

ABSTRACT
Triple-negative breast cancer (TNBC) is the most challenging breast cancer subtype to treat due to the lack of effective targeted therapies. Transmembrane (TMEM) proteins represent attractive drug targets for cancer therapy, but biological functions of most members of the TMEM family remain unknown. Here, we report for the first time that TMEM63A (transmembrane protein 63A), a poorly characterized TMEM protein with unknown functions in human cancer, functions as a novel oncogene to promote TNBC cell proliferation, migration, and invasion in vitro and xenograft tumor growth and lung metastasis in vivo. Mechanistic investigations revealed that TMEM63A localizes in endoplasmic reticulum (ER) and lysosome membranes, and interacts with VCP (valosin-containing protein) and its cofactor DERL1 (derlin 1). Furthermore, TMEM63A undergoes autophagy receptor TOLLIP-mediated autophagic degradation and is stabilized by VCP through blocking its lysosomal degradation. Strikingly, TMEM63A in turn stabilizes oncoprotein DERL1 through preventing TOLLIP-mediated autophagic degradation. Notably, pharmacological inhibition of VCP by CB-5083 or knockdown of DERL1 partially abolishes the oncogenic effects of TMEM63A on TNBC progression both in vitro and in vivo. Collectively, these findings uncover a previously unknown functional and mechanistic role for TMEM63A in TNBC progression and provide a new clue for targeting TMEM63A-driven TNBC tumors by using a VCP inhibitor.
Abbreviations: ATG16L1, autophagy related 16 like 1; ATG5, autophagy related 5; ATP5F1B/ATP5B, ATP synthase F1 subunit beta; Baf-A1, bafilomycin A1; CALCOCO2/NDP52, calcium binding and coiled-coil domain 2; CANX, calnexin; DERL1, derlin 1; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; HSPA8, heat shock protein family A (Hsp70) member 8; IP, immunoprecipitation; LAMP2A, lysosomal associated membrane protein 2; NBR1, NBR1 autophagy cargo receptor; OPTN, optineurin; RT-qPCR, reverse transcription-quantitative PCR; SQSTM1/p62, sequestosome 1; TAX1BP1, Tax1 binding protein 1; TMEM63A, transmembrane protein 63A; TNBC, triple-negative breast cancer; TOLLIP, toll interacting protein; VCP, valosin containing protein
KEYWORDS: Macroautophagic degradation, proteostasis, selective autophagy receptor, transmembrane protein, triple-negative breast cancer
Introduction
Triple-negative breast cancer (TNBC) is characterized by the lack of expression of ESR (estrogen receptor), PGR (progesterone receptor), and ERBB2/HER2 (erb-b2 receptor tyrosine kinase 2), and accounts for approximately 15–20% of all breast cancers [1]. Compared with other subtypes of breast cancer, TNBC has an extremely aggressive clinical course with earlier age of onset, higher probability of recurrence and distant metastasis, poorer overall survival, and the lack of validated targeted therapies [2]. The discovery of targetable molecular targets that drive TNBC progression is absolutely imperative.
Transmembrane (TMEM) proteins are defined by the presence of one or more transmembrane domains, and constitute a large family of proteins spanning the entirety of the lipid bilayer of biological membrane [3]. Due to the amphiphilic nature of TMEM proteins, they form either α-helical or β-barrel structures while embedded in the membrane [4]. Accumulating evidence shows that TMEM proteins play vital roles in cellular functions by functioning as ion channels, transporters, signal receptors, and enzymes [3,5]. Consequently, dysregulation of TMEM proteins has been linked with cancer progression and therapeutic resistance [6,7]. Moreover, TMEM proteins, such as EGFR (epidermal growth factor receptor) and ERBB2/HER2, represent attractive drug targets for cancer therapy. Therefore, identifying actionable TMEM proteins that contribute to cancer progression remains important areas of investigation.
Although approximately 20–30% of human genes are predicted to encode TMEM proteins [4,8], the structure, function, and mechanism of action of most members of the TMEM family remain unknown due to the technique difficulty in expression and purification of these proteins and the diversity in their characteristics and subcellular localization [6,8,9]. These TMEM proteins with unknown structure and function are classified into the TMEM protein family. A case in point is TMEM63A (transmembrane protein 63A), which contains 10 transmembrane domains [10,11] and is a poorly characterized member of the TMEM protein family. Available evidence shows that TMEM63A serves as an osmolarity sensitive ion channel for the osmoreception [12], and affects the proliferation and migration of goat peripheral blood mononuclear cells [13]. In addition, mutations in TMEM63A have recently been observed in patients with hypomyelination during infancy [10,14], global developmental delay [15], and hypomyelinating leukodystrophy [16]. However, the biological function and related mechanism of TMEM63A in human cancers have not yet been explored.
The endoplasmic reticulum (ER) is a multifunctional organelle essential for the biosynthesis, folding, stabilization, maturation, and trafficking of secretory and TMEM proteins [17]. Disruption of ER homeostasis by internal and external stimuli results in the accumulation of misfolded/unfolded and misassembled proteins, thus leading to the induction of ER stress [18]. To overcome this problem, cells have evolved two guardian pathways to maintain cellular protein homeostasis either by ER-associated degradation (ERAD) or by autophagy [19,20].
In ERAD, misfolded or unassembled proteins are retrotranslocated to the cytosol to be degraded by the proteasome [21]. VCP/p97 (valosin containing protein) and its cofactor DERL1 (derlin 1) are two core components of the ERAD pathway [22–24]. VCP is bound to ER membrane and contributes to extraction of misfolded and ubiquitinated proteins to the cytosol for degradation by the proteasome or through autophagy [25,26]. DERL1 forms a protein channel to mediate retrotranslocation of misfolded or misassembled proteins across the ER membrane to the cytosol for degradation by the proteasome [23,27,28]. Accumulating evidence shows that both VCP and DERL1 are overexpressed in multiple types of human cancer and contribute to tumor growth and metastasis [29–34], thus marking them as potential therapeutic targets [35]. Indeed, antibodies against DERL1 can suppress tumor growth [34], and several VCP inhibitors exert antitumor activity in multiple cancer model systems [36,37]. Despite their functional importance in cancer, the precise molecular mechanisms of VCP and DERL1 in human cancer remain to be characterized.
Autophagy is an evolutionarily conserved pathway for the degradation of cytoplasmic materials through the lysosomal machinery [38,39], and is generally classified into nonselective and selective autophagy [38]. In selective autophagy, cytoplasmic components are selected and tagged before being sequestered into an autophagosome by means of selective autophagy receptors, such as SQSTM1 (sequestosome 1), NBR1 (NBR1 autophagy cargo receptor), OPTN (optineurin), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), ATG16L1 (autophagy related 16 like 1), TOLLIP (toll-interacting protein), or TAX1BP1 (Tax1 binding protein 1) [40]. These receptors recognize ubiquitinated substrates via their ubiquitin-binding domains and tether them to the phagophore membranes by their LC3-interacting regions (LIRs) [41–44].
In this study, we provide the first evidence that TMEM63A plays a pivotal role in TNBC progression. Mechanistic investigations revealed that TMEM63A is a downstream effector of VCP and an upstream regulator of DERL1 through TOLLIP-mediated autophagic degradation pathway. Moreover, pharmacological inhibition of VCP or depletion of DERL1 abolishes the oncogenic effects of TMEM63A on TNBC progression both in vitro and in vivo. Taken together, these findings reveal a previously unknown biological function and regulatory mechanism of TMEM63A in TNBC progression and provide potential therapeutic option for TMEM63A-driven TNBC progression with VCP inhibitors.
Results
TMEM63A is overexpressed in TNBC and promotes the proliferation, migration and invasion of TNBC cells
To identify potential TMEM proteins contributing to TNBC progression, we recently carried out iTRAQ-based quantitative proteomic analysis on 90 cases of TNBC tissues and 72 cases of matched adjacent normal tissues, and found that TMEM63A was detected in 12.2% (11/90) TNBC tissues and 0% (0/72) of adjacent normal breast tissues (Figure 1A). We were particularly interested in this protein because analysis of Gene Expression Omnibus (GEO) public datasets revealed that TMEM63A was one of 47 genes that were commonly upregulated in clinical breast cancer brain metastases samples relative to primary breast tumors (GEO accession number: GSE100534 [45]) (Fig. S1A) and in MDA-231-derived brain metastasis cells (MDA-231 BrM) compared to parental MDA-231 cells (GEO accession number: GSE12237 [46]) (Fig. S1B) [47]. In addition, TMEM63A was also upregulated in MDA-231-derived lung metastasis cells (LM2-4142, −4173, −4175, and −4180) compared to parental MDA-231 cells (GEO accession number: GSE2603 [48]) (Fig. S1C). These results indicate TMEM63A may be relevant to breast cancer progression, but the biological functions and related molecular mechanism of TMEM63A in human cancer have not yet been explored.
Figure 1.
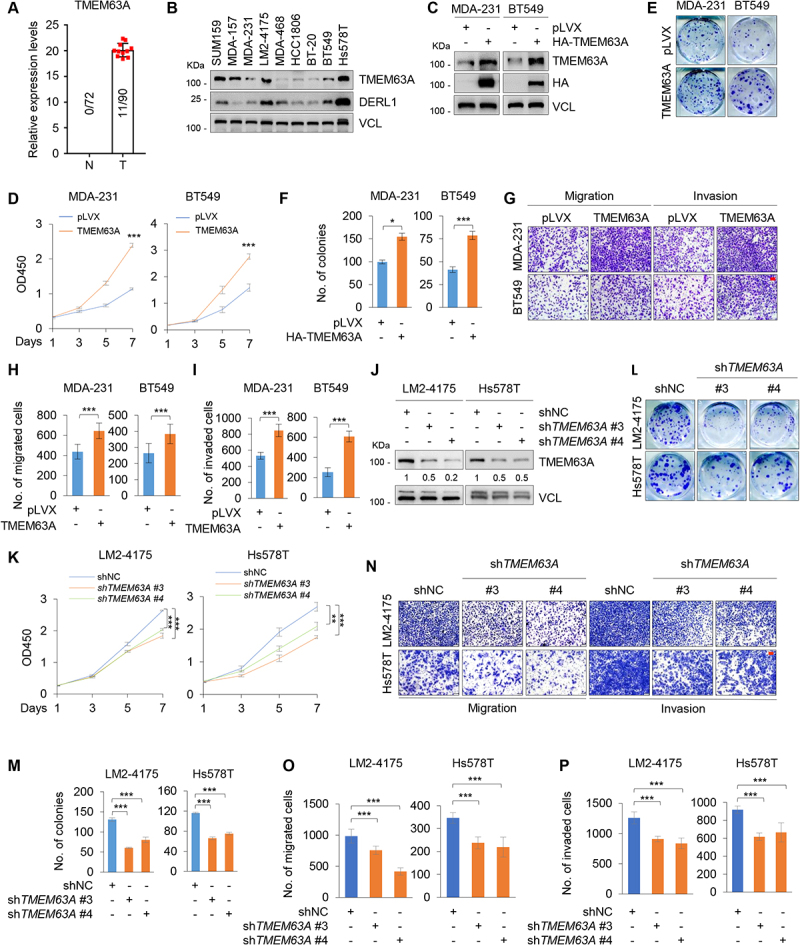
TMEM63A is upregulated in TNBC and promotes TNBC cell proliferation, migration, and invasion in vitro. (A) Detection of TMEM63A expression status in 90 cases of TNBC tissues and 72 cases of matched adjacent normal tissues by iTRAQ-based quantitative proteomic analysis. (B) Immunoblotting analysis of TMEM63A and DERL1 expression levels in 9 TNBC cell lines. (C) Establishment of stable MDA-231 and BT549 cells expressing empty vector pLVX and HA-TMEM63A by lentiviral infection. The expression status of TMEM63A in these resultant cell lines was validated by immunoblotting. (D-F) MDA-231 and BT549 cells stably expressing pLVX and HA-TMEM63A were subjected to CCK-8 assays (D) and colony formation assays (E-F). Representative images of survival colonies (E) and corresponding quantitative results (F) are shown. (G-I) MDA-231 and BT549 cells stably expressing pLVX and HA-TMEM63A were subjected to Transwell migration and invasion assays. Representative images of migrated and invaded cells are shown in G, and corresponding quantitative results are shown in H and I, respectively (scale bar:100 μm). (J) Immunoblotting analysis of TMEM63A expression in LM2-4175 and Hs578T cells stably expressing shNC and shTMEM63A (#3 and #4) with the indicated antibodies. Densitometric quantitation of western blots was performed using ImageJ. (K-M) LM2-4175 and Hs578T stably expressing shNC and shTMEM63A (#3 and #4) were subjected to CCK-8 assays (K) and colony formation assays (L-M). Representative images of survival colonies (L) and corresponding quantitative results (M) are shown. (N-P) LM2-4175 and Hs578T stably expressing shNC and shTMEM63A (#3 and #4) were subjected to Transwell migration and invasion assays. Representative images of migrated and invaded cells are shown in NN, and corresponding quantitative results are shown in 0Oand P P, respectively (scale bar: 100 μm). *, **, and *** indicate statistically significant at p < 0.05, p < 0.01, and p < 0.001 level, respectively.
To address the biological functions of TMEM63A in TNBC progression, we first examined the expression levels of TMEM63A in 9 TNBC cell lines by immunoblotting. Results showed that SUM159, MDA-157, LM2-4175, and Hs578T cell lines expressed relatively higher levels of TMEM63A than BT20, MDA-231, MDA-468, HCC1806 and BT549 cells did (Figure 1B). In addition, we noticed that the expression levels of TMEM63A were higher in LM2-4175 cell line, a highly lung metastatic variant of MDA-231 cell line [48], than in its parental MDA-231 cell line (Figure 1B). Based on the expression levels of TMEM63A in and biological characteristics of these TNBC cell lines, we next stably expressed pLVX and HA-TMEM63A in MDA-231 and BT549 cell lines by lentiviral infection. The expression status of TMEM63A in these resultant cell lines was validated by immunoblotting (Figure 1C). Cell proliferation assays using CCK-8 kit and colony formation assays showed that ectopic expression of TMEM63A enhanced the proliferation (Figure 1D) and colony formation (Figure 1E,F) of MDA-231 and BT549 cells. Transwell invasion and migration assays revealed that MDA-231 and BT549 cells expressing TMEM63A had enhanced migratory and invasive potential compared to control cells expressing empty vector (Figure 1G-I).
To further verify the aforementioned results, we next knocked down endogenous TMEM63A in LM2-4175 and Hs578T cells by lentiviral infection with expression vectors encoding two independent shRNAs targeting TMEM63A (shTMEM63A #3 and #4) and negative control shRNA (shNC). The effectiveness of shRNA-mediated knockdown of TMEM63A was confirmed by immunoblotting (Figure 1J). Cell function assays as described above demonstrated that knockdown of TMEM63A attenuated the proliferation (Figure 1K), colony growth (Figure 1L,M), and migratory and invasive potential (Figure 1N-P) of LM2-4175 and Hs578T cells. Taken together, these results suggest that TMEM63A contributes to the malignant characteristics of TNBC cells.
TMEM63A is a novel binding partner of VCP
To examine the upstream regulatory factors of TMEM63A in TNBC cells, we established stable HEK293T cell lines expressing pCDH and Flag-TMEM63A (Figure 2A). Then, total cellular lysates from these established cell lines were subjected to IP analysis using an anti-Flag antibody (Fig. S2A), and the IP complex was subjected to electrospray ionization-tandem mass spectrometry (ESI-MS/MS) analysis to identify binding partners of TMEM63A. By this approach, we found that 191 proteins were specifically associated with Flag-TMEM63A according to the identified unique peptides more than 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that these TMEM63A-interacting proteins were associated with ribosome, proteasome, protein processing in endoplasmic reticulum, and others (Fig. S2B). The top 10 TMEM63A-interacting proteins based on the number of unique peptides identified by mass spectrometry are shown in Figure 2B. IP and immunoblotting analyses with the indicated antibodies further demonstrated that TMEM63A indeed interacted with the top three TMEM63A-interacting proteins, including VCP (valosin containing protein), CANX (calnexin), and ATP5F1B/ATP5B (ATP synthase F1 subunit beta) (Figure 2C).
Figure 2.
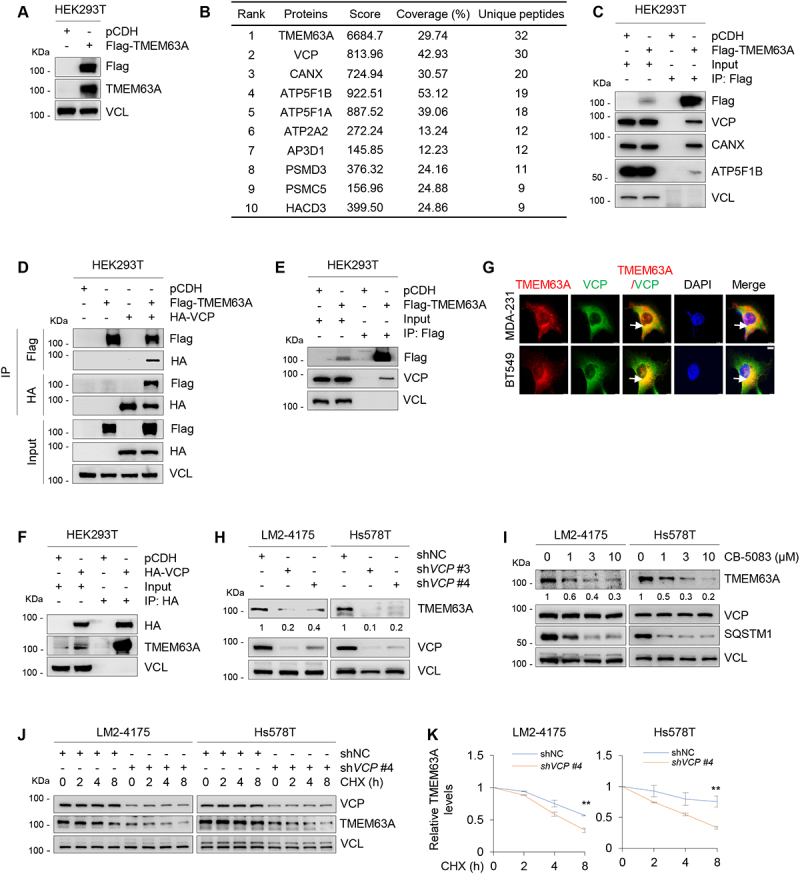
TMEM63A is a novel binding partner of VCP and is stabilized by VCP. (A) Immunoblotting analysis of TMEM63A expression in HEK293T cells stably expressing pCDH and Flag-TMEM63A with the indicated antibodies. (B) The top 10 TMEM63A-interacting proteins based on the number of identified unique peptides by mass spectrometry. (C) Total cellular lysates from HEK293T cells stably expressing pCDH and Flag-TMEM63A were subjected to IP assays with an anti-Flag antibody, followed by immunoblotting with the indicated antibodies. (D) HEK293T cells were transfected with pCDH, Flag-TMEM63A, and HA-VCP alone or in combination. After 48 h of transfection, IP and immunoblotting assays were performed with the indicated antibodies. (E-F) HEK293T cells were transfected with pCDH, Flag-TMEM63A (E) or HA-VCP (F). After 48 h of transfection, IP and immunoblotting assays were performed with the indicated antibodies. (G) MDA-231 and BT549 cells stably expressing pLVX and Flag-TMEM63A were subjected to immunofluorescent staining with an anti-Flag (red) or anti-VCP (green) antibody. DNA was counterstained with DAPI (blue). Typical colocalization between Flag-TMEM63A and VCP (yellow) is indicated by white arrows (scale bar: 7.5 μm). (H) LM2-4175 and Hs578T cells stably expressing shNC and shVCP (#3 and #4) were subjected to immunoblotting analysis with the indicated antibodies. (I) LM2-4175 and Hs578T cells were treated with or without increasing doses of VCP inhibitor CB-5083 for 8 h, and then subjected to immunoblotting with the indicated antibodies. (J-K) LM2-4175 and Hs578T cells stably expressing shNC and shVCP #4 were treated with or without 100 μg/mL CHX for the indicated times, and then subjected to immunoblotting analysis with the indicated antibodies (J). Relative TMEM63A protein levels (TMEM63A: VCL) are shown in K. **, p < 0.01. In panels H-J, densitometric quantitation of Western blots was performed using ImageJ.
As VCP ranks among the top one in TMEM63A-interacting proteins and plays a key role in the control of protein homeostasis and cancer aggressiveness [30], we thereafter focused on addressing the biological significance of the noted interaction between TMEM63A and VCP. To further validate whether TMEM63A interacts with VCP, HEK293T cells were transfected with Flag-TMEM63A, HA-VCP alone or in combination, and subjected to reciprocal IP assays with an anti-Flag or anti-HA antibody. Immunoblotting analysis with the indicated antibodies revealed that Flag-TMEM63A and HA-VCP pulled down each other only when both were co-expressed (Figure 2D). Moreover, Flag-TMEM63A interacted with endogenous VCP (Figure 2E), while HA-VCP interacted with endogenous TMEM63A (Figure 2F) in HEK293T cells. Immunofluorescent staining showed that TMEM63A partially co-localized with VCP in MDA-231 and BT549 cells (Figure 2G). Together, these results suggest that TMEM63A is a novel binding partner of VCP.
VCP stabilizes TMEM63A in TNBC cells
As VCP plays a central role in maintaining protein homeostasis [30], we next determined whether VCP and TMEM63A regulate each other. Notably, either ectopic expression of TMEM63A in MDA-231 and BT549 cells (Fig. S2C) or shRNA-mediated knockdown of endogenous TMEM63A in LM2-4175 and Hs578T cells (Fig. S2D) did not markedly affect the expression levels of VCP. Conversely, knockdown of VCP in LM2-4175 and Hs578T cells significantly downregulated the protein levels of TMEM63A (Figure 2H). As a positive control, the expression levels of SQSTM1, a known substrate of VCP [36], were decreased following treatment of LM2-4175 and Hs578T cells with VCP inhibitor CB-5083 [25]. Consistently, treatment of LM2-4175 and Hs578T cells with CB-5083 resulted in a decrease in TMEM63A protein levels in a dose-dependent manner (Figure 2I). RT-qPCR assays showed that depletion or pharmacological inhibition of VCP did not significantly affect TMEM63A mRNA levels (Fig. S2E and S2F). These results suggest the regulation of TMEM63A by VCP to be post-transcriptional. In support of this notion, cycloheximide (CHX)-chase assays showed that knockdown of VCP shortened the half-life of TMEM63A protein (Figure 2J,K). Collectively, these results suggest that VCP is a stabilizer of TMEM63A.
VCP blocks TOLLIP-mediated autophagic degradation of TMEM63A
The ubiquitin-proteasome system and autophagy are two major cellular degradation systems in eukaryotic cells [49]. To date, the molecular mechanism of TMEM63A degradation in eukaryotic cells remains unknow. To examine whether TMEM63A is degraded through ubiquitin-proteasome system, we treated MDA-231 and BT549 cells with the proteasome inhibitor MG-132 for the indicated times. Immunoblotting assays showed no significant change in TMEM63A protein levels following MG-132 treatment (Fig. S3A). As a positive control, MG-132 treatment resulted in a significant increase in the protein levels of CDKN1A/p21, a known substrate of the ubiquitin-proteasome system [50]. In contrast, incubation of MDA-231 and BT549 cells with the autophagy inhibitors bafilomycin A1 (Baf-A1) (Figure 3A) and ammonium chloride (NH4Cl) (Figure 3B) led to a remarkable accumulation of TMEM63A in a time-dependent manner. Conversely, the protein levels of TMEM63A were decreased in MDA-231 and BT549 cells following treatment with the autophagy inducer rapamycin (Rapa) in a time-dependent manner (Figure 3C). These results suggest that TMEM63A is degraded mainly through the autophagy-lysosome but not the ubiquitin-proteasome pathway.
Figure 3.
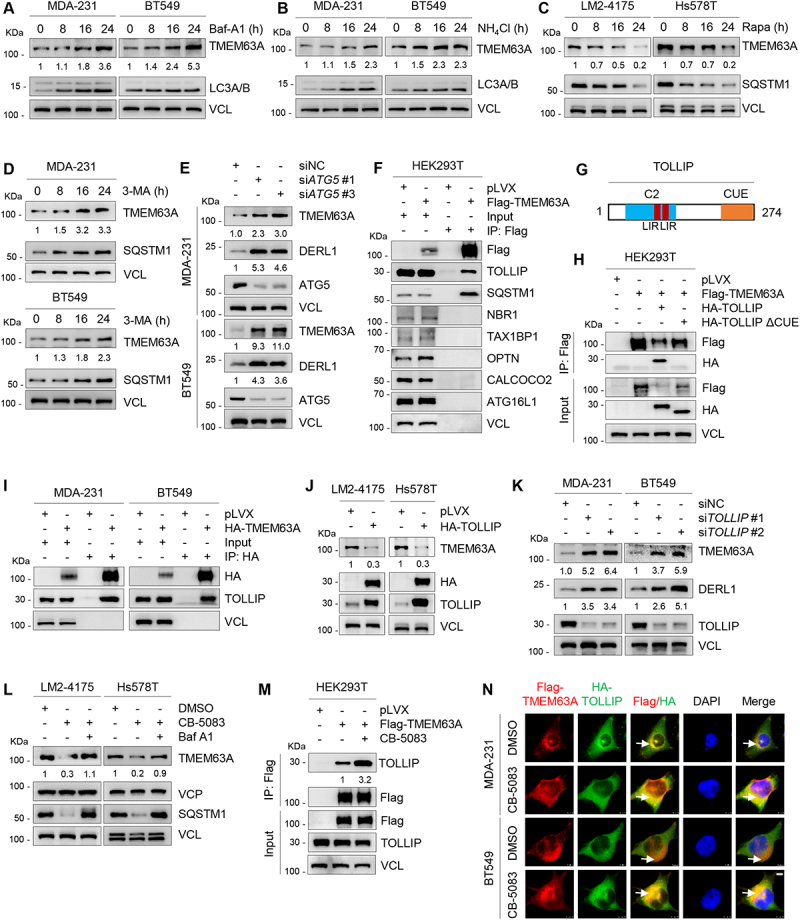
VCP blocks TOLLIP-mediated autophagic degradation of TMEM63A. (A-B) MDA-231 and BT549 cells were treated with or without 200 nM Baf-A1 (A) or 20 mM NH4Cl (B) for the indicated times, and then subjected to immunoblotting analysis with the indicated antibodies. (C) LM2-4175 and Hs578T cells were treated with or without 1 μM rapamycin (Rapa) for the indicated times, and then subjected to immunoblotting analysis with the indicated antibodies. (D) MDA-231 and BT549 cells were treated with or without 1 mM 3-MA for the indicated times, and then subjected to immunoblotting analysis with the indicated antibodies. (E) MDA-231 and BT549 cells were transfected with siNC and two independent siRNAs targeting ATG5. After 48 h of transfection, total cellular lysates were subjected to immunoblotting analysis with the indicated antibodies. (F) HEK293T cells were transfected with empty vector pLVX and Flag-TMEM63A, and were subjected to IP and immunoblotting analysis with the indicated antibodies after 48 h of transfection. (G) Schematic presentation of functional domains of TOLLIP [40]. LIR, LC3-interacting region; CUE, coupling of ubiquitin conjugation to ER degradation domain; C2, C2 domain. (H) HEK293T cells were transfected with the indicated expression vectors, and then subjected to IP and immunoblotting analysis with the indicated antibodies after 48 h of transfection. (I) MDA-231 and BT549 cells stably expressing pLVX and HA-TMEM63A were subjected to IP and immunoblotting analysis with the indicated antibodies. (J) LM2-4175 and Hs578T cells stably expressing pLVX and HA-TOLLIP were subjected to immunoblotting analysis with the indicated antibodies. (K) MDA-231 and BT549 cells were transfected with siNC and two independent siRNAs targeting TOLLIP. After 48 h of transfection, total cellular lysates were subjected to immunoblotting analysis with the indicated antibodies. (L) LM2-4175 and Hs578T were treated with the or without VCP inhibitor CB-5083 alone or in combination with Baf-A1 for 8 h and then subjected to immunoblotting analysis with the indicated antibodies. (M) HEK293T cells stably expressing empty vector pLVX and Flag-TMEM63A were treated with or without 3 μM VCP inhibitor CB-5083 for 4 h, and then subjected to IP and immunoblotting analysis with the indicated antibodies. (N) MDA-231 and BT549 cells stably expressing Flag-TMEM63A and HA-TOLLIP were treated with DMSO or 3 μM VCP inhibitor CB-5083 for 4 h. Immunofluorescence staining was performed with an anti-Flag (red) or anti-HA (green) antibodies. DNA was counterstained with DAPI (blue). Typical colocalization between Flag-TMEM63A and HA-TOLLIP (yellow) is indicated by white arrows (scale bar: 7.5 μm). In panels A-E, J-M, densitometric quantitation of Western blots was performed using ImageJ.
In mammals, three types of autophagy have been documented, including macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [51]. To determine whether TMEM63A is degraded through CMA pathway, we knocked down HSPA8 (heat shock protein family A (Hsp70) member 8) and LAMP2A (lysosomal associated membrane protein 2), two core components of the CMA machinery [52], using two specific siRNAs, and found no remarkable changes in the protein levels of TMEM63A after depletion of either HSPA8 or LAMP2A (Fig. S3B and S3C). In line with these observations, induction of CMA through prolonged nutrient starvation did not significantly alter TMEM63A protein levels (Fig. S3D). To determine whether macroautophagy contributes to TMEM63A degradation, MDA-231 and BT549 cells were treated with 1 mM 3-methyladenine (3-MA), a selective inhibitor of macroautophagy through blocking autophagosome formation [53]. As shown in Figure 3D, TMEM63A protein levels were increased in the presence of 3-MA. It has been previously reported that ATG5 (autophagy related 5) is an essential gene for mammalian macroautophagy [54]. We next knocked down endogenous ATG5 gene in MDA-231 and BT549 cells using two independent siRNAs targeting ATG5 (siATG5 #1 and #3), and then examined the expression levels of TMEM63A by immunoblotting. Results showed that knockdown of ATG5 resulted in a significant increase in protein levels of TMEM63A compared to its control counterpart (Figure 3E). These results collectively indicate that TMEM63A is degraded mainly through the macroautophagy pathway.
As macroautophagy is mediated by selective autophagy receptors, such as SQSTM1, NBR1, OPTN, CALCOCO2/NDP52, ATG16L1,TAX1BP1, and TOLLIP [55,56], we next screened autophagy receptors that bind to TMEM63A by IP assays. As shown in Figure 3F, TMEM63A bound to the autophagy receptors SQSTM1 and TOLLIP, but not NBR1, OPTN, CALCOCO2/NDP52, ATG16L1 and TAX1BP1. Previous studies have shown that SQSTM1 binds to ubiquitinated substrates through its C-terminal UBA (ubiquitin-associated) domain [57], whereas TOLLIP protein contains a CUE (coupling of ubiquitin to ER degradation) domain at its C terminus that interacts with ubiquitin or ubiquitinated proteins [58]. To investigate whether SQSTM1 mediates TMEM63A degradation, we deleted the C-terminal UBA domain of SQSTM1 (SQSTM1 ΔUBA), and found that both wild-type and ΔUBA mutant SQSTM1 enabled to interact with TMEM63A (Fig. S3E and S3F). Additionally, knockdown of SQSTM1 did not change the protein expression levels and half-life of TMEM63A (Fig. S3G and S3H), indicating that SQSTM1 is not required for TMEM63A degradation. In contrast, deletion of the CUE domain of TOLLIP (TOLLIP ΔCUE) significantly attenuated the interaction between TOLLIP and TMEM63A (Figure 3G,H). The interaction between TMEM63A and TOLLIP was further verified in MDA-231 and BT549 cells stably expressing HA-TMEM63A (Figure 3I). Moreover, ectopic expression of TOLLIP in LM2-4175 and Hs578T cells significantly downregulated the protein levels of TMEM63A (Figure 3J). Consistently, knockdown of TOLLIP significantly upregulated the protein levels of TMEM63A (Figure 3K). These results suggest that TOLLIP is involved in autophagic degradation of TMEM63A. We then investigated whether VCP is involved in autophagic degradation of TMEM63A and found that the reduced expression of TMEM63A caused by CB-5083 was rescued by Baf-A1(Figure 3L). These results indicate that VCP inhibits the autophagic degradation of TMEM63A. Furthermore, ectopic expression of HA-VCP reduced the ubiquitination levels of TMEM63A (Fig. S3I), and the interaction between TMEM63A and TOLLIP was significantly enhanced following treatment with VCP inhibitor CB-5083 [25] (Figure 3M). Immunofluorescence staining also demonstrated that CB-5083 treatment enhanced the intracellular co-localization of TMEM63A and TOLLIP in MDA-231 and BT549 cells (Figure 3N). Collectively, these results suggest that VCP blocks TOLLIP-mediated autophagic degradation of TMEM63A.
TMEM63A regulates oncoprotein DERL1 at the protein level
As TMEM63A is a predicted transmembrane protein [10,11], we next examined its subcellular localization by immunofluorescent staining with different antibodies against lysosome membrane marker LAMP2A [59], endoplasmic reticulum membrane marker CANX [60], and plasma membrane marker EGFR (epidermal growth factor receptor) [61]. As shown in Figure 4A, TMEM63A partially co-localized with LAMP2A (left) and CANX (middle) but not EGFR (right). As lysosome and endoplasmic reticulum play critical roles in the maintenance of cellular proteostasis [62], we employed iTRAQ-based quantitative proteomics technology to identify downstream proteins of TMEM63A using total cellular lysates from LM2-4175 cells stably expressing shNC and two shRNAs targeting TMEM63A (shTMEM63A #3 and #4). According to the cutoff value of 1.5-fold change, 222 proteins were upregulated, whereas 26 proteins were downregulated, in cells expressing shTMEM63A (#3 and #4) when compared to cells expressing shNC (Figure 4B). KEGG pathway analysis revealed that these differentially expressed proteins in cells expressing shTMEM63A and shNC were associated with protein processing in endoplasmic reticulum, ribosome, and others (Figure 4C). The top 10 downregulated proteins and 10 upregulated proteins after knockdown of TMEM63A in LM2-4175 cells are shown in Figure 4D,E, respectively.
Figure 4.
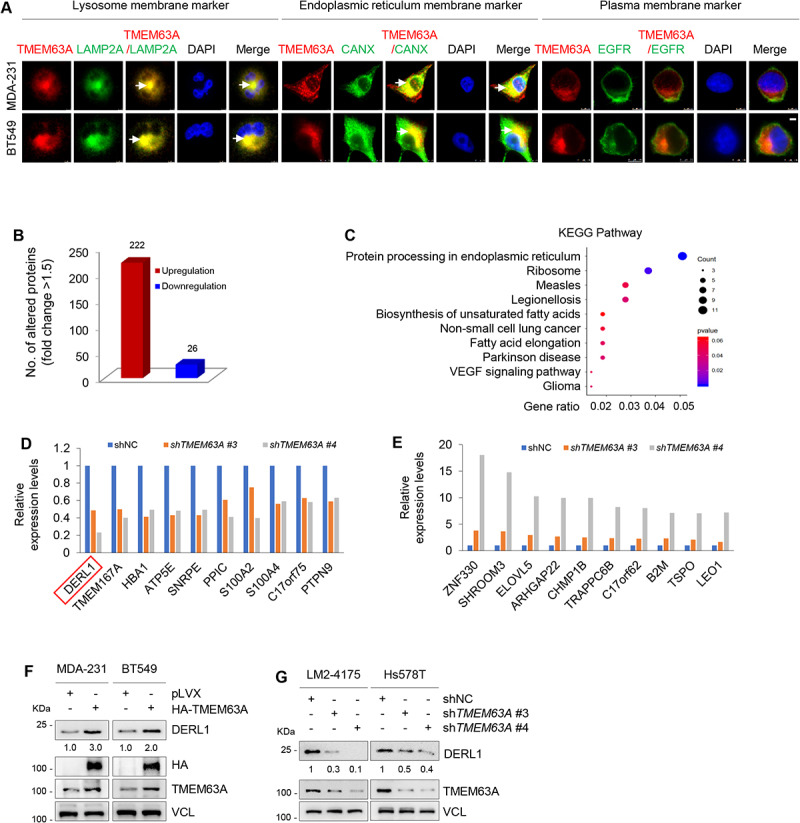
TMEM63A regulates oncoprotein DERL1 at the protein level. (A) MDA-231 and BT549 cells stably expressing pLVX and Flag-TMEM63A were subjected to immunofluorescent staining with antibodies against Flag (red), LAMP2A (green, left), CANX (green, middle), and EGFR (green, right). DNA was counterstained with DAPI (blue). Typical colocalization between Flag-TMEM63A and LAMP2A or CANX (yellow) is indicated by white arrows (scale bar:7.5 μm). (B) The number of differentially expressed proteins between cells expressing shTMEM63A (#3 and #4) and shNC based on the cutoff value of 1.5-fold change. (C) KEGG pathway analysis of differentially expressed proteins between cells expressing shTMEM63A (#3 and #4) and shNC. (D-E) The top 10 downregulated proteins (D) and 10 upregulated proteins (E) after knockdown of TMEM63A in LM2-4175 cells. (F) MDA-231 and BT549 cells stably expressing pLVX and HA-TMEM63A were subjected to immunoblotting analysis with the indicated antibodies. (G) LM2-4175 and Hs578T stably expressing shNC and shTMEM63A (#3 and #4) were subjected to immunoblotting analysis with the indicated antibodies. In panels F and G, densitometric quantitation of western blots was performed using ImageJ.
As DERL1 ranks first among the downregulated proteins after knockdown of TMEM63A and functions as an oncogene to promote tumor progression in different cancers [63–65], it was selected for further analysis. Consistent with quantitative proteomic results, immunoblotting analysis revealed that ectopic expression of TMEM63A in MDA-231 and BT549 cells upregulated (Figure 4F), whereas knockdown of TMEM63A in LM2-4175 and Hs578T cells downregulated (Figure 4G), DERL1 protein levels. RT-qPCR assays demonstrated that overexpression or knockdown of TMEM63A did not affect the mRNA levels of DERL1 (Fig. S4A and S4B). In addition, there was a correlation between protein levels between TMEM63A and DERL1 in TNBC cell lines (Figure 1B). Together, these results suggest that TMEM63A regulates DERL1 at the protein level.
TMEM63A blocks TOLLIP-mediated autophagic degradation of DERL1
To address the underlying mechanism by which TMEM63A regulates DERL1, we treated MDA-231 and BT549 cells with the proteasome inhibitor MG-132, the autophagy inhibitor Baf-A1, or the autophagy inducer rapamycin (Rapa). Immunoblotting analysis showed that DERL1 protein levels were increased following treatment with Baf-A1 (Figure 5A), but were decreased in the presence of rapamycin (Rapa) (Figure 5B). In contrast, treatment of cells with MG-132 did not significantly affect the protein levels of DERL1 (Fig. S4C). More importantly, pharmacological inhibition of lysosomal degradation by Baf-A1 rescued the downregulation of DERL1 caused by TMEM63A knockdown (Figure 5C). These results suggest that TMEM63A regulates DERL1 through the autophagy-lysosome pathway.
Figure 5.
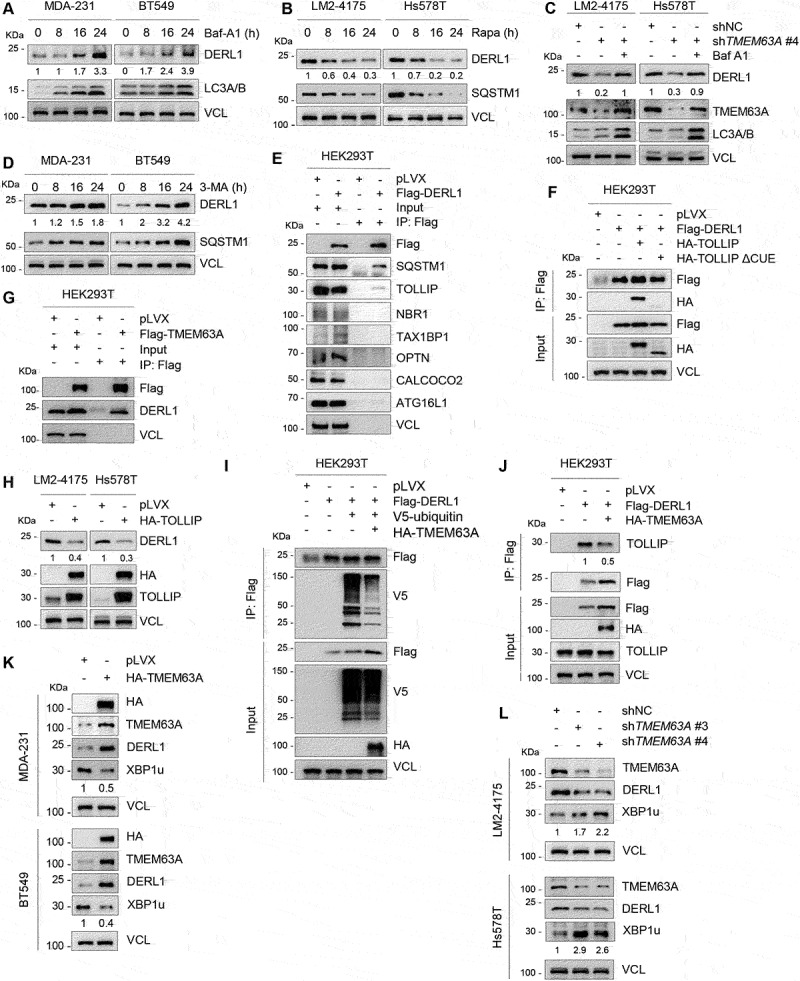
TMEM63A stabilizes DERL1 through blocking TOLLIP-mediated autophagic degradation of DERL1. (A) MDA-231 and BT549 cells were treated with or without 200 nM Baf-A1 for the indicated times and then subjected to immunoblotting analysis with the indicated antibodies. (B) LM2-4175 and Hs578T cells were treated with or without 1 μM rapamycin (Rapa) for the indicated times and then subjected to immunoblotting analysis with the indicated antibodies. (C) LM2-4175 and Hs578T cells stably expressing shNC and shTMEM63A #4 were treated with or without 200 nM Baf-A1 for 24 h and then subjected to immunoblotting analysis with the indicated antibodies. (D) MDA-231 and BT549 cells were treated with or without 1 mM 3-MA for the indicated times and then subjected to immunoblotting analysis. (E-F) HEK293T cells were transfected with the indicated expression vectors, and then subjected to IP and immunoblotting analysis with the indicated antibodies after 48 h of transfection. (G) HEK293T cells stably expressing pLVX and Flag-TMEM63A were subjected to IP and immunoblotting analysis with the indicated antibodies. (H) Immunoblotting analysis of LM2-4175 and Hs578T stably expressing pLVX and HA-TOLLIP with the indicated antibodies. (I-J) HEK293T cells were transfected with the indicated plasmids, and then subjected to IP and immunoblotting analysis with the indicated antibodies after 48 h of transfection. (K) MDA-231 and BT549 cells stably expressing pLVX and HA-TMEM63A were subjected to immunoblotting analysis. (L) LM2-4175 and Hs578T stably expressing shNC and shTMEM63A (#3 and #4) were lysed for immunoblotting analysis. In panels A-D, H, and J-L, densitometric quantitation of western blots was performed using ImageJ.
To assess whether CMA pathway mediates DERL1 degradation, we knocked down LAMP2A and HSPA8, two essential components of the CMA machinery [52], in MDA-231 and BT549 cells by two independent siRNAs. As shown in Fig. S4D and S4E, depletion of either LAMP2A or HSPA8 had no effects on DERL1 protein levels. IP assays also demonstrated that DERL1 did not interact with HSPA8 and LAMP2A (Fig. S4F). These results suggest that CMA is not responsible for lysosomal degradation of DERL1. In contrast, treatment of MDA-231 and BT549 cells with macroautophagy inhibitor 3-MA [53] resulted in an accumulation of DERL1 (Figure 5D). Consistently, knockdown of ATG5, an essential gene for mammalian macroautophagy [54], resulted in a significant increase in protein levels of DERL1 compared to the control counterpart (Figure 3E). These results suggest that DERL1 may be degraded by macroautophagy.
Then, we attempted to identify the autophagy receptor for DERL1 by IP assays, and found that DERL1 bound to the autophagy receptors SQSTM1 and TOLLIP, but not NBR1, OPTN, CALCOCO2/NDP52, ATG16L1 and TAX1BP1 (Figure 5E). Interestingly, deletion of the C-terminal UBA domain of SQSTM1 (SQSTM1 ∆UBA) did not affect the interaction between SQSTM1 with DERL1 (Fig. S4G). Moreover, knockdown of SQSTM1 using two independent siRNAs targeting SQSTM1 (siSQSTM1 #2 and #3) did not affect DERL1 protein levels (Fig. S4H). These results indicate that SQSTM1 is not the autophagy receptor for DERL1 degradation. In contrast, wild-type but not CUE domain-deficient TOLLIP interacted with DERL1 (Figure 5F). Moreover, Flag-TMEM63A interacted with endogenous DERL1 in HEK293T cells (Figure 5G), and ectopic expression of TOLLIP significantly reduced the protein levels of DERL1 in LM2-4175 and Hs578T cells (Figure 5H). Moreover, TOLLIP knockdown resulted in an increase in the protein levels of DERL1 (Figure 3K). These results suggest that TOLLIP may mediate autophagic degradation of DERL1. In support of this notion, ectopic expression of HA-TMEM63A reduced the ubiquitination levels of Flag-DERL1 (Figure 5I) and compromised the interaction of DERL1 with TOLLIP (Figure 5J). It has been reported that DERL1 can degrade the unspliced form of XBP1 (X-box binding protein 1; XBP1u) by binding to its C terminus [66]. We found that overexpression of TMEM63A in MDA-231 and BT549 cells led to DERL1 upregulation and XBP1u downregulation (Figure 5K). In contrast, knockdown of TMEM63A in LM2-4175 and Hs578T cells decreased DERL1 levels and increased XBP1u levels (Figure 5L). Together, these data suggests that TMEM63A blocks TOLLIP-mediated autophagic degradation of DERL1.
Pharmacological inhibition of VCP or depletion of DERL1 impairs TMEM63A-mediated TNBC progression both in vitro and in vivo
To investigate whether TMEM63A exerts oncogenic effects on TNBC cells through regulating DERL1, we knocked down DERL1 in MDA-231 and BT549 cells stably expressing HA-TMEM63A and performed functional rescue assays as described in Figure 1 (Fig. S5A). Results showed that knockdown of DERL1 partially abolished TMEM63A-induced cell proliferation (Figure 6A) and colony formation (Figure 6B,C) of MDA-231 and BT549 cells. Additionally, depletion of DERL1 also partially abrogated the pro-migration and pro-invasion effects of TMEM63A (Figure 6D-F).
Figure 6.
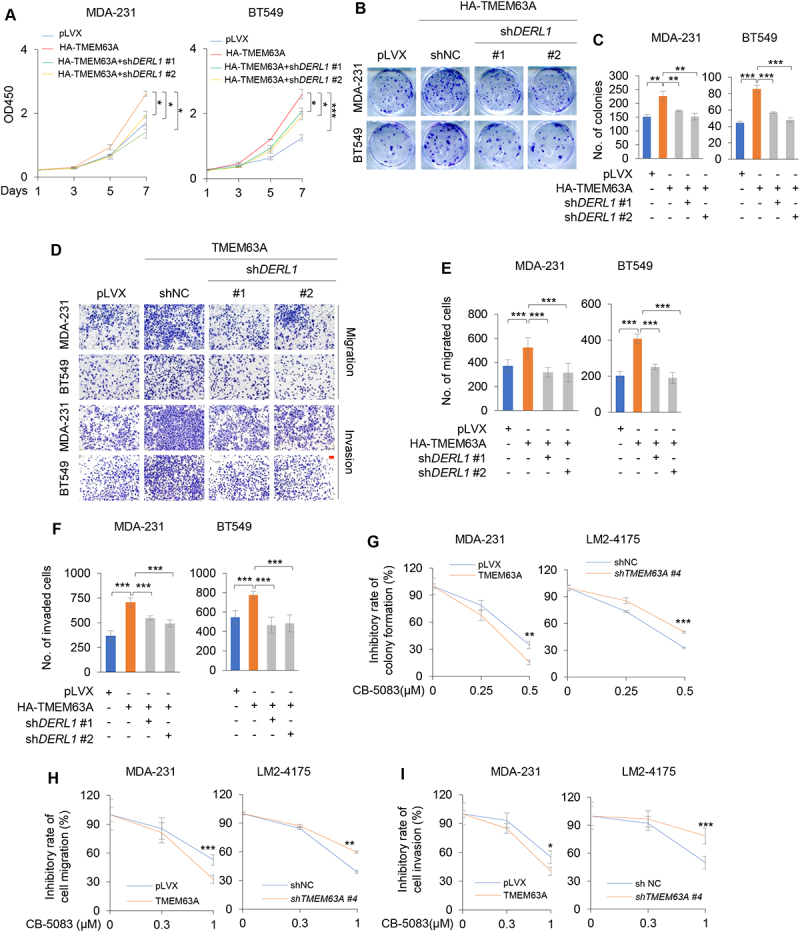
Pharmacological inhibition of VCP or depletion of DERL1 impairs TMEM63A-mediated TNBC cell proliferation, migration, and invasion in vitro. (A-F) MDA-231 and BT549 cells stably expressing HA-TMEM63A were infected with lentiviral vectors encoding shDERL (#1 and #2), and then subjected to CCK-8 (A), colony formation (B-C), migration (D-E) and invasion (D and F) assays (scale bar:100 μm). Representative images of survival colonies and corresponding quantitative results are shown in B and C, respectively. Representative images of migrated and invaded cells are shown in D, and corresponding quantitative results are shown in E and F, respectively. (G) MDA-231 cells stably expressing pLVX and HA-TMEM63A (left) and LM2-4175 cells stably expressing shNC and shTMEM63A #4 cells (right) were treated with or without increasing doses of VCP inhibitor CB-5083 and then subjected to colony formation assays. Representative images of survival colonies and the relative inhibitory rate of CB-5083 on colony formation ability of MDA-231 and LM2-4175 cells are shown in Supplementary Fig. S5B and Figure 6G, respectively. (H-I) MDA-231 cells stably expressing pLVX and HA-TMEM63A (left) and LM2-4175 cells stably expressing shNC and shTMEM63A #4 cells (right) were treated with or without increasing doses of VCP inhibitor CB-5083 and then subjected to Transwell migration and invasion assays. Representative images of migrated and invaded cells are shown in Supplementary Fig. S5C and S5D. The relative inhibitory rate of CB-5083 on the migration and invasion of MDA-231 and LM2-4175 cells are shown in H and I, respectively. *, **, and *** indicate statistically significant at p < 0.05, p < 0.01, and p < 0.001 level, respectively.
As VCP is an upstream regulator of TMEM63A and VCP inhibitors exerts broad antitumor activity in a range of both hematologic and solid tumor models [67,68], we next examined the effects of VCP inhibitor CB-5083 on TMEM63A-mediated TNBC progression. In vitro function assays revealed that the inhibitory effects of CB-5083 on cell viability, migration and invasion were more pronounced in MDA-231 cells stably expressing TMEM63A compared to cells expressing empty vector pLVX (Figure 6G-I and Fig. S5B-S5D, left panel). The opposite results were obtained from LM2-4175 cells stably expressing shNC and shTMEM63A # 4 (Figure 6G-I and Fig. S5B-S5D, right panel).
We next investigated the effects of TMEM63A on tumor growth and lung metastasis of TNBC cells and whether pharmacological inhibition of VCP or depletion of DERL affects TMEM63A-mediated tumorigenic ability and lung metastasis. To achieve this aim, MDA-231 cells stably expressing empty vector pLVX, HA-TMEM63A, and HA-TMEM63A in combination with shDERL1 #1 were injected into the mammary fat pad (tumorigenesis) and the tail vein (lung metastasis) of nude mice. After one week of injection, mice were administrated with vehicle or VCP inhibitor CB-5083. In concordance with the in vitro findings, ectopic expression of TMEM63A in MDA-231 cells markedly enhanced tumor volume and weight (Figure 7A,B, and Fig. S6A) and lung metastasis (Figure 7C,D, and Fig. S6B and S6C). Notably, pharmacological inhibition of VCP by CB-5083 or knockdown of DERL1 compromised the noted oncogenic effects of TMEM63A (Figure 7A-D, and S6A-S6C). Moreover, tumors overexpressing TMEM63A were more sensitive to CB-5083 treatment compared to controls (Figure 7A-D, and S6A-S6C). These results suggest that TMEM63A promotes TNBC progression in vivo through, at least in part, regulating DERL1, and that VCP inhibitors have therapeutic potential for TNBC tumors with high expression of TMEM63A.
Figure 7.
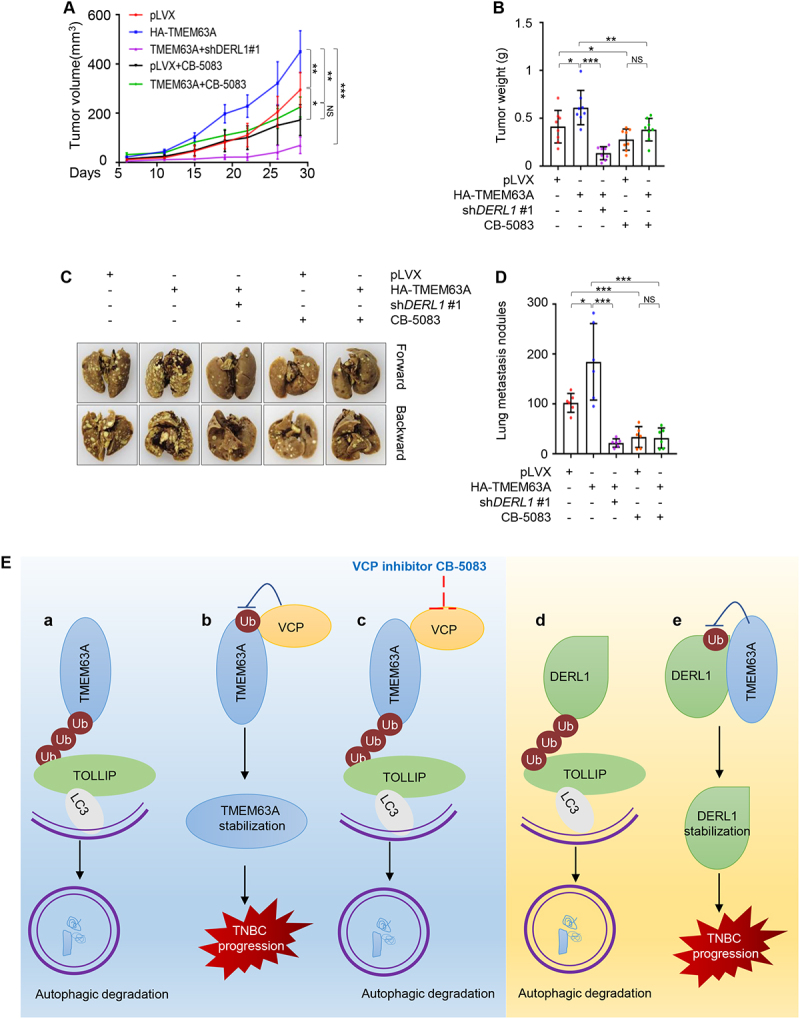
Pharmacological inhibition of VCP or depletion of DERL1 impairs TMEM63A-mediated xenograft tumor growth and lung metastasis of TNBC cells in vivo. (A-B) MDA-231 cells stably expressing pLVX, HA-TMEM63A, HA-TMEM63A in combination with shDERL1 #1 were inoculated into the mammary fat pad of 6-8-week-old female BALB/c nude mice (n = 8). After one week of incubation, mice were administrated with vehicle or CB-5083 (50 mg/kg) by oral gavage. After 4 weeks of treatment, mice were euthanized, xenograft tumors were removed and weighed. Images of removed xenograft tumors from mice are shown in Supplementary Fig. S6A. Tumor volumes and weights are shown in A and B, respectively. (C-D) MDA-231 cells stably expressing pLVX, HA-TMEM63A, HA-TMEM63A in combination with shDERL1 #1 were injected into 6-8-week-old female BALB/c nude female mice (n = 6) through tail vein. After one week of injection, mice were administrated with vehicle or CB-5083 (50 mg/kg) by oral gavage. After 4 weeks of administration, mice were euthanized, the lung tissues were removed, and the number of metastatic lung nodules were counted. Representative images of metastatic lung nodules and quantitative results of metastatic lung nodules is shown in C and D, respectively. Images of metastatic lung nodules and HE staining of lung tissues are shown in Supplementary Fig. S6B and S6C, respectively. (E) The proposed working model. TMEM63A undergoes TOLLIP-mediated autophagic degradation (a), and this process is blocked by VCP leading to TMEM63A stabilization (b). VCP inhibitor CB-5083 enhances the interaction of TMEM63A with TOLLIP, thus promoting TMEM63A degradation (c). In addition, DERL1 is also subjected to TOLLIP-mediated autophagic degradation (d), and TMEM63A stabilizes DERL1 through preventing it from TOLLIP-mediated autophagic degradation (e).
Discussion
In this study, we report several interesting findings concerning a previously unappreciated functional and mechanistic role of the VCP-TMEM63A-DERL1 signaling axis in TNBC progression (Figure 7E).
First, TMEM63A functions as a novel oncogene to promote TNBC progression. Emerging evidence shows that abnormal expression or mutation of some TMEM proteins (such as TMEM16A and TMEM88) play key roles in regulating cancer cell proliferation, invasion, metastasis, and sensitivity to anticancer agents, and that some TMEMs (such as TMEM48) can be used as potential prognostic biomarkers [6,7,69–72]. In this context, it has been shown that TMEM17 is upregulated in breast tumor tissues and promotes malignant progression of breast cancer cells by activating AKT-GSK3B/GSK3β pathway [73], and that TMEM45A promotes hypoxia-induced chemoresistance of breast cancer cells [74]. In addition, TMEM25 may be a good prognostic marker for patients with breast cancer [75] . In view of the key role of TMEM proteins in human cancer and their potential as anticancer drug targets, in-depth investigations of their functions and molecular mechanisms in cancer development and progression could provide new clues for the discovery of new therapeutic targets for cancer therapy.
TMEM63A is a member of the TMEM63 family of mechanically activated ion channels [12,76], and is required to constitute a hyperosmolarity activated ion channel [12]. Interestingly, mutations in TMEM63A have been shown to be associated with several developmental disorders [14–16], but biological function of TMEM63A in human cancer has not been explored to date. In this study, we provide the first evidence that TMEM63A was highly expressed in TNBC tumors and promoted TNBC cell proliferation, migration, and invasion in vitro (Figure 1) and xenograft tumor growth and lung metastasis in vivo (Figure 7). These results suggest that TMEM63A represents a potential therapeutic target for TNBC. In supported of our findings, mechanically activated ion-channel proteins, such as PIEZO1, have been implicated in the development and progression of multiple types of human cancer through various mechanisms [77,78].
Second, VCP stabilizes TMEM63A through blocking its autophagic degradation mediated by autophagy receptor TOLLIP. VCP exerts diverse biological functions mostly through modulating proteasomal degradation of client proteins, such as RNF8 (RING finger protein 8) [79] and mutant p53 [80]. In addition, VCP plays key roles in regulation of both selective and nonselective autophagy, and is required for autophagic degradation of its substrates, such as empty AGO1 [81–83]. Consequently, loss of VCP results in compromised protein degradation via the proteasome and the autophagy machinery [84]. In this study, we found that TMEM63A was subjected to autophagic degradation, and VCP stabilized TMEM63A through blocking TOLLIP-mediated macroautophagic degradation of TMEM63A (Figures 2–3 and Figs. S2 and S3). Previous studies have demonstrated that TOLLIP acts as a selective autophagy receptor for autophagic degradation of protein aggregates (termed aggrephagy) [58]. Furthermore, the oncogenic effects of TMEM63A on TNBC progression were suppressed by VCP inhibitor CB-5083 both in vivo and in vitro (Figures 6G-I, 7A-7D, Figs. S5B-S5D, and S6).
Third, TMEM63A stabilizes DERL1 through preventing it from TOLLIP-mediated autophagic degradation. Accumulating evidence shows that DERL1 is highly expressed in multiple types of human cancer, including breast [31,33,34], cervix [85], lung [34,86–88], colon [64,89], bladder [90,91], esophagus [91], liver [92], and pancreas [34], and functions as an oncogene contributing to malignant phenotypes of these tumors [31,32,85,86,89,91,92]. In addition, DERL1 is a biomarker for predicting prostate cancer aggressiveness and lethal outcome [93], hepatocellular carcinoma metastasis [92], and poor prognosis of patients with breast cancer [31,32], non-small cell lung cancer [86,88], muscle invasive bladder cancer [91], and bladder cancer [90]. Consequently, targeting DERL1 by antibodies significantly suppresses colon tumor growth in mice [34]. Despite its functional importance in human cancer, the regulatory mechanism of DERL1 in cancer still remains elusive. In this study, we found that DERL1 underwent autophagic degradation, and TMEM63A enhanced the stability of DERL1 through blocking TOLLIP-mediated autophagic degradation (Figures 4 and 5). Functional rescue assays further demonstrated that knockdown of DERL1 in TMEM63A-overexpressing cells attenuated TMEM63A-mediated cell proliferation, migration, and invasion in vitro and tumor growth and lung metastasis in vivo (Figures 6 and 7). These results suggest that TMEM63A promotes TNBC progression through, at least in part, stabilizing DERL1.
Finally, emerging evidence shows that the expression levels of TOLLIP are downregulated in gastric adenocarcinoma in comparison to normal mucosa [94], indicating that TOLLIP may act as a tumor suppressor in gastric cancer. In contrast, TOLLIP promotes colitis-associated cancer via chronic inflammatory responses and lymphocyte accumulation [95]. Similarly, TOLLIP promotes colorectal cancer progression through reducing immune surveillance by increasing STAT5 and PD-L1 and reducing STAT1 [96]. These findings indicate that TOLLIP has a complicated role in human cancer in a context dependent manner. To address the potential role of TOLLIP in breast cancer cell proliferation, migration, and invasion, we knocked down of TOLLIP in MDA-231 cells using two independent siRNAs targeting TOLLIP. Results showed that depletion of TOLLIP did not significantly affect the proliferation, migration, and invasion of MDA-231 cells (unpublished data). Given that TOLLIP has many substrates with various functions, this phenotype may be due to the compensation effects among different substrates. In addition, we cannot rule out the possibility that the role TOLLIP in tumor progression may be context dependent.
In summary, findings presented in this study suggest that TMEM63A acts as a novel oncogene to promote TNBC progression, and that TOLLIP-mediated autophagic degradation pathway links the VCP-TMEM63A-DERL1 signaling axis to TNBC progression. Moreover, pharmacological inhibition of VCP impairs TMEM63A-mediated TNBC progression. Thus, TMEM63A may serve as a potential therapeutic target for TNBC tumors.
Materials and methods
Cell culture and reagents
Human TNBC cell lines, BT20 (CBP60354), BT549 (TCHu 93), HCC1806 (CBP60373), Hs578T (TCHu127), MDA-MB-231 (hereinafter referred to as MDA-231 for brevity) (TCHu227), MDA-MB-157 (MDA-157) (CBP60381), MDA-MB-468 (MDA-468) (TCHu136) and human embryonic kidney 293T (HEK293T) (GNHu17) cell lines were obtained from Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and Shanghai Key Laboratory of Breast Cancer (Fudan University, Shanghai, China). MDA-MB-231-derived LM2-4175 cells were kindly provided by Guohong Hu (University of Chinese Academy of Sciences, Shanghai, China). SUM159 cell lines were kindly provided by Suling Liu (Fudan University, Shanghai, China). All cell lines were authenticated through short tandem repeat (STR) profiling and were used for less than six months within 15–20 passages. All cell lines were cultured in DMEM (BasalMedia, L110) supplemented with 10% fetal bovine serum (FBS; ExCell Biol, FSP500) and 1% penicillin-streptomycin (BasalMedia, S110B). All cells were cultured in a 5% CO2 incubator at 37°C. MG-132, CB-5083, bafilomycin A1 (Baf-A1), 3-methyladenine (3-MA), rapamycin (Rapa), and puromycin were purchased from Selleck Chemicals. Cycloheximide (CHX) was obtained from Cell Signaling Technology. Other compounds were purchased from Sigma-Aldrich unless otherwise noted. Detailed information for all chemical inhibitors is listed in Table S1.
Cell viability and colony formation assays
Cells were counted after digestion and seeded into each well of 96-well plates (1000 cells per well). Cell viability assays were carried out using CCK-8 kit (Yeasen, 40203ES92*) by adding 10 μL CCK-8 solution to each well, and absorbance at 450 nm (OD450) was measured at the indicated times. Colony formation assays were carried out in a six-well plate (1000 cells per well). The medium was changed every three days. After 14 days of culture, cells were fixed with methanol, stained with crystal violet, and counted.
Cell migration and invasion assays
Cells were counted after digestion, and a total of 2–5 × 104 cells in serum-free medium were plated in the upper chamber coated with (invasion) or without (migration) matrigel (Corning, 354,480 and 353,097, respectively). The lower chamber was covered with medium containing 10% FBS. After 24 h of incubation, cells were fixed with methanol, stained with crystal violet, and counted.
Tumorigenicity and lung metastasis assays
All procedures were in accordance with institutional guidelines for the Care and Use of Laboratory Animals and were approved by the Animal Experiments Committee of Fudan University. To perform tumorigenesis assays, 3.5 × 106 cells were inoculated into the mammary fat pad of 6-8-week-old BALB/c nude female mice (n = 8). CB-5083 was dissolved in 0.5% sodium carboxymethyl cellulose (Selleck, S6703). After one week of injection, mice were administrated with vehicle or CB-5083 (50 mg/kg) by oral gavage. Oral gavage was performed once a day for five consecutive days and then suspended for 2 days (qd5/2 off) for 4 weeks. Tumor volume was measured twice a week and calculated by the formula of (length × width2)/2. After mice were euthanized, xenograft tumors were removed and weighed. For experimental lung metastasis assays, 1 × 106 cells were injected into 6-8-week-old BALB/c nude female mice (n = 6) through tail vein. Vehicle or CB-5083 (50 mg/kg) was administrated into mice by oral gavage after one week of injection. Oral gavage was performed once a day for five consecutive days and then suspended for 2 days (qd5/2 off) for 4 weeks. After mice were euthanized, the lung tissues were removed and the number of metastatic lung nodules were counted under a microscope.
DNA constructs, siRNAs, shRNA, and transfection
The plasmids encoding TMEM63A (CH802067), VCP (CH897720), and DERL1 (CH823019) were purchased from Vigene Bioscience, and subcloned into lentiviral vectors pCDH-CMV-MCS-EF1-Puro (System Biosciences, CD510B-1) or pLVX-IRES-NEO (Biofeng, 632,181). Short hairpin RNA (shRNA) sequences targeting TMEM63A, VCP, and DERL1 were obtained from BLOCK-iT™ RNAi Designer (http://rnaidesigner.thermofisher.com/rnaiexpress/design.do) and ligated into the lentiviral vector pLKO.1-TRC (Addgene,10,878, deposited by David Root). Small interfering RNAs (siRNAs) targeting SQSTM1 and DERL1 were purchased from GenePharma (Shanghai, China). All cDNA plasmids and sequences are listed in Tables S2-S5.
DNA constructs and shRNAs were transfected into HEK293T cells using Neofect DNA transfection reagent (Tengyi Biol, TF201201). The supernatants were collected after 48 h of transfection, filtered, and used to infect cells in the presence of 8 μg/mL polybrene (Sigma, H9268). After 48 h of infection, cells were selected using 2 μg/mL puromycin (Selleck, S7417) or 0.5 mg/mL G418 (Sangon Biotech, A600958-0005) for 1–2 weeks. The siRNAs were transfected into cells using Lipofectamine 2000 transfection reagent (Invitrogen, 11,668,019).
RNA extraction and RT-qPCR
Total RNA from cultured cells and tissue samples was extracted using TRIzol reagent (Invitrogen, 15,596,018). Reverse transcription was conducted to generate cDNA from the isolated RNA using HiScript III RT SuperMix (Vazyme, R323-01). qPCR assays were performed using Chamq Universal SYBR qPCR Master Mix (Vazyme, Q711-03). Relative gene expression levels between different samples were calculated using the 2−ΔΔCT method. The primers used in this study are listed in Table S6.
Antibodies, immunoblotting, and immunoprecipitation
The antibodies used in this study are listed in Table 1. For immunoblotting, cultured cells were washed with PBS and lysed with RIPA buffer (Yeasen, 20101ES60). After BCA protein quantification, equal quantities of protein samples were used for SDS-PAGE. The proteins were transferred to PVDF membrane (Millipore, IPVH00010) and incubated with primary and secondary antibodies after blocking the membrane with 5% BSA (Sigma, V900933-1 KG). Proteins were detected using an enhanced chemiluminescence detection kit (Yeasen, 36208ES80). For immunoprecipitation (IP) assays, cultured cells were washed with PBS and lysed with NP-40 buffer (Beyotime, P0013F). After BCA protein quantification, equal quantities of protein samples were mixed with the corresponding antibodies and incubated overnight in a rotating shaker at 4°C. On the second day, protein A/G beads (Bimake, B23202) were added and then washed after 3 h of incubation. The samples were then subjected to immunoblotting.
Table 1.
Antibodies used in this study.
| Antibodies | Vendors | Cat# | Hosts | Working concentration |
|---|---|---|---|---|
| TMEM63A | Abcam | ab80342 | Rabbit | 1:1000 |
| VCL/vinculin | Sigma | V9131 | Mouse | 1:5000 |
| HA | CST | 3724S | Rabbit | 1:3000 /1:200 (IF) |
| Flag | Sigma | F1804 | Mouse | 1:3000 /1:200 (IF) |
| VCP | Abcam | ab109240 | Rabbit | 1:5000 /1:200 (IF) |
| CANX | Abcam | ab133615 | Rabbit | 1:1000 |
| CANX | Abcam | ab92573 | Rabbit | 1:500 (IF) |
| ATP5F1B/ATP5B | Abcam | ab170947 | Rabbit | 1:1000 |
| SQSTM1 | Abcam | ab109012 | Rabbit | 1:10,000 |
| V5 | CST | 13202S | Rabbit | 1:1000 |
| LC3 | CST | 12741 | Rabbit | 1:1000 |
| TOLLIP | Proteintech | 11,315-1-AP | Rabbit | 1:1000 |
| NBR1 | Proteintech | 16,004-1-AP | Rabbit | 1:1000 |
| TAX1BP1 | Abcam | ab176572 | Rabbit | 1:1000 |
| OPTN | Abcam | ab213556 | Rabbit | 1:1000 |
| CALCOCO2/NDP52 | Proteintech | 12,229-1-AP | Rabbit | 1:1000 |
| ATG16L1 | Abcam | ab187671 | Rabbit | 1:1000 |
| CDKN1A/p21 | CST | 2947S | Rabbit | 1:1000 |
| LAMP2A | Abcam | ab125068 | Rabbit | 1:2000 /1:200 (IF) |
| HSPA8 | Abcam | ab51052 | Rabbit | 1:3000 |
| DERL1 | Abcam | ab176732 | Rabbit | 1:2000 |
| XBP1 | Abcam | ab220783 | Rabbit | 1:1000 |
| EGFR | CST | 4267P | Rabbit | 1:200 (IF) |
| ATG5 | Abcam | ab108327 | Rabbit | 1:3000 |
Note: IF, immunofluorescent staining.
Immunofluorescent staining
Cells were digested and then counted. Each disc was plated with 80,000 cells. On next day, after cells adhered to the walls on the disc, the culture medium was discarded, the cells washed with PBS and fixed with 4% paraformaldehyde (Sangon Biotech, E672002-0500). After treatment with 0.1% Triton X-100 (Sigma, 93,443) and blocked with 5% BSA, cells were incubated overnight at 4°C with the primary antibody and then with the secondary fluorescent antibody. Cells were then placed on slides and stained with DAPI solution (Abcam, ab104139). Images were taken using a Leica fluorescence confocal microscope.
Proteomics analysis
HEK293T cells stably expressing pCDH and Flag-TMEM63A were lysed in NP-40 buffer. Equal quantities of protein samples were subjected to IP assays using an anti-Flag antibody, and separated by SDS-PAGE. The gel was stained with Coomassie Brilliant Blue and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. For identifying downstream proteins of TMEM63A, LM2-4175 cells stably expressing two independent shRNAs targeting TMEM63A (shTMEM63A #3 and #4) and negative control shRNA (shNC) were lysed using RIPA buffer, and equal quantities of protein samples were used for isobaric tags for relative and absolute quantification (iTRAQ) analysis.
Statistical analyses
All experiments were repeated three times for each group. Results are presented as means ± standard deviation (SD). Statistical analysis of two groups was conducted using Student’s t-test or Chi-square test. P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We sincerely acknowledge members in the Li laboratory for their technical assistance and helpful advice.
Funding Statement
This work is supported, in whole or in part, by the National Natural Science Foundation of China (82072918 to FLZ; 81572584, 81772805, and 82173275 to DQL) and by the National Key R&D Program of China (2017YFC0908400 and 2018YFE0201600 to DQL).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethics approval and consent to participate
All procedures for animal experiment were in accordance with institutional guidelines for the Care and Use of Laboratory Animals and were approved by the Animal Experiments Committee of Fudan University.
Data availability
All data supporting the findings of this study is available from the corresponding authors on reasonable request.
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2103992
References
- [1].Foulkes W, Smith I, Reis-Filho J.. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. [DOI] [PubMed] [Google Scholar]
- [2].Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019. Feb;9(2):176–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schmit K, Michiels C. TMEM Proteins in Cancer: a Review. Front Pharmacol. 2018;9:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ryu H, Fuwad A, Yoon S, et al. Biomimetic membranes with transmembrane proteins: state-of-the-art in transmembrane protein applications. Int J Mol Sci. 2019;20(6):1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang S, Dai H, Li W, et al. TMEM116 is required for lung cancer cell motility and metastasis through PDK1 signaling pathway. Cell Death Dis. 2021 Nov 16 12(12):1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schmit K, Michiels C. TMEM Proteins in Cancer: a Review. Front Pharmacol. 2018;9:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marx S, Dal Maso T, Chen JW, et al. Transmembrane (TMEM) protein family members: poorly characterized even if essential for the metastatic process [review]. Semin Cancer Biol. 2020. Feb;60:96–106. [DOI] [PubMed] [Google Scholar]
- [8].Lluis M, Godfroy J, Yin H. Protein engineering methods applied to membrane protein targets. Protein Eng Des Sel. 2013;26(2):91–100. [DOI] [PubMed] [Google Scholar]
- [9].Marx S, Dal Maso T, Chen J, et al. Transmembrane (TMEM) protein family members: poorly characterized even if essential for the metastatic process. Semin Cancer Biol. 2020;60:96–106. [DOI] [PubMed] [Google Scholar]
- [10].Yan H, Helman G, Murthy S, et al. Heterozygous variants in the mechanosensitive ion channel tmem63a result in transient hypomyelination during infancy. Am J Hum Genet. 2019;105(5):996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yan H, Helman G, Murthy SE, et al. Heterozygous variants in the mechanosensitive ion channel TMEM63A result in transient hypomyelination during infancy. Am J Hum Genet. [2019 Nov 7];105(5):996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao X, Yan X, Liu Y, et al. Co-expression of mouse TMEM63A, TMEM63B and TMEM63C confers hyperosmolarity activated ion currents in HEK293 cells. Cell Biochem Funct. 2016. Jun;34(4):238–241. [DOI] [PubMed] [Google Scholar]
- [13].Yuan C, Zhang H, Wang W, et al. Transmembrane protein 63A is a partner protein of haemonchus contortus galectin in the regulation of goat peripheral blood mononuclear cells [research support, Non-U.S. Gov’t]. Parasit Vectors. 2015 Apr 9 8(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tonduti D, Mura E, Masnada S, et al. Spinal cord involvement and paroxysmal events in “Infantile Onset Transient Hypomyelination” due to TMEM63A mutation. J Hum Genet. 2021. Oct;66(10):1035–1037. [DOI] [PubMed] [Google Scholar]
- [15].Fukumura S, Hiraide T, and Yamamoto A, et al. A novel de novo TMEM63A variant in a patient with severe hypomyelination and global developmental delay. Brain Dev. 2022. Feb;44(2):178–183. [DOI] [PubMed] [Google Scholar]
- [16].Yan H, Ji H, Kubisiak T, et al. Genetic analysis of 20 patients with hypomyelinating leukodystrophy by trio-based whole-exome sequencing. J Hum Genet. 2021. Aug;66(8):761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017 Dec 12; 61(6):625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moon HW, Han HG, Jeon YJ. Protein quality control in the endoplasmic reticulum and cancer. Int J Mol Sci. 2018 Oct 3; 19(10):3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oikonomou C, Hendershot LM. Disposing of misfolded ER proteins: a troubled substrate’s way out of the ER. Mol Cell Endocrinol. 2020 Jan 15; 500:110630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lipatova Z, Gyurkovska V, Zhao SF, et al. Characterization of constitutive ER-phagy of excess membrane proteins. PLoS Genet. 2020. Dec;16(12):e1009255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lipatova Z, Segev N. A role for Macro-ER-Phagy in ER quality control. PLoS Genet. 2015. Jul;11(7):e1005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ye Y, Shibata Y, Yun C, et al. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004 Jun 24 429(6994):841–847. [DOI] [PubMed] [Google Scholar]
- [23].Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004 Jun 24; 429(6994):834–840. [DOI] [PubMed] [Google Scholar]
- [24].Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001 Dec 6; 414(6864):652–656. [DOI] [PubMed] [Google Scholar]
- [25].Zhou HJ, Wang J, Yao B, et al. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J Med Chem. 2015 Dec 24 58(24):9480–9497. [DOI] [PubMed] [Google Scholar]
- [26].Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003 Jul 7; 162(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kadowaki H, Nagai A, Maruyama T, et al. Pre-emptive quality control protects the er from protein overload via the proximity of ERAD components and SRP. Cell Rep. 2015;13(5):944–956. [DOI] [PubMed] [Google Scholar]
- [28].Rao B, Li S, Yao D, et al. The cryo-EM structure of an ERAD protein channel formed by tetrameric human Derlin-1. Sci Adv. 2021. Mar;7(10). 10.1126/sciadv.abe8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li C, Huang Y, Fan Q, et al. p97/VCP is highly expressed in the stem-like cells of breast cancer and controls cancer stemness partly through the unfolded protein response. Cell Death Dis. 2021 Mar 17 12(4):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fessart D, Marza E, Taouji S, et al. P97/CDC-48: proteostasis control in tumor cell biology. Cancer Lett. 2013 Aug 28 337(1):26–34. [DOI] [PubMed] [Google Scholar]
- [31].Liu Y, Wang Z, Liu H, et al. Derlin-1 functions as a growth promoter in breast cancer. Biol Chem. 2020 Feb 25 401(3):377–387. [DOI] [PubMed] [Google Scholar]
- [32].Zeng J, Tian Q, Zeng Z, et al. Derlin-1 exhibits oncogenic activities and indicates an unfavorable prognosis in breast cancer. Cell Biol Int. 2020. Feb;44(2):593–602. [DOI] [PubMed] [Google Scholar]
- [33].Wang J, Hua H, Ran Y, et al. Derlin-1 is overexpressed in human breast carcinoma and protects cancer cells from endoplasmic reticulum stress-induced apoptosis. BCR. 2008;10(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ran Y, Hu H, Hu D, et al. Derlin-1 is overexpressed on the tumor cell surface and enables antibody-mediated tumor targeting therapy. Clin Cancer Res off J Am Assoc Cancer Res. 2008 Oct 15 14(20):6538–6545. [DOI] [PubMed] [Google Scholar]
- [35].Costantini S, Capone F, Polo A, et al. Valosin-Containing protein (VCP)/p97: a prognostic biomarker and therapeutic target in cancer. Int J Mol Sci. 2021 Sep 21 22(18):10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Anderson D, Le Moigne R, Djakovic S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28(5):653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Magnaghi P, D’Alessio R, Valsasina B, et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol. 2013. Sep;9(9):548–556. [DOI] [PubMed] [Google Scholar]
- [38].Kim BW, Kwon DH, Song HK. Structure biology of selective autophagy receptors. BMB Rep. 2016. Feb;49(2):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018. Mar;20(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen W, Shen T, Wang L, et al. Oligomerization of selective autophagy receptors for the targeting and degradation of protein aggregates. Cells. 2021 Aug 5 10(8):1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fracchiolla D, Sawa-Makarska J, Martens S. Beyond Atg8 binding: the role of AIM/LIR motifs in autophagy. Autophagy. 2017 May 4; 13(5):978–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vainshtein A, Grumati P. Selective autophagy by close encounters of the ubiquitin kind. Cells. 2020 Oct 24; 9(11):2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014 Jan 23 53(2):167–178. [DOI] [PubMed] [Google Scholar]
- [44].Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–624. [DOI] [PubMed] [Google Scholar]
- [45].Schulten HJ, Bangash M, Karim S, et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J Transl Med. 2017 Dec 29 15(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009 Jun 18 459(7249):1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chang G, Shi L, Ye Y, et al. YTHDF3 induces the translation of m(6)A-Enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell. 2020 Dec 14 38(6):857–871 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung [research support, N.I.H., extramural research support, Non-U.S. Gov’tResearch support, U.S. Gov’t, Non-P.H.S.Research support, U.S. Gov’t, P.H.S.]. Nature. 2005 Jul 28 436(7050):518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019 Nov 15; 366(6467):818–822. [DOI] [PubMed] [Google Scholar]
- [50].Bloom J, Amador V, Bartolini F, et al. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003 Oct 3 115(1):71–82. [DOI] [PubMed] [Google Scholar]
- [51].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014. Jan;24(1):24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014. Jan;24(1):92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982. Mar;79(6):1889–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. [DOI] [PubMed] [Google Scholar]
- [55].Kirkin V, Rogov V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019;76(2):268–285. [DOI] [PubMed] [Google Scholar]
- [56].Conway O, Akpinar H, Rogov V, et al. Selective autophagy receptors in neuronal health and disease. J Mol Biol. 2020;432(8):2483–2509. [DOI] [PubMed] [Google Scholar]
- [57].Lee Y, Weihl CC. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy. 2017 Sep 2; 13(9):1615–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014 Jul 31; 158(3):549–563. [DOI] [PubMed] [Google Scholar]
- [59].Issa AR, Sun J, Petitgas C, et al. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy. 2018;14(11):1898–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wada I, Rindress D, Cameron PH, et al. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991 Oct 15 266(29):19599–19610. [PubMed] [Google Scholar]
- [61].Sinclair JKL, Walker AS, Doerner AE, et al. Mechanism of Allosteric Coupling into and through the Plasma Membrane by EGFR. Cell Chem Biol. 2018 Jul 19 25(7):857–870 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dong Z, Cui H. The Autophagy-Lysosomal pathways and their emerging roles in modulating proteostasis in tumors. Cells. 2018 Dec 20; 8(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zeng J, Tian Q, Zeng Z, et al. Derlin-1 exhibits oncogenic activities and indicates an unfavorable prognosis in breast cancer. Cell Biol Int. 2020;44(2):593–602. [DOI] [PubMed] [Google Scholar]
- [64].Tan X, He X, Jiang Z, et al. Derlin-1 is overexpressed in human colon cancer and promotes cancer cell proliferation. Mol Cell Biochem. 2015;408(1–2):205–213. [DOI] [PubMed] [Google Scholar]
- [65].Mao M, Zhang J, Jiang J. Overexpression of Derlin-1 is associated with poor prognosis in patients with non-small cell lung cancer. Ann Clin Lab Sci. 2018;48(1):29–34. [PubMed] [Google Scholar]
- [66].Chen C, Malchus N, Hehn B, et al. Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J. 2014;33(21):2492–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Le Moigne R, Aftab BT, Djakovic S, et al. The p97 Inhibitor CB-5083 Is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther. 2017. Nov;16(11):2375–2386. [DOI] [PubMed] [Google Scholar]
- [68].Anderson DJ, Le Moigne R, Djakovic S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. [2015 Nov 9];28(5):653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Duvvuri U, Shiwarski DJ, Xiao D, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’tResearch Support, U.S. Gov’t, Non-P.H.S.]. Cancer Res. 2012 Jul 1 72(13):3270–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shiwarski DJ, Shao C, Bill A, et al. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN [research support, N.I.H., extramural research support, Non-U.S. Gov’t research support, U.S. Gov’t, Non-P.H.S.]. Clin Cancer Res off J Am Assoc Cancer Res. 2014 Sep 1 20(17):4673–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang H, Zou L, Ma K, et al. Cell-specific mechanisms of TMEM16A Ca(2+)-activated chloride channel in cancer [review research support, Non-U.S. Gov’t]. Mol Cancer. 2017 Sep 11 16(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang X, Yu X, Jiang G, et al. Cytosolic TMEM88 promotes invasion and metastasis in lung cancer cells by binding DVLS [research support, Non-U.S. Gov’t]. Cancer Res. 2015 Nov 1 75(21):4527–4537. [DOI] [PubMed] [Google Scholar]
- [73].Zhao Y, Song K, Zhang Y, et al. TMEM17 promotes malignant progression of breast cancer via AKT/GSK3beta signaling. Cancer Manag Res. 2018;10:2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Flamant L, Roegiers E, Pierre M, et al. TMEM45A is essential for hypoxia-induced chemoresistance in breast and liver cancer cells [research support, Non-U.S. Gov’t]. BMC Cancer. 2012 Sep 6 12(1):391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Doolan P, Clynes M, Kennedy S, et al. TMEM25, REPS2 and Meis 1: favourable prognostic and predictive biomarkers for breast cancer [research support, Non-U.S. Gov’t]. Tumour Biol. 2009;30(4):200–209. [DOI] [PubMed] [Google Scholar]
- [76].Murthy S, Dubin A, Whitwam T, et al. OSCA/TMEM63 are an evolutionarily conserved family of mechanically activated ion channels. eLife. 2018;7. 10.7554/eLife.41844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dombroski JA, Hope JM, Sarna NS, et al. Channeling the force: piezo1 mechanotransduction in cancer metastasis. Cells. 2021 Oct 20 10(11):2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yu JL, Liao HY. Piezo-type mechanosensitive ion channel component 1 (Piezo1) in human cancer. Biomed Pharmacothe. 2021. Aug;140:111692. [DOI] [PubMed] [Google Scholar]
- [79].Singh AN, Oehler J, Torrecilla I, et al. The p97-Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF8. EMBO J. 2019 Oct 4 38(21):e102361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wang J, Chen Y, Huang C, et al. Valosin-Containing protein stabilizes mutant p53 to promote pancreatic cancer growth. Cancer Res. 2021 Aug 1 81(15):4041–4053. [DOI] [PubMed] [Google Scholar]
- [81].Kobayashi H, Shoji K, Kiyokawa K, et al. VCP machinery mediates autophagic degradation of empty argonaute. Cell Rep. 2019 Jul 30 28(5):1144–1153 e4. [DOI] [PubMed] [Google Scholar]
- [82].Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009 Dec 14 187(6):875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kobayashi H, Tomari Y. Identification of an AGO (Argonaute) protein as a prey of TER94/VCP. Autophagy. 2020. Jan;16(1):190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kustermann M, Manta L, Paone C, et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy. 2018;14(11):1911–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Li L, Liu M, Zhang Z, et al. Derlin1 functions as an oncogene in cervical cancer via AKT/mTOR signaling pathway. Biol Res. 2019 Feb 27 52(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dong QZ, Wang Y, Tang ZP, et al. Derlin-1 is overexpressed in non-small cell lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated up-regulation of MMP-2 and MMP-9. Am J Pathol. 2013. Mar;182(3):954–964. [DOI] [PubMed] [Google Scholar]
- [87].Yang F, Wei K, Qin Z, et al. MiR-598 suppresses invasion and migration by negative regulation of derlin-1 and epithelial-mesenchymal transition in non-small cell lung cancer. Cell Physiol Biochem. 2018;47(1):245–256. [DOI] [PubMed] [Google Scholar]
- [88].Mao M, Zhang J, Jiang J. Overexpression of Derlin-1 is associated with poor prognosis in patients with non-small cell lung cancer. Ann Clin Lab Sci. 2018. Jan;48(1):29–34. [PubMed] [Google Scholar]
- [89].Tan X, He X, Jiang Z, et al. Derlin-1 is overexpressed in human colon cancer and promotes cancer cell proliferation. Mol Cell Biochem. 2015. Oct;408(1–2):205–213. [DOI] [PubMed] [Google Scholar]
- [90].Wu Z, Wang C, Zhang Z, et al. High expression of derlin-1 is associated with the malignancy of bladder cancer in a chinese han population. PloS one. 2016;11(12):e0168351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dong Q, Fu L, Zhao Y, et al. Derlin-1 overexpression confers poor prognosis in muscle invasive bladder cancer and contributes to chemoresistance and invasion through PI3K/AKT and ERK/MMP signaling. Oncotarget. 2017 Mar 7 8(10):17059–17069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fan J, Tian L, Huang S, et al. Derlin-1 promotes the progression of human hepatocellular carcinoma via the activation of AKT pathway. Onco Targets Ther. 2020;13:5407–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Shipitsin M, Small C, Choudhury S, et al. Identification of proteomic biomarkers predicting prostate cancer aggressiveness and lethality despite biopsy-sampling error. Br J Cancer. 2014 Sep 9 111(6):1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, et al. Helicobacter pylori induces increased expression of Toll-like receptors and decreased Toll-interacting protein in gastric mucosa that persists throughout gastric carcinogenesis. Helicobacter. 2013;18(1):22–32. [DOI] [PubMed] [Google Scholar]
- [95].Begka C, Pattaroni C, Mooser C, et al. Toll-Interacting protein regulates immune cell infiltration and promotes colitis-associated cancer. iScience. 2020;23(3):100891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang Y, Lee C, Geng S, et al. Enhanced tumor immune surveillance through neutrophil reprogramming due to Tollip deficiency. JCI Insight. 2019;4(2). 10.1172/jci.insight.122939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study is available from the corresponding authors on reasonable request.