

ABSTRACT
Phage display is an established method for the in vitro selection of recombinant antibodies and other proteins or peptides from gene libraries. Here we describe SpyDisplay, a phage display method in which the display is achieved via SpyTag/SpyCatcher protein ligation instead of genetically fusing the displayed protein to a phage coat protein. In our implementation, SpyTagged antibody antigen-binding fragments (Fabs) are displayed via protein ligation on filamentous phages carrying SpyCatcher fused to the pIII coat protein. A library of genes encoding Fab antibodies was cloned in an expression vector containing an f1 replication origin, and SpyCatcher-pIII was separately expressed from a genomic locus in engineered E. coli. We demonstrate the functional, covalent display of Fab on phage, and rapidly isolate specific high-affinity clones via phage panning, confirming the robustness of this selection system. SpyTagged Fabs, the direct outcome of the panning campaign, are compatible with modular antibody assembly using prefabricated SpyCatcher modules and can be directly tested in diverse assays. Furthermore, SpyDisplay streamlines additional applications that have traditionally been challenging for phage display: we show that it can be applied to N-terminal display of the protein of interest and it enables display of cytoplasmically folding proteins exported to periplasm via the TAT pathway.
KEYWORDS: SpyTag/SpyCatcher, antibody phage display, SpyDisplay, Fab antibody library
Introduction
The in vitro selection of peptides and proteins with desired properties from large gene libraries is a powerful approach that has been used extensively for the discovery of binding molecules, including antibodies.1,2 All in vitro selection technologies require the physical linkage of genotype and phenotype. Phage display is the oldest and most widely used method due to its robustness and favorable properties, such as speed, simplicity, and accommodation of large libraries. To couple genotype to phenotype, the proteins to be selected are displayed on the surface of engineered filamentous M13 phages, which contain the genetic information of the presented proteins. In conventional phage display, the displayed protein is genetically fused to a coat protein of the phage, in most cases to the minor coat protein pIII. Instead of a full phage genome, smaller plasmid derivatives called phagemids are commonly used in phage display, as they simplify cloning and allow the creation of larger libraries.3,4 Such phagemids contain the genes for the displayed proteins fused to the gene encoding the phage coat protein used for display, antibiotic resistance genes, and genetic elements required for plasmid-like replication as well as for replication of ssDNA and its packaging in phage capsid. Phage display with phagemids necessitates the use of helper phage, which provides the remaining structural and regulatory phage proteins that are not present in the phagemid and thus allows phage assembly after superinfection of phagemid-containing bacteria. After several rounds of phage display, the selected genes are typically subcloned into a suitable expression plasmid for screening and further analysis.5
The SpyTag/SpyCatcher protein ligation technology is a versatile method to covalently link two proteins.6 The SpyTag, a short peptide of 13 amino acids, reacts spontaneously with SpyCatcher protein (12.3 kDa) to form an isopeptide bond between an aspartic acid residue in the tag and a lysine residue in the Catcher, crosslinking the two (Figure 1a: SpyTag/SpyCatcher system). The reaction is fast, specific and has been further optimized in the form of the SpyTag2/SpyCatcher2 and SpyTag3/SpyCatcher3 systems.8,9 This technology has been used in many different applications, for example in the production of vaccine nanoparticles or stabilized enzymes.10 Furthermore, it has recently been applied for modular antibody assembly and site-specific labeling of antibodies.11,12
Figure 1.
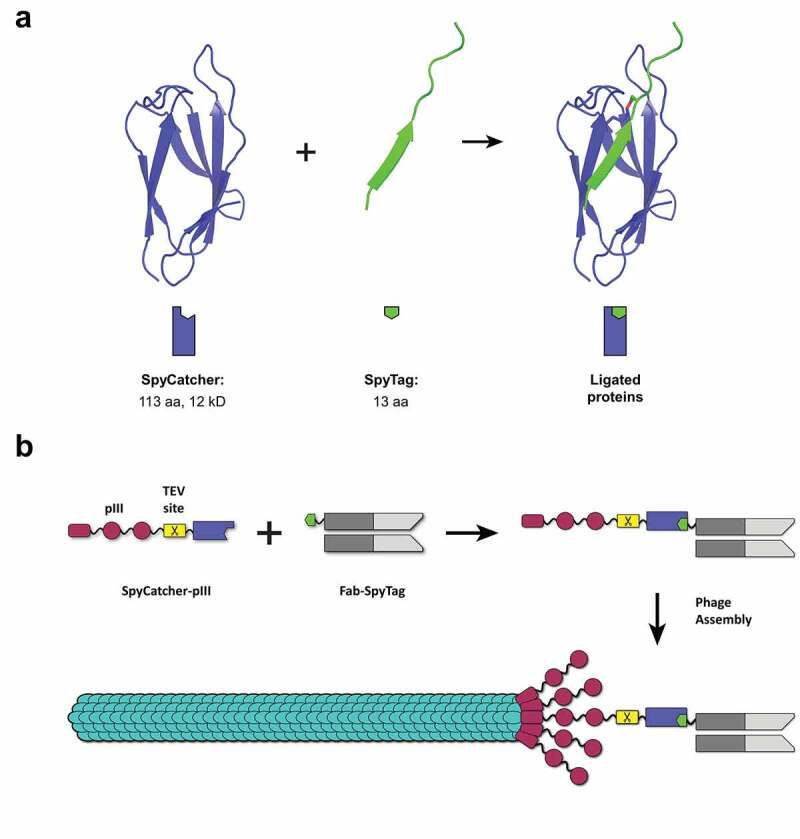
SpyTag/SpyCatcher and the concept of SpyDisplay.
(a) SpyCatcher (violet) and SpyTag (green) react spontaneously to form an isopeptide bond (red). Structures from PDB 4MLI.7 (b) In SpyDisplay of antibodies, SpyCatcher-pIII reacts with SpyTagged Fab antibody fragments in the periplasm before (or concomitant with) phage assembly, resulting in phages displaying antibodies (shown here for monovalent display).
In this work, we establish a phage display method based on SpyTag/SpyCatcher technology that we refer to as SpyDisplay. We show that SpyTagged antigen binding fragments (Fabs) expressed in E. coli react in vivo with coexpressed SpyCatcher-pIII, resulting in Fab-SpyCatcher-pIII fusion proteins that are incorporated into phage particles, thus enabling phage display (Figure 1b). In SpyDisplay, the library and display system are separated by using a phagemid-based expression vector which encodes the SpyTagged Fab, and a separate genomically integrated and inducible gene encoding SpyCatcher-pIII. This design avoids any subcloning steps for the expression of free Fab and allows the use of smaller phagemids without the pIII gene. We demonstrate the selection of high-affinity Fabs from an antibody library using SpyDisplay. Furthermore, we show that SpyDisplay is a straightforward way for N-terminal phage display (where the protein of interest is displayed with a free C-terminus) and is also compatible with the display of cytoplasmic folding proteins, exported to bacterial periplasm via the twin arginine translocase (TAT) pathway.
Results
Generation of a SpyCatcher-pIII expressing E. coli strain and production of SpyCatcher-displaying phages
To present antibodies on the phage surface via SpyDisplay, SpyCatcher needs to be fused to M13 minor coat protein pIII. We decided to use the improved version SpyCatcher2/SpyTag2,8 referred to below as SpyCatcher/SpyTag. A TEV protease cleavage site was placed between SpyCatcher and pIII to allow mild and affinity-independent elution of bound phages by proteolytic cleavage.13 The SpyCatcher-TEV-pIII expression cassette was integrated into the E. coli genome to avoid using an additional plasmid. The SpyCatcher-pIII gene under control of the arabinose promotor was integrated at the araBAD locus of TG1 E. coli (Figure 2a), replacing the endogenous araBAD genes required for arabinose metabolism.14 In this new bacterial strain, termed SK25, arabinose could be used to induce the expression of SpyCatcher-pIII, as assessed by western blotting (Figure 2b)
Figure 2.
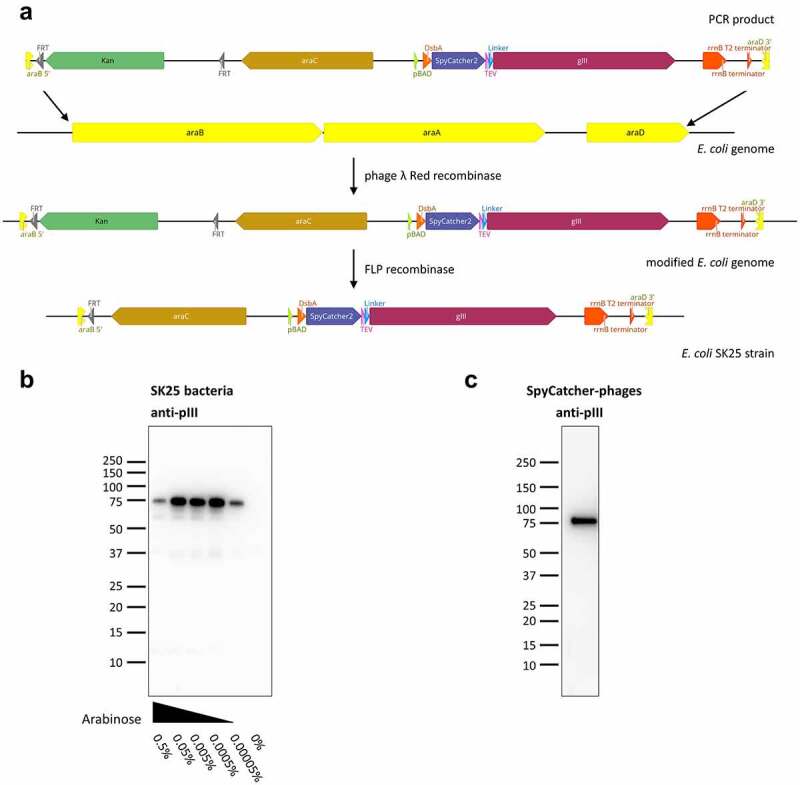
Creation of SpyCatcher-pIII expressing E. coli strain SK25.
(a) Phage λ Red recombinase A was used to replace the araBAD genes of TG1 E. coli with a cassette consisting of a kanamycin resistance gene, araC, and SpyCatcher-pIII under control of the pBAD promotor. In the second step, the kanamycin resistance cassette was excised via FLP recombinase. (b) Expression of SpyCatcher-pIII in SK25 bacteria analyzed by immunoblotting. SK25 cells were grown overnight at 22°C in presence of varying concentrations of arabinose and equal numbers of bacteria were lysed. Lysates were probed with anti-M13-pIII followed by sheep anti-mouse IgG (H/L):HRP. For SpyCatcher-pIII, the apparent molecular weight by SDS PAGE of 75 kDa is higher than the calculated molecular weight of 57 kDa. (c) Immunoblot of phages produced by infecting SK25 bacteria with Hyperphage. PEG-precipitated phages were separated electrophoretically, immunoblotted, and detection was performed as described in B.
To test the incorporation of SpyCatcher-pIII into the phage capsid, we produced SpyCatcher phages by infecting SK25 cells with Hyperphage, a helper phage lacking pIII.15 Western blotting of these phages with anti-pIII antibody yielded a single, strong band for SpyCatcher-pIII (Figure 2c). SpyCatcher-pIII migrated at a slightly higher molecular weight than calculated, which has also been observed for wildtype pIII.16
Production of Fab-phages in E. coli SK25
To establish antibody phage display via SpyTag/SpyCatcher, Fabs of two therapeutic antibodies, anti-tumor necrosis factor (TNF) adalimumab and anti-ErbB2 trastuzumab, were cloned into the expression vector pBBx2-F-Spy2-H,11 which contains an f1 phage replication origin and can therefore be used as a phagemid. In this phagemid, Fab expression is under control of the lac promotor and the heavy chain of the Fab is C-terminally fused to three consecutive peptide tags: FLAG-tag, SpyTag2, and His6-tag. The phagemids were transformed into E. coli SK25 cells and the cells were subsequently infected with VCSM13 helper phage. Monovalent Fab-phages displaying not more than one antibody Fab per phage particle were produced using arabinose to induce expression of SpyCatcher-pIII and isopropyl β-D-1-thiogalactopyranoside (IPTG) to induce expression of the Fab. In parallel, polyvalent Fab-phages were produced using Hyperphage and analogous induction of expression with arabinose and IPTG. Phages were purified and concentrated by polyethylene glycol (PEG) precipitation from the supernatants of overnight cultures.
Initially, phage titers of mono- and polyvalent Fab-phages measured by spot titration differed by three orders of magnitude and were 3.1 × 1013 colony-forming units (cfu)/mL (SD = 0.5 × 1013 cfu/mL) for monovalent Fab-phages and 6.5 × 109 cfu/mL (SD = 1.9 × 109 cfu/mL) for the polyvalent ones. It has been described that fusions to the N-terminus of pIII decrease phage infectivity and we suspected this to be the explanation for the apparent low titer of the polyvalent Fab-phages.17 Therefore, to allow better comparison of titers, Fab-phage particles were digested with TEV protease prior to spot titration, cleaving the Fab-SpyCatcher fusion from the pIII protein. After treatment with TEV protease, the titer of the monovalent phages was unchanged (2.8 × 1013 cfu/mL; SD = 0.8 × 1013 cfu/mL), while the polyvalent titer was 100-fold higher (7.2 × 1011 cfu/mL; SD = 1.5 × 1011 cfu/mL) than before protease treatment. The remaining difference of two orders of magnitude between regular helper phage and Hyperphage titer has been observed before17 and is likely caused by steric hindrance of the modified pIII proteins during phage assembly.
Western blots of monovalent and polyvalent SpyDisplay Fab-phages confirmed that Fab-SpyCatcher-pIII fusions were successfully inserted into the phage particles (Figure 3a). As wildtype pIII is incorporated faster into the phage coat than modified pIII, monovalent phages had a much lower ratio of Fab-pIII to wildtype pIII (about 4%, estimated by densitometry) than the polyvalent phages (Figure 3a). Since an estimated 4% of all pIII proteins carry a Fab and there are 5 copies of pIII per phage particle, approximately 20% of all phages display Fab on their surface in the monovalent setup, a range similar to that of other monovalent phage display libraries.16 As expected, polyvalent Fab-phages generated in the absence of wildtype pIII exhibited a much higher Fab display rate. However, even in polyvalent display, only roughly 80% of the SpyCatcher-pIII was coupled to a full-length Fab, whereas the remaining SpyCatcher-pIII was coupled to what we presume to be a short degradation product of the Fab, caused by cleavage within the CH1 domain of the Fab heavy chain.18 We further evaluated the phages by ELISA with anti-pVIII-horseradish peroxidase (HRP) as detection reagent. By coating cognate and irrelevant antigens, we confirmed that the displayed Fabs were functional and specific (Figure 3b). To test the elution of SpyDisplay phages by proteolysis, phages bound to immobilized antigen were incubated with TEV protease or control buffer for 30 minutes, and residual bound phages after washing were quantified by ELISA, confirming the effectiveness of our elution protocol (Figure 3c, Suppl. Fig. S1).
Figure 3.
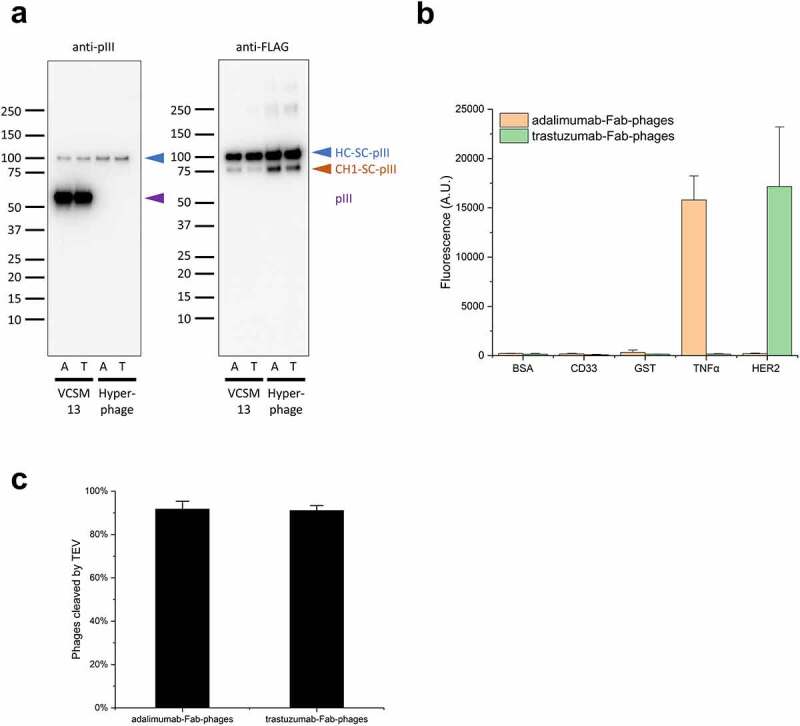
Production of Fab-phages.
(a) Immunoblots of SpyDisplay phages displaying Fabs of adalimumab (A) or trastuzumab (T) and produced with VCSM13 or Hyperphage as helper phage. Detection was performed with anti-M13-pIII followed by sheep anti-mouse IgG (H/L):HRP or anti-FLAG-HRP. Bands corresponding to the heavy chain fusion, degraded heavy chain fusion, and wildtype pIII are marked. (b) ELISA with polyvalent Fab-phages on cognate and irrelevant antigens, detection with anti-pVIII-HRP. (c) Fractions of monovalent Fab-phages eluted from MaxiSorp plates after treatment with TEV Protease for 30 minutes, relative to buffer control.
SpyTag/SpyCatcher-based Fab-phage assembly occurs intracellularly
For coupling of phenotype to genotype, it is essential that the ligation of SpyCatcher-pIII to the SpyTagged Fab takes place inside the bacterial cell. If phage particles presenting free SpyCatcher-pIII were secreted into the supernatant, ligation with Fabs released from other clones in the Fab-phage production culture could occur. This would lead to phages presenting antibodies different from the ones encoded by their phagemid. These scenarios can be tested by spiking experiments, in which SK25 bacteria expressing an antibody A are mixed in large excess with bacteria expressing an antibody B, and Fab-phages are produced in the mixed culture. For example, at 100:1 mixing ratio of A and B, and assuming equal Fab and phage production rates, the ratio of phagemids encoding antibodies A and B in the phage population is expected to be A:B = 100:1, and the ratio of free antibody released to the medium during phage production would also be A:B = 100:1.
If coupling occurred exclusively intracellularly, 100% of the phages displaying antibody B would carry the correct genotype B. On the other end of the spectrum, if coupling occurred exclusively in the medium, one would expect the following distribution: 98.01% of phages displaying A and encoding A (A-A), 0.99% displaying B and encoding A (B-A), 0.99% displaying A and encoding B (A-B), 0.01% displaying B and encoding B (B-B). Therefore, 99% of phages displaying antibody B (B-A and B-B) would carry the wrong genotype A (0.99%: 0.01% of total phages). Identifying the genotype of phages displaying the underrepresented antibody B is thus a sensitive method to assess the degree of extracellular coupling of Fab to phage.
We therefore mixed exponentially growing cultures of SK25 cells expressing Fabs of therapeutic antibodies adalimumab and trastuzumab in ratios of either 100:1 or 1:100 immediately before superinfection with VCSM13 helper phage. Fab-phages were produced overnight and used for one round of panning on the cognate antigen of the underrepresented antibody. As control for unspecific binding, pure adalimumab and trastuzumab Fab-phages were produced separately, quenched with SpyTag3 peptide to saturate potentially free SpyCatcher sites, and also mixed in volume ratios of 100:1 and 1:100. Phages eluted after the panning round were rescued in SK25 cells and plated. Ninety-five clones were picked from each condition and sequenced. Selection of trastuzumab phages on ErbB2 yielded 100% correct clones (95/95), while selection of adalimumab-phages on TNF resulted in 96% correct clones (91/95). In the corresponding controls, 99% and 100% correct clones were found, respectively (94/95; 95/95).
The results of both variations of the experiment are in strong agreement with a vast preponderance of intracellular ligation of Fabs to SpyCatcher-pIII. The small number of incorrect genotypes found is similar to the control and is therefore most likely due to unspecific phage binding not unusual in phage display.19
SpyDisplay enables N-terminal display and display mediated via the TAT pathway
To display a protein N-terminally on phages, i.e., with a free and unmodified C-terminus, it should be sufficient to attach the SpyTag to the N-terminus of the displayed protein, as SpyTag-SpyCatcher ligation is agnostic toward the position of the tag.20 To test the efficiency of N-terminal SpyDisplay, we added SpyTag to the N-terminus of maltose binding protein (MBP) and of a well characterized anti-fluorescein single-chain (scFv) antibody.21 Western blotting of phages produced with Hyperphage showed good display rates, with about 95% of all pIII proteins carrying the displayed protein (Figure 4a, left). Furthermore, we confirmed correct folding of the scFv antibody by testing its capacity to bind its antigen via ELISA (Figure 4a, right). These data confirm that N-terminal display with SpyDisplay is possible and indeed well-working.
Figure 4.
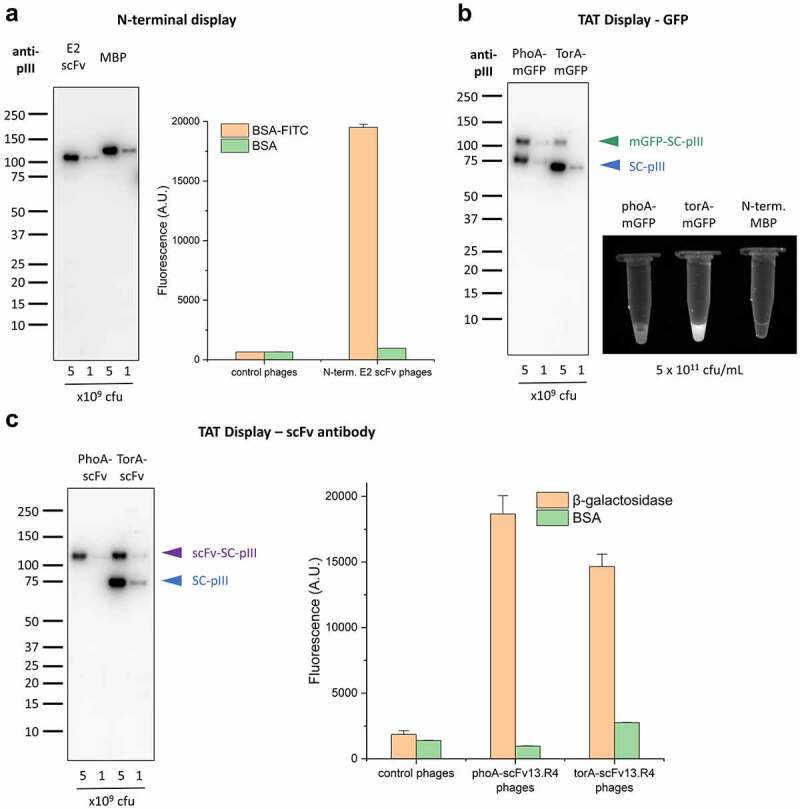
Versatility of display setups with SpyDisplay.
(a) Left: Immunoblot analysis of polyvalent SpyDisplay phages displaying the E2 scFv or MBP with an N-terminal SpyTag. Detection was performed with anti-M13-pIII followed by sheep anti-mouse IgG (H/L):HRP. Right: ELISA of polyvalent phages displaying N-terminally SpyTagged E2 scFv on control (BSA) or cognate antigen (BSA-FITC), in comparison to control phages (displaying N-terminally SpyTagged MBP). (b) Left: Immunoblot analysis of polyvalent SpyDisplay phages displaying mGFP-SpyTag with a PhoA or TorA leader peptide. Detection performed with anti-M13-pIII followed by sheep anti-mouse IgG (H/L):HRP. Right: Fluorescence image of GFP-phages and control phages at equal concentrations (5 × 1011 cfu/mL) in microcentrifuge tubes. (c) Left: Immunoblot analysis of polyvalent SpyDisplay phages displaying scFv13.R4-SpyTag with a PhoA or TorA leader peptide. Detection performed with anti-M13-pIII followed by sheep anti-mouse IgG (H/L):HRP. Right: ELISA of polyvalent phages displaying scFv13.R4 with on control (BSA) or cognate antigen (b-galactosidase), in comparison to control phages.
Another benefit of SpyDisplay could be the possibility to display proteins that fold in the cytoplasm. In addition to the Sec-dependent protein translocation pathways, where the proteins fold in the periplasm, bacteria possess the TAT pathway.22 In this pathway, proteins fold in the cytoplasm and are transported in folded state across the plasma membrane into the periplasm. However, using the TAT pathway to display proteins fused to full-length pIII has been challenging in the past, as periplasmic export of full-length pIII is incompatible with this translocation pathway, possibly due to multiple internal disulfide bonds.23 We expressed two proteins with a TAT pathway signal sequence, monomeric green fluorescent protein (mGFP) and an antibody that can fold both in the cytoplasm or periplasm (scFv13.R4) against β-galactosidase.24 GFP has been shown to only efficiently mature its fluorophore when expressed in the cytoplasm, but not in the periplasm.25 Indeed, when mGFP-SpyTag equipped with a TorA TAT signal sequence was expressed in SK25 cells superinfected with Hyperphage, strongly fluorescent phages were produced, whereas, at the same phage particle concentration, control PhoA-mGFP-SpyTag (Sec pathway) phages showed only weak fluorescence (Figure 4b, right), demonstrating the efficacy of TAT pathway-driven SpyDisplay. When analogously expressed in SK25 superinfected with Hyperphage, intracellularly folding TorA-scFv13.R4-SpyTag showed similar activity in ELISA as periplasmatically folding PhoA-scFv13.R4-SpyTag (Figure 4c). Western blot analysis (Figures 4B, 4c) revealed relatively modest display rates via the TAT pathway (about 10% for mGFP and 30% for the scFv in relation to total pIII). This is probably caused by the TAT pathway not exporting sufficient protein into the periplasm to fully occupy all SpyCatcher sites on the polyvalent phages. The apparent free SpyCatcher sites in the case of PhoA-mGFP display are in fact coupled to a small peptide containing FLAG and SpyTag, caused by degradation of mGFP in the periplasm. This degradation does not occur when mGFP is displayed using the TAT pathway (Suppl. Fig. S2 and slight band shift in Figure 4b). The presence of excess free SpyCatcher with TAT-based display suggests that the TAT pathway may have lower efficiency than Sec-based export, at least during phage production. Therefore, monovalent display might be best suited for TAT-based display.
Selection of antibodies from a SpyDisplay Fab-phage library
To test the efficiency of SpyDisplay for selecting high-affinity antibodies, we used a subset of our in-house constructed human Fab library termed Pioneer. The antibodies in this library were of the germlines IGHV1-69 and IGLV3-1, with all six complementarity-determining regions (CDRs) diversified. This Fab library was cloned in the pBBx2-F-Spy2-H expression vector and was transformed into SK25 cells, yielding 1.3 × 1011 transformants. Monovalent Fab-phages were produced by infection of library transformants with VCSM13.
We performed SpyDisplay pannings against mGFP and against the paratope of the therapeutic antibody sarilumab to generate anti-idiotype antibodies. Monovalent display was used for all panning rounds with the aim of selecting high-affinity antibodies. Both antigens were immobilized on MaxiSorp plates in two different ways, by passive adsorption and by using biotinylated antigens on preimmobilized streptavidin/neutravidin, thus yielding four independent panning setups. After three rounds of panning, the panning output was transformed into E. coli SK13, a bacterial strain optimized for the expression of SpyTagged Fabs by removal of two proteases which cleave SpyTag2 in the periplasm.11
To screen for antigen-binding clones, 368 randomly picked single colonies from each panning (i.e., 736 colonies per antigen) were grown in 384-well plates, and Fab antibodies were expressed overnight. The next day, bacteria were lysed, and antibody-containing lysates were tested by ELISA. Four hundred and seventy-two clones (64.1%) from the mGFP pannings and 550 clones (74.7%) from the sarilumab pannings showed ELISA signals of at least tenfold over background (Figure 5a). For passively adsorbed antigens, 30 clones with the highest ELISA signal were sequenced for each target. From pannings on biotinylated antigens, 95 clones with the highest ELISA signals were further screened for a low koff-rate via biolayer interferometry (BLI),26 and for each antigen the 20 clones with the lowest koff-rate were sequenced. For mGFP, sequencing a total of 50 clones resulted in 34 unique antibodies (68%), whereas for sarilumab, 23 unique antibodies (46%) were found. Next, all unique Fabs were expressed in 50 mL cultures, purified,27 and their monovalent affinities were measured by BLI on immobilized antigen. As mGFP-coated sensors could not be regenerated, anti-mGFP antibodies were first measured at a single concentration, and only for the top 20 antibodies a full kinetic measurement was performed. 98% of all antibodies measured (20 anti-mGFP and 23 anti-sarilumab) had affinities lower than 10 nM, and 37% had affinities lower than 1 nM. The best antibodies against mGFP and sarilumab had affinities of 40 pM and 24 pM, respectively (Figure 5b, Supp. Fig S3: Sensorgrams of all antibodies found). The measured affinities demonstrate the suitability of SpyDisplay for selection of high-affinity antibodies.
Figure 5.
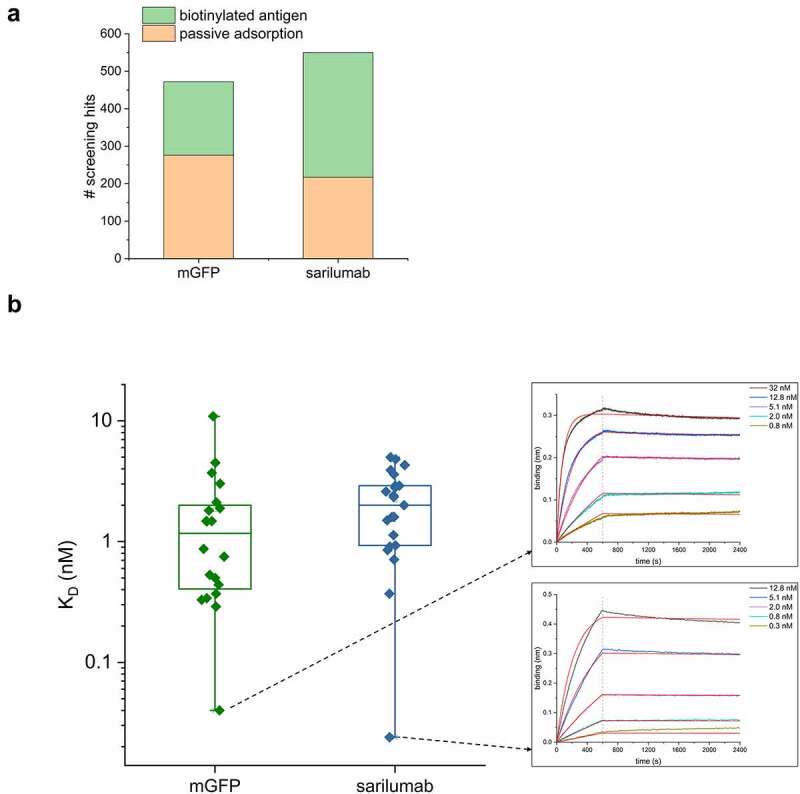
SpyDisplay selections of antibodies from a Fab library. (a) Number of positive hits (>10-fold over background in ELISA on cognate antigen) from screening Fabs generated against mGFP or the paratope of sarilumab (out of 736 tested antibodies per antigen). (b) Left: Distribution of KD values of sequenced and purified antibodies against mGFP and sarilumab. Right: BLI sensorgrams of the highest affinity antibodies against each antigen.
Discussion
In this study, we have established SpyDisplay as a new phage display method with some critical differences to traditional phage display. A key distinction between SpyDisplay and traditional antibody phage display is the separate folding of antibody and coat protein, which may be beneficial for the display of correctly folded antibodies. We have not found a simple way to prove this experimentally, as essentially all antibodies we work with have already gone through a selection by phage display, and as our libraries are already optimized for expression in E. coli. With our assumption, which is derived from first principles, we hope to encourage researchers who have struggled with phage display of difficult proteins to evaluate the suitability of SpyDisplay for their proteins of interest. In practical terms, the use of the library phagemid as expression plasmid saves time by eliminating a molecular cloning step before or after screening, which is laborious, especially if many pannings are performed in parallel. Alternative methods for avoiding subcloning, such as using an amber stop codon between Fab and pIII, have been shown to deliver sufficient free Fab for screening and small-scale expressions. However, due to low expression yields, production of larger amounts of Fab still necessitates the use of a dedicated expression vector.28 Having a phagemid without pIII has a further advantage during library construction because, as transformation efficiency inversely correlates with plasmid size, larger libraries can be generated by using the smaller SpyDisplay phagemid which lacks the pIII gene (1230 bp).29 Additionally, our optimized SpyDisplay protocol allows one panning round to be performed in a single day without the need for plating the output, which is especially important for high throughput antibody selections. Finally, SpyDisplay enables selection of antibodies with high affinity against folded proteins and antibody paratopes. The suitability of SpyDisplay for selection of antibodies against challenging targets like unstructured proteins will have to be shown in the future, while bearing in mind that the success of selections is also contingent upon the quality of the antibody library.
Monovalent and polyvalent phage display are commonly implemented by switching the type of helper phage. Monovalent display is crucial for the selection of high-affinity antibodies, whereas polyvalent display provides avidity. The efficiency of monovalent SpyDisplay for the selection of high-affinity antibodies is demonstrated by the selection of antibodies in the low pM range in our test pannings. As for regular phage display, use of Hyperphage enables polyvalent SpyDisplay and indeed showed high display rates. Phage titers were lower than for monovalent SpyDisplay yet still adequate for panning with standard library sizes. We envision that, in the context of Fab library panning, polyvalent SpyDisplay will facilitate the selection of antibodies against challenging targets like carbohydrates.30 Affinities of such antibodies can later be improved by an optional affinity maturation process.
We have shown that the displayed antibody and the anchoring SpyCatcher-pIII protein, which are translated independently of each other, form a fusion protein exclusively in the periplasm. Optimized periplasmic secretion signal peptides for each polypeptide chain can be used. In this implementation of antibody SpyDisplay, three different signal peptides are used: OmpA and PhoA for light and heavy antibody chains, and DsbA for the SpyCatcher-pIII fusion. Furthermore, we showed that the TAT pathway is compatible with SpyDisplay, enabling the cytoplasmic folding of the displayed proteins. The display rate via the TAT pathway could potentially be increased through optimization of the TAT signal peptide. We anticipate that TAT-based display will be useful for selecting intrabodies, antibodies that can fold in the cytoplasm, without the need for specialized intrabody libraries.
Mazor et al. also achieved a separation of the expression of antibody and coat protein by forming a complex between an IgG and Fc-binding ZZ-domain-pIII fusion.30 Similarly, the Jun/Fos leucine zipper has been used to link the displayed protein to pIII.31 Since these methods are non-covalent, the complex might dissociate during stringent washing steps required for the selection of high-affinity antibodies. However, it is possible to stabilize the leucine zipper by introducing a disulfide bond between the dimerized proteins.32 CysDisplay is another phage display method to express the antibody separately from pIII and relies on the spontaneous formation of disulfide bonds between engineered free cysteines at the C-terminus of Fab heavy chain and the N-terminus of pIII. While this method has proven powerful and became the basis of the HuCAL platform,16 significant side reactions in this setup are the formation of pIII homodimers as well as Fab homodimers, making the control of the display rate much less straightforward compared to the specific SpyTag-SpyCatcher reaction.
In a previous study,11 we showed that efficient periplasmic expression of SpyTagged Fabs is only possible in a protease knockout strain of E. coli, termed SK13. However, the proteases responsible for SpyTag cleavage do not seem to harm the assembly of Fab-SpyCatcher-pIII-phages. This suggests that the ligation of SpyTagged Fab to SpyCatcher-pIII occurs sufficiently fast in vivo and that SpyTag is protected from cleavage by becoming part of the structured Ig-like fold of the reconstituted SpyTag-SpyCatcher fusion. This is consistent with the greatly enhanced thermal stability of SpyCatcher after coupling to SpyTag.11 Therefore, the SK25 (an F‘ strain without protease knock-outs) can be used for all SpyDisplay panning steps. For screening and purification, the selected phagemids are transformed into SK13 to express high concentrations of soluble SpyTagged Fabs. SK13 is an F- strain and thus also avoids unintended infection with contaminating phages, which could lead to the expression of other antibodies in parallel. The resulting Fabs are fully compatible with SpyTag-based modular antibody assembly, enabling rapid site-specific labeling and change of oligomeric state using prefabricated SpyCatcher modules.11 This enables screening in mono- and bivalent format in parallel and greatly accelerates further antibody characterization, thus providing a substantial economic benefit.
We have established SpyDisplay here mainly for the selection of antibodies, but we do not expect any limitations regarding the displayable proteins, provided they contain a SpyTag and can be transported to the periplasm. The SpyTag of the displayed protein can be inserted at any accessible position within a protein,6 providing manifold options for the topology of protein display on the phage. In particular, SpyTag fused N-terminally to the displayed protein enables selection of proteins that require an unmodified C-terminus, which is not possible with the standard pIII-fusion approach. SpyTag/SpyCatcher seems furthermore well suited to display proteins in other selection systems. For example, both yeast display33 and bacterial display34,35 via SpyTag/SpyCatcher have been demonstrated. The physical separation of the SpyTagged library from the other elements of the display system as shown here enables switching between display methods, provided the vector stays compatible with the host expression system. By utilizing a dual expression vector compatible with both E. coli and yeast expression,36 it would be possible to seamlessly switch from phage to yeast display simply by transforming the output of phage display into a SpyDisplay-compatible yeast strain displaying SpyCatcher. Similarly, a compatible plasmid would allow for a transition from phage display to mammalian display.37 Such an approach could combine the benefits of these popular protein selection systems, namely the large library size of phage display and the ability to perform eukaryotic expression and flow cytometry sorting of the selection output in yeast or mammalian cells.
Other protein ligation technologies besides SpyTag/SpyCatcher have been established and could be used to establish analogous ‘covalent capture’ display methods. Such technologies include SnoopTag/SnoopCatcher,38 DogTag/DogCatcher,39 SilkTag/SilkCatcher,40 split inteins,41 or enzyme-based ligation technologies such as ones based on sortase,42 butelase,43 or peptiligase.44
In vitro selections are often hindered by biases such as the accumulation of truncated sequences or selection not based on affinity.45 The high screening hit rates, high diversities, and subnanomolar affinities of the antibodies selected by SpyDisplay suggest that SpyDisplay is resilient against such biases. This study demonstrates that SpyDisplay has the potential to significantly improve selection campaigns.
Materials and methods
Plasmids
pBBx2-F-Spy2-H,11 pKD46,46 pCP20,46 pKD13,46 pET28a_SpyCatcher2,11 pACYC17747
Oligonucleotides
161_SKE, 175_SKE, 185_SKE, 186_SKE, 164_SKE, 165_SKE, 178_SKE, 179_SKE, 180_SKE, 181_SKE, 182_SKE, 183_SKE (all this study, nucleotide sequences shown in the supplementary information)
Strains and phages
E. coli TG1, E. coli SK25 (this study), E. coli SK1311
VCSM13 (Agilent), Hyperphage (Progen)
Antibodies
Anti-M13-pIII monoclonal antibody (E8033S, New England Biolabs), sheep anti-mouse IgG (H/L):HRP (AAC10P, Bio-Rad), anti-FLAG-Tag (M2) IgG-HRP (A8592, Sigma), anti-M13 bacteriophage coat protein g8p (ab9225, Abcam), goat anti-human IgG F(ab’)2 (STAR126, Bio-Rad), anti-M13-pVIII-HRP (Cytiva 27–9421-01, Sigma)
E. coli cultivation
E. coli strains were cultivated in 2xYT medium supplemented with glucose (Glc, varying concentrations), arabinose (Ara, varying concentrations), isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.25 mM), kanamycin (Kan, 50 µg/mL), and/or chloramphenicol (Cam, 34 µg/mL). Cells were grown on orbital shakers in Erlenmeyer flasks at 250 rpm (Fab-phage production) or in 24 deep-well blocks at 400 rpm (panning) at temperatures between 22°C and 37°C. For generation of SK25, plates were incubated at 42°C for plasmid curing. For selection of the F plasmid in TG1 derivatives, cells were plated on M9 agar.
Plasmid cloning
For selection and soluble expression of selected binders from SpyDisplay pannings, pBBx2-F-Spy2-H11 was used.
Human antibody phage display library
A subset of the Pioneer library (Bio-Rad) was used for phage display, which contained human Fab antibodies with the heavy chain germline IGHV1-69 and the light chain germline IGLV3-1. The CDR diversity was generated by gene synthesis and CDRs were cloned into the plasmid pBBx2-F-Spy2-H in successive steps.
Generation of E. coli SK25
The method used for generation of E. coli SK25 is based on the λ-Red-recombinase system described by Datsenko and Wanner.46 First, the PCR-product (sequence in supplementary information) consisting of the SpyCatcher2-pIII expression cassette with the FRT-Kan-FRT cassette and araC was amplified with oligonucleotides 161_SKE and 175_SKE using the template for SK25 generation. PCR-products of correct size were gel-purified using Wizard SV Gel and PCR Clean-Up Kit (Promega). E. coli TG1 cells harboring the helper plasmid pKD46 were made electrocompetent (in presence of 50 mM arabinose instead of 1 mM arabinose) and transformed with the PCR product as described.46 Mutants were identified by colony PCR using the oligos 185_SKE and 186_SKE. After helper plasmid curing, the KanR mutant was transformed with pCP20 for excision of the KanR cassette via Flp-FRT recombination. After curing of pCP20, correct excision of the KanR cassette was analyzed by colony PCR using primers 164_SKE and 165_SKE. Final verification of the correct integration of the SpyCatcher2-pIII expression cassette was performed by sequencing of the final colony PCR product with primers 164_SKE, 165_SKE, 178_SKE, 179_SKE, 180_SKE, 181_SKE, 182_SKE, and 183_SKE. Maintenance of the F plasmid was controlled after each mutagenesis step by plating on M9 agar.
Colony PCR
Fresh colonies were picked and directly resuspended in 16 µL of the PCR mastermix. PCR was performed with hot start Taq DNA polymerase (New England Biolabs) according to the manufacturer’s instructions. For generation of E. coli SK25, wildtype E. coli TG1 was always tested side-by-side as control.
Fab-phage production
2xYT/1%Glc/Cam medium was inoculated from a glycerol stock of E. coli SK25 harboring the antibody sublibrary to an OD600 of 0.1 and cultivated at 37°C and 220 rpm in 2 L shake flasks. At OD600 of 0.5 to 0.8, helper phages were added (VCSM13 in monovalent SpyDisplay, Hyperphage in the polyvalent setup), followed by incubation at 37°C for 45 min without shaking and another 45 min at 220 rpm. After helper phage infection, medium was changed (2xYT/Kan/Cam/0.25 mM IPTG/0.002% arabinose) for overnight production of Fab-phages at 22°C and 220 rpm. For small-scale preparations (<2 L), phage-containing supernatants were harvested by centrifugation (6,000 x g, 30 min, 4°C). After filtration (0.22 µm) phages were precipitated with ¼th volume of 20% (w/v) PEG 6000/2.5 M NaCl for 60 min on ice, followed by centrifugation (13,000 x g, 30 min, 4°C). The phage pellet was resuspended in phosphate-buffered saline (PBS)/20% glycerol and stored at −80°C. For large-scale preparations (>2 L), phages were filtered and concentrated using a SartoJet Membrane Pump (Sartorius) with Sartocon Slice Hydrosart cassettes (0.45 µm for cell removal and 100 kDa cutoff for phage concentration; Sartorius), followed by the final precipitation with PEG/NaCl as described above.
Phage ELISA
96-well plates (Maxisorp F96, Thermo Fisher Scientific) were coated with anti-M13 bacteriophage coat protein g8p antibody or goat anti-human IgG F(ab’)2 in PBS overnight at 4°C. Plates were washed five times with TBST (TBS with 0.05% (v/v) Tween 20) after coating and blocking with 33% (v/v) ChemiBLOCKER (Merck Millipore) in TBST for 1 h at room temperature (RT). Next, serial dilutions of Fab-phages in 33% (v/v) ChemiBLOCKER/TBST were transferred to the blocked plates and incubated for 1 h at RT. After washing (ten times with TBST), bound Fab-phages were detected with anti-M13-pVIII-HRP diluted 1:5,000 in 33% (v/v) ChemiBLOCKER/TBST (1 h, RT), followed by a final wash (ten times with TBST) and addition of QuantaBlu fluorogenic peroxidase substrate (Thermo Fisher Scientific). Fluorescence (excitation at 320 ± 25 nm, emission at 430 ± 35 nm) was measured with an Infinite 200 microplate reader (Tecan). To measure the efficiency of phage elution by TEV proteolysis, serially diluted phages were bound on cognate antigens, followed by incubation with TEV Buffer (1 µg/mL TEV protease in 50 mM Tris pH 8.0, 0.5 mM EDTA, 1 mM DTT, 0.5% (w/v) BSA) or control buffer (same buffer but without TEV protease). After washing, an ELISA with anti-pVIII-HRP was performed to quantify the remaining bound phages after elution. The linear regions of the digest and corresponding control curves were fitted with a linear equation with shared Y-axis intercept (Suppl Fig S1). To determine the efficiency of cleavage, the slope of the TEV digestion condition was divided by the slope of the corresponding control and fit errors were propagated.
Immunoblotting
Western blots were performed as described.11 Briefly, different dilutions of phage preparations were run on SDS-PAGE and afterward blotted onto PVDF membranes via semi-dry transfer. For detection of pIII and derivatives, blots were incubated with anti-M13-pIII monoclonal antibody, followed by washing and incubation with sheep anti-mouse IgG (H/L):HRP. Alternatively, anti-FLAG-Tag (M2) IgG-HRP was used for detection of FLAG-tagged proteins. Clarity ECL substrate (Bio-Rad) was used as HRP substrate and a Chemidoc MP imager (Bio-Rad) was used for signal detection.
Spot titration
Titer of phage preparations was determined by spot titration. (Fab)-phage preparations were serially ten-fold diluted in 2xYT-medium in a 96-well microtiter plate (ThermoFisher Scientific) in triplicates. Equal amounts of freshly grown E. coli SK25 with an OD600 of 0.5 to 0.8 were added and the mixture was incubated for 30 min at 37°C. Afterward, 5 µL of each dilution was spotted on LB/Cam/Glc agar plates. Titers (colony-forming units (cfu)/mL) were determined after overnight incubation at 37°C.
Antibody selection with SpyDisplay
Antigens or streptavidin/neutravidin were immobilized overnight on polystyrene plates (MaxiSorp, Thermo Fisher Scientific). Plate blocking was performed with 5% (w/v) milk powder or 5% (w/v) BSA in PBST (PBS with 0.05% (v/v) Tween 20) for 1 hour at RT, followed by incubation with biotinylated antigen where applicable. Blocked Fab-phages were transferred onto the antigen-coated plates and incubated for 3 hours (first round) or 2 hours (second and third round) at RT at 400 rpm. After washing (5 times with TBST), selected Fab-phages were eluted with TEV-buffer (1 µg/mL TEV in 50 mM Tris pH 8.0, 0.5 mM EDTA, 1 mM DTT, 0.5% (w/v) BSA) for 30 min at RT and transferred to 3.5 mL E. coli SK25 cells (OD600 = 0.6–0.8) in 24-deep-well blocks. To allow phage infection of bacteria, the cultures were incubated at 37°C, 400 rpm for 1 h. Afterward, chloramphenicol was added to a final concentration of 34 µg/mL, followed by further incubation at 37°C and 400 rpm for 30 min. VCSM13 was added to a final concentration of 109 cfu/mL, followed by incubation at 37°C and 400 rpm for 60 min. Following infection, cells were centrifuged at 2,200 x g at RT for 5 min and resuspended in a larger volume (25 mL Fab-phage expression medium: 2xYT/Cam/Kan/0.25 mM IPTG/0.005% Ara) to ensure the efficacy of the antibiotics (Supp. Fig. S4). Fab-phages were expressed overnight at 30°C and 350 rpm in an orbital shaker (Multitron, Infors HT). The next panning round was performed with Fab-phage-containing supernatants of the overnight cultures. After three rounds of panning, the plasmids of the panning output were isolated via DNA preparation (PureYield Plasmid Miniprep System, Promega) and transformed into chemically competent E. coli SK13 cells.11
Identification of antigen-binding clones via primary screening ELISA
Identification of antigen binding clones was performed as described previously.48 Briefly, 368 single clones were picked, Fabs were expressed overnight in 384-well microtiter plate (ThermoFisher Scientific), and crude bacterial lysates were used for ELISA screening. 384-well plates (MaxiSorp black, ThermoFisher Scientific) were coated with antigens and controls overnight in PBS at 4°C. Plates were washed five times with PBST after coating and blocking with 5% milk-PBST or 5% BSA-PBST for 1 h at RT. Next, blocked lysates (diluted 1:2 in PBST) were transferred onto the antigen coated plates and incubated for 1 h at RT. After washing (ten times with PBST), bound Fabs were detected with anti-FLAG-tag (M2) IgG-HRP diluted 1:20,000 in 0.5% milk-PBST (1 h, RT), followed by washing (ten times with PBST) and addition of QuantaBlu Fluorogenic Peroxidase Substrate (Thermo Fisher Scientific). Fluorescence (excitation at 320 ± 25 nm, emission at 430 ± 35 nm) was measured with an Infinite 200 microplate reader (Tecan). Antibodies which showed a signal greater than ten-fold over background and did not recognize the negative control proteins were considered as antigen-binding clones. For pannings on passively adsorbed antigen, 30 clones with the highest ELISA signals were sequenced and unique clones were expressed and purified as described below. For pannings on biotinylated antigens, a Koff ranking was performed with 95 top hits from primary screening.
Koff ranking via BLI
For clones derived from pannings on biotinylated antigens, off-rate screening was performed on Octet RED384 and Octet HTX instruments (Sartorius) as described previously.26 Lysates of 95 antibodies showing the strongest signal in primary screening ELISA were tested. Biotinylated antigens were immobilized on streptavidin sensors (SA, Sartorius) with typical immobilization levels of 5 ± 0.5 nm and 1.5 ± 0.5 nm for biotinylated mGFP (lot-specific variations) and 5 ± 0.5 nm for biotinylated sarilumab. Baseline was measured in mock lysates without target-specific antibodies for 5 min, followed by the association phase (7.5 min) in specific lysates. Dissociation rate was again measured in mock lysates (7.5 min). Data were analyzed using a 1:1 interaction model with Octet Analysis Studio software 12.2. Curves with poor fits (R2 < 0.96) were excluded from analysis.
Protein expression and purification
Fabs were expressed and purified as described previously.27 In short, 2xYT/0.1%Glc/Cam medium was inoculated from an overnight culture with E. coli SK13 containing the expression plasmids and cultivated at 30°C and 250 rpm until the OD600 reached about 0.5, followed by induction with 0.8 mM IPTG and overnight incubation at 27.5°C. Bacterial pellets were lysed with BugBuster (Merck KGaA), supplemented with 20 units/mL Benzonase (Merck KGaA), 2 mg/mL lysozyme (Merck KGaA) and protease inhibitors (Complete EDTA free; Roche) and loaded on Ni-NTA agarose (Qiagen). After washing, Fab fragments were eluted with imidazole-containing buffer (250 mM imidazole, 500 mM NaCl, 20 mM NaH2PO4 pH 7.4), followed by buffer exchange to PBS via PD10 columns (GE Healthcare). TEV protease was expressed and purified as described.49
Affinity determination
Affinities of purified Fab fragments were determined on Octet RED384 and Octet HTX instruments (Sartorius) as described previously.26 Briefly, biotinylated antigens were immobilized on streptavidin sensors (SA, Sartorius). Kinetic parameters were determined with purified Fab fragments at five concentrations ranging from 200 nM to 0.13 nM in running buffer (PBS, 0.1% (w/v) BSA, 0.02% (v/v) Tween 20). The association was measured for 600 s and the dissociation for 300 to 1,800 s, depending on the binding strength. For sarilumab, before each single cycle of the measurement, biosensor surface was regenerated with 10 mM glycine, pH 3.0. In case of mGFP, each Fab dilution was measured on a separate sensor without regeneration. Data were analyzed using a 1:1 interaction model with Octet Analysis Studio software 12.2.
Supplementary Material
Acknowledgments
We thank our colleagues in the Fab production and assay teams for antibody purification and quality control and our colleagues in lab support for production of the library phages. We thank Melissa Wich for critical reading of the manuscript.
Funding Statement
The authors reported there is no funding associated with the work featured in this article.
Abbreviations
CDR: complementarity-determining region; cfu: colony-forming unit; Fab: antigen binding fragment of an antibody; FRT: flippase recognition target; HRP: horseradish peroxidase; IgG: immunoglobulin G; IPTG: isopropyl β-D-1-thiogalactopyranoside; κ: kappa light chain of an antibody; λ: lambda light chain of an antibody; MBP: maltose-binding protein; mGFP: monomeric green fluorescent protein; pIII: filamentous phage protein III; TAT: twin arginine translocase; TEV: tobacco etch virus; ELISA: enzyme-linked immunosorbent assay; BLI: biolayer interferometry; PBS: phosphate-buffered saline; TBS: tris-buffered saline; BSA: bovine serum albumin; PEG: polyethylene glycol; scFv: single-chain variable fragment; SD: standard deviation
Contributions
S.-J.K., H.H., M.C., C.H., and M.P. performed experiments. S.-J.K., C.H., M.P., and F.Y. designed experiments and analyzed the data. A.K. and F.Y. conceived the project. C.H., S.-J.K., M.P., A.K., and F.Y. wrote the manuscript.
Disclosure statement
All authors are employees of Bio-Rad AbD Serotec GmbH. Bio-Rad Laboratories, Inc. filed patent applications on technologies described herein, on which F.Y. is listed as inventor.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2177978
References
- 1.Jijakli K, Khraiwesh B, Fu W, Luo L, Alzahmi A, Koussa J, Chaiboonchoe A, Kirmizialtin S, Yen L, Salehi-Ashtiani K.. The in vitro selection world. Methods. 2016;106:3–13. doi: 10.1016/j.ymeth.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12(1):433–55. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 3.Qi H, Lu H, Qiu HJ, Petrenko V, Liu A. Phagemid vectors for phage display: properties, characteristics and construction. J Mol Biol. 2012;417(3):129–43. doi: 10.1016/j.jmb.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 4.Breitling F, Dubel S, Seehaus T, Klewinghaus I, Little M. A surface expression vector for antibody screening. Gene. 1991;104(2):147–53. doi: 10.1016/0378-1119(91)90244-6. [DOI] [PubMed] [Google Scholar]
- 5.Dubel S, Breitling F, Fuchs P, Braunagel M, Klewinghaus I, Little M. A family of vectors for surface display and production of antibodies. Gene. 1993;128(1):97–101. doi: 10.1016/0378-1119(93)90159-Z. [DOI] [PubMed] [Google Scholar]
- 6.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A. 2012;109(12):E690–697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Fierer JO, Rapoport TA, Howarth M. Structural analysis and optimization of the covalent association between spycatcher and a peptide tag. J Mol Biol. 2014;426(2):309–17. doi: 10.1016/j.jmb.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keeble AH, Banerjee A, Ferla MP, Reddington SC, Anuar I, Howarth M. Evolving accelerated amidation by spytag/spycatcher to analyze membrane dynamics. Angew Chem Int Ed Engl. 2017;56(52):16521–25. doi: 10.1002/anie.201707623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keeble AH, Turkki P, Stokes S, Khairil Anuar INA, Rahikainen R, Hytonen VP, Howarth M. Approaching infinite affinity through engineering of peptide-protein interaction. Proc Natl Acad Sci U S A. 2019;116(52):26523–33. doi: 10.1073/pnas.1909653116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keeble AH, Howarth M. Power to the protein: enhancing and combining activities using the spy toolbox. Chem Sci. 2020;11(28):7281–91. doi: 10.1039/D0SC01878C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hentrich C, Kellmann SJ, Putyrski M, Cavada M, Hanuschka H, Knappik A, Ylera F. Periplasmic expression of spytagged antibody fragments enables rapid modular antibody assembly. Cell Chem Biol. 2021;28(6):813–824 e816. doi: 10.1016/j.chembiol.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Alam MK, Gonzalez C, Hill W, El-Sayed A, Fonge H, Barreto K, Geyer CR. Synthetic modular antibody construction by using the spytag/spycatcher protein-ligase system. Chembiochem. 2017;18(22):2217–21. doi: 10.1002/cbic.201700411. [DOI] [PubMed] [Google Scholar]
- 13.Ward RL, Clark MA, Lees J, Hawkins NJ. Retrieval of human antibodies from phage-display libraries using enzymatic cleavage. J Immunol Methods. 1996;189(1):73–82. doi: 10.1016/0022-1759(95)00231-6. [DOI] [PubMed] [Google Scholar]
- 14.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pbad promoter. J Bacteriol. 1995;177(14):4121–30. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rondot S, Koch J, Breitling F, Dubel S. A helper phage to improve single-chain antibody presentation in phage display. Nat Biotechnol. 2001;19(1):75–78. doi: 10.1038/83567. [DOI] [PubMed] [Google Scholar]
- 16.Rothe C, Urlinger S, Lohning C, Prassler J, Stark Y, Jager U, Hubner B, Bardroff M, Pradel I, Boss M, et al. The human combinatorial antibody library hucal gold combines diversification of all six cdrs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol. 2008;376(4):1182–200. doi: 10.1016/j.jmb.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Loset GA, Kristinsson SG, Sandlie I. Reliable titration of filamentous bacteriophages independent of piii fusion moiety and genome size by using trypsin to restore wild-type piii phenotype. Biotechniques. 2008;44(4):551–552, 554. doi: 10.2144/000112724. [DOI] [PubMed] [Google Scholar]
- 18.Robinson MP, Ke N, Lobstein J, Peterson C, Szkodny A, Mansell TJ, Tuckey C, Riggs PD, Colussi PA, Noren CJ, et al. Efficient expression of full-length antibodies in the cytoplasm of engineered bacteria. Nat Commun. 2015;6(1):8072. doi: 10.1038/ncomms9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miersch S, Li Z, Hanna R, McLaughlin ME, Hornsby M, Matsuguchi T, Paduch M, Saaf A, Wells J, Koide S, et al. Scalable high throughput selection from phage-displayed synthetic antibody libraries. J Vis Exp. 2015;95:51492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang WB, Sun F, Tirrell DA, Arnold FH. Controlling macromolecular topology with genetically encoded spytag-spycatcher chemistry. J Am Chem Soc. 2013;135(37):13988–97. doi: 10.1021/ja4076452. [DOI] [PubMed] [Google Scholar]
- 21.Honegger A, Spinelli S, Cambillau C, Pluckthun A. A mutation designed to alter crystal packing permits structural analysis of a tight-binding fluorescein-scfv complex. Protein Sci. 2005;14(10):2537–49. doi: 10.1110/ps.051520605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer T, Berks BC. The twin-arginine translocation (tat) protein export pathway. Nat Rev Microbiol. 2012;10(7):483–96. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- 23.Speck J, Arndt KM, Muller KM. Efficient phage display of intracellularly folded proteins mediated by the tat pathway. Protein Eng Des Sel. 2011;24(6):473–84. doi: 10.1093/protein/gzr001. [DOI] [PubMed] [Google Scholar]
- 24.Fisher AC, Kim JY, Perez-Rodriguez R, Tullman-Ercek D, Fish WR, Henderson LA, DeLisa MP. Exploration of twin-arginine translocation for expression and purification of correctly folded proteins in escherichia coli. Microb Biotechnol. 2008;1(5):403–15. doi: 10.1111/j.1751-7915.2008.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (tat) pathway in escherichia coli. Mol Microbiol. 2001;39(1):47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 26.Ylera F, Harth S, Waldherr D, Frisch C, Knappik A. Off-rate screening for selection of high-affinity anti-drug antibodies. Anal Biochem. 2013;441(2):208–13. doi: 10.1016/j.ab.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 27.Knappik A, Brundiers R. Recombinant antibody expression and purification. In: Walker JM, editor. The protein protocols handbook. Totowa (NJ): Humana Press; 2009. p. 1929–43. [Google Scholar]
- 28.Chasteen L, Ayriss J, Pavlik P, Bradbury AR. Eliminating helper phage from phage display. Nucleic Acids Res. 2006;34(21):e145. doi: 10.1093/nar/gkl772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan D. Studies on transformation of escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–80. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 30.Mazor Y, Van Blarcom T, Carroll S, Georgiou G. Selection of full-length iggs by tandem display on filamentous phage particles and escherichia coli fluorescence-activated cell sorting screening. FEBS J. 2010;277(10):2291–303. doi: 10.1111/j.1742-4658.2010.07645.x. [DOI] [PubMed] [Google Scholar]
- 31.Paschke M, Hohne W. A twin-arginine translocation (tat)-mediated phage display system. Gene. 2005;350(1):79–88. doi: 10.1016/j.gene.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Strauch E-M, Georgiou G. Mechanistic challenges and engineering applications of protein export in e. Coli. In: Lee SY, editor. Systems biology and biotechnology of escherichia coli. Netherlands: Springer Dordrecht; 2009. p. 327–49. [Google Scholar]
- 33.Kajiwara K, Aoki W, Koike N, Ueda M. Development of a yeast cell surface display method using the spytag/spycatcher system. Sci Rep. 2021;11(1):11059. doi: 10.1038/s41598-021-90593-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallus S, Peschke T, Paulsen M, Burgahn T, Niemeyer CM, Rabe KS. Surface display of complex enzymes by in situ spycatcher-spytag interaction. Chembiochem. 2020;21(15):2126–31. doi: 10.1002/cbic.202000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallus S, Mittmann E, Rabe KS. A modular system for the rapid comparison of different membrane anchors for surface display on escherichia coli. Chembiochem. 2022;23(2):e202100472. doi: 10.1002/cbic.202100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel CA, Wang J, Wang X, Dong F, Zhong P, Luo PP, Wang KC. Parallel selection of antibody libraries on phage and yeast surfaces via a cross-species display. Protein Eng Des Sel. 2011;24(9):711–19. doi: 10.1093/protein/gzr034. [DOI] [PubMed] [Google Scholar]
- 37.Tesar D, Hotzel I. A dual host vector for fab phage display and expression of native igg in mammalian cells. Protein Eng Des Sel. 2013;26(10):655–62. doi: 10.1093/protein/gzt050. [DOI] [PubMed] [Google Scholar]
- 38.Veggiani G, Nakamura T, Brenner MD, Gayet RV, Yan J, Robinson CV, Howarth M. Programmable polyproteams built using twin peptide superglues. Proc Natl Acad Sci U S A. 2016;113(5):1202–07. doi: 10.1073/pnas.1519214113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keeble AH, Yadav VK, Ferla MP, Bauer CC, Chuntharpursat-Bon E, Huang J, Bon RS, Howarth M. Dogcatcher allows loop-friendly protein-protein ligation. Cell Chem Biol. 2022;29(2):339–350 e310. doi: 10.1016/j.chembiol.2021.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan R, Hakanpää J, Elfving K, Taberman H, Linder MB, Aranko AS. Biomolecular Click Reactions Using a Minimal pH-Activated Catcher/Tag Pair for Producing Native-Sized Spider-Silk Proteins. Angew Chem Int Ed Engl. 2023;e202216371. doi: 10.1002/anie.202216371. [DOI] [PubMed] [Google Scholar]
- 41.Vila-Perello M, Muir TW. Biological applications of protein splicing. Cell. 2010;143(2):191–200. doi: 10.1016/j.cell.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmohl L, Schwarzer D. Sortase-mediated ligations for the site-specific modification of proteins. Curr Opin Chem Biol. 2014;22:122–28. doi: 10.1016/j.cbpa.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen GK, Wang S, Qiu Y, Hemu X, Lian Y, Tam JP. Butelase 1 is an asx-specific ligase enabling peptide macrocyclization and synthesis. Nat Chem Biol. 2014;10(9):732–38. doi: 10.1038/nchembio.1586. [DOI] [PubMed] [Google Scholar]
- 44.Toplak A, Nuijens T, Quaedflieg PJLM, Wu B, Janssen DB. Peptiligase, an enzyme for efficient chemoenzymatic peptide synthesis and cyclization in water. Advanced Synthesis & Catalysis. 2016;358(13):2140–47. doi: 10.1002/adsc.201600017. [DOI] [Google Scholar]
- 45.Plessers S, Van Deuren V, Lavigne R, Robben J. High-throughput sequencing of phage display libraries reveals parasitic enrichment of indel mutants caused by amplification bias. Int J Mol Sci. 2021;22(11):11. doi: 10.3390/ijms22115513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in escherichia coli k-12 using pcr products. Proc Natl Acad Sci U S A. 2000;97(12):6640–45. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the p15a cryptic miniplasmid. J Bacteriol. 1978;134(3):1141–56. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarutat T, Frisch C, Nickels C, Merz H, Knappik A. Isolation and comparative characterization of ki-67 equivalent antibodies from the hucal phage display library. Biol Chem. 2006;387(7):995–1003. doi: 10.1515/BC.2006.123. [DOI] [PubMed] [Google Scholar]
- 49.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble his(6)-tagged tev protease. Methods Mol Biol. 2009;498:297–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.