHODS
Amino acid mapping
Five amino acid residues of ELOVL4 that are substituted in SCA34 were mapped onto the crystal structure of human ELOVL7 (PDB ID: 6Y7F) using PyMOL software (version 2.5.2; Schrödinger, New York, NY, USA).
Plasmids
The mammalian expression vectors pEFh-3 × FLAG-156 and pCE-puro 3 × FLAG-157 were used for the expression of N-terminally 3 × FLAG-tagged proteins, while the mammalian expression vector pCE-puro HA-152 was used for the expression of N-terminally HA-tagged protein. The pCE-puro 3 × FLAG-CERS3 and pCE-puro HA-ELOVL2 plasmids have been described previously.52,58 The pEFh-3 × FLAG-ELOVL4 plasmid was constructed by transferring ELOVL4 from pCE-puro 3 × FLAG-ELOVL459 to the pEFh-3 × FLAG-1 vector. The ELOVL4 mutants were generated via overlap extension PCR using pCE-puro 3 × FLAG-ELOVL4 plasmid as a template and primers (Table 1), followed by cloning into the pEFh-3 × FLAG-1 vector to produce pEFh-3 × FLAG-mutated ELOVL4 plasmids.
TABLE 1.
Oligonucleotides used for the generation of mutated ELOVL4 plasmids
| Primer name | Sequence |
|---|---|
| ELOVL4_F | 5′-GGATCCATGGGGCTCCTGGACTCGGAGCCGG-3′ (BamHI) |
| ELOVL4_R | 5′-TTAATCTCCTTTTGCTTTTCCATTTTTC-3′ |
| E4_L168F-1 | 5′-TGTTTACCTTCTGGTGGATTGGAATTAAGTG-3′ |
| E4_L168F-2 | 5′-CAATCCACCAGAAGGTAAACATCGTACAGTG-3′ |
| E4_I171T-1 | 5′-TTGTGGTGGACTGGAATTAAGTGGGTTGCAG-3′ |
| E4_I171T-2 | 5′-CTTAATTCCAGTCCACCACAAGGTAAACATC-3′ |
| E4_Q180P-1 | 5′-GCAGGAGGACCAGCATTTTTTGGAGCCCAGTTG-3′ |
| E4_Q180P-2 | 5′-AAAAAATGCTGGTCCTCCTGCAACCCACTTAATTC-3′ |
| E4_T233M-1 | 5′-ATTGGGCACATGGCACTGTCTCTTTACACTG-3′ |
| E4_T233M-2 | 5′-AGACAGTGCCATGTGCCCAATGGTCACATGG-3′ |
| E4_W246G-1 | 5′-CTTCCCCAAAGGGATGCACTGGGCTCTAATTG-3′ |
| E4_W246G-2 | 5′-CAGTGCATCCCTTTGGGGAAGGGGCAGTCAG-3′ |
| E4_R216X-1 | 5′-TTGGTGGAAATGATACCTGACTATGTTGCAAC-3′ |
| E4_R216X-2 | 5′-GTCAGGTATCATTTCCACCAAAGATATTTCTG-3′ |
The restriction enzyme BamHI used for cloning into the expression vector is noted in parentheses, with its sequence underlined.
The all-in-one CRISPR/Cas9 vector pYU751 consists of a WT Cas9 nuclease, a guide RNA (gRNA) cloning cassette, EGFP, and the gene that encodes puromycin N-acetyltransferase. This vector was constructed from the GeneArt CRISPR Nuclease Vector with the OFP plasmid (Thermo Fisher Scientific, Waltham, MA, USA) by incorporating the puromycin N-acetyltransferase gene and substituting the ORF reporter with EGFP. To generate Q180P and W246G Elovl4 KI mouse ES cells, a pair of oligonucleotides (Table 2) encoding gRNAs targeting exon 4 (for Q180P) and exon 6 (for W246G) of Elovl4 were annealed and cloned into the BaeI site of pYU751, generating the plasmids pUKA27 and pUKA16, respectively.
TABLE 2.
Oligonucleotides used for the generation of CRISPR/Cas9 plasmids
| Oligonucleotide name | Sequence | Plasmid name |
|---|---|---|
| mElovl4_Q180P-gRNA_F1 | 5′-GAAAGGTGGGTCCCTCACCTGTTTT-3′ | pUKA27 |
| mElovl4_Q180P-gRNA_R1 | 5′-AGGTGAGGGACCCACCTTTCCGGTG-3′ | |
| mElovl4_W246G-gRNA_F1 | 5′-AGAGCCCAGTGCATCCACTTGTTTT-3′ | pUKA16 |
| mElovl4_W246G-gRNA_R1 | 5′-AAGTGGATGCACTGGGCTCTCGGTG-3′ |
Cell culture and transfection
HEK 293 T cells (RCB2202; Riken BioResouce Research Center, Tsukuba, Japan) and HeLa cells (RCB0007; Riken BioResouce Research Center) were cultured in Dulbecco’s Modified Eagle’s Medium containing 4500 mg/L glucose (D6429; Merck, Darmstadt, Germany) and 1000 mg/L glucose (D6046; Merck), respectively, supplemented with 10% FBS, 100 units/mL penicillin, and 100 µg/mL streptomycin. HEK 293 T cells were grown on collagen-coated dishes. E14tg2a mouse ES cells (AES0135; Riken BioResouce Research Center) were cultured in Glasgow’s MEM (Thermo Fisher Scientific) that was supplemented with 10% FBS, 0.1 mM MEM non-essential amino acids solution (Thermo Fisher Scientific), 1 mM sodium pyruvate (Thermo Fisher Scientific), 0.1 mM 2-mercaptoethanol (FUJIFILM Wako Pure Chemical, Osaka, Japan), 1000 units/mL mouse Leukemia Inhibitory Factor (FUJIFILM Wako Pure Chemical), 100 units/mL penicillin, and 100 µg/mL streptomycin, and were grown on a 0.1% gelatin-coated dish. All cells were cultured at 37 °C in 5% CO2 and used in less than 20 passages. Transfections were performed using Lipofectamine transfection reagent with PLUS reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Indirect immunofluorescence microscopy
HeLa cells and differentiated ES cells were subjected to indirect immunofluorescence microscopy as previously described.60 Rabbit anti-FLAG polyclonal antibody (1:2000 dilution),61 mouse anti-calnexin monoclonal PM060 antibody (RRID: AB_10597560; 1:400 dilution; Medical & Biological Laboratories, Tokyo, Japan), and mouse anti-class III β-tubulin monoclonal MAB1195 antibody (RRID: AB_357520; 1:500 dilution; R&D Systems, Minneapolis, USA) were used as primary antibodies. Alexa Fluor 488-conjugated anti-mouse IgG antibody (RRID: AB_2534069) and Alexa Fluor 594-conjugated anti-rabbit IgG antibody (RRID: AB_2534073; each 1:200 dilution; Thermo Fisher Scientific) were used as secondary antibodies, and DAPI (1 µg/mL) was added simultaneously. Cover slips were mounted with ProLong gold antifade reagent (Thermo Fisher Scientific) and observed using a Leica DM5000B microscope (Leica Microsystems, Wetzlar, Germany).
Immunoblotting
Immunoblotting was performed as previously described,60 using rabbit anti-FLAG polyclonal antibody (1:1000 dilution)61 and mouse anti-α-tubulin monoclonal T9026 antibody (RRID: AB_477593; 1:1000 dilution; Merck) as primary antibodies and anti-rabbit and anti-mouse IgG, HRP-linked F(ab′)2 fragments (each 1:7500 dilution; GE Healthcare Life Sciences, Little Chalfont, UK) as secondary antibodies. Chemiluminescence detection was performed using chemiluminescence solution; this solution consisted of 100 mM Tris-HCl (pH 8.5), 0.2 mM p-coumaric acid (Merck), 2.5 mM luminol (FUJIFILM Wako Pure Chemical), and 0.002% hydrogen peroxide.
Generation of SCA34 KI ES cells
For homology-directed repair, the single-stranded oligodeoxynucleotides (ssODNs) mElovl4_Q180P_ssODN and mElovl4_W246G_ssODN were designed. Each ssODN contained the mutant sequence and bilateral homology arms for homology-directed repair in ES cells (Table 3). For each ssODN, the nontarget strand, which was not complementary to the gRNA, was chosen to avoid heteroduplex formation with gRNA.49 In the mutant sequence, mElovl4_Q180P_ssODN and mElovl4_W246G_ssODN contained ApaI and SphI restriction enzyme sites, respectively; these were introduced to examine the presence of mutations at the Elovl4 locus. To obtain Elovl4 Q180P KI cells and W246G KI cells, ES cells were transfected with pUKA27 and mElovl4_Q180P_ssODN (for Q180P KI cells), pUKA16 and mElovl4_W246G_ssODN (for W246G KI cells), or empty pYU751 vectors (as controls). Twenty-four hours after transfection, the cells were treated with 2 µg/mL puromycin and incubated for another 24 h to eliminate untransfected cells. The cells that survived were subsequently diluted and cultured in the absence of puromycin for 9 days. Single colonies were isolated using cloning rings, then expanded and stored. Genomic DNAs were extracted from the cells, and the DNA fragments containing the target sites were amplified by PCR. The amplified DNA fragments were digested with restriction enzymes (ApaI for Q180P and SphI for W246G) to examine the presence of the mutations. Positive clones identified by restriction enzyme analysis were subjected to sequence confirmation by Sanger sequencing.
TABLE 3.
ssODNs used for homology-directed repair
| Elovl4 mutation | Sequence |
|---|---|
| Q180P | 5′-CTAGGTAAACCTACTCCCACCCACATGGGGGTTCAGACAGAAAGGTGGGTCCCTCACCGGGCCCTCCAGCCACCCACTTGATTCCA ATCCACCACAGAGTGAACATGGTGCAGTGGTGGT-3′ (ApaI) |
| W246G | 5′-GTAGAAGTTGAGGAAGAGGAAGATGAAGCTGATGGCGTAGGCGATCAGAGCCCAGTGCATGCCCTTGGGGAAGGGGCAGTC GGTGTAGAGAGACAGTGCTGTGTGTCCGATGGTCACGTGGAA-3′ (SphI) |
The restriction enzymes used for genotyping KI cells are noted in parentheses, with their sequences underlined.
Neuronal differentiation of ES cells
Embryoid bodies were formed by growing ES cells in embryoid body formation medium; this mixture consisted of equal volumes of advanced DMEM/F12 (Thermo Fisher Scientific) and Neurobasal Medium (Thermo Fisher Scientific), supplemented with 10% KnockOut serum replacement (Thermo Fisher Scientific), 2 mM L-glutamine (Thermo Fisher Scientific), 100 units/mL penicillin, 100 µg/mL streptomycin, and 0.1 mM 2-mercaptoethanol. Two days later, the embryoid bodies were cultured in embryoid body formation medium that was supplemented with 5 µM retinoic acid. Three days later, differentiation of the embryoid bodies into neurons was induced by replacing the medium with neuronal differentiation medium. This medium consisted of a mixture of equal volumes of Advanced DMEM/F12 and Neurobasal Medium, supplemented with 2% B-27 Supplement (Thermo Fisher Scientific), 2 mM L-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin. On the following day, the embryoid bodies were digested with 0.25% Trypsin/EDTA (Merck), dispersed in Leibovitz’s L-15 Medium (Thermo Fisher Scientific) supplemented with 0.2 mg/mL DNase I (Merck), and seeded onto a poly-D-lysine/laminin-coated dish in the neuronal differentiation medium for an additional 4 days.
Lipid analyses
For the analysis of ceramides and PCs, HEK 293 T cells were transfected with the following combinations of plasmids: pEFh-3 × FLAG-ELOVL4 (WT or one of the mutants), pCE-puro 3 × FLAG-CERS3, and pCE-puro HA-ELOVL2 plasmids for analysis of ceramides; pEFh-3 × FLAG-ELOVL4 (WT or one of the mutants) and pCE-puro HA-ELOVL2 plasmids for analysis of PCs. Forty-eight hours after transfection, cells were collected and subjected to lipid extraction.
For the extraction of ceramides from HEK 293 T cells, the cells were suspended in 100 μL of water and mixed successively with 375 μL of chloroform/methanol/12 M formic acid solution (100:200:1, v/v), 125 μL of chloroform, and 125 μL of water. After centrifugation, the organic phase was recovered and treated with 71 μL of 0.5 M NaOH for 1 h at 37 °C to hydrolyze ester bonds in the glycerolipids. The reaction mixture was neutralized via the addition of 35.5 μL of 1 M formic acid and mixed successively with 135 μL of methanol and 210 μL of water. After centrifugation, the organic phase was recovered and dried. For the extraction of PCs from HEK 293 T or ES cells, the cells were suspended in 100 μL of water and mixed successively with 375 μL of chloroform/methanol/12 M formic acid solution (100:200:1, v/v), 0.5 pmol of seven deuterium (d7)-labeled internal standard (15:0/18:1-d7-PC; Avanti Polar Lipids, Alabaster, USA), 125 μL of chloroform, and 125 μL of water. After centrifugation, the organic phase was recovered and dried.
The dried lipids were suspended in a chloroform/methanol solution (1:2, v/v), and LC-MS/MS analyses of ceramides and PCs were performed using an LC-coupled triple quadrupole mass spectrometer Xevo TQ-S (Waters, Milford, MA, USA). Ceramides and PCs extracted from HEK 293 T cells were separated by LC using an ACQUITY UPLC CSH C18 column (1.7 μm particle size, 2.1 × 100 mm; Waters), and PCs extracted from ES cells were separated by LC using a YMC-Triart C18 metal-free column (1.9 μm particle size, 2.1 × 50 mm; YMC, Kyoto, Japan). Lipid separation by LC was performed at a flow rate of 0.3 mL/min for 25 min using a gradient system, in which mobile phase A and mobile phase B were mixed as follows: 0 min, 40% B; 0–18 min, gradient to 100% B; 18–23 min, 100% B; 23 min, return to 40% B; 23–25 min, 40% B. Mobile phase A consisted of an acetonitrile/water solution (3:2, v/v) that contained 5 mM ammonium formate, while mobile phase B consisted of an acetonitrile/2-propanol solution (1:9, v/v) that contained 5 mM ammonium formate. Separated lipids were ionized by electrospray ionization and analyzed in positive ion mode. Quantitative analyses were performed in the multiple reaction monitoring mode of the MS/MS using selected m/z values and appropriate collision energies and cone voltages (Tables 4 and 5). Data analyses were performed using MassLynx software (Waters). Ceramides were quantified using the external standard curve generated from C30:0 ceramide (Cayman Chemical, Ann Arbor, MI, USA) and expressed as the proportion of each ceramide species to the sum of all measured ceramide species listed in Table 4. PCs were quantified by calculating the ratio of the peak area of each PC species to that of the internal standard (15:0/18:1-d7-PC).
TABLE 4.
Selected m/z values and collision energies for the detection of ceramide species in the LC–MS/MS analysis
| Acyl chain | Precursor ions (Q1, m/z) |
Product ion (Q3, m/z) | Collision energy (eV) | |
|---|---|---|---|---|
| [M − H2O + H]+ | [M + H]+ | |||
| C18:0 | 548.6 | 566.6 | 264.3 | 20 |
| C20:0 | 576.6 | 594.6 | 264.3 | 20 |
| C22:0 | 604.6 | 622.6 | 264.3 | 25 |
| C24:0 | 632.6 | 650.6 | 264.3 | 30 |
| C26:0 | 660.7 | 678.7 | 264.3 | 30 |
| C28:0 | 688.7 | 706.7 | 264.3 | 30 |
| C30:0 | 716.7 | 734.7 | 264.3 | 35 |
| C32:0 | 744.8 | 762.8 | 264.3 | 40 |
| C34:0 | 772.8 | 790.8 | 264.3 | 40 |
| C18:1 | 546.6 | 564.6 | 264.3 | 20 |
| C20:1 | 574.6 | 592.6 | 264.3 | 20 |
| C22:1 | 602.6 | 620.6 | 264.3 | 25 |
| C24:1 | 630.6 | 648.6 | 264.3 | 30 |
| C26:1 | 658.7 | 676.7 | 264.3 | 30 |
| C28:1 | 686.7 | 704.7 | 264.3 | 30 |
| C30:1 | 714.7 | 732.7 | 264.3 | 35 |
| C32:1 | 742.8 | 760.8 | 264.3 | 35 |
| C34:1 | 770.8 | 788.8 | 264.3 | 40 |
| C36:1 | 798.8 | 816.8 | 264.3 | 40 |
| C38:1 | 826.8 | 844.8 | 264.3 | 40 |
| C24:4 | 624.6 | 642.6 | 264.3 | 30 |
| C26:4 | 652.7 | 670.7 | 264.3 | 30 |
| C28:4 | 680.7 | 698.7 | 264.3 | 30 |
| C30:4 | 708.7 | 726.7 | 264.3 | 35 |
| C32:4 | 736.8 | 754.8 | 264.3 | 35 |
| C34:4 | 764.8 | 782.8 | 264.3 | 40 |
| C36:4 | 792.8 | 810.8 | 264.3 | 40 |
| C38:4 | 820.9 | 838.9 | 264.3 | 40 |
| C30:5 | 706.7 | 724.7 | 264.3 | 35 |
| C32:5 | 734.7 | 752.7 | 264.3 | 35 |
| C34:5 | 762.8 | 780.8 | 264.3 | 40 |
| C36:5 | 790.8 | 808.8 | 264.3 | 40 |
| C38:5 | 818.8 | 836.8 | 264.3 | 40 |
| C30:6 | 704.7 | 722.7 | 264.3 | 35 |
| C32:6 | 732.7 | 750.7 | 264.3 | 35 |
| C34:6 | 760.8 | 778.8 | 264.3 | 40 |
| C36:6 | 788.8 | 806.8 | 264.3 | 40 |
| C38:6 | 816.8 | 834.8 | 264.3 | 40 |
TABLE 5.
Selected m/z values for the detection of PC species in the LC-MS/MS analysis
| Sum of two acyl chains | Precursor ion (Q1, m/z) | Product ion (Q3, m/z) |
|---|---|---|
| C33:1-d7 | 753.5 | 184.0 |
| C44:5 | 892.8 | 184.0 |
| C44:6 | 890.8 | 184.0 |
| C44:7 | 888.7 | 184.0 |
| C46:5 | 920.8 | 184.0 |
| C46:6 | 918.8 | 184.0 |
| C46:7 | 916.8 | 184.0 |
| C48:5 | 948.8 | 184.0 |
| C48:6 | 946.8 | 184.0 |
| C48:7 | 944.8 | 184.0 |
| C50:5 | 976.9 | 184.0 |
| C50:6 | 974.9 | 184.0 |
| C50:7 | 972.8 | 184.0 |
| C52:5 | 1004.9 | 184.0 |
| C52:6 | 1002.9 | 184.0 |
| C52:7 | 1000.9 | 184.0 |
| C54:5 | 1032.9 | 184.0 |
| C54:6 | 1030.9 | 184.0 |
| C54:7 | 1028.9 | 184.0 |
| C56:5 | 1061.0 | 184.0 |
| C56:6 | 1059.0 | 184.0 |
| C56:7 | 1056.9 | 184.0 |
Collision energy and cone voltage were 30 eV and 15 V, respectively.
Real-time quantitative RT-PCR
Total RNAs were isolated from ES cells using the NucleoSpin RNA Kit (Takara Bio, Kusatsu, Japan), and cDNAs were synthesized from total RNAs using the PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio). Real-time quantitative RT-PCR was performed using the KOD SYBR qPCR Mix (TOYOBO, Osaka, Japan), cDNAs, and specific forward and reverse primers for the respective genes (Table 6). The reaction was performed on a CFX96 Touch real-time PCR detection system (Bio-Rad, Hercules, CA, USA). The mRNA levels were normalized to that of Actb.
TABLE 6.
Primers used for real-time quantitative RT-PCR
| Primer name | Sequence |
|---|---|
| mElovl4-rt_F1 | 5′-ACGTGATCATGTACTCCTACTATGG-3′ |
| mElovl4-rt_R1 | 5′-CCGTTCGATGAGATACCATTCGTGG-3′ |
| mTubb3-rt_F3 | 5′-GAGGCCTCCTCTCACAAGTATGTGC-3′ |
| mTubb3-rt_R3 | 5′-GTTGCCAGCACCACTCTGACCAAAG-3′ |
| mElovl2-rt_F | 5′-GCTGGTCATCCTGTTCTTAAACTTC-3′ |
| mElovl2-rt_R | 5′-TTATTGAGCCTTCTTGTCCGTCATG-3′ |
| mElovl5-rt_F2 | 5′-TCGGGTGGCTGTTCTTCCAGATTGG-3′ |
| mElovl5-rt_R | 5′-AGGGAAGCTGTTGGTGTGTCCGTTG-3′ |
| Mouse_Actb_primer | Mouse Housekeeping Gene Primer Set (Takara Bio) |
Quantification and statistical analysis
Data are presented as means + SD or ± SD. Tests for normality of the data and outliers were not performed. Differences between groups were evaluated using Dunnett’s test in JMP 13 software (SAS Institute, Cary, NC, USA), and P values of less than 0.05 were considered to be statistically significant.
Funding Statement
This work was supported by KAKENHI grants: grant [numbers JP22H04986 (to A.K.) and JP22H02757 (to T.S.)] from the Japan Society for the Promotion of Science (JSPS) and by a Grant for Basic Science Research Projects (to T.S.) from the Sumitomo Foundation.
DISCLOSURE STATEMENT
The authors report there are no competing interests to declare.
REFERENCES
- 1.Robinson KJ, Watchon M, Laird AS.. Aberrant cerebellar circuitry in the spinocerebellar ataxias. Front Neurosci. 2020;14:707. doi: 10.3389/fnins.2020.00707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan R, Yau WY, O’Connor E, Houlden H.. Spinocerebellar ataxia: an update. J Neurol. 2019;266:533–544. doi: 10.1007/s00415-018-9076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corral-Juan M, Casquero P, Giraldo-Restrepo N, Laurie S, Martinez-Piñeiro A, Mateo-Montero RC, Ispierto L, Vilas D, Tolosa E, Volpini V, et al. New spinocerebellar ataxia subtype caused by SAMD9L mutation triggering mitochondrial dysregulation (SCA49). Brain Commun. 2022;4:fcac030. doi: 10.1093/braincomms/fcac030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kihara A. Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res. 2016;63:50–69. doi: 10.1016/j.plipres.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Sassa T, Kihara A.. Metabolism of very long-chain fatty acids: genes and pathophysiology. Biomol Ther (Seoul). 2014;22:83–92. doi: 10.4062/biomolther.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernert JT, Jr., Sprecher H.. An analysis of partial reactions in the overall chain elongation of saturated and unsaturated fatty acids by rat liver microsomes. J Biol Chem. 1977;252:6736–6744. doi: 10.1016/S0021-9258(17)39911-8. [DOI] [PubMed] [Google Scholar]
- 7.Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, Kihara A.. ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U S A. 2010;107:18439–18444. doi: 10.1073/pnas.1005572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaudin M, Sellami L, Martel C, Touzel-Deschênes L, Houle G, Martineau L, Lacroix K, Lavallée A, Chrestian N, Rouleau GA, et al. Characterization of the phenotype with cognitive impairment and protein mislocalization in SCA34. Neurol Genet. 2020;6:e403. doi: 10.1212/NXG.0000000000000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bourassa CV, Raskin S, Serafini S, Teive HA, Dion PA, Rouleau GA.. A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol. 2015;72:942–943. doi: 10.1001/jamaneurol.2015.0888. [DOI] [PubMed] [Google Scholar]
- 10.Bourque PR, Warman-Chardon J, Lelli DA, LaBerge L, Kirshen C, Bradshaw SH, Hartley T, Boycott KM.. Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet. 2018;4:e263. doi: 10.1212/NXG.0000000000000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, Martin C, Meloche C, Gravel M, Drouin CA, Rouleau GA, Nguyen DK, Cossette P.. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol. 2014;71:470–475. doi: 10.1001/jamaneurol.2013.6337. [DOI] [PubMed] [Google Scholar]
- 12.Haeri G, Hajiakhoundi F, Alavi A, Ghiasi M, Munhoz RP, Rohani M.. Congenital ichthyosis in a case of spinocerebellar ataxia type 34: a novel presentation for a known mutation. Mov Disord Clin Pract. 2021;8:275–278. doi: 10.1002/mdc3.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukherjee S, Roy M, Ghosh S, Guha G, Prasad Saha S, Dalal A.. Rare mutation in ELOVL4 gene in SCA34 and cognitive affection: expounding the role of cerebellum. Clin Neurol Neurosurg. 2021;210:106983. doi: 10.1016/j.clineuro.2021.106983. [DOI] [PubMed] [Google Scholar]
- 14.Ozaki K, Ansai A, Nobuhara K, Araki T, Kubodera T, Ishii T, Higashi M, Sato N, Soga K, Mizusawa H, et al. Prevalence and clinicoradiological features of spinocerebellar ataxia type 34 in a Japanese ataxia cohort. Parkinsonism Relat Disord. 2019;65:238–242. doi: 10.1016/j.parkreldis.2019.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, Yamane K, Irioka T, Ishiura H, Doi K, et al. A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol. 2015;72:797–805. doi: 10.1001/jamaneurol.2015.0610. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Lin Z, Wang H.. Progressive symmetric erythrokeratodermia with spinocerebellar ataxia due to ELOVL4 mutation in a Chinese family. Indian J Dermatol Venereol Leprol. 2021;88:132–132. doi: 10.25259/ijdvl_488_20. [DOI] [PubMed] [Google Scholar]
- 17.Xiao C, Binkley EM, Rexach J, Knight-Johnson A, Khemani P, Fogel BL, Das S, Stone EM, Gomez CM.. A family with spinocerebellar ataxia and retinitis pigmentosa attributed to an ELOVL4 mutation. Neurol Genet. 2019;5:e357. doi: 10.1212/nxg.0000000000000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagaraja RY, Sherry DM, Fessler JL, Stiles MA, Li F, Multani K, Orock A, Ahmad M, Brush RS, Anderson RE, et al. W246G mutant ELOVL4 impairs synaptic plasticity in parallel and climbing fibers and causes motor defects in a rat model of SCA34. Mol Neurobiol. 2021;58:4921–4943. doi: 10.1007/s12035-021-02439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hopiavuori BR, Anderson RE, Agbaga MP.. ELOVL4: very long-chain fatty acids serve an eclectic role in mammalian health and function. Prog Retin Eye Res. 2019;69:137–158. doi: 10.1016/j.preteyeres.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein PS, Tammur J, Singh N, Hutchinson A, Dixon M, Pappas CM, Zabriskie NA, Zhang K, Petrukhin K, Leppert M, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001;42:3331–3336. [PubMed] [Google Scholar]
- 21.Maugeri A, Meire F, Hoyng CB, Vink C, Van Regemorter N, Karan G, Yang Z, Cremers FP, Zhang K.. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci. 2004;45:4263–4267. doi: 10.1167/iovs.04-0078. [DOI] [PubMed] [Google Scholar]
- 22.Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 23.Grayson C, Molday RS.. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem. 2005;280:32521–32530. doi: 10.1074/jbc.m503411200. [DOI] [PubMed] [Google Scholar]
- 24.Karan G, Yang Z, Howes K, Zhao Y, Chen Y, Cameron DJ, Lin Y, Pearson E, Zhang K.. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol Vis. 2005;11:657–664. [PubMed] [Google Scholar]
- 25.Okuda A, Naganuma T, Ohno Y, Abe K, Yamagata M, Igarashi Y, Kihara A.. Hetero-oligomeric interactions of an ELOVL4 mutant protein: implications in the molecular mechanism of Stargardt-3 macular dystrophy. Mol Vis. 2010;16:2438–2445. [PMC free article] [PubMed] [Google Scholar]
- 26.Vasireddy V, Vijayasarathy C, Huang J, Wang XF, Jablonski MM, Petty HR, Sieving PA, Ayyagari R.. Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol Vis. 2005;11:665–676. [PubMed] [Google Scholar]
- 27.Esteve-Rudd J, Hazim RA, Diemer T, Paniagua AE, Volland S, Umapathy A, Williams DS.. Defective phagosome motility and degradation in cell nonautonomous RPE pathogenesis of a dominant macular degeneration. Proc Natl Acad Sci U S A. 2018;115:5468–5473. doi: 10.1073/pnas.1709211115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuny S, Cho WJ, Dimopoulos IS, Sauvé Y.. Early onset ultrastructural and functional defects in RPE and photoreceptors of a Stargardt-like macular dystrophy (STGD3) transgenic mouse model. Invest Ophthalmol Vis Sci. 2015;56:7109–7121. doi: 10.1167/iovs.15-17567. [DOI] [PubMed] [Google Scholar]
- 29.Aldahmesh MA, Mohamed JY, Alkuraya HS, Verma IC, Puri RD, Alaiya AA, Rizzo WB, Alkuraya FS.. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet. 2011;89:745–750. doi: 10.1016/j.ajhg.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diociaiuti A, Martinelli D, Nicita F, Cesario C, Pisaneschi E, Macchiaiolo M, Rossi S, Condorelli AG, Zambruno G, El Hachem M.. Two Italian patients with ELOVL4-related neuro-ichthyosis: expanding the genotypic and phenotypic spectrum and ultrastructural characterization. Genes. 2021;12:343. doi: 10.3390/genes12030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mir H, Raza SI, Touseef M, Memon MM, Khan MN, Jaffar S, Ahmad W.. A novel recessive mutation in the gene ELOVL4 causes a neuro-ichthyotic disorder with variable expressivity. BMC Med Genet. 2014;15:25. doi: 10.1186/1471-2350-15-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherry DM, Hopiavuori BR, Stiles MA, Rahman NS, Ozan KG, Deak F, Agbaga MP, Anderson RE.. Distribution of ELOVL4 in the developing and adult mouse brain. Front Neuroanat. 2017;11:38. doi: 10.3389/fnana.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett LD, Brush RS, Chan M, Lydic TA, Reese K, Reid GE, Busik JV, Elliott MH, Anderson RE.. Effect of reduced retinal VLC-PUFA on rod and cone photoreceptors. Invest Ophthalmol Vis Sci. 2014;55:3150–3157. doi: 10.1167/iovs.14-13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harkewicz R, Du H, Tong Z, Alkuraya H, Bedell M, Sun W, Wang X, Hsu YH, Esteve-Rudd J, Hughes G, et al. Essential role of ELOVL4 protein in very long chain fatty acid synthesis and retinal function. J Biol Chem. 2012;287:11469–11480. doi: 10.1074/jbc.m111.256073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Sandhoff R, Kono M, Zerfas P, Hoffmann V, Ding BC, Proia RL, Deng CX.. Depletion of ceramides with very long chain fatty acids causes defective skin permeability barrier function, and neonatal lethality in ELOVL4 deficient mice. Int J Biol Sci. 2007;3:120–128. doi: 10.7150/ijbs.3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasireddy V, Uchida Y, Salem N, Jr., Kim SY, Mandal MN, Reddy GB, Bodepudi R, Alderson NL, Brown JC, Hama H, et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (≥C28) and the unique ω-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabionet M, Bayerle A, Jennemann R, Heid H, Fuchser J, Marsching C, Porubsky S, Bolenz C, Guillou F, Gröne HJ, et al. Male meiotic cytokinesis requires ceramide synthase 3-dependent sphingolipids with unique membrane anchors. Hum Mol Genet. 2015;24:4792–4808. doi: 10.1093/hmg/ddv204. [DOI] [PubMed] [Google Scholar]
- 38.McMahon A, Lu H, Butovich IA.. A role for ELOVL4 in the mouse meibomian gland and sebocyte cell biology. Invest Ophthalmol Vis Sci. 2014;55:2832–2840. doi: 10.1167/iovs.13-13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyamoto M, Sassa T, Sawai M, Kihara A.. Lipid polarity gradient formed by ω-hydroxy lipids in tear film prevents dry eye disease. eLife. 2020;9:e53582. doi: 10.7554/elife.53582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otsuka K, Sawai-Ogawa M, Kihara A.. Formation of fatty alcohols-components of meibum lipids-by the fatty acyl-CoA reductase FAR2 is essential for dry eye prevention. FASEB J. 2022;36:e22216. doi: 10.1096/fj.202101733r. [DOI] [PubMed] [Google Scholar]
- 41.Sassa T, Tadaki M, Kiyonari H, Kihara A.. Very long-chain tear film lipids produced by fatty acid elongase ELOVL1 prevent dry eye disease in mice. FASEB J. 2018;32:2966–2978. doi: 10.1096/fj.201700947r. [DOI] [PubMed] [Google Scholar]
- 42.Sawai M, Watanabe K, Tanaka K, Kinoshita W, Otsuka K, Miyamoto M, Sassa T, Kihara A.. Diverse meibum lipids produced by Awat1 and Awat2 are important for stabilizing tear film and protecting the ocular surface. iScience. 2021;24:102478. doi: 10.1016/j.isci.2021.102478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulos A, Sharp P, Johnson D, Easton C.. The occurrence of polyenoic very long chain fatty acids with greater than 32 carbon atoms in molecular species of phosphatidylcholine in normal and peroxisome-deficient (Zellweger’s syndrome) brain. Biochem J. 1988;253:645–650. doi: 10.1042/bj2530645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson BS, Johnson DW, Poulos A.. Unique molecular species of phosphatidylcholine containing very-long-chain (C24–C38) polyenoic fatty acids in rat brain. Biochem J. 1990;265:763–767. doi: 10.1042/bj2650763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nie L, Pascoa TC, Pike ACW, Bushell SR, Quigley A, Ruda GF, Chu A, Cole V, Speedman D, Moreira T, et al. The structural basis of fatty acid elongation by the ELOVL elongases. Nat Struct Mol Biol. 2021;28:512–520. doi: 10.1038/s41594-021-00605-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizutani Y, Sun H, Ohno Y, Sassa T, Wakashima T, Obara M, Yuyama K, Kihara A, Igarashi Y.. Cooperative synthesis of ultra long-chain fatty acid and ceramide during keratinocyte differentiation. PLoS One. 2013;8:e67317. doi: 10.1371/journal.pone.0067317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto M, Sassa T, Kyono Y, Uemura H, Kugo M, Hayashi H, Imai Y, Yamanishi K, Kihara A.. Comprehensive stratum corneum ceramide profiling reveals reduced acylceramides in ichthyosis patient with CERS3 mutations. J Dermatol. 2021;48:447–456. doi: 10.1111/1346-8138.15725. [DOI] [PubMed] [Google Scholar]
- 48.Kim M, Habiba A, Doherty JM, Mills JC, Mercer RW, Huettner JE.. Regulation of mouse embryonic stem cell neural differentiation by retinoic acid. Dev Biol. 2009;328:456–471. doi: 10.1016/j.ydbio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE.. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 50.Deák F, Anderson RE, Fessler JL, Sherry DM.. Novel cellular functions of very long chain-fatty acids: insight from ELOVL4 mutations. Front Cell Neurosci. 2019;13:428. doi: 10.3389/fncel.2019.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeboah GK, Lobanova ES, Brush RS, Agbaga MP.. Very long chain fatty acid-containing lipids: a decade of novel insights from the study of ELOVL4. J Lipid Res. 2021;62:100030. doi: 10.1016/j.jlr.2021.100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikeda M, Kanao Y, Yamanaka M, Sakuraba H, Mizutani Y, Igarashi Y, Kihara A.. Characterization of four mammalian 3-hydroxyacyl-CoA dehydratases involved in very long-chain fatty acid synthesis. FEBS Lett. 2008;582:2435–2440. doi: 10.1016/j.febslet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 53.Borroni B, Di Gregorio E, Orsi L, Vaula G, Costanzi C, Tempia F, Mitro N, Caruso D, Manes M, Pinessi L, et al. Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38). Parkinsonism Relat Disord. 2016;28:80–86. doi: 10.1016/j.parkreldis.2016.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Gregorio E, Borroni B, Giorgio E, Lacerenza D, Ferrero M, Lo Buono N, Ragusa N, Mancini C, Gaussen M, Calcia A, et al. ELOVL5 mutations cause spinocerebellar ataxia 38. Am J Hum Genet. 2014;95:209–217. doi: 10.1016/j.ajhg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gazulla J, Orduna-Hospital E, Benavente I, Rodríguez-Valle A, Osorio-Caicedo P, Alvarez-de Andrés S, García-González E, Fraile-Rodrigo J, Fernández-Tirado FJ, Berciano J.. Contributions to the study of spinocerebellar ataxia type 38 (SCA38). J Neurol. 2020;267:2288–2295. doi: 10.1007/s00415-020-09840-1. [DOI] [PubMed] [Google Scholar]
- 56.Jojima K, Edagawa M, Sawai M, Ohno Y, Kihara A.. Biosynthesis of the anti-lipid-microdomain sphingoid base 4,14-sphingadiene by the ceramide desaturase FADS3. FASEB J. 2020;34:3318–3335. doi: 10.1096/fj.201902645r. [DOI] [PubMed] [Google Scholar]
- 57.Kihara A, Anada Y, Igarashi Y.. Mouse sphingosine kinase isoforms SPHK1a and SPHK1b differ in enzymatic traits including stability, localization, modification, and oligomerization. J Biol Chem. 2006;281:4532–4539. doi: 10.1074/jbc.m510308200. [DOI] [PubMed] [Google Scholar]
- 58.Ohno Y, Nakamichi S, Ohkuni A, Kamiyama N, Naoe A, Tsujimura H, Yokose U, Sugiura K, Ishikawa J, Akiyama M, et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci U S A. 2015;112:7707–7712. doi: 10.1073/pnas.1503491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohno Y, Kamiyama N, Nakamichi S, Kihara A.. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω-O-acylceramide. Nat Commun. 2017;8:14610. doi: 10.1038/ncomms14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitamura T, Takagi S, Naganuma T, Kihara A.. Mouse aldehyde dehydrogenase ALDH3B2 is localized to lipid droplets via two C-terminal tryptophan residues and lipid modification. Biochem J. 2015;465:79–87. doi: 10.1042/bj20140624. [DOI] [PubMed] [Google Scholar]
- 61.Kitamura T, Seki N, Kihara A.. Phytosphingosine degradation pathway includes fatty acid α-oxidation reactions in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2017;114:E2616–E2623. doi: 10.1073/pnas.1700138114. [DOI] [PMC free article] [PubMed] [Google Scholar]