

ABSTRACT
Macroautophagy (hereafter “autophagy”) is a membrane-mediated biological process that involves engulfing and delivering cytoplasmic components to lysosomes for degradation. In addition to autophagy’s pro-survival effect during nutrient starvation, excessive activation of autophagy machinery can also cause regulated cell death, especially iron-dependent ferroptosis. Here, we report a key role of TMEM164 (transmembrane protein 164) in selectively mediating ATG5 (autophagy related 5)-dependent autophagosome formation during ferroptosis, rather than during starvation. In contrast, the membrane protein ATG9A (autophagy-related 9A) is dispensable for the formation of autophagosomes during ferroptosis. TMEM164-mediated autophagy degrades ferritin, GPX4 (glutathione peroxidase 4), and lipid droplets to increase iron accumulation and lipid peroxidation, thereby promoting ferroptotic cell death. Consequently, the loss of TMEM164 limits the anticancer activity of ferroptosis-mediated cytotoxicity in mice. High TMEM164 expression is associated with improved survival and increased immune cell infiltration in patients with pancreatic cancer. These findings establish a new mode of autophagy-dependent ferroptosis.
KEYWORDS: Autophagy, cell death, ferroptosis, membrane protein, tumor immunity
Introduction
Ferroptosis is a type of iron-dependent regulated cell death that plays a context-dependent role in health and disease [1,2]. Modulating the activities of ferroptosis holds promise for cancer treatment [3]. In addition to mammalian cells, ferroptosis has also been detected in plants following heat stress [4], indicating that this cell death modality may be a conserved process. Generally, the induction of ferroptosis involves an imbalance between oxidative stress and antioxidant defense, which ultimately leads to lipid peroxidation, plasma membrane damage, and the release of danger/damage-associated molecular patterns (DAMPs) [5,6]. The classical ferroptosis inducers are inhibitors of the antioxidant SLC7A11 (solute carrier family 7 member 11)-GPX4 (glutathione peroxidase 4) axis [7]. Several regulated cell death effectors, such as caspases, MLKL (mixed lineage kinase domain like pseudokinase) and GSDMD (gasdermin D), are not necessary for ferroptosis [8], highlighting the uniqueness of the execution of this pathway.
Macroautophagy (hereafter “autophagy”) is a lysosomal-dependent degradation process, by which the recycling of cellular material can promote survival under various environmental stresses, especially nutritional deprivation [9,10]. In contrast, unrestricted autophagy may lead to cell death by selectively degrading survival-promoting molecules or organelles. Autophagy is regulated and executed by a group of conserved proteins called ATG (autophagy related) proteins, which form different complexes to shape membrane dynamics to engulf and degrade the cargoes [11]. Recently, ferroptosis was recognized as a form of autophagy-dependent cell death and the depletion of core components of autophagic machinery, such as ATG5, ATG7, and BECN1/Vps30/Atg6 (beclin 1), inhibits ferroptotic cell death [12–21]. Autophagy-induced signaling during ferroptosis is associated with oxidative stress and iron accumulation, and excessive autophagy can feedback accelerate these signals understood [22,23]. Although these advances in knowledge are significant, the selective process and regulation of autophagy-dependent ferroptosis are still poorly.
TMEM164 (transmembrane protein 164) is a member of the transmembrane protein (TMEM) family that spans various biological membranes, including the plasma membrane and the membrane of organelles [24]. The Xq22.3q23 microdeletion containing the TMEM164 gene is associated with Alport syndrome, intellectual disability, midfacial hypoplasia, and elliptocytosis [25,26]. The changes in the TMEM164 gene and other TMEM members are independent factors affecting the prognosis of lung adenocarcinoma [27]. Although TMEM164 is a highly conserved protein, its function remains unknown.
In this study, we analyzed the data of a clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 (CRISPR-associated protein 9) library screening from a previous study that used the small molecule compound ML210 to trigger ferroptosis in human kidney cancer cells [28]. This genetic screen indicated that TMEM164 is one of the top candidate genes for promoting ferroptosis. Our function and mechanism studies further revealed that TMEM164 plays a key role in promoting autophagy-dependent ferroptosis by sustaining autophagosome formation. In contrast, TMEM164 is dispensable for classical starvation-induced autophagosome formation.
Results
TMEM164 acts as a promoter of ferroptosis
CRISPR-Cas9 genetic screening provides a valuable approach to reveal the new genetic dependence of ferroptosis. We analyzed a genome-scale CRISPR-Cas9 knockout screening study in 786-O cells (a human clear cell, renal carcinoma cell line) during ferroptosis triggered by the chemical ML210 [28]. In addition to the well-characterized ferroptosis promoters ACSL4 (acyl-CoA synthetase long chain family member 4) [29–31] and POR (cytochrome p450 oxidoreductase) [32,33], TMEM164 was identified as one of top hits (Figure 1A), highlighting a potential role of TMEM164 in regulating ferroptosis.
Figure 1.

TMEM164 acts as a promoter of ferroptosis. (A) Partial volcano plots highlighting ACSL4, TMEM164, and POR as the top hits in a previous genome-wide screen using CRISPR-Cas9 in ML210-induced 786-O cells [28]. (B) Western blot analysis of TMEM164 expression in the indicated TMEM164-knockdown HT1080 and PANC1 cells. (C) Cell death assay in the indicated HT1080 and PANC1 cells following treatment with erastin (5 μM), sulfasalazine (500 μM), sorafenib (10 μM), RSL3 (0.5 μM), ML162 (0.5 μM), ML210 (5 μM), staurosporine (200 nM), or CCT137690 (5 μM) for 24 h (data are shown in a heat map as the means of 3 biologically independent samples). (D) Western blot analysis of TMEM164 expression in control PANC1, RSL3-resistant PANC1 (PANC1R), or TMEM164-overexpressed PANC1R (PANC1R164) cells. (E) qPCR analysis of TMEM164 mRNA in PANC1R cells following treatment with brusatol (100 nM) for 6–24 h (n = 3 biologically independent samples; *P < 0.05, one-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (F) Cell viability in the indicated PANC1 cells following treatment with RSL3 (0.25–10 μM) for 24 h (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (G-I) Analysis of intracellular Fe2+ (G), intracellular MDA (H), and extracellular HMGB1 (I) in indicated control and TMEM164-knockdown PANC1 cells following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 24 h (n = 3 biologically independent samples; *P < 0.05 versus control shRNA group, one-tailed t test; data are presented as means ± SD).
To determine whether TMEM164 is required for ferroptosis, we utilized shRNA-mediated RNA interference (RNAi) to inhibit the expression of TMEM164 in HT1080 cells (a human fibrosarcoma cell line) and PANC1 cells (a human pancreatic ductal adenocarcinoma cell line), which are widely used in the study of ferroptosis mechanisms [34–37]. The suppression of TMEM164 expression by two different shRNAs (Figure 1B) blocked SLC7A11 inhibitors (e.g., erastin, sulfasalazine, and sorafenib)- or GPX4 inhibitors (e.g., RSL3, ML162, and ML210)-induced cell death (Figure 1C). In contrast, the loss of TMEM164 had no significant effects on cell death due to the apoptosis inducer staurosporine or the necroptosis trigger CCT137690 (Figure 1C) [38]. Thus, TMEM164 appears be a specific regulator of ferroptosis, but not apoptosis and necroptosis.
To further examine the effects of TMEM164 in ferroptosis, we established an RSL3-resistant PANC1 cell line (termed PANC1R) using a method involving stepwise dose escalation and the limiting of single-cell dilution. Western blot analysis revealed that the expression of TMEM164 was reduced in PANC1R cells (Figure 1D). We also examined other key ferroptosis regulators, such as GPX4 and SLC7A11, accordingly. The expression of SLC7A11 in PANC1R cells was not significantly changed compared to parental cells (Figure 1D). In contrast, GPX4 expression was upregulated in PANC1R cells compared to parental cells (Figure 1D). Ferroptosis-resistant cells have aberrant expression of many genes [39], mainly regulated by the antioxidant transcription factor NFE2L2/NRF2 (NFE2 like BZIP transcription factor 2) [40,41]. Brusatol, an NFE2L2 inhibitor [42], inhibited TMEM164 mRNA expression in PANC1R cells (Figure 1E), supporting the role of NFE2L2 in negatively regulating TMEM164 gene expression.
Compared to the parental cell line, PANC1R cells were capable of maintaining viability with continuous exposure to RSL3 at 10 μM (Figure 1F). In contrast, the gene transfection-mediated overexpression of TMEM164 restored the sensitivity of PANC1R cells (termed PANC1R164 cells; Figure 1D) to RSL3, and this process was inhibited by the ferroptosis inhibitor liproxstatin-1 (Figure 1F). The overexpression of TMEM164 reduced GPX4 expression in PANC1R cells (Figure 1D), suggesting a negative expression correlation between TMEM164 and GPX4 in PANC1R cells.
We next examined the effects of TMEM164 on characteristic biochemical events of ferroptosis, including iron accumulation and lipid peroxidation. Quantitative analysis of intracellular ferrous ions (Fe2+) and malondialdehyde (MDA, an endogenous genotoxic product of lipid peroxidation) revealed that the knockdown of TMEM164 inhibited erastin- or RSL3-induced Fe2+ accumulation and lipid peroxidation in PANC1 cells (Figure 1G,H). As expected, the release of a prototypical DAMP, namely the molecule HMGB1 (high mobility group box 1) [43], during ferroptosis was also inhibited by the knockdown of TMEM164 (Figure 1I). Altogether, these findings demonstrate that TMEM164 is a positive regulator of ferroptosis.
TMEM164 promotes ferroptosis by activating autophagy
To study the mechanism and action of TMEM164 in ferroptotic cell death, we determined the effect of TMEM164 silencing on the expression of proteins responsible for the repair of lipid peroxides, including GPX4 [44], AIFM2/FSP1 (apoptosis inducing factor mitochondria associated 2) [45,46], and DHODH (dihydroorotate dehydrogenase (quinone)) [47]. Compared to AIFM2 and DHODH, the protein expression of GPX4 was reduced by erastin or RSL3 in PANC1 cells (Figure 2A). The loss of TMEM164 reduced the downregulation of GPX4 protein during ferroptosis (Figure 2A). TMEM164 had no significant effects on the protein expression of AIFM2 and DHODH (Figure 2A) as well as the mRNA of GPX4 (Figure 2B). These data indicate that TMEM164 selectively regulates GPX4 protein degradation during ferroptosis.
Figure 2.
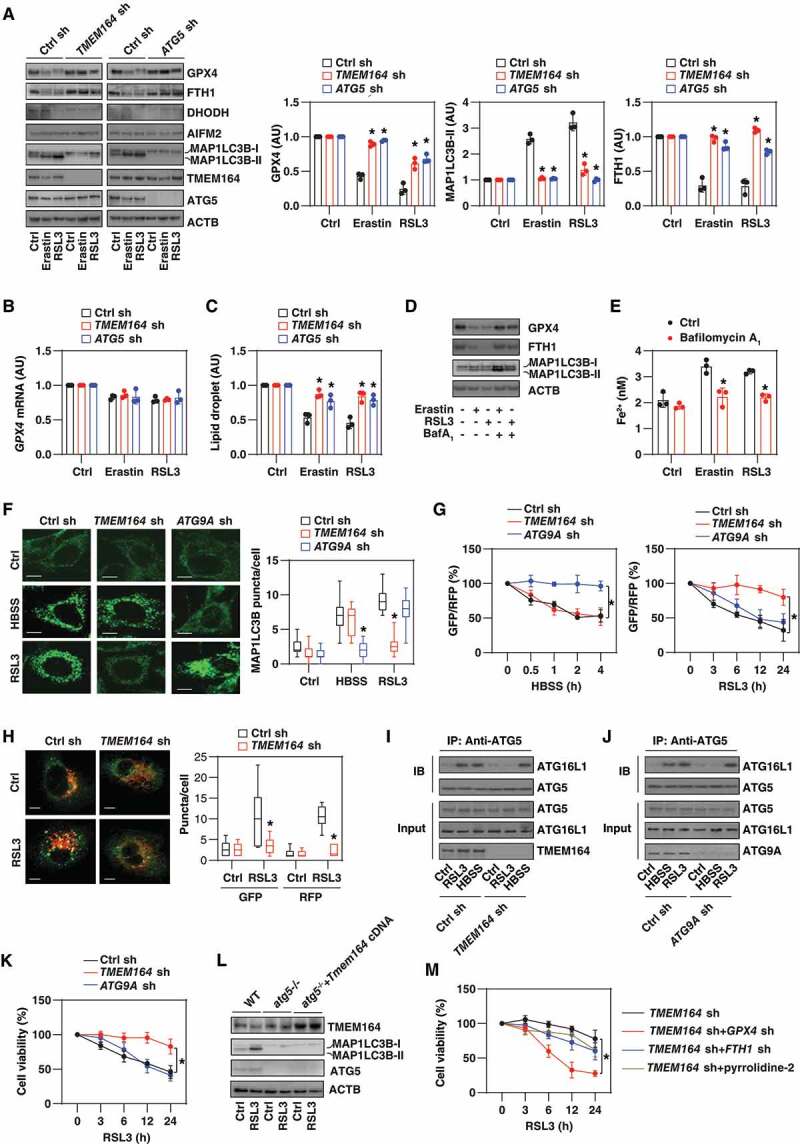
TMEM164 promotes ferroptosis by activating autophagy. (A) Western blot analysis of the indicated protein expression in TMEM164-knockdown or ATG5-knockdown PANC1 cells following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 24 h. Relative protein quantification is shown in the right panel (n = 3 biologically independent samples; *P < 0.05 versus control shRNA group, one-tailed t test; data are presented as means ± SD). (B, C) Analysis of GPX4 mRNA and lipid droplet levels in the indicated gene knockdown PANC1 cells following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 24 h (n = 3 biologically independent samples; *P < 0.05 versus control shRNA group, one-tailed t test; data are presented as means ± SD). (D) Western blot analysis of the indicated protein expression in PANC1 cells following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 24 h in the absence or presence of bafilomycin A1 (BafA1; 20 nM) for 24 h. (E) Analysis of intracellular Fe2+ in PANC1 cells following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 24 h in the absence or presence of bafilomycin A1 (20 nM) for 24 h (n = 3 biologically independent samples; *P < 0.05 versus control group, one-tailed t test; data are presented as means ± SD). (F) Image analysis of MAP1LC3B puncta in the indicated gene knockdown PANC1 cells following treatment with HBSS (2 h) or RSL3 (0.5 μM, 12 h). The data are presented as box-and-whisker plots from 10 fields. Boxes represent the median and the 25th and 75th percentiles. *P < 0.05 versus control shRNA group, one-tailed t test; bar: 15 μm. (G) Analysis of the time course of relative GFP:RFP ratio in the indicated PANC1 cells expressing GFP-LC3-RFP-LC3ΔG in response to HBSS or RSL3 (0.5 µm; n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (H) Image analysis of GFP/RFP puncta in indicated PANC1 cells following treatment with RSL3 (0.5 μM) for 12 h. The data are presented as box-and-whisker plots from 10 fields. Boxes represent the median and the 25th and 75th percentiles. *P < 0.05 versus control shRNA group, one-tailed t test; bar: 15 μm. (I, J) Immunoprecipitation (IP) analysis of the interaction between ATG5 and ATG16L1 in the indicated PANC1 cells following treatment with HBSS or RSL3. (K) Cell viability assay of indicated PANC1 cells following treatment with RSL3 (0.5 µm) for 3–24 h (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (L) Western blot analysis of protein expression in indicated MEFs following treatment with RSL3 (0.2 μM) for 12 h. (M) Cell viability assay of indicated PANC1 cells following treatment with RSL3 (0.5 µm) for 3–24 h in the absence or presence of 5 nM pyrrophenone (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD).
Because GPX4 is a degradative substrate of autophagy in ferroptosis [36,48], we evaluated whether other autophagic substrates are also affected by TMEM164. We focused on ferritin and lipid droplets because the autophagic degradation of ferritin by ferritinophagy [12,13] or lipid droplets by lipophagy [49] favors iron accumulation and/or lipid peroxidation during ferroptosis. The knockdown of TMEM164 prevented erastin- or RSL3-induced downregulation of the protein expression of FTH1 (ferritin heavy chain 1) (Figure 2A). The measurement of lipid droplets also revealed that erastin- or RSL3-induced lipid droplet degradation requires TMEM164 (Figure 2C). Similar to the knockdown of TMEM164, shRNA-mediated ATG5 RNAi prevented the degradation of GPX4, FTH1, and lipid droplets in PANC1 cells after treatment with erastin or RSL3 (Figure 2A,C). Bafilomycin A1, an inhibitor of autophagosome-lysosome fusion [50], also increased FTH1 and GPX4 expression, as well as decreased Fe2+ accumulation in response to erastin or RSL3 (Figure 2D,E).
Because the degradation of GPX4, FTH1, and lipid droplets requires different autophagy receptors [12,13,48,49], we hypothesized that TMEM164 may affect the upstream formation of autophagosomes rather than by directly recognizing cargoes. In fact, the knockdown of TMEM164 inhibited the formation of MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3 beta)-II based on immunoblotting measurement (Figure 2A) or MAP1LC3B puncta assessed by immunofluorescence analysis (Figure 2F) following RSL3 treatment, but not in response to Hanks’ balanced salt solution (HBSS)-induced starvation. As a control, knockdown of ATG9A, a multi-spanning membrane protein essential for autophagosome formation during starvation [51–53], had the reverse effect, inhibiting MAP1LC3B puncta formation in response to HBSS, but not RSL3 (Figure 2F). An autophagy flux assay using a GFP-LC3-RFP-LC3ΔG probe [54] relies on the stable fluorescence of RFP, but quenching of GFP, within the lysosome following autophagosome-lysosome fusion. This microplate reader assay similarly revealed that the knockdown of TMEM164 or ATG9A impaired only RSL3-induced or HBSS-induced autophagic flux, respectively (Figure 2G). Confocal microscopy assays further confirmed that the knockdown of TMEM164 inhibited RSL3-induced accumulation of LC3 puncta in PANC1 cells by the GFP-LC3-RFP-LC3ΔG construct (Figure 2H).
ATG5 is essential for autophagosome formation through the formation of the ATG12–ATG5-ATG16L1 complex [55,56]. The loss of TMEM164 reduced the binding between ATG5 and ATG16L1 in response to RSL3, but not the stimulation from HBSS (Figure 2I). In contrast, the shRNA-mediated knockdown of ATG9A had no effect on the RSL3-stimulated increase in ATG5-ATG16L1 complex formation (Figure 2J). In contrast to the knockdown of TMEM164, the silencing of ATG9A failed to block RSL3-induced cell viability inhibition (Figure 2K). The overexpression of Tmem164 failed to restore MAP1LC3B-II expression in atg5-/- MEFs in the absence or presence of RSL3 (Figure 2L). Collectively, these findings underscore a special upstream role of TMEM164 (but not ATG9A) in the production of ATG5-dependent autophagosomes for ferroptosis.
Next, we examined which autophagy substrate is critical for TMEM164-mediated ferroptosis. The knockdown of GPX4 restored the sensitivity of TMEM164-knockdown PANC1 cells to ferroptosis more than the knockdown of FTH1 or the administration of pyrrophenone (Figure 2M, an inhibitor of lipid droplet formation [57]). Thus, GPX4 may be the most important autophagic degradation substrate for TMEM164-mediated ferroptosis, although other substrates also contribute to this process.
TMEM164 mediates the anticancer activity of RSL3 in vivo
To evaluate the effects of TMEM164-mediated ferroptosis in vivo, we used a PANC1-derived xenograft model in immunodeficient NOD SCID mice. Compared with the control group, the anticancer activity of RSL3 was limited in the TMEM164-knockdown (TMEM164 KD) PANC1 group (Figure 3A). Subsequent analysis of ferroptosis markers (including the mRNA of PTGS2 [prostaglandin-endoperoxide synthase 2] or ACSL4) (Figure 3B,C), iron accumulation (Figure 3D), MDA levels (Figure 3E), and ACSL4 production 5-hydroxyeicosatetraenoic acid (5-HETE; Figure 3F) confirmed the role of TMEM164 in promoting RSL3-induced ferroptosis in isolated tumors. In contrast, the activity of CASP3 (caspase 3) was not changed in the TMEM164 KD group in the absence or presence of RSL3 (Figure 3G). Consistent with the finding that TMEM164 is required for autophagy-dependent ferroptosis in vitro (Figure 2), western blot analysis confirmed that RSL3-induced MAP1LC3B-II expression in isolated tumors was inhibited in the TMEM164 KD group (Figure 3H).
Figure 3.
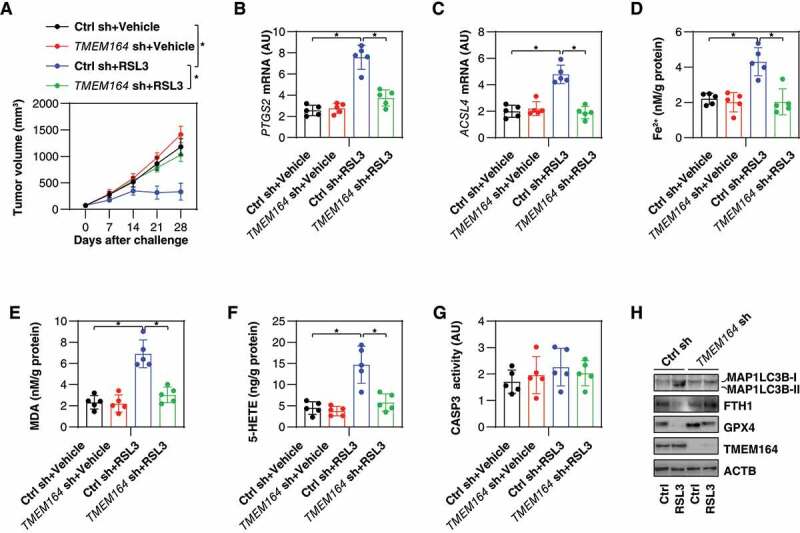
TMEM164 mediates the anticancer activity of RSL3 in vivo. (A) NOD SCID mice were injected subcutaneously with the indicated PANC1 cells (5 × 106/mouse) and treated with RSL3 (100 mg/kg, once every other day, intratumoral injection) at day 7 for 2 weeks. Tumor volume was calculated weekly (n = 5 mice/group; *P < 0.05, ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (B-H) in parallel, the activity or levels of PTGS2 mRNA (B), ACSL4 mRNA (C), Fe2+ (D), MDA (E), 5-HETE (F), CASP3 activity (G), and the indicated protein expression (H) in isolated tumors at day 28 were assayed (n = 5 mice/group; *P < 0.05, ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). .
The role of TMEM164 expression in human pancreatic ductal adenocarcinomas
To determine clinical relevance of the experimental findings, the prognostic value of TMEM164 was evaluated in patients with pancreatic cancer using The Cancer Genome Atlas (TCGA) database. TMEM164 mRNA was significantly upregulated in PDACs compared with normal pancreas tissues (Figure 4A). High levels of TMEM164 mRNA were associated with improved survival (Figure 4B), suggesting that TMEM164 may exert a tumor suppressor effect in human pancreatic cancer. We next assayed the localization of TMEM164 protein expression in human pancreatic ductal adenocarcinomas from The Human Protein Atlas platform. Immunohistochemistry staining showed that the expression of TMEM164 was mainly confined to ductal cells in certain PDAC patients (Figure 4C). In addition, the Tumor IMmune Estimation Resource (TIMER) showed that the mRNA expression level of TMEM164 was positively related to the infiltration of a variety of immune cells, especially cytotoxic CD8+ T cells, antigen-presenting dendritic cells, and cancer-associated fibroblasts (Figure 4D). In contrast, TMEM164 was negatively related to the infiltration of cancer-associated fibroblasts (Figure 4D). These analyses suggest that TMEM164 plays a role in modulating the immune microenvironment of human pancreatic ductal adenocarcinomas.
Figure 4.

The role of TMEM164 in human pancreatic tumors. (A) Analysis of the gene expression of TMEM164 in pancreas from tumor patients and normal controls using datasets from the Cancer Genome Atlas (TCGA; one-tailed t test). The data are presented as box-and-whisker plots. Boxes represent the median and the 25th and 75th percentiles. (B) Prognostic value of TMEM164 in human pancreatic cancer cohorts from the TCGA database. (C) Immunohistochemistry stains of TMEM164 in pancreas from tumor patients and normal controls from the Human Protein Atlas platform (bar=100 µm). (D) Analysis of the relationship between the gene expression of TMEM164 and the immune cell infiltration in the pancreas of tumor patients using datasets from the Tumor IMmune Estimation Resource.
Discussion
Autophagy is a highly integrated process that maintains cell homeostasis by promoting cell survival or causing cell death. Here, we demonstrated a key role of TMEM164 in driving autophagy-dependent ferroptotic cell death by promoting autophagosome formation during lipid peroxidation (Figure 5). These findings not only provide insights for understanding the degradation regulation mechanism of ferroptosis, but also suggest an alternative strategy to trigger antitumor immunity [58].
Figure 5.

A model illustrating the role of TMEM164 in mediating autophagy-dependent cell death. ATG9A is required for starvation-induced autophagosome formation, which leads to programmed survival through non-selective degradation of cytosolic components. In contrast, a ferroptosis activator, such as RSL3, induces TMEM164-dependent autophagosome formation resulting in the selective degradation of anti-ferroptosis regulators (such as GPX4, FTH1, and lipid droplets).
Dysregulated autophagy is related to various cell death modalities [59]. Historically, the phrase “autophagic cell death” was used to morphologically define a type of cell death that accompanies large-scale autophagic vacuolization of the cytoplasm [60]. Today, a related phrase, “autophagy-dependent cell death”, is used to functionally define a type of cell death that mechanistically relies on the autophagic machinery (or its components) [61]. Our knowledge of ferroptosis as an autophagy-dependent form of cell death provides an example for understanding the selective degradation mechanism that promotes cell death [62]. For example, the autophagic degradation of ferritin [12,13], the circadian clock protein BMAL1/ARNTL (basic helix-loop-helix ARNT like 1) [63], lipid droplets [49], the iron transporter SLC40A1/ferroportin (solute carrier family 40 member 1) [62], GPX4 [36,48], or CDH2 (cadherin 2) [64] enhances iron accumulation and/or reactive oxygen species/ROS production, leading to ferroptosis in a context-dependent manner.
Previous studies have shown that two important subtypes of autophagy (macroautophagy and chaperone-mediated autophagy) promote GPX4 degradation and ferroptosis [36,48], suggesting that subtypes of autophagic degradation of GPX4 may depend on cell type or stimulation. Our data suggest that autophagic degradation plays a major role in regulating GPX4 expression in PANC1 cells during ferroptosis. The knockdown of ATG5 or the administration of bafilomycin A1 restored erastin- or RSL3-induced GPX4 protein degradation to 80% compared to controls. Although we ruled out the possibility of regulating GPX4 at the transcriptional level, there are other possibilities (e.g., pre-transcriptional, post-transcriptional, pre-translational, translational and post-translational levels) that regulate GPX4 expression during ferroptosis [65,66]. In addition to different autophagy receptors responsible for ferroptosis [22], our current study shows that the regulation of autophagosome formation by TMEM164 during ferroptosis is distinct from classic starvation-induced autophagy.
Autophagy is a membrane-driven dynamic process involving the assembly, formation, and maturation of intracellular membrane structures, including phagophores, autophagosomes, and autolysosomes [11]. In mammalian cells, ATG9A is the transmembrane protein of the ATG family and has been proposed to play a key role in guiding the membranes of donor organelles to form autophagosomes during starvation [67]. Our current study shows that unlike ATG9A-dependent membrane acquisition, the transmembrane protein TMEM164 selectively participates in the ATG5-dependent autophagosome assembly process during ferroptosis. Although there are still many questions to be answered about the precise role of TMEM164 in controlling membrane dynamics, these new findings provide clues for elucidating the difference in autophagy mechanisms between starvation and ferroptosis.
Like the dysregulation of the autophagic-lysosomal pathway, ferroptosis plays a context-dependent role in cancer, and interventions to stimulate or inhibit ferroptosis have been proposed as cancer treatment or prevention [3]. For example, chronic inflammation caused by ferroptotic death can promote KrasG12D-driven pancreatic tumorigenesis in mice [68]. In established tumors, drug-induced ferroptosis combined with other chemotherapy, radiotherapy, or targeted therapy has become an emerging anticancer strategy in preclinical tumor models [69–73]. The Xq22.3q23 microdeletion carrying the TMEM164 and ACSL4 genes is related to Alport syndrome and intellectual disability in humans [25,26], although it is not clear whether ferroptosis is related to these neurogenetic diseases. Our TCGA database analysis further supports the role of highly expressed TMEM164 in maintaining the anti-tumor immune environment of PDAC.
Together, our results reveal a previously unrecognized molecular link between autophagy and ferroptosis through the transmembrane protein TMEM164. Further understanding of the TMEM164-dependent autophagic process may guide the development of improved approaches and/or drugs for the treatment of ferroptosis-related diseases.
Materials and methods
Reagents
Erastin (S7242), sulfasalazine (S1576), sorafenib (S7397), RSL3 (S8155), CCT137690 (S2744), bafilomycin A1 (S1413), brusatol (S7956), and liproxstatin-1 (S7699) were purchased from Selleck Chemicals. ML162 (20455), ML210 (23282), pyrrophenone (13294), and staurosporine (25096) were purchased from Cayman Chemical. The antibodies to FTH1 (4393), MAP1LC3B (3868), ATG5 (2630), ATG16L1 (8089), ATG9A (13509), SLC7A11 (12691) and ACTB (3700) were purchased from Cell Signaling Technology. The antibodies to TMEM164 (NBP1-93939) were purchased from Novus. The antibodies to GPX4 (ab125066) were purchased from Abcam. The antibodies to AIFM2 (H00084883-D01P) and DHODH (14877-1-AP) were purchased from Thermo Fisher Scientific.
Cell culture
The HT1080 (CCL-121) and PANC1 (CRL-1469) cell lines were obtained from the American Type Culture Collection. The mouse pancreatic ductal adenocarcinoma cancer cell line KPC was a gift from David Tuveson (Cold Spring Harbor Laboratory). Wild-type and atg5-/- embryonic fibroblasts (MEFs) were a gift from Noboru Mizushima (Tokyo Medical and Dental University). Cell lines were cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, 11995073) supplemented with 10% heat-inactivated fetal bovine serum (Millipore, TMS-013-B) and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070-063) at 37°C, 95% humidity, and 5% CO2. The identity of the cell line was verified by short tandem repeat analysis, and routine mycoplasma testing was negative for contamination.
Animal models
Wild-type or TMEM164 KD PANC1 cells (5 × 106 cells) were injected subcutaneously into the dorsal side of NOD SCID mice (female, 8–10 weeks old). On the 7th day, these mice were given RSL3 (100 mg/kg, once every other day, intratumoral injection) for 2 weeks as previously described [44]. The diameter of the tumor was measured twice a week with a caliper, and the tumor volume was calculated using the following formula: length × width2 × π/6. At 14 days after the injection, the mice were euthanized, and the xenograft solid tumors were collected.
All mice were maintained under specific pathogen-free conditions on a regular 12-h light and dark cycle (7:00-19:00 light period; room temperature 20°–25°C; relative humidity: 40%–60%). Food and water were available ad libitum. All animal experiments were conducted in accordance with the institutional ethics guidelines related to animal care and were approved by an institutional animal health and use committee.
Cytotoxicity assay
The level of cell death was assayed using a LIVE/DEAD cell viability/cytotoxicity assay kit (Thermo Fisher Scientific, L3224) according to the manufacturer’s protocol.
Biochemical assay
Commercially available ELISA kits were used to measure the concentrations or activity of HMGB1 (Shino Test Corporation, ST51011), iron (Sigma-Aldrich, MAK025), MDA (Sigma-Aldrich, MAK085), lipid droplets (Cayman Chemical, 500001), 5-HETE (Abbexa, abx251443), and CASP3 (Cell Signaling Technology, 5723) in the indicated samples.
Autophagic flux analysis
We employed a GFP-LC3-RFP-LC3ΔG probe that was a gift from Noboru Mizushima (Addgene, 84572), which provides a simple and quantitative method to evaluate autophagic flux in vitro or in vivo [54]. In brief, cells (5000 cells/well) expressing GFP-LC3-RFP-LC3ΔG in a black 96-well plate with a clear bottom (Corning, 3904) were treated with RSL3 (0.5 µM) at the indicated times, and the signals of GFP and RFP were analyzed using a microplate reader (Tecan).
RNAi and gene transfection
The following human shRNAs were obtained from Sigma-Aldrich in a lentiviral format: human TMEM164-1 (TRCN0000122719), human TMEM164-2 (TRCN0000322570), mouse Tmem164 (TRCN0000248887), human ATG5 (TRCN0000151963), human GPX4 (TRCN0000046249), human FTH1 (TRCN0000029432), and human ATG9A (TRCN0000244083). We seeded 1 × 105 cells in each well of a 12-well plate in 500 μl of complete medium that were transduced by lentiviral vectors at a multiplicity of infection of 10:1. Transduction was carried out in the presence of polybrene (8 μg/ml; Thermo Fisher Scientific, TR1003 G) in an antibiotic-free medium. After recovering with complete culture medium, puromycin (5 μg/ml; Thermo Fisher Scientific, A1113802) was used for the selection of transduced cells. TMEM164/Tmem164 cDNA was obtained from OriGene (SC105570 and MC207858) and gene transfection was performed by Lipofectamine 3000 (Thermo Fisher Scientific, L3000001).
qPCR
According to the manufacturer’s instructions, we used a QIAGEN RNeasy Plus Micro Kit (74034) and iScript cDNA Synthesis Kit (Bio-Rad, 1708890) to extract total RNA and synthesize first-strand cDNA, respectively. Then, 20-μl reactions were prepared by combining 4 μl of iScript select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20×, 2 μM), and 12 μl of RNA diluted in RNase-free water. Quantitative real-time PCR (qPCR) was carried out using synthesized cDNA, primers, and SsoFast EvaGreen supermix (Bio-Rad, 172–5204). The primers were used as below: human GPX4: 5’-ACAAGAACGGCTGCGTGGTGAA-3’ and 5’-GCCACACACTTGTGGAGCTAGA-3’; human PTGS2: 5’-CGGTGAAACTCTGGCTAGACAG-3’ and 5’-GCAAACCGTAGATGCTCAGGGA-3’; human ACSL4: 5’-GCTATCTCCTCAGACACACCGA-3’ and 5’-AGGTGCTCCAACTCTGCCAGTA-3’; human TMEM164: 5’-AAGTTCGCCACCAAGACCGTCA-3’ and 5’-GCTTGAAGACGACGATAGCTCC-3’; human ACTB: 5’-CACCATTGGCAATGAGCGGTTC-3’ and 5’-AGGTCTTTGCGGATGTCCACGT-3’. The data were normalized to ACTB RNA, and the fold change was calculated via the 2−ΔΔCt method [20]. Based on the untreated group, the relative concentration of mRNA was expressed in arbitrary units, and its assigned value was 1.
Western blot analysis
After treatment, whole cells were harvested and lysed at 4°C in ice-cold Cell Lysis Buffer (Cell Signaling Technology, 9803) containing a protease inhibitor cocktail (Thermo Fisher Scientific, 78429) [74]. A bicinchoninic acid (BCA) assay was used to detect protein concentration, and then 30 µg of protein in each lysate sample was subjected to SDS-PAGE (Bio-Rad, 3450124) at 100–120 V for 90 min, and then transferred to PVDF membrane (Bio-Rad, 1704273). The PVDF membrane was blocked with 5% nonfat dry milk (Cell Signaling Technology, 9999), and then incubated with the indicated primary antibody (1:500-1:1000) at 4°C overnight. The membrane was washed 3 times in tris-buffered saline Tween 20 (Cell Signaling Technology, 9997) buffer, and then incubated with an HRP-linked anti-mouse IgG secondary antibody (Cell Signaling Technology, 7076; 1:1000) or HRP-linked anti-rabbit IgG secondary antibody (Cell Signaling Technology, 7074; 1:1000) for 1 h at room temperature. After enhanced chemiluminescence exposure, a ChemiDoc Touch Imaging System (Bio-Rad, 1708370) or X-ray films were used for visualization and analysis of protein expression. ACTB was used as a housekeeping control for whole cell lysates.
Immunoprecipitation analysis
After treatment, the cells were lysed at 4°C using an ice-cold radio-immunoprecipitation assay lysis buffer (Millipore, 20–188), and the cell insoluble matter was removed by centrifugation (12,000 g, 10 min) [75]. BCA was used to detect the protein concentration, and then 300 μg of protein in each lysate sample was pre-cleared for 3 h with protein A/G Sepharose beads (Abcam, ab193262) at 4°C. Then, in the presence of protein A/G Sepharose beads, the sample with control IgG or a specific antibody (4 µg/mL) was gently shaken overnight at 4°C. After incubation, the protein A/G Sepharose beads were washed thoroughly with PBS, and the protein was eluted by boiling in 2 × Laemmli sample buffer (Bio-Rad, 161-0737) before SDS-PAGE electrophoresis.
Immunofluorescence analysis
Cells were cultured on glass coverslips and fixed in 3% formaldehyde for 30 min at room temperature prior to detergent extraction with 0.1% Triton X-100 (Cell Signaling Technology, 39487) for 10 min at 25°C. Coverslips were saturated with 2% bovine serum albumin (Cell Signaling Technology, 9998) in PBS for 1 h at room temperature and processed for immunofluorescence with MAP1LC3B antibody (1:200; Cell Signaling Technology, 3868), followed by Alexa Fluor 488-conjugated IgG (1:500; Thermo Fisher Scientific, A21206). Images were taken with a ZEISS LSM 800 confocal microscope. Relative quantitative colocalization analysis was performed from 10 random fields.
Bioinformatics analysis
The TCGA datasets, including gene expression and clinical outcomes data, were obtained from level 3 datasets at FireBrowse (http://gdac.broadinstitute.org; 2016 January). GEPIA (http://gepia.cancer-pku.cn/index.html), a developed interactive web server for analyzing the TCGA data, was used to separate the TCGA cohorts into groups with high/low expression of selected genes, which were then used for the prognostic signature validation. TIMER2.0 (http://timer.cistrome.org/), a web resource for systematic evaluations of the clinical impact of different immune cells in diverse cancer types, was used to assay the impact of the expression of TMEM164 on immune cell infiltration in patients with pancreatic ductal adenocarcinomas. The analysis of protein expression in human pancreatic cancer was based on The Human Protein Atlas online platform (http://www.proteinatlas.org/). In brief, the immunohistochemical staining of the indicated protein was used to yield a protein expression profile by the Human Protein Atlas consortium.
Statistical analysis
GraphPad Prism 8.4.3 was used to collect and analyze data. Unpaired Student’s t tests were used to compare the means of two groups. A one-way or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used for comparison among the different groups. A P value of <0.05 was considered statistically significant.
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript. This work was partially supported by grants from the National Natural Science Foundation of China (81872323 and 82073299 to C.L.; 82172656 to X.Y.; 81830048 to J.L.).
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. DOI: 10.1038/s41422-020-00441-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. DOI: 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–296. DOI: 10.1038/s41571-020-00462-0 [DOI] [PubMed] [Google Scholar]
- [4].Distefano AM, Martin MV, Cordoba JP, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216(2):463–476. DOI: 10.1083/jcb.201605110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kuang F, Liu J, Kang R, et al. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. DOI: 10.3389/fcell.2020.586578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. DOI: 10.1080/15548627.2020.1810918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. DOI: 10.1038/cdd.2015.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. DOI: 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Klionsky DJ, Emr SD.. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. DOI: 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- [11].Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45. DOI: 10.4161/15548627.2014.984267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. DOI: 10.1080/15548627.2016.1187366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rong Y, Fan J, Ji C, et al. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2022;29(6):1164–1175. DOI: 10.1038/s41418-021-00907-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang Z, Guo M, Li Y, et al. RNA-Binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16(8):1482–1505. DOI: 10.1080/15548627.2019.1687985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xie Y, Kuang F, Liu J, et al. DUSP1 blocks autophagy-dependent ferroptosis in pancreatic cancer. J Pancreatology. 2020;3(3):154–160. DOI: 10.1097/JP9.0000000000000054. [DOI] [Google Scholar]
- [18].Li C, Zhang Y, Liu J, et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17(4):948–960. DOI: 10.1080/15548627.2020.1739447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu K, Huang J, Liu J, et al. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. 2022;13(6):521. DOI: 10.1038/s41419-022-04974-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu J, Zhu S, Zeng L, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy. 2021:1–14. DOI: 10.1080/15548627.2021.2008692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kang R, Zhu S, Zeh HJ, et al. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14(12):2173–2175. DOI: 10.1080/15548627.2018.1513758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435. DOI: 10.1016/j.chembiol.2020.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. DOI: 10.1016/j.semcancer.2019.03.002 [DOI] [PubMed] [Google Scholar]
- [24].Schmit K, Michiels C. TMEM proteins in cancer: a review. Front Pharmacol. 2018;9:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gazou A, Riess A, Grasshoff U, et al. Xq22.3-Q23 deletion including ACSL4 in a patient with intellectual disability. Am J Med Genet A. 2013;161(4):860–864. DOI: 10.1002/ajmg.a.35778 [DOI] [PubMed] [Google Scholar]
- [26].Poreau B, Ramond F, Harbuz R, et al. Xq22.3q23 microdeletion harboring TMEM164 and AMMECR1 genes: two case reports confirming a recognizable phenotype with short stature, midface hypoplasia, intellectual delay, and elliptocytosis. Am J Med Genet A. 2019;179(4):650–654. DOI: 10.1002/ajmg.a.61057 [DOI] [PubMed] [Google Scholar]
- [27].Fan T, Liu Y, Liu H, et al. Transmembrane Protein-based risk model and H3K4me3 modification characteristics in lung adenocarcinoma. Front Oncol. 2022;12:828814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zou Y, Palte MJ, Deik AA, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. DOI: 10.1038/s41467-019-09277-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. DOI: 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. DOI: 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. DOI: 10.1016/j.bbrc.2016.08.124 [DOI] [PubMed] [Google Scholar]
- [32].Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309. DOI: 10.1038/s41589-020-0472-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2020. DOI: 10.1016/j.molcel.2020.11.024. [DOI] [PubMed] [Google Scholar]
- [34].Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52(3):1011–1022. [DOI] [PubMed] [Google Scholar]
- [35].Eling N, Reuter L, Hazin J, et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu Y, Wang Y, Liu J, et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021;28(1–2):55–63. DOI: 10.1038/s41417-020-0182-y [DOI] [PubMed] [Google Scholar]
- [37].Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064–2077. DOI: 10.1158/0008-5472.CAN-16-1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xie Y, Zhu S, Zhong M, et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153(5):1429–43 e5. DOI: 10.1053/j.gastro.2017.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dai C, Chen X, Li J, et al. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27(9):645–656. DOI: 10.1038/s41417-020-0170-2 [DOI] [PubMed] [Google Scholar]
- [40].Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. DOI: 10.1002/hep.28251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Olayanju A, Copple IM, Bryan HK, et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015;78:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wen Q, Liu J, Kang R, et al. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278–283. DOI: 10.1016/j.bbrc.2019.01.090 [DOI] [PubMed] [Google Scholar]
- [44].Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. DOI: 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. DOI: 10.1038/s41586-019-1707-0 [DOI] [PubMed] [Google Scholar]
- [46].Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. DOI: 10.1038/s41586-019-1705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mao C, Liu X, Zhang Y, et al. DHODH-Mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. DOI: 10.1038/s41586-021-03539-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. 2019;116(8):2996–3005. DOI: 10.1073/pnas.1819728116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. DOI: 10.1016/j.bbrc.2018.12.039 [DOI] [PubMed] [Google Scholar]
- [50].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (1). Autophagy. 2021;17:1–382. DOI: 10.1080/15548627.2020.1797280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Judith D, Jefferies HBJ, Boeing S, et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J Cell Biol. 2019;218(5):1634–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Noda T, Kim J, Huang WP, et al. Apg9p/cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol. 2000;148(3):465–480. DOI: 10.1083/jcb.148.3.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lang T, Reiche S, Straub M, et al. Autophagy and the cvt pathway both depend on AUT9. J Bacteriol. 2000;182(8):2125–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. DOI: 10.1016/j.molcel.2016.09.037 [DOI] [PubMed] [Google Scholar]
- [55].Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–398. DOI: 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- [56].Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20(21):5971–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang I, Cui Y, Amiri A, et al. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur J Pharm Biopharm. 2016;100:66–76. [DOI] [PubMed] [Google Scholar]
- [58].Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6):e20210518. DOI: 10.1084/jem.20210518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jung S, Jeong H, Yu SW. Autophagy as a decisive process for cell death. Exp Mol Med. 2020;52(6):921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;17(11):3361–3374. DOI: 10.1080/15548627.2021.1872241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. DOI: 10.1126/sciadv.aaw2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen X, Song X, Li J, et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy. 2022;1–21. DOI: 10.1080/15548627.2022.2059170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589. DOI: 10.1038/s41467-021-21841-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Han L, Bai L, Fang X, et al. SMG9 drives ferroptosis by directly inhibiting GPX4 degradation. Biochem Biophys Res Commun. 2021;567:92–98. DOI: 10.1016/j.bbrc.2021.06.038 [DOI] [PubMed] [Google Scholar]
- [67].Guardia CM, Christenson ET, Zhou W, et al. The structure of human ATG9A and its interplay with the lipid bilayer. Autophagy. 2020;16(12):2292–2293. DOI: 10.1080/15548627.2020.1830522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. DOI: 10.1038/s41467-020-20154-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lang X, Green MD, Wang W, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673–1685. DOI: 10.1158/2159-8290.CD-19-0338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. DOI: 10.1038/nature14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. DOI: 10.1038/s41586-019-1170-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature. 2019;572(7769):402–406. DOI: 10.1038/s41586-019-1426-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lei G, Zhang Y, Koppula P, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–162. DOI: 10.1038/s41422-019-0263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069–2083. DOI: 10.1080/15548627.2020.1714209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. DOI: 10.1083/jcb.200911078 [DOI] [PMC free article] [PubMed] [Google Scholar]