

ABSTRACT
Macroautophagy/autophagy proteins have been linked with the development of immune-mediated diseases including lupus, but the mechanisms for this are unclear due to the complex roles of these proteins in multiple immune cell types. We have previously shown that a form of noncanonical autophagy induced by ITGAV/alpha(v) integrins regulates B cell activation by viral and self-antigens, in mice. Here, we investigate the involvement of this pathway in B cells from human tissues. Our data reveal that autophagy is specifically induced in the germinal center and memory B cell subpopulations of human tonsils and spleens. Transcriptomic analysis show that the induction of autophagy is related to unique aspects of activated B cells such as mitochondrial metabolism. To understand the function of ITGAV/alpha(v) integrin-dependent autophagy in human B cells, we used CRISPR-mediated knockdown of autophagy genes. Integrating data from primary B cells and knockout cells, we found that ITGAV/alpha(v)-dependent autophagy limits activation of specific pathways related to B cell responses, while promoting others. These data provide new mechanistic links for autophagy and B-cell-mediated immune dysregulation in diseases such as lupus.
KEYWORDS: Autophagy, B cells, cell-cycle, mitophagy, non-canonical autophagy, TLR signaling
Introduction
Conventionally, autophagy is an evolutionarily conserved intracellular process for recycling of organelles and degradation of unwanted cytoplasmic cargo. Canonical autophagy (known as macroautophagy) is activated during cell stress or starvation and is involved in various fundamental cellular processes such as growth and differentiation [1,2]. During macro-autophagy, interactions between a series of autophagy-related proteins lead to lipidation of the ubiquitin-like protein LC3 and its recruitment to double-membrane vesicles, called autophagosomes, which deliver cytoplasmic contents to lysosomes for degradation or recycling. Additionally, autophagy-related proteins and LC3 are also involved in other intracellular trafficking events related to signaling and processing of pathogen-derived materials via the recruitment of LC3 to vesicles that are not autophagosomes [3–5]. These processes are referred to by different autophagy-related terms for more noncanonical roles of autophagy proteins. Genome-wide association studies (GWASs) have implicated autophagy proteins in immune-mediated diseases such as inflammatory bowel diseases and systemic lupus erythematosus (SLE), making them attractive targets for the modulation of immune response [6–8]. However, the mechanisms by which autophagy proteins are involved in these diseases are not clear.
Multiple functions have been described for autophagy proteins in different immune cell populations [9,10]. In B cells of the immune system, macroautophagy has been shown to be involved in differentiation and self-renewal of the B1-B cell subset, antibody production by plasma cells, and maintenance of plasma cells as well as memory B cells [11–14]. More recently, the noncanonical form of autophagy has also been shown to be activated in B cells [15]. Similarly, one of the core autophagy proteins, ATG5, has been shown to be involved in B cell receptor (BCR) trafficking related to antigen presentation, probably via a noncanonical autophagy mechanism [16]. However, the exact functions of noncanonical autophagy in B cells and its mode of activation have remained unclear. We have previously shown that signaling through the ITGAV/αv family of integrins can activate autophagy proteins, which then regulate the rate and duration of TLR (toll-like receptor) signaling in B cells [17]. Specifically, we have found that upon B cell stimulation by TLR ligands, ITGAV integrins are activated, leading to activation of SRC and SYK kinases. SYK activation then leads to activation of autophagy proteins and LC3 lipidation, via a mechanism requiring reactive oxygen species (ROS) production. The LC3 lipidation and recruitment to TLR-containing endosomes is required for the TLRs and their ligands to transition from NFKB signaling endosomes to IRF7 signaling late endosomes. Thus, LC3 recruitment to TLR containing endo-lysosomal compartments regulates the outcome of signaling by regulating the trafficking of TLRs and their ligands. LC3 recruitment also eventually leads to the termination of TLR signaling in lysosomal compartments. Loss of ITGAV/alpha(v) integrins or autophagy proteins specifically in B cells disrupts this process of TLR trafficking and signaling, resulting in enhanced NFKB signaling and altered IRF7 signaling, particularly the loss of autophagy proteins leads to the absence of IRF7 signaling. As a result, these B cells show increased responses to TLR ligands in vitro [17]. Overall, these results indicate that ITGAV integrins and autophagy proteins regulate B cell TLR responses by curtailing NFKB signaling while promoting IRF7 signaling. Confirming the importance of this function of integrin-induced autophagy pathway in vivo, we showed that B-cell-specific deletion of the itgav gene led to enhanced B cell response to TLR ligand containing viral antigens and accelerated self-reactive B cell responses related to lupus-like autoimmunity, in mice [18,19]. This role of autophagy proteins in B cells does not require the formation of double-membrane autophagosomes and is distinct from the role of macroautophagy in plasma cell function. We have termed this as TLR-induced autophagy (TIA) or activation-induced autophagy related to B cell activation by TLR or BCR ligands and “triggered through αv integrin.” Furthermore, other groups have also shown the importance of autophagy proteins in regulating TLR signaling in macrophages and plasmacytoid dendritic cells [20,21].
This function of autophagy proteins in fine-tuning the rate and duration of TLR signaling and coordinating signaling through other cell surface receptors such as the integrins begins to shed light on how these proteins might be involved in regulating immune activation in diseases such as lupus. B cell activation is known to be the key initial driver for immune activation in SLE [22,23], and based on our mouse studies, we were keen to assess the role of autophagy proteins in B cell activation during SLE. However, before assessing the status of this pathway in B cells from a disease micro-environment, it is essential to establish how these proteins are functioning in human B cell populations. B cells from the bone marrow enter circulation as transitional B cells, which differentiate into mature naïve B cells. Naïve B cells encounter antigens through their BCR and other receptors such as the TLRs also get activated during this process of antigenic stimulation. This leads to the generation of more activated mature B cell subsets such as germinal center (GC) B cells, memory B cells, and plasma cells. These activated mature B cell subsets each have varying capacity to respond to antigenic stimulation and inherent differences in their signaling capacities ensure the most appropriate B cell response to antigens. To obtain an overall picture of the role of autophagy proteins in different human B cell subsets, we utilized the human tonsil tissue, which allows us to isolate different B cell subpopulations (transitional, naïve, GC, and memory B cells) [24]. Analysis of autophagy pathway in the B cell subsets revealed striking differences among the subsets, with induction of the pathway seen mainly in activated B cell subsets. This led to the questions of why this was the case and how this relates to the function of these proteins in human B cells?
We hypothesized that the autophagy pathway is upregulated in activated B cell populations because autophagy proteins have important roles in these B cell subsets both through macroautophagy and noncanonical forms of autophagy like TLR-induced autophagy (TIA). While macroautophagy could play an important role in regulating functions such as protein secretion by plasma cells, we predict that activation-induced autophagy plays a role in adjusting the threshold of response of activated B cells. Alterations in these types of adjustment mechanisms could lead to the aberrant activation of B cells seen in diseases such as lupus. To elucidate these functions of activation-induced autophagy in more detail, we leveraged the differences between B cell subsets in the induction of autophagy pathway, to identify genes and pathways related to activation of autophagy. We then combine this with an analysis of genes and pathways specifically regulated by αv integrin-dependent autophagy in an activated B cell line, to define specifically the consequences of activation-induced autophagy in human B cells.
Results
B cell subsets from human tissues show distinct differences in activation of autophagy pathway
We have previously shown that TIA preferentially occurs in activated B cell subsets in mice, including B-1, marginal zone (MZ), and GC B cells. To determine whether human B cells show similar differences in TIA, we used flow cytometry to sort human tonsil B cell subsets. We isolated tonsil transitional, naïve, GC, and memory B cells (Figure 1A) and measured LC3-II levels [17,19,25] following stimulation with ligands to TLR7 and TLR9 (R848 and CpG DNA, respectively). Naïve B cells expressed low levels of LC3-I and LC3-II, either with or without TLR stimulation (Figure 1B). This subset also did not produce high levels of LC3-II after stimulation with the macro-autophagy inducer rapamycin. Although the levels of LC3-II varied among donors, naïve B cells expressed the lowest levels compared to other subsets. We therefore concluded that naïve cells show little capacity for TIA or macroautophagy. Transitional B cells expressed LC3-I without stimulation, and treatment with rapamycin or treatment with chloroquine (which blocks LC3 degradation, allowing the measurement of basal flux through the autophagy pathway) both caused accumulation of low levels of LC3-II, indicating that these cells are capable of macroautophagy and that this occurs at low levels in unstimulated cells. However, transitional cells did not undergo TIA, as they showed no increase in LC3-II after TLR treatment (Figure 1B).
Figure 1.
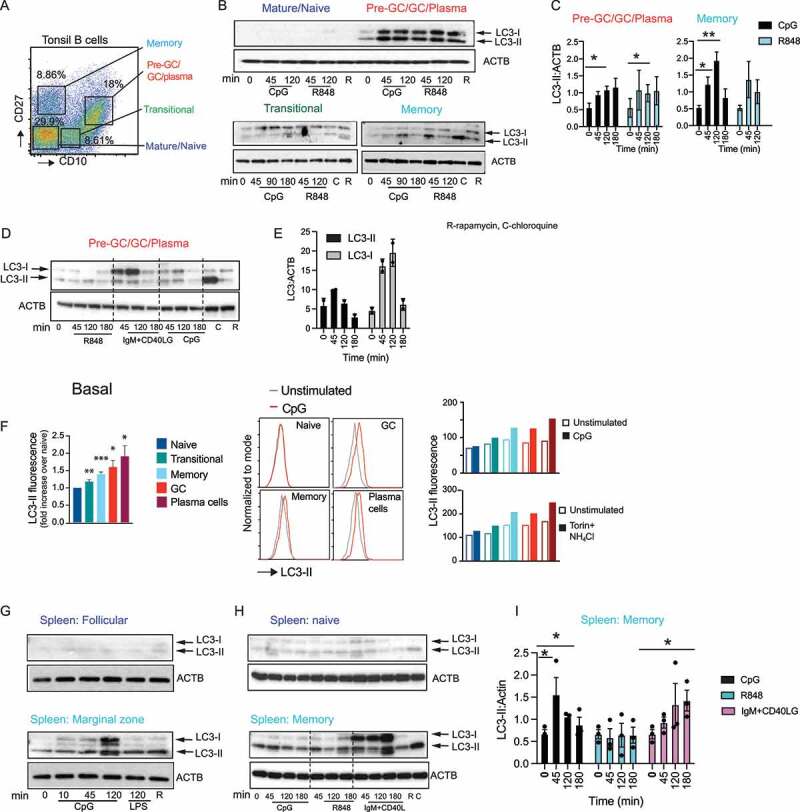
Autophagy proteins are specifically induced in activated B cell subsets from human tissue. (A) Flow cytometry gating for fluorescence activated cell sorting (FACS) of the different tonsil B cell subsets (CD19+). (B) Western blot analysis for LC3 expression and processing in sorted B cell subsets from human tonsils is shown. B cell subsets were sorted based on CD19, CD10, and CD27 expression and additional analysis of these subsets was used to delineate them as: naïve B cells (CD19+ CD10 low CD27 low, IgM ++, IgD+), transitional B cells (CD19+, MME/CD10++, CD27 low, IgM++, IgD+), memory B cells (CD19+, MME/CD10low CD27++, IgM++, IgD negative), and pre-GC/GC/plasma cells (CD19+, CD10++++, CD27++, IgM±, IgD negative). Sorted B cells (200,000–250,000 per condition) were stimulated for the indicated times with CpG DNA (3 μM) or R848 (5 μg/ml) or rapamycin (R) (0.6 μM) or chloroquine (C) (0.1 mM) for indicated time (mins) and cytoplasmic extracts from cell lysates were used to stain for LC3. ACTB is shown as the loading control. (C) Quantification of LC3 lipidation as measured by changes in the LC3-II band and ACTB by densitometry. (D) LC3 processing upon stimulation with anti-IgM (10 μg/mL) and CD40LG (CD40 ligand; 5 μg/mL). (E) Quantification of either LC3-I or LC3-II compared to the amount of ACTB in the pre-GC/GC/plasma cell subset after anti-IgM and CD40LG stimulation based on western blots from two different donors. (F) Flow cytometry data for the measurement of LC3-II on unsorted tonsil cells left unstimulated to calculate basal LC3-II levels or cells stimulated with different ligands at indicated time points and processed for surface staining and LC3-II staining. Cells are gated on surface markers to identify the different tonsil subsets. Naïve B cells (IgD+ CD27 negative, MME/CD10 negative), transitional (IgD+, MME/CD10+++, CD27 negative), memory (IgD negative, MME/CD10 negative, CD27+), GC (IgD-neg, MME/CD10+++ CD27+ CD38 mid), and plasma cells (IgD-neg, MME/CD10+++, CD27high CD38high) were analyzed for LC3-II expression using mean fluorescent intensity (MFI). LC3-II levels on unstimulated B cell subsets (basal) shown as MFI for each subset relative to MFI from naïve B cells to allow for a combination of data from multiple donors. Data are presented as mean ± s.e.m. *, p < 0.05, **, p < 0.01, ***p < 0.001, Student’s t-test from four different tonsil samples. LC3-II levels in the tonsil B cell subsets from one donor after combined stimulation with torin (1 μM; macroautophagy inducer) and NH4Cl (15 mM; for blockade of LC3 degradation) or with CpG (3 μM) is shown as MFI. Examples of histogram overlays are shown for some of the B cell population with CpG stimulation. (G-H) Analysis of LC3 processing in B cell subsets marginal zone (CD19+ CD27+ CD23 negative), follicular (CD19+, CD27 negative CD23+), naïve (CD19+, MME/CD10neg CD27neg), and memory (CD19+, MME/CD10 negative CD27+), sorted from human spleens. (I) Quantification of LC3-II band compared to ACTB by densitometry in spleen memory B cell subset. Data show representative blots based on blots from eight different tonsil donor samples and five different spleen donor samples. Quantification of data from 3–5 individual donors are shown, and significance indicated is indicated by * (p <0.05) or ** (p <0.01). p-values calculated using Student’s t-test.
TLR stimulation caused increases in LC3-II levels in both memory and GC B cells (Figure 1B,C). These cell subsets also showed increases in LC3-II levels after treatment with rapamycin or chloroquine (Figure 1B-D). We therefore conclude that these subsets are capable of both TIA and macroautophagy. In memory B cells, levels of LC3-II peaked 2 h after stimulation with TLRs and had dropped by 3 h (Figure 1B,C). This is likely due to reduction in the generation of LC3-II as TLR signaling ends, and LC3-II gets degraded. However, we were unable to definitively show this as the treatments used to prevent LC3-II degradation, such as chloroquine, bafilomycin A1, and NH4Cl, also block endosomal TLR signaling and TIA. High levels of basal autophagy and autophagic flux in the GC B cell subset have been reported by others in mice [15], and we found that basal levels of LC3-II varied in the GC subsets from different donors. Donors with low basal LC3-II showed more robust activation upon stimulation with TLR ligands. Stimulation of GC B cells through BCR cross-linking and co-stimulation through CD40 also caused an increase in LC3-II induction, but this occurred in combination with a more robust increase in LC3-I levels (Figure 1D,E), likely reflecting an increase in LC3 expression or preprocessing. This LC3-I increase was not seen to the same extent after TLR stimulation, suggesting that signaling from the BCR may activate distinct or additional autophagy pathways to those activated downstream of TLRs.
To more accurately compare LC3-II generation between B cell subsets, we used a flow cytometry (FACS) approach that specifically measures LC3-II [12,26]. LC3-II was first measured in individual cell populations from the same culture without any stimulation. This approach confirmed that GC and memory B cells expressed higher levels of LC3-II than naïve or transitional B cells (Figure 1F). Stimulation with a combination of the autophagy promoter torin together with NH4Cl to measure macroautophagy or stimulation with CpG DNA to measure TIA confirmed our findings from the western blot analysis that memory and GC B cells induce macroautophagy, and they are also the main subsets robustly inducing TIA (Figure 1F). Furthermore, this analysis allowed us to further divide the pre-GC/GC/plasma cell subset into GC cells and plasma cells and assess autophagy in plasma cells, which are not present in sufficient numbers in the tonsil samples for sorting and western blot analysis. This cell subset also had high basal levels of LC3-II, and this increased after stimulation with CpG DNA or torin and NH4Cl stimulation (Figure 1F).
We observed similar results in human splenic B cells. Naïve (CD27neg MME/CD10 negative) and follicular (CD27negative FCER2/CD23+) spleen B cells showed very little expression of LC3-I or induction of autophagy after stimulation with TLR ligands, stimulation of the BCR, or treatment with rapamycin. In contrast, memory B cells and marginal zone-like B cells had higher levels of LC3-II without stimulation, and levels increased after stimulation with CpG DNA or BCR-CD40 cross-linking (Figure 1G-I). Thus, based on these data, we concluded that “activated” B cells, including memory, GC, plasma cells, and marginal zone-like B cells, are the principal B cells that can undergo autophagy and do so in response to both TLR stimulation and activation through the BCR and CD40.
In mouse B cells, TIA promotes a transition from NFKB to IRF7 activation downstream of TLR7 and TLR9 [17,19,21]. To determine whether a similar transition is associated with TIA in humans, we measured nuclear NFKB and IRF7 in tonsil and spleen B cell subsets after stimulation. NFKB was activated by either TLR and BCR and CD40 stimulation in all cells tested, and we quantified changes in the kinetics of activation in subsets where we had cells for multiple donors (Fig S1). Naïve cells showed robust activation of NFKB mainly with BCR and CD40 stimulation compared to TLR stimulation, while the GC cells showed robust activation of NFKB mainly with CpG stimulation. IRF7 activation, however, was only seen at high levels in tonsil GC and memory cells and in spleen marginal zone-like cells (Fig S1). In GC cells, IRF7 activation was highest with CpG stimulation, while some activation was also seen with BCR and CD40 stimulation. In memory B cells, IRF7 activation was already high basally before any stimulation. However, quantification of results from different donors indicated IRF7 was activated upon CpG and R848 stimulation in the memory cells (Fig S1C). Transitional cells also showed some level of IRF7 activation upon TLR stimulation; however, we could not isolate enough cells from multiple donors for detailed analysis. Although low levels of IRF7 activation were seen in naïve B cells, these were not consistent among donors. Hence, as we have seen in mouse B cells, activation of autophagy corresponds to a switch from NFKB to IRF7 signaling in activated human B cell subsets.
ITGAV signaling is activated in GC and memory B cells subsets
We have previously shown that TIA in mouse B cells requires ITGAV/αv integrins and involves ITGAV-ITGB3/αvβ3 integrin activation and internalization. We therefore next determined whether changes in αv integrin expression or activation were associated with TIA in human B cell subsets. Using flow cytometry, we observed that ITGAV-ITGB3/αvβ3 and the related heterodimer ITGAV-ITGB5/αvβ5 were expressed on the surface of all tonsil B cell subsets. We observed slight changes in ITGAV-ITGB5 expression in transitional B cells compared to other subsets, and it is not clear whether this is related to TIA. However, there were no major differences in ITGAV-ITGB3 expression between the cell subsets (Figure 2A). Integrin activity is often regulated by associated proteins, including members of the tetraspanin family, which promote activation and internalization of integrins [27]. Notably, the tetraspanin CD9, which forms a complex with ITGAV/αv integrins [28], and tetraspanin CD82, which also binds integrins, were upregulated in tonsil GC and memory B cells, respectively, suggesting that these cells may be primed for activation of ITGAV-ITGB3 and internalization of ITGAV-ITGB3 (Figure 2B).
Figure 2.
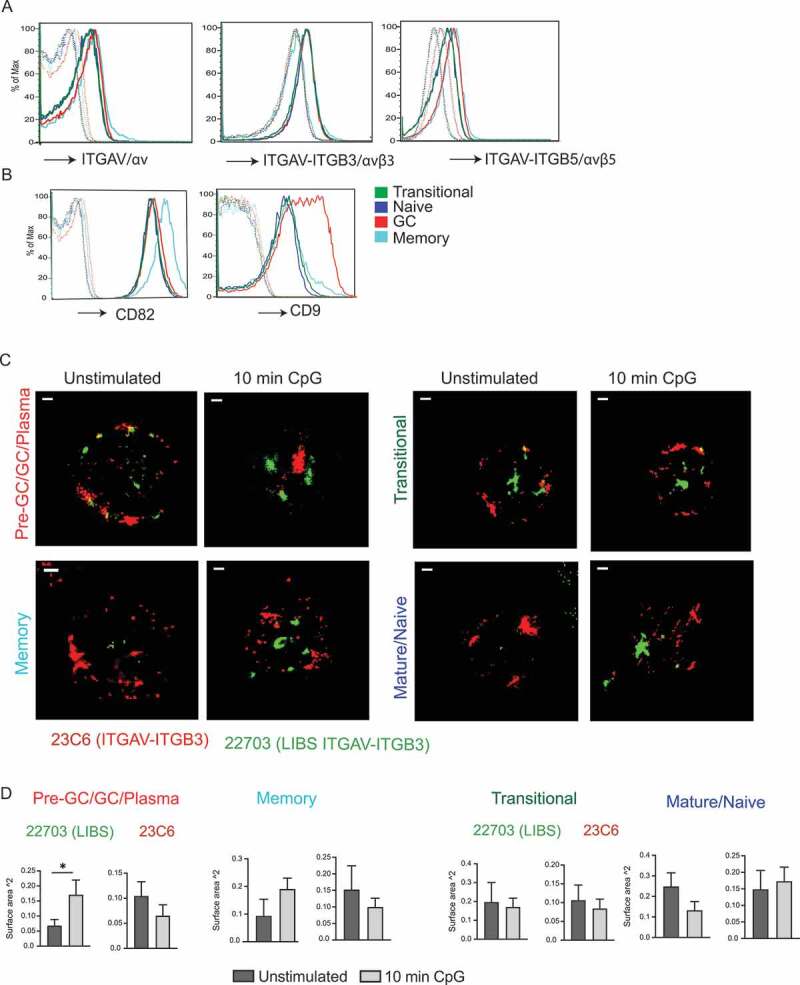
ITGAV integrin expression and activation in tonsil B cell subsets. (A-B) Flow cytometry for staining of integrin heterodimers (A) or tetraspanins CD9 and CD82 (B) on the tonsil B cell subsets. Tonsil B cell subsets were distinguished as in Fig 1A. Dotted lines indicate cells without the specific integrin heterodimer or tetraspanin stain or cells stained with streptavidin APC alone. (C) STORM images for staining of sorted tonsil B cell subsets with anti-ITGAV-ITGB3 antibody 23C6 and anti- ITGAV-ITGB3 LIBS antibody, with or without CPG stimulation. (D) Graphs represent quantification of the surface area of red or green aggregate staining with 23C6 or LIBS antibody based on a set threshold to remove background. Bar graphs represent mean of individual data points with s.e.m.; each data point is based on one aggregate structure staining, and multiple aggregate structures were analyzed over multiple cells. Scale bars: 1 μm. Significance is indicated by * p <0.05 based on Student’s t-test.
To test whether TIA in GC B cells was associated with ITGAV-ITGB3 integrin activation, we analyzed ITGAV-ITGB3 localization and activation in sorted tonsil B cells using super-resolution microscopy, to allow better resolution of integrin localization. Antibody 23C6, which preferentially binds non-activated ITGAV-ITGB3 heterodimer, stained unstimulated GC and memory cells uniformly on what appeared to be the plasma membrane, consistent with flow cytometry analysis of surface ITGAV-ITGB3 (Figure 2C). Following stimulation of GC cells with CpG DNA, 23C6 stained intracellular compartments, consistent with our previous findings that TLR signaling promotes the internalization of ITGAV-ITGB3 to endo-lysosomal vacuoles in mouse B cells [17]. TLR stimulation also caused the formation of intracellular ITGAV-ITGB3 aggregates that stained strongly with a different ITGAV-ITGB3 antibody, 22,703, which specifically recognizes the ligand-induced binding site (LIBS) on activated ITGAV-ITGB3 [29], confirming that TLR stimulation promoted the activation of this integrin (Figure 2C). Staining of ITGAV-ITGB3 with these two antibodies did not strongly colocalize, consistent with activated, ligand-bound ITGAV-ITGB3 (stained with LIBS antibody 22,703) trafficking separately from non-ligand bound ITGAV-ITGB3. Interestingly, in the GC cells, there was also some staining with the LIBS antibody in the unstimulated cells, indicating that some integrin might already be in activated state in these cells, but CpG stimulation led to formation of larger clusters of these activated integrins. CpG treatment of memory B cells also led to large clusters staining with the LIBS antibody, indicating that TLR signaling activates ITGAV-ITGB3 in these cells. Transitional and mature/naïve B cells did not show clear changes in staining with either of the antibodies after TLR stimulation (Figure 2C). Therefore, we quantified these aggregates by measuring the size of each of the red (23C6 stained) or green (LIBS stained) clusters. This showed that in GC and memory B cells stimulation led to an increase in the size of clusters stained with the LIBS antibody, but this was not seen in transitional and naïve B cells (Figure 2D). Thus, we conclude that ITGAV-ITGB3 activation and internalization to large aggregates correlates with induction of autophagy in human GC and memory B cells, consistent with our previous observations in mouse MZ and GC B cells.
Transcriptional analysis identifies a role for mitochondria-related genes in activation of autophagy proteins in GC B cells
To investigate which pathways are related to induction of autophagy in activated B cell subsets, we identified genes differentially expressed in the sorted tonsil B cell subsets by RNA-Sequencing (RNA-Seq) (Figure 3A). GC B cells showed the most distinct differences in gene expression compared to other subsets. Many of the genes expressed at higher levels in GC cells reflect known differences in their functional properties, including genes involved in cell proliferation, metabolism, cytoskeleton, and DNA repair (Figure 3A). We then focused on comparisons of gene and functional modules [30–32] between GC B cells, which undergo autophagy and naïve B cells, which do not show high levels of basal or TLR-induced autophagy.
Figure 3.
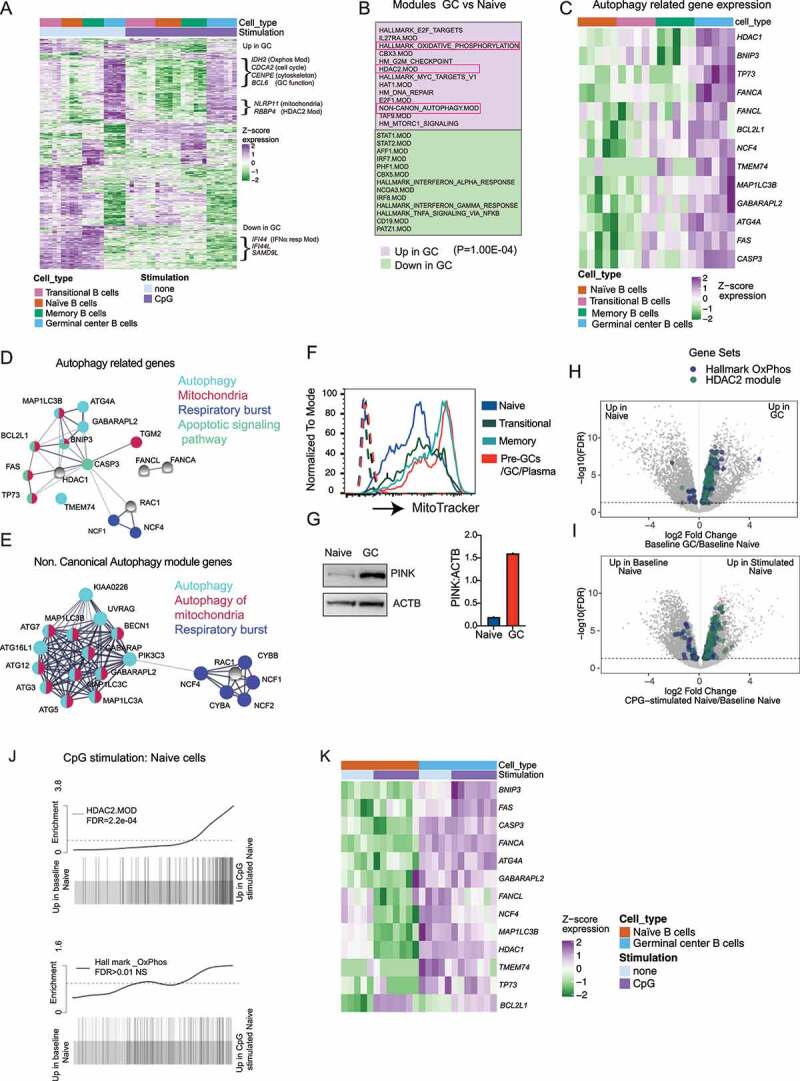
Expression and activation of autophagy pathway in activated B cells are related to the upregulation of mitochondrial proteins. Sorted B cell subsets (as in Fig 1A) with or without stimulation (3 h) with CpG DNA were used for RNA-Seq. Cells were stimulated with either CpG 2395 or CpG 2006, and data include results from both stimulations. (A) Gene expression heat map generated using the 350 most significantly DE genes from each subset compared to other subsets. Genes were hierarchically clustered using Euclidean distance and complete linkage; colors represent scaled gene expression values. Genes upregulated or downregulated in GC cells are highlighted. (B) Modules upregulated or downregulated in GC cells compared to naïve cells, based on mroast analysis. 13 modules from each subset are shown, and P-value from mroast analysis is indicated. (C) Heat map of selected autophagy genes’ scaled expression in the different B cell subsets. (D) String network analysis for protein–protein interactions (PPI) among autophagy-related genes that are enriched in GC cells. Proteins are presented as circular nodes and known interactions from STRING are shown as gray lines. Thickness of the line indicates strength of data support. (E) String network analysis of the genes from the Non.Canonical Autophagy module for PPI interactions. (F) Flow cytometry analysis of mitochondrial mass in the tonsil subsets using MitoTracker dye. Histogram overlays show each tonsil B cell subset, and dotted histograms show each B cell subset without the MitoTracker dye stain. (G) Western blots for mitophagy-related protein PINK in sorted naïve and GC B cell subset from one donor. Quantification is presented for PINK based on the amount of ACTB. (H-I) Volcano plots showing differentially expressed genes in naive cells compared to GC cells or naïve cells with and without CpG stimulation. The genes belonging to Oxidative phosphorylation and HDAC2 modules from gene set enrichment analysis are highlighted. (J) Barcode plots showing enrichment of HDAC2 or Hallmark Oxphos module genes in naïve cells upon CpG stimulation. FDR (false discovery rate) is indicated in the plot. (K) Heat map of selected autophagy genes’ scaled expression in GC and naïve B cell subsets with or without CpG stimulation. Gene expression data are based on RNA-Seq from five different donor tonsil samples.
From the analysis of modules highly expressed in the GC cells compared to naïve B cells, we found that GC B cells had significantly upregulated expression of one of the previously identified autophagy modules, the noncanonical autophagy module [4] (Figure 3B). Moreover, analysis of individual genes also showed that genes related to autophagy were upregulated in GC B cells, including the MAP1LC3B/LC3 and ATG4A genes (Figure 3C). These autophagy-related genes showed a similar pattern of expression as LC3 lipidation (lower in naïve and higher in memory and GC cells) (Figure 3C). Notably, the majority of the genes showing this pattern of expression and genes from the noncanonical autophagy modules are implicated in a specific form of autophagy, termed mitophagy [33,34], or are related to respiratory burst, which is involved in noncanonical autophagy (Figure 3D,E). In particular, genes such as NCF1 and NCF4 which are related to ROS production are involved specifically in the non-canonical autophagy pathway. Supporting these gene expression data, we found that both GC and memory B cells show increased mitochondrial mass compared to naïve and transitional cells, and GC cells also show significant upregulation of markers of mitophagy such as PINK1 (Figure 3F,G). Moreover, gene modules related to mitochondrial metabolisms such as oxidative phosphorylation (Oxphos) and HDAC2 modules were also significantly elevated in GC B cells compared to naïve cells (Figure 3B,H,I). Genes involved in mitochondrial metabolism have previously been linked with autophagy [35,36], and in our previous work we have shown that ROS production is required for TIA in mouse B cells [17]. Therefore, we predict that the expression of genes related to mitochondrial metabolism, mitophagy, and ROS production lead to specific activation of the autophagy pathway in activated B cell subsets such as GC B cells. Although both the HDAC2 and OxPhos modules showed a trend toward upregulation upon stimulation in naïve B cells (Figure 3J), the autophagy-related genes themselves did not show upregulation upon stimulation in the naïve cells (Figure 3K), suggesting that the activation of autophagy does not simply reflect a response to increased metabolic activity after cell activation but instead reflect phenotypic changes acquired as B cells mature.
Mitochondria-related genes are involved in TLR-induced autophagy in B cells
To further investigate the role of autophagy in human B cells, we turned to a genetically tractable system, the human B cell lymphoma line HBL-1. These cells resemble activated GC B cells and respond strongly to TLR9 stimulation [37]. HBL-1 cells undergo both basal autophagy, revealed by ongoing LC3-II conversion and turnover, and TLR-induced autophagy after treatment with CpG DNA (Figure 4A). CRISPR deletion of either of the core autophagy gene ATG5 or ATG7 blocked both basal and TLR-induced autophagy, as seen by the complete loss of the LC3-II band in western blot, after treatment with TLR ligands, rapamycin or chloroquine (Figure 4A). In contrast, deletion of the gene for integrin αv (ITGAV) (which causes complete loss of ITGAV-ITGB3/αvβ3 integrin) blocked TLR-induced LC3-II conversion but had no effect on basal autophagy as seen by LC3 lipidation in response to chloroquine or NH4Cl stimulation (Figure 4B,C and Fig S2A-C). Deletion of ATG5, ATG7, and ITGAV also affected TLR signaling, increasing basal NFKB activation and prolonging activation after stimulation but preventing activation of IRF7 (Figure 4D). Hence, these data show that αv is required for TIA and that TIA regulates NFKB and IRF7 activation in HBL-1 cells, recapitulating our findings in B cells from equivalent atg5 and itgav gene knockouts in mice.
Figure 4.
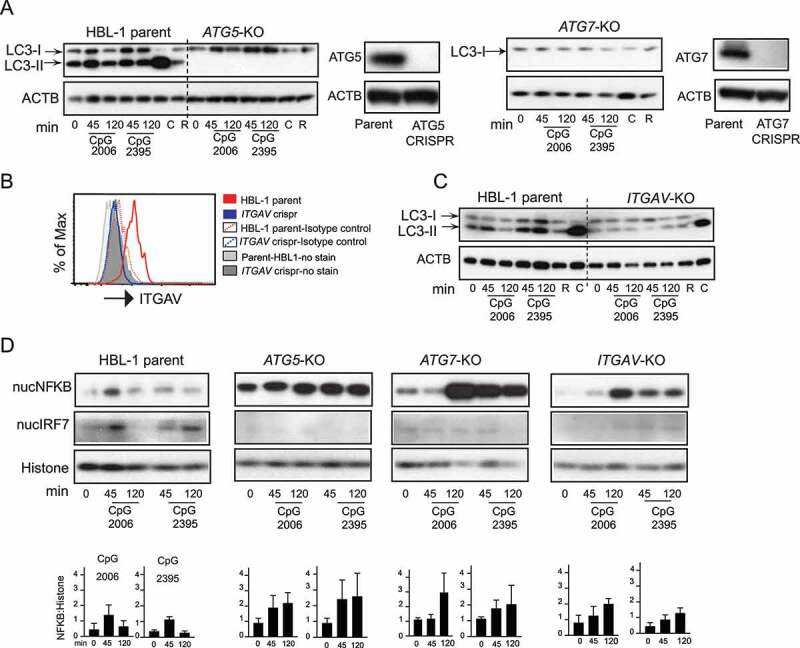
Integrin-dependent autophagy regulates TLR signaling in activated human B cell line. (A) Western blot analysis of LC3 processing or LC3 lipidation in parental HBL-1 cells or cells with CRISPR of autophagy genes (ATG5 or ATG7) labeled as ATG5-KO and ATG7-KO. Parental cells or CRISPR cells were stimulated with either CpG 2006 or CpG 2395 or with autophagy modulators rapamycin (R) or chloroquine (C) for indicated times, and cytoplasmic extracts were used to assess LC3. ACTB is shown as loading control. Western blot analysis of changes in the protein levels of ATG5 or ATG7 in the CRISPR cells compared to parental cells are also shown. (B) Flow cytometry analysis for changes in protein levels of ITGAV integrin after CRISPR of the ITGAV gene. (C) LC3 lipidation in parental HBL-1 cells or HBL-1 cells with CRISPR of ITGAV gene. (D) Nuclear fraction from the stimulated parental cells (HBL-1 parent) or cells with CRISPR of ATG5 gene (ATG5-KO) or ATG7 gene (ATG7-KO) or ITGAV gene (ITGAV-KO) were analyzed for NFKB and IRF7 activation. RELA/p65 for NFKB and IRF7 levels are shown by western blot. Histone is shown as nuclear loading control. Graphs show quantification for NFKB upon either CpG 2006 or CpG 2395 stimulation in relation to loading controls. Quantification is based on densitometry from at least three independent experiments. In all cases, representative blots from at least three independent experiments are shown.
We next used HBL-1 cells to test whether mitophagy is required for TIA, by deleting the BCL-family member BNIP3, which is critical for initiation of mitophagy (33, 34) and is upregulated in GC B cells (Figure 3C). Knockout of the BNIP3 gene in HBL-1 cells did not affect basal autophagy, as shown by LC3 lipidation in response to chloroquine or NH4Cl (Figure 5A-B and Fig S2B-C). However, LC3-II did not increase after stimulation with TLR ligands, similar to the effects of ITGAV-deletion (Figure 5A-B, Fig S2A). Similar to ITGAV knockout, BNIP3 knockout also resulted in sustained NFKB activation and loss of IRF7 activation (Figure 5C-D). These data were consistent with a role for BNIP3 and mitophagy in the activation of the autophagy machinery downstream of TLR signaling, but we were unable to measure how loss of BNIP3 or ITGAV integrin would affect autophagic flux, as the inhibitors commonly used to assess LC3 degradation (chloroquine, bafilomycin A1, or NH4Cl) all block TLR signaling. Addition of these inhibitors together with TLR ligands or after addition of TLR ligands did not lead to an increase in the accumulation of LC3-II above what was seen with the inhibitors alone, indicating that these inhibitors prevent TLR-signaling-induced LC3 lipidation (Fig S2D-I). As an alternative assay of TIA, we measured LC3-II by flow cytometry. This assay reliably detects LC3-II upon TLR stimulation or chloroquine stimulation as seen by LC3-II accumulation in parental cells, which is lost in the ATG5-knockout cells (Fig S2J-K). This assay confirmed that loss of BNIP3 and ITGAV integrin specifically inhibited TLR-induced LC3 lipidation (Fig S2K). Deletion of BNIP3 caused a slight increase in mitochondrial mass as seen by flow cytometry, but this was not seen by microscopy (Fig S2 L and M). However, consistency with the role of BNIP3 in mitophagy deletion of BNIP3 led to a decrease in mitophagy (Fig S2N). Curiously, ITGAV-knockout cells showed an increase in mitochondrial mass through both flow cytometry and microscopy measurements (Fig S2 L and M) and these cells also showed a decrease in mitophagy, suggesting that ITGAV-ITGB3 may be involved in mitophagy (Fig S2N). Hence, these data demonstrate that TIA in B cells involves pathways shared with mitophagy and regulates TLR activation of NFKB and IRF7 transcription factors (Fig S3).
Figure 5.
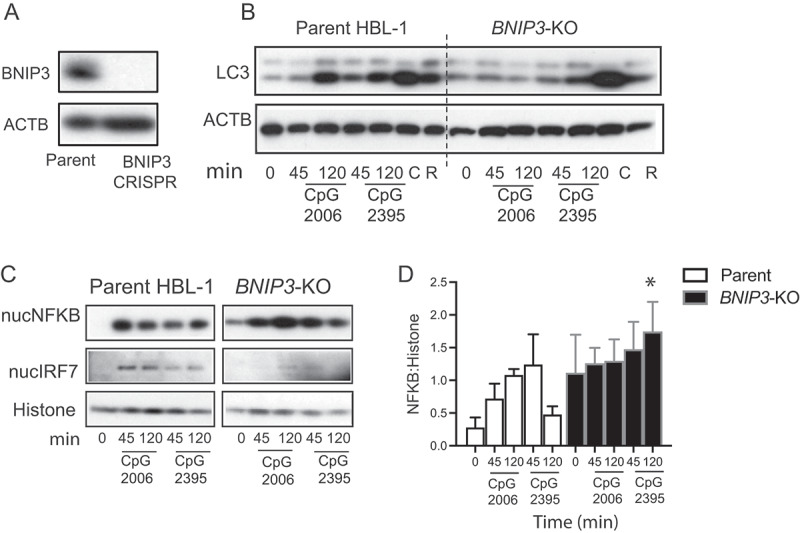
BNIP3 is involved in regulating TLR-induced autophagy in human B cells. (A) Western blot analysis shows changes in the protein levels for BNIP3 in the CRISPR cells compared to parental cells. (B) Western blot analysis of LC3 processing in parental HBL-1 cells or cells with CRISPR of the mitochondrial gene BNIP3 (BNIP3-KO). Parental cells or CRISPR cells were stimulated with CpG or with autophagy modulators rapamycin (R) or chloroquine (C) for indicated times, and cytoplasmic extracts were used to assess LC3. ACTB is shown as loading control. (C) Nuclear fraction of the stimulated cells was analyzed for NFKB and IRF7 activation by western blot. RELA/p65 and IRF7 levels are shown. Histone is shown as nuclear loading control for nuclear extracts. (D) Quantification of the amount NFKB compared to loading control histone by densitometry. Representative blots based on multiple independent experiments are shown. Quantification is based on combined 3–4 experiments, and data are presented as mean ± s.e.m. *, p <0.05, is based on Student’s t-test and indicates significance compared to parental sample with the corresponding stimulation.
Activation-induced autophagy regulates the transcriptional response to stimulation in GC B cells
To determine whether activation-induced autophagy is associated with changes in transcriptional responses to stimulation, we compared gene expression in response to stimulation in naïve and GC B cells. Cells were left unstimulated or stimulated with ligands for TLR7 (R848) and TLR9 (CpG DNA) or with a combination of BCR ligand (anti-IgM) and CD40LG (CD40-ligand) and then subject to RNA-Seq. A greater number of differentially expressed genes were induced in naïve B cells after stimulation (6717 genes compared with 4423 genes in stimulated GC B cells, respectively). Naïve B cells also showed greater variation between responses to three stimuli (CpG DNA, R848, or BCR and CD40) than GC B cells, which showed broadly similar transcriptional changes to all stimulations (Fig S4A). To overcome some heterogeneity in responses between cell types, we analyzed the expression of gene modules, curated by common function or co-expression patterns. The majority of gene modules differentially expressed after stimulation in GC cells were also altered in stimulated naïve cells (Table S1 and Fig S4B). Interestingly, these modules were induced less strongly in GC B cells when measured as fold-changes in expression after stimulation (Figure 6A and Fig S4B). In many cases, this was due to higher basal expression in GC B cells compared to naïve cells, rather than reduced expression after stimulation, as shown for the modules MYC target version 2, HDAC2, and Unfolded Protein Response in Figure 6A. We did not identify any modules that were significantly upregulated by stimulation only in GC cells, but two modules related to cell cycle and proliferation (hallmark G2-M checkpoint and E2F modules) were significantly downregulated by all stimuli only in GC cells (Fig S4B).s
Figure 6.
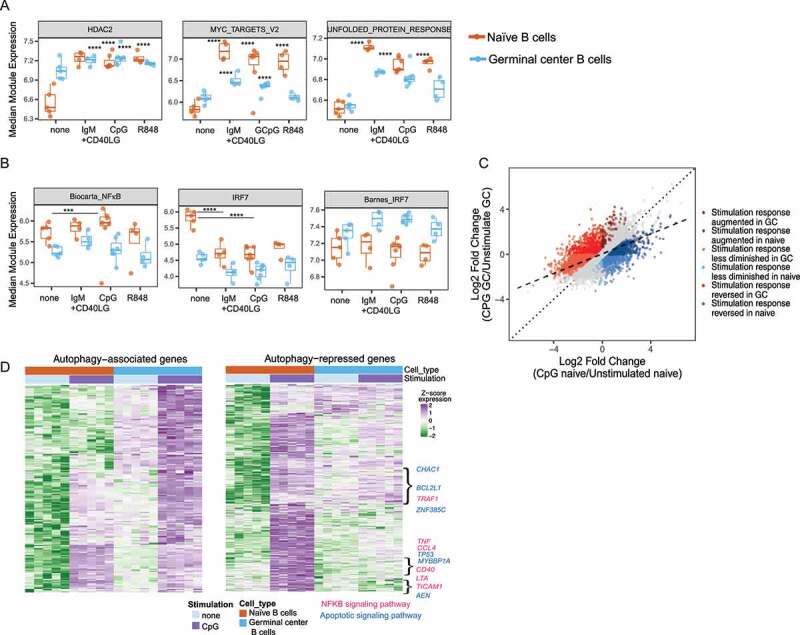
Autophagy proteins limit TLR responses in activated human B cells. Sorted B cells with or without stimulation (3 h) with either CpG or R848 or anti-IgM +CD40LG were used for RNA-sequencing. Data for CpG include both cells stimulated with CpG 2395 or with CpG 2006. (A) Graphs showing log2 TMM-normalized median module expression of gene sets in naïve and GC cells using mroast analysis. Box indicates interquartile range. (B) Median module expression for NFKB- and IRF7-related modules in GC vs. naïve cells with or without stimulation. P-values from mroast gene set tests are indicated as ***(p <0.001) or ****(p = 1X10 −4). (C) Graph showing gene expression changes upon CpG stimulation in GC cells compared to naïve cells. Dotted line represents perfect correspondence between CpG response in naïve and GC cells. Dashed line is fit to observed data. Non-gray dots highlight genes exhibiting significantly different stimulation responses between naïve and GC cells: in dark red are genes which increase to a greater degree in GC cells upon stimulation than in Naïve cells and in dark blue are genes which increase to a greater degree in naïve cells than in GC cells. In light red are genes that decrease to a greater degree in naïve cells upon stimulation than in GC cells, and in light blue are genes that decrease to a greater degree in GC cells than naïve cells. In medium red are genes that show increased expression in GC cells upon stimulation but show decrease in expression in naïve cells, and in medium blue are genes that show increase in expression in naïve cells upon stimulation but show the opposite response in GC cells. (D) Heat map of genes showing increased basal increase in GC cells and induced upon CpG stimulation (GC_0 > N_0 and GC_CPG > GC_0) is shown as the autophagy-associated set, and heat map of genes induced upon CpG stimulation in the naïve cells that show increased stimulation response in the naïve cells compared to GC cells (N_CPG > N_0 and CPG_resp_N > CPG_resp_GC) is shown as the autophagy-repressed set. Genes were hierarchically clustered using Euclidean distance and complete linkage; colors represent scaled gene expression values. Data are derived from RNA-Seq on samples from five different donors. Genes showing the association with specific networks based on STRING analysis for protein interactions are highlighted.
Since autophagy is associated with changes in NFKB and IRF7 activation, we analyzed the expression of gene modules connected with these transcription factor pathways. NFKB-responsive genes showed reduced response in stimulated GC B cells compared with naïve B cells (Figure 6B). Among the IRF7 modules analyzed one module, identified from a combination of immune cells and known to comprise mainly of type I interferon-related genes [32] was expressed at higher levels in naïve cells than in GC cells, and expression was suppressed after stimulation in both cell types. However, another module based on genes identified from a B cell line (Barnes IRF7 module) [38] showed a trend toward upregulation in stimulated GC cells, in agreement with our measurements of activated IRF7 in stimulated GC B cells (Figure 6B). This reduced activation of NFKB modules with increase in activation of IRF7 is in agreement with our hypothesis that autophagy limits induction of NFKB-related genes but promotes expression of IRF7-related genes.
We then wanted to determine whether changes observed from the module analysis were also seen at the level of individual genes. To simplify this analysis, we focused on transcriptional responses to only one stimulation and chose CpG, as both subsets responded to this stimulation. We identified 2475 genes with differential responses to stimulation in the two cell subsets, represented by the colored dots (Figure 6C). Of the genes that were upregulated in response to stimulation in both cell subsets, the majority (586/664) showed a stronger response in naïve cells (dark blue dots), whereas only 78 showed stronger induction in GC cells (dark red dots). A similar pattern was seen for downregulated genes, with the majority showing stronger responses in naïve (coral dots) than GC cells (light blue dots) (Figure 6C).
Taken together, analyses of both genes and modules indicated that GC B cells generally showed smaller induction or suppression of gene expression after stimulation compared to naïve cells. We predict that activation-induced autophagy mediated by αv integrins is in part responsible for this suppression of transcriptional responses to TLR stimulation in GC B cells. Thus, we generated a list of genes that showed reduced responses to CpG stimulation in GC cells compared to naïve cells and hypothesize that these include genes potentially repressed by activation-induced autophagy (autophagy repressed genes) (Figure 6D). We further identified a smaller set of genes that were preferentially upregulated after stimulation in GC cells, which we speculate may represent genes whose expression is promoted by autophagy (autophagy associated genes) (Figure 6D). String analysis showed that while both these gene sets included genes associated with mitochondria, metabolism, and exosomes, the autophagy repressed set included additional genes associated with the NFKB signaling and apoptotic signaling pathway (Figure 6D).
Integrin-dependent autophagy limits specific pathway while promoting others
To confirm whether the gene expression changes identified from the tonsil B cells are due to activation-induced autophagy mediated by ITGAV integrins, we made use of the HBL-1 cells in which we could disrupt autophagy by gene deletion. We profiled gene expression in ATG5, ATG7, or ITGAV-knockout HBL-1 cell lines without activation or after stimulation through TLR9 with CpG DNA. All three knockout lines showed significant changes in gene expression compared with parental HBL-1 cells (Fig S5A). To identify gene expression changes specifically related to integrin-dependent autophagy, we focused on genes that showed concordant significant expression changes in all three knockout cell lines (pooled knockouts) compared to parental HBL-1 cells. Based on our model that integrin-dependent autophagy represses GC B cell response to TLR stimulation, we first asked whether the disruption of integrin-dependent autophagy in the knockout cell lines led to an increase in expression of TLR-responsive genes upon CpG stimulation. Interestingly, TLR-responsive genes identified from the parental cells were basally upregulated in unstimulated knockout cells. This is shown for TLR stimulation-upregulated genes in ATG5 knockout cells in Figure 7A, and we observed a similar pattern in all three knockout cells (Figure 7B). Although many of these genes were further upregulated after stimulation in knockout cells, this induction was less pronounced than in control cells. Genes exhibiting this pattern included genes related to B cell activation (AICDA) and TLR signaling (NFKB1, RELA) (Figure 7B, Fig S5B) as well as other genes that have not been well characterized in B cells (such as SIRPA and RASGRP1) (Figure 7B, Fig S5C, and Fig S6A).
Figure 7.
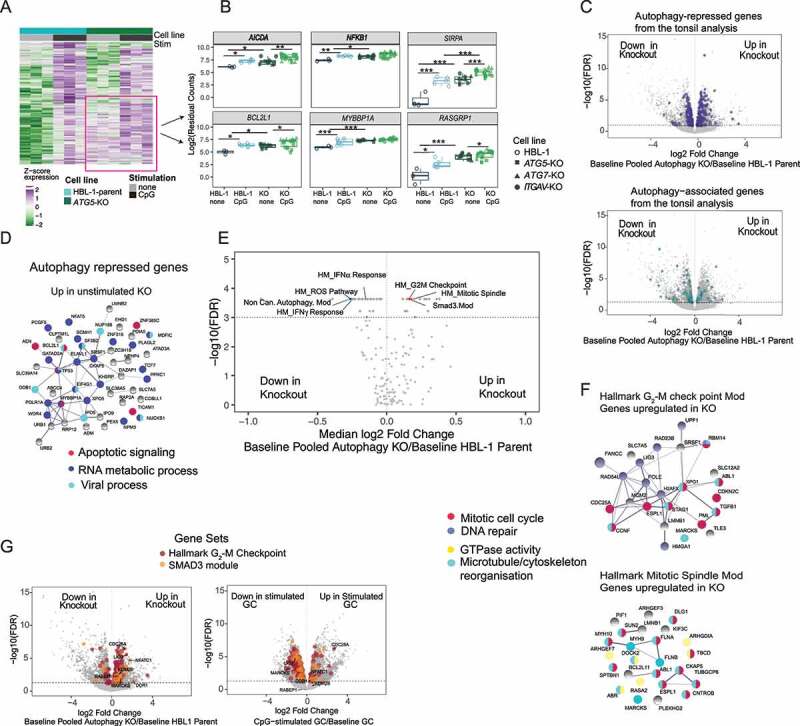
Integrin-dependent autophagy limits TLR responses in human B cells. HBL-1 parental cell line and cell lines with ATG5-CRISPR, ITGAV-CRISPR, or ATG7-CRISPR were stimulated with either CpG 2395 or 2006 for 3 h or left unstimulated and subjected to RNA-Seq. Data are all from three technical replicates for each condition: unstimulated or stimulated for each cell line, used for RNA-Seq experiment. Stimulation data include samples stimulated with CpG 2395 and CpG 2006. (A) Heat map of genes induced upon stimulation in the parental cells and/or the ATG5 knockout cells. Genes were hierarchically clustered using Euclidean distance and complete linkage; colors represent scaled gene expression values. Data generated from three replicates of unstimulated or stimulated parental cells or knockout cells. (B) Individual gene plots showing batch corrected and normalized log2 counts for gene expression in parental or knockout cell lines with or without stimulation. Each dot represents the gene expression from a single sample from each cell line and data from n = 3 experiments are plotted p-values are based on multiple t tests corrected for using Benjamini and Hochberg’s methods and presented as * (p <0.05) or ** (p <0.01) ***(p <0.001). (C) Volcano plots showing differentially expressed genes between data from combined unstimulated parental cells and combined data from knockout cells (data pooled from ATG-CRISPR, ATG7-CRISPR, and ITGAV-CRISPR cells). Genes from the autophagy-associated or autophagy-regulated sets from the tonsil analysis that also showed significant changes in the combined unstimulated knockout cells are highlighted. (D) Genes from the autophagy repressed set that are significantly upregulated by 0.5-fold in the unstimulated knockout cells are plotted for PPI network, and the known functions of these networks are highlighted based on the string pathway analysis tool. (E) Volcano plot showing changes in gene module expression between combined unstimulated parental lines and combined unstimulated knockout cell lines (data pooled for the three knockouts). Dashed line represents significance threshold FDR <0.001; modules of interest are highlighted. (F) Genes significantly upregulated (undefined.5-fold) in the unstimulated pooled knockout cells compared to parental cells, from G2-M checkpoint and mitotic spindle gene modules are plotted for PPI network, and the known functions of these networks are highlighted based on the string pathway analysis tool. (G) Volcano plots showing differentially expressed genes between combined unstimulated parental cells and combined knockout cells (pooled knockout) or the genes differentially expressed in GC B cells upon stimulation compared to unstimulated cells. The genes from the G2-M checkpoint module and the SMAD3 module are highlighted. Data are representative of replicates from each cell lines or five different donor tonsil samples.
Many of the putative autophagy repressed genes identified from primary GC B cells were also upregulated in this manner in the knockout HBL-1 cells, including genes such as BCL2L1, MYBBP1A, as well as genes involved in NFKB signaling (RELA, TNF) (Figure 7B, Fig S5B). We performed further analysis on the expression changes of genes from this set in the parental and knockout cells (Figure 7C). String analysis of the 49 most upregulated genes (undefined.5-fold) from this set in the knockout cells showed associations with common cellular processes including apoptotic signaling (Figure 7D). Whereas the smaller number of genes from this set downregulated (undefined.5-fold) in knockout cells, they were not associated with any specific cellular process. Meanwhile, 21 genes from the potential autophagy associated gene set (Figure 7C) were downregulated (undefined.5-fold) in knockout HBL-1 cells, including UPP1, CD86, SLC41A2 and SLC7A11, confirming that these genes are associated with autophagy (Fig S6B). To confirm that these gene expression changes were not due to loss of macroautophagy, we analyzed the expression of a subset of these differentially expressed genes by real-time quantitative PCR (RT-qPCR) in knockout and parental cells after induction of macroautophagy by treatment with rapamycin. The results confirmed the changes in gene expression seen by RNA-Seq and showed that expression of these genes was not altered by rapamycin treatment (Fig S6C and D). Thus, these data confirm the role of integrin-dependent autophagy in limiting activation of NFKB and apoptotic signaling pathways upon TLR signaling in GC B cells and allow us to identify sets of autophagy repressed and associated genes (Fig S6A and S6B). Furthermore, the increased basal expression of TLR responsive genes in the knockout HBL-1 cells suggests that a major role for integrin-dependent autophagy may be in regulating the basal expression of TLR-responsive genes (Fig S3). Based on this, we focus much of our subsequent analysis on differentially expressed genes in unstimulated conditions.
Although the analysis of individual genes supported the role of integrin-dependent autophagy in regulating gene expression, many of the putative autophagy-regulated genes identified in primary B cells were not strongly expressed in HBL-1 cells. To better identify the corresponding patterns of gene regulation between these cell systems and cellular processes regulated by integrin-dependent autophagy, we analyzed gene modules that were differentially expressed in the three HBL-1 knockout cell lines compared to the parental HBL-1 cell line. Among the gene modules significantly upregulated or downregulated in HBL-1 knockout cells, several were related to genes and modules differentially expressed in GC B cells (Figure 7E and Table S2). A number of gene modules related to cell proliferation and growth were upregulated in the knockout HBL-1 cells. These included two modules related to cell division, the hallmark G2-M checkpoint and mitotic spindle modules, and the upregulation of these modules was driven primarily by genes involved in mitosis and microtubule/cytoskeleton reorganization (Figure 7F). A SMAD3-regulated module that contained genes involved in cell proliferation was also upregulated in knockout HBL-1 cells. Notably, the G2-M checkpoint and SMAD3 modules were both downregulated following TLR stimulation in GC B cells, supporting a model in which these modules are repressed by integrin-dependent autophagy induced by TLR signaling (Figure 7E,G). Significantly changed genes from these modules in the cell lines and in GC cells are highlighted in Figure 7G. Additional gene modules differentially expressed in GC B cells were also upregulated in one or more of the knockout HBL-1 cells, identifying further potential autophagy-regulated modules. Specifically, the MYC target module was upregulated in both ITGAV-KO and ATG7-KO cells, indicating that this module is repressed by integrin-dependent autophagy, while the UPR module was only upregulated in the ATG7-KO cells, indicating that this module is probably regulated by macroautophagy (Fig S5D).
Several gene modules were downregulated in autophagy-knockout HBL-1 cells (Figure 7E and Table S2), including type-I and type-II IFN response modules. Genes significantly changed in these modules are consistent with the role of autophagy in the activation of IRF7 as well as antigen presentation (Fig S5E). Noncanonical autophagy and ROS modules were also downregulated in knockout cells and included many mitochondrial and respiratory burst genes that were shown to be upregulated in primary GC cells (Figure 3D), suggestive of a positive feedback loop, in which integrin-dependent autophagy increases expression of genes involved in the noncanonical autophagy pathway. Together, these data indicate that integrin-dependent autophagy limits activation of genes involved in GC B cell growth, proliferation, and apoptosis, while reinforcing the pathway of noncanonical autophagy.
Discussion
Through biochemical and transcriptional analysis of primary B cell populations, here we show that human memory and GC B cells acquire the ability to activate autophagy after stimulation. This is associated with increased expression of genes involved in noncanonical autophagy, mitophagy, and activation of integrin ITGAV-ITGB3. Complementary experiments on the genetically tractable B cell line HBL-1 demonstrate that TLR-induced autophagy in human B cells is triggered by ITGAV integrin and occurs through a mitophagy-related pathway (Fig S3). Furthermore, we show that this pathway regulates TLR signaling and transcriptional responses, limiting activation of genes related to cell proliferation and apoptosis while promoting expression of genes related to antigen presentation and interferon signaling. These data therefore provide important insights into the complex role of autophagy proteins in human B cells.
Human memory and GC B cells were the main B cell subpopulations to exhibit LC3 lipidation in response to TLR stimulation. These data are in agreement with our studies from mouse B cells and studies from others, which showed that activated mouse B cell populations including MZ and GC B cells preferentially activate LC3 after stimulation [15,17,19]. The small percentage of plasma cells we found in the tonsils expressed LC3-II and induced LC3 lipidation upon both macroautophagy-related and TLR stimulation, but we could not perform a detailed analysis of TLR stimulation on this subset due to the low number of cells. We also observed low levels of autophagy induction in transitional B cells. Again, this is consistent with our findings in mouse spleen B cells and suggests that B cells utilize this pathway during development and maturation in the bone marrow. However, this pathway appears to be downregulated as the cells transition to naïve B cells state. In mouse B cells, we have shown that during TLR stimulation, ITGAV-ITGB3 integrin promotes LC3 recruitment to TLR-containing endosomes, through a noncanonical autophagy pathway involving ROS [17]. Autophagy in human GC B cells also requires integrin ITGAV-ITGB3 and is associated with increased expression of genes associated with noncanonical autophagy and ROS production, indicating the same integrin-dependent autophagy mechanism in humans. We hypothesize that the increase in mitochondrial proteins together with increase in ROS production leads to the increased capacity of activated human B cells to specifically undergo TLR-induced autophagy. These conclusions are supported by previous studies including our own showing important roles for ROS in TLR-induced autophagy as well as recent studies showing a link between mitochondria and autophagy in B cells. Mitochondrial proteins have been shown to be prominent in activated B cells from mice [39,40], and the switch to noncanonical autophagy in mouse GC B cells is also linked with changes in mitochondrial homeostasis [15]. This increase in capacity to undergo TLR-induced autophagy due to increase in mitochondrial proteins is likely related to the reliance of mitochondria on the mitophagy pathway. In support of this, we identified a key role for components of the mitophagy pathway in regulating TLR-induced autophagy in human B cells. Many of these mitochondrial genes were also upregulated in naïve cells after stimulation, although this did not appear to be sufficient to allow upregulation of autophagy-related genes in naïve cells in our hands. Therefore, we predict that this ability to activate TLR-induced autophagy is acquired over time as the cells transition to GC B cell stage.
We have previously shown that the ITGAV integrin-dependent noncanonical activation of autophagy proteins in response to TLR stimulation regulates B cell responses by promoting endo-lysosomal trafficking of TLRs, which restricts TLR signaling through NFKB but promotes activation of IRF7. A prediction from this model is that cells undergoing activation-induced autophagy will show reduced expression of the majority of TLR-response genes (those activated by NFKB) but will activate the expression of a new subset of genes (related to IRF7 activation). Comparisons of the transcriptome of naïve and GC B cells after stimulation follow this prediction, allowing the identification of potential autophagy repressed and activated genes, and further supporting our model, where some of the autophagy-repressed genes correspond to known NFKB targets. GC B cells also showed increased expression of one set of reported IRF7 gene signatures upon stimulation compared with naïve cells, in agreement with our model. However, we did not observe strong activation of these genes after stimulation, and the autophagy-activated genes did not correspond closely with known IRF7-activated genes. We hypothesize that this is due to the timing of gene expression measurement. However, it is also possible that autophagy promotes activation of additional IRFs or other transcription factors that are responsible for autophagy-activated genes.
The analysis of differentially expressed gene modules revealed cellular processes that were differentially activated in naïve and GC B cells, and the comparison of these results against results from autophagy-knockout HBL-1 cells provided supporting evidence for the role of autophagy in regulating several of these. In particular, loss of integrin-dependent autophagy increased activation of genes involved in microtubule reorganization, cell division, apoptosis, and transcriptional regulation, suggesting that this form of autophagy limits the ability of GC B cells to divide and proliferate following antigenic stimulation. Conversely, loss of integrin-dependent autophagy led to a decrease in expression of genes involved in antigen presentation, cytokine signaling, and IFN signaling, consistent with the role of autophagy in activation of IRF transcription factors and type I IFN production [20]. Autophagy-knockout HBL-1 cells also provide additional insights into the role of integrin-dependent autophagy in GC B cell function. First, we identified additional autophagy-regulated genes. Notably, genes involved in somatic hypermutation such as AICDA and TLR signaling genes were upregulated in knockout cells, in agreement with our findings in B-cell-specific itgav-knockout mice [19]. We also observed increased expression of other genes including SIRPA, RASGRP1, and MYBBP1A which have not been studied in B cells. Second, it was notable that the major effect of deletion of autophagy components was to increase the expression of TLR-responsive genes in unstimulated cells, rather than to increase their activation after treatment with exogenous TLR ligands, and this was accompanied by increased basal NFKB activation. We speculate that a major role for ITGAV integrin-dependent autophagy pathway is to suppress this basal activation of TLR signaling in B cells that are poised to respond to antigens including self-reactive B cells. HBL-1 cells harbor mutations that can cause activation of MYD88 and the BCR and TLR9 are already colocalized in these cells in the absence of exogenous ligands [37]. Therefore, this cell line may be particularly sensitive to this mechanism of basal regulation. However, similar low-level basal activation of self-reactive B cells has also been reported in culture, probably due to the activation by cellular debris containing endogenous TLR ligands [41]. Furthermore, we consistently observed basal NFKB activation and proliferation in B cells from itgav- and atg5-knockout mice, both in culture and directly after isolation, supporting the idea that integrin-dependent autophagy regulates basal TLR activation in primary nontransformed cells [17,19]. Moreover, basal activation of TLR pathway has been reported in immune cells from SLE subjects, and another family of integrin has also been implicated in basal suppression of TLR signaling [42,43]. Thus, loss of this basal suppression mechanism could contribute to autoimmunity, by lowering the threshold for self-reactive B cells to escape tolerance or undergo class-switching (Fig S3).
HBL-1 cells are activated B-cell-like (ABC) diffuse large B cell lymphoma (DLBCL) cell line that arise from post germinal center B cells [44] and retain some properties of these cells. Relevant to our studies, they have been shown to undergo self-antigen-induced BCR signaling and grow in the presence of cellular debris [45]. Despite the caveat of high basal NFKB activation in these cells, robust induction of NFKB and lipidation of LC3 upon stimulation indicated that they were appropriate for our experiments. Furthermore, their growth in cell debris proved to be particularly advantageous for highlighting mechanisms of regulation of self-reactive B cells. A significant overlap in gene expression changes in ITGAV-KO cells with ATG7-KO cells further established ITGAV as an important component of the TLR-induced autophagy pathway. However, we were surprised to find significant differences in the ATG5 and ATG7-KO cells from the gene expression analysis (Fig S5A-B). It remains to be determined whether this is an effect seen only in the cell line or if it is relevant for specific function of ATG5 and ATG7.
Genetic variants of autophagy genes, including ATG5, are associated with an increased risk of SLE [8,46], and disruption of autophagy has been reported in a range of autoimmune diseases [47,48]. However, the complex role of autophagy in B cells and other immune cell types has made it difficult to identify the mechanisms by which autophagy may protect or contribute to disease. Our data highlight the advantages of studying specific autophagy pathways in a single cell type and across distinct subpopulations in understanding this complex problem. In B-cell-focused experiments in mice, we have previously shown that ITGAV integrin-dependent noncanonical autophagy plays a critical role in regulating autoreactive responses, and disruption of integrin-dependent autophagy only in B cells is sufficient to increase generation of autoantibodies and accelerate lupus-like autoimmunity [17,18]. Our current data confirm that this integrin-dependent autophagy pathway is active in human B cells and regulates transcriptional responses to stimulation. This provides a potential mechanism by which genetic variants of autophagy genes may promote SLE, via altered activation-induced autophagy. Although the full contribution of macroautophagy and activation-induced autophagy pathways to autoimmune disease development is likely to be complex, our data specify processes that can be investigated to unravel such complexities in terms of B cell dysregulation during development of lupus.
Materials and methods
Purification of B lymphocytes from human tissue
Tonsils were obtained from patients undergoing tonsillectomies, and spleens were obtained from patients undergoing pancreatectomy in accordance with an IRB approved protocol. Tissues were harvested and processed in RPMI (Gibco, 11,875,119) (10% fetal bovine serum (Sigma, F0926), 2 mM glutamine (Gibco, 35,050,061) 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, 15,070,063), and 50 mM 2-mercaptoethanol (Gibco, 21,985,023)) to generate single-cell suspensions, which were stored as frozen aliquots.
Antibodies, reagents, and cell lines
Anti-human antibodies used for flow cytometry include antibodies against CD19-PEcy7 (HIB19; BD Biosciences, 560,728), CD24-BV421 (ML5; BioLegend, 311,122), MME/CD10-APC (HI10; BioLegend, 312,210), CD27-BV605 (L128; BD Biosciences, 562,655), CD38-PerCP-Cy5.5 (HIT2; BD Biosciences, 303,522), IgD-BV510 (IA6–2; BD Biosciences, 563,034), ITGAV/CD51-PE (NKI-M9; BioLegend, 327,910), and IgG2a, Isotype Control-PE (G155–178; BD Biosciences, 554,648). Type C CPG ODN 2395, Type B CPG ODN 2006, and R848 were from Invivogen (tlrl-2006, tlrl-2395, and tlrl-r848). Antibodies anti-RELA/NFKB p65 (D14E12; 8242), anti-KDM1A/LSD1 (2139), anti-histone (D1H2; 4499), and horseradish peroxidase conjugated anti-rabbit IgG (7074) were from Cell Signaling Technology. Anti-LC3B (L7543) and anti-ACTB/actin (AC-74; A5316), chloroquine (C6628), and bafilomycin A1 (B1793) were from Sigma-Aldrich. Rapamycin was from Selleck Chemicals (S1039). Anti-SQSTM1/p62 antibody (03-GP62-C) was from American Research Products. Anti-IRF7 antibodies (H-246; sc-9083) and (AHP1180) were from Santa Cruz Biotechnology Inc and Bio-Rad Laboratories Inc, respectively. Anti-IgM (109-006-129) was from Jackson Immuno Research Laboratories, and CD40LG/sCD40 ligand (310–02) was from Peprotech. Anti-LC3 antibody (4E12; FCCH100171) was from Luminex Corp. The human diffuse large B cell lymphoma (HBL-1) cell line was provided by Dr Richard James (James Lab, Seattle Children’s Research Institute, Seattle, WA, USA).
Flow cytometry and cell sorting
Frozen aliquots of single-cell suspensions from tonsils or spleens were harvested in PBS (Gibco, 14,190–144) with 0.5% BSA (Sigma, A2153) and 2 mM EDTA (Invitrogen, 15,575–038). Single-cell suspensions were blocked with Fc Block (Biolegend, 422,302) and stained with fluorochrome-tagged antibodies for surface markers at 4°C for 30 min. Samples were acquired using the LSRII flow cytometer (Becton and Dickinson) and analyzed using the FlowJo software (Tree Star Inc.). For FACS of tonsil or spleen B cell populations, after B cell enrichment with the negative selection cocktail (Stem Cell Technologies, 19,054), cells were labeled with anti-CD19-PEcy7, anti-CD24-BV421, anti-MME/CD10-APC, anti-CD27-BV605, anti-CD38-PerCPcy5.5, and anti-IgD-BV510 antibodies and then sorted at 4°C with FACS Aria (BD Bioscience). Post-sort purity was checked, and cells were rested for 1 h at 37°C with 5% CO2 in RPMI media before being used for stimulation or RNA extraction. For expansion of clones, transfected HBL-1 cell mixes were single cell sorted into multiple 96-well plates containing 200 μl of complete IMDM with GlutaMAX media (Gibco, 31,980,097) supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin, and 50 μM 2-mercaptoethanol and cultured at 37°C with 5% CO2.
For flow-cytometry-based staining of LC3-II, single-cell suspension of tonsil cells or cell lines were plated on 96-well plate and stimulated for different time points with either CpG, chloroquine, torin 2 (Cell Signaling Technology, 14,385), or ammonium chloride (NH4Cl) and after surface antibody labeling cells were washed with PBS containing 0.05% saponin (Sigma, 47,036) and stained with LC3 antibody using the Guava LC3 assay kit (Luminex, FCCH100171). For mitochondrial staining, single-cell suspension of cells was stained with MitoTracker CMXros (Thermo Fisher, M46752) and Green FM (Thermo Fisher, M46750) dyes according to manufacturer’s protocol.
RNA sequencing
Bulk RNA-sequencing
The indicated populations were isolated by flow cytometry, and after stimulation cells were resuspended in SMARTer v4 lysis reagents (Takara, 634,894). Cells were lysed, and reverse transcription was performed followed by PCR amplification to generate full-length amplified cDNA. For the cell lines, stimulated or unstimulated cells were used for RNA extraction with the QIAGEN RNeasy Mini kit (74106), and 0.5 ng was used as input for the SMARTseq kit. Sequencing libraries were constructed using the NexteraXT DNA sample preparation kit (Illumina, FC-131-1096) to generate Illumina-compatible barcoded libraries. Libraries were pooled and quantified using a Qubit® Fluorometer (Life Technologies). Dual-index, single-read sequencing of the pooled libraries was carried out on a HiSeq2500 sequencer (Illumina) with 58-base reads, using HiSeq v4 Cluster (GD-401-4001) and SBS kits (Illumina, FC-401-4002) with a target depth of 5 million raw reads per sample.
RNA-sequencing pipeline analysis
Raw RNA-Seq reads were processed using a local Galaxy server. A minimum quality score of 30 was enforced for all reads by trimming bases from 5“and 3” ends using the FASTQ Quality Trimmer tool in Galaxy. Trimmed reads were then aligned to the GRCh38 (release 77) reference genome using STAR. Raw gene counts were generated using htseq-count (v.0.5.4p3), based on the union of exons for each gene as described in the Ensembl gene model GTF file for GRCh38. Quality metrics for aligned reads were obtained using the Picard (v.1.56) suite of tools (http://picard.sourceforge.net). To ensure the highest data quality, we sequentially examined distributions of unrelated Picard quality control metrics (PF_ALIGNED_BASES and MEDIAN_CV_COVERAGE) and eliminated outlier libraries. Counts were normalized using the trimmed mean of M values (TMM). Genes were included in analyses if they had greater than 1 count per million in at least 10% of libraries. Differential expression of individual genes was determined with limma-voom. Study subject ID was used as a blocking variable when comparing multiple treatments or cell subsets taken from the same individuals. Raw P values were corrected for multiple testing. Gene set analyses were performed using mroast or camera. PPI interactions were determined using STRING (string-db.org).
QR-TPCR analysis
HBL-1 parental or knockout cell lines were left unstimulated or stimulated with rapamycin for 3 h. RNA was prepared using QIAGEN RNeasy mini kit (74104) and converted into cDNA by reverse transcription (Applied Biosystems, 4368814). Real-time PCR was performed with SYBR Green (Applied Biosystems, 436759) using the following primers:
SIRPA: GCAAGAAGGATCAGGTCAGC (Forward)
GGCATTGGGTCTCGATAAGA (Reverse)
RASGRP1: CTGGGCTTCTGGAGTGTCTC (Forward)
ATGAGGCCTTCCCTGTCTTT (Reverse)
PLXNA1: GACAGACATCCACGAGCTGA (Forward)
GTGTCAGCGACTTCTCCACA (Reverse)
NFATC1: CCAGGGGTTAAGTCCTCTCC (Forward)
GAGAAAGGTCGTGGAGCTTG (Reverse)
CD86: ACTAGCACAGACACACGGATG (Forward)
CTTCAGAGGAGCAGCACCAGA (Reverse)
Upp1: CCTGGAGCATTGCGTTTGTC (Forward)
TTCTCTGGGCCCACTCGG (Reverse)
ADGRG5: GTTTGTGCTTGGCAATCTGGAG (Forward)
GAGTTCTTCCCATGTCTCTGTTGT (Reverse)
LILRB1: ACTGGACATCGACCCAGAGA (Forward)
GTGTCCATCTCCACCCCATC (Reverse)
Western Blots
Nuclear and cytoplasmic extracts were prepared by lysing cells in hypotonic nuclear extraction buffer (1 M HEPES, pH 7.5, 5 M NaCl, 0.5 M EDTA, pH 8, 50% glycerol, 10% Igepal [Sigma, 18,896], and 10% Triton X-100 [Sigma, 93,443]) supplemented with protease inhibitor cocktail (Pierce, A32961) for 10 min followed by centrifugation at 1500 g for 5 min at 4°C to pellet the nuclei and nuclei were resuspended in RIPA (Pierce, 89,901) buffer. Lysates were centrifuged for 10 min at 4°C at 14,000 g, and supernatant was collected as nuclear fraction, separated by electrophoresis using NuPage 4–12% Bis-Tris gels (Invitrogen, NP0336BOX) or NuPage 12% Bis-Tris gels (Invitrogen, NP0342BOX) and blotted onto PVDF membranes. Nonspecific binding was blocked with 5% BSA (Sigma, A2153) in TBS-Tween (0.1%; Takara, T9142) followed by incubation with primary antibodies (1:1000 dilution) overnight at 4°C and secondary antibody horseradish peroxidase conjugated antibodies (1:5000 dilution) for 1 h at room temperature. Membranes were washed thoroughly with TBS-Tween (0.1%) after antibody incubations and developed using ECL reagents (Millipore, WBKLS0500). For re-probing, blots were stripped for 30 min at 37°C with Restore PLUS stripping buffer (Thermo Scientific, 46,430).
CRISPR targeting
CRISPR crRNAs targeting BNIP3, ATG5, ATG7, and ITGAV were identified using the Broad Institute single guide RNA design tool and synthesized (IDT) containing phosphorothioate linkages and 2’ O methyl modifications. crRNAs and trans-activating crRNA (tracrRNA; IDT, 1,072,532) hybrids were mixed with Cas9 nuclease (IDT, 1,081,058) at the ratio of 1.2:1 and transfected into cells by Neon electroporation (Thermo Fisher Scientific, MPK1025). Transfected cell mixture was single cell sorted, and total genomic DNA was extracted from the single-cell clones using Quick-DNA Microprep Kit (ZymoResearch, D3021). To assess the knockout efficiency, guide target genomic regions were amplified using PrimeSTAR GXL DNA Polymerase (Takara Bio, R050A) with primers about 250–350 bp away from the guide target site. PCR products were purified using the GeneJet PCR Purification Kit (Thermo Fisher Scientific, K0701) and analyzed by Sanger Sequencing and Inference of CRISPR Edits (ICE; ice.synthego.com). Flow cytometry or western blot was used to validate knockdown on a protein level.
| Guide designation | Protospacer | tracrRNA binding sequence |
| ATG5 | CCUUAGAUGGACAGUGCAGA | GUUUUAGAGCUAUGCU |
| ATG7 | CUUGAAAGACUCGAGUGUGU | |
| ITGAV | AGCCGAAGUAACUUCCCUCG | |
| BNIP3 | UUCAGCAAUAAUGGGAACGG |
Super-resolution microscopy
Super-resolution microscopy on tonsil B cell subsets was performed as previously described [17,49]. Briefly, FACs sorted tonsil B cell subsets were seeded on to poly-L-lysine coated chamber slides (Nunc, 155411PK) stimulated, fixed, and stained as previously described [17]. Integrin staining was performed using primary ITGAV-ITGB3/αvβ3 antibody (23C6; Biolegend, 304,412) or LIBS ITGAV-ITGB3/αvβ3 antibody (22703), which was a gift from Dr Simon Goodman at Merk laboratories. Anti-rabbit atto-488 antibody (Hypermol, Germany,2302) or streptavidin 647 (Invitrogen, S32357) was used as the secondary reagent. Samples were imaged at room temperature using a customized Nikon N-STORM with 60X oil immersion and a “Perfect Focus” system. Imaging was performed in an extracellular solution containing reducing and oxygen scavengers, as specified by dSTORM protocols [50]. After activation, the fluorochromes were converted into a desired density of single molecules per frame and imaged continuously at 10,000–30,000 frames. Localization was reconstructed using Nikon Elements Imaging software. Quantification of ITGAV-ITGB3 (23C6) or LIBS ITGAV-ITGB3 (22703) staining was performed by transforming images into 8 bits and using 3D Object counter plugin from FIJI. The surface area of ITGAV-ITGB3 (23C6) staining was obtained by setting the intensity threshold at 15 without size limit, and the surface area of LIBS ITGAV-ITGB3 (22703) staining was obtained by setting the intensity threshold at 40 without size limit.
Mitophagy assay and quantification
Parental HBL-1, BNIP3-knockout, and ITGAV-knockout cells were seeded on 0.01% poly-L-lysine coated chamber slides (Nunc, 155,411) and plated overnight. Cells were washed with HBSS (Gibco, 2,402,017) twice, incubated with 100 nM Mtphagy dye (Dojindo, MD01) at 37°C for 30 min, and washed with HBSS twice again. Cells were then stained with 100 nM MitoTracker Green FM (Thermo Fisher, M46750) for another 30 min in media and imaged on a × 63 oil objectives (aperture 1.4) on Leica TCS SP5 confocal microscope. Images were stacked using FIJI software, and the relative mean fluorescence intensity per cell was obtained by subtracting extracellular background from intracellular staining measures in FIJI.
Statistical methods
GraphPad prism software or R-package was used for statistical analysis. In all cases, well-established statistical tests with default settings were used. Specific tests used to derive p values and software used for each statistical analysis are described in the text. Unless otherwise stated, we used 2-tailed tests and considered a P value of less than 0.05 as significant.
Supplementary Material
Acknowledgements
We thank all the members of the Acharya, Lacy-Hulbert, and James’ laboratories for their assistance and advice on this project. We thank the flow cytometry and genomics core facilities at Benaroya Research Institute (BRI) for expert assistance and Dr Simon Goodman at Merk laboratories for integrin antibodies. We thank Dr Hannah DeBerg at BRI for additional assistance on data organization and upload. Graphical abstract was generated using Biorender.com
Funding Statement
The work was supported by the Lupus Research Alliance [519405].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
RNA-sequencing data associated with the paper is deposited in GEO (gene expression omnibus) database with accession number GSE209755.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2113296
References
- [1].Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. 2019;19(3):170–183. [DOI] [PubMed] [Google Scholar]
- [2].Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Galluzzi L, Green DR. Autophagy-Independent functions of the autophagy machinery. Cell. 2019;177(7):1682–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martinez J, Malireddi RK, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906. DOI: 10.1038/ncb3192 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [5].Tam JM, Mansour MK, Acharya M, et al. The role of autophagy-related proteins in Candida albicans infections. Pathogens. 2016;5(2):34. DOI: 10.3390/pathogens5020034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bentham J, Morris DL, Graham DSC, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464. DOI: 10.1038/ng.3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Conway KL, Kuballa P, Song JH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145(6):1347–1357. DOI: 10.1053/j.gastro.2013.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou XJ, Lu XL, Lv JC, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–1337. DOI: 10.1136/ard.2010.140111 [DOI] [PubMed] [Google Scholar]
- [9].Kuballa P, Nolte WM, Castoreno AB, et al. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611–646. [DOI] [PubMed] [Google Scholar]
- [10].Metur SP, Klionsky DJ. Adaptive immunity at the crossroads of autophagy and metabolism. Cell Mol Immunol. 2021;18(5):1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen M, Hong MJ, Sun H, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014;20(5):503–510. DOI: 10.1038/nm.3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Clarke AJ, Riffelmacher T, Braas D, et al. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J Exp Med. 2018;215(2):399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Conway KL, Kuballa P, Khor B, et al. ATG5 regulates plasma cell differentiation. Autophagy. 2013;9(4):528–537. DOI: 10.4161/auto.23484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14(3):298–305. DOI: 10.1038/ni.2524 [DOI] [PubMed] [Google Scholar]
- [15].Martinez-Martin N, Maldonado P, Gasparrini F, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. 2017;355(6325):641–647. DOI: 10.1126/science.aal3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Arbogast F, Arnold J, Hammann P, et al. ATG5 is required for B cell polarization and presentation of particulate antigens. Autophagy. 2019;15(2):280–294. DOI: 10.1080/15548627.2018.1516327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Acharya M, Sokolovska A, Tam JM, et al. Alphav Integrins combine with LC3 and atg5 to regulate Toll-like receptor signalling in B cells. Nat Commun. 2016;7:10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Acharya M, Raso F, Sagadiev S, et al. B cell alphav integrins regulate TLR-Driven autoimmunity. J Immunol. 2020;205(7):1810–1818. DOI: 10.4049/jimmunol.1901056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raso F, Sagadiev S, Du S, et al. Alphav integrins regulate germinal center B cell responses through noncanonical autophagy. J Clin Invest. 2018;128(9):4163–4178. DOI: 10.1172/JCI99597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997. DOI: 10.1016/j.immuni.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee HK, Lund JM, Ramanathan B, et al. Autophagy-Dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315(5817):1398–1401. [DOI] [PubMed] [Google Scholar]
- [22].Jenks SA, Cashman KS, Zumaquero E, et al. Distinct effector B cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49(4):725–39 e6. DOI: 10.1016/j.immuni.2018.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Teichmann LL, Schenten D, Medzhitov R, et al. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38(3):528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giltiay NV, Shu GL, Shock A, et al. Targeting CD22 with the monoclonal antibody epratuzumab modulates human B-cell maturation and cytokine production in response to Toll-like receptor 7 (TLR7) and B-cell receptor (BCR) signaling. Arthritis Res Ther. 2017;19(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17(1):1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Eng KE, Panas MD, Karlsson Hedestam GB, et al. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010;6(5):634–641. [DOI] [PubMed] [Google Scholar]
- [27].van Spriel AB, de Keijzer S, van der Schaaf A, et al. The tetraspanin CD37 orchestrates the alpha(4)beta(1) integrin-Akt signaling axis and supports long-lived plasma cell survival. Sci Signal. 2012;5(250):ra82. DOI: 10.1126/scisignal.2003113 [DOI] [PubMed] [Google Scholar]
- [28].Chang Y, Finnemann SC. Tetraspanin CD81 is required for the alpha v beta5-integrin-dependent particle-binding step of RPE phagocytosis. J Cell Sci. 2007;120(Pt 17):3053–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goodman SL, Grote HJ, Wilm C. Matched rabbit monoclonal antibodies against alphav-series integrins reveal a novel alphavbeta3-LIBS epitope, and permit routine staining of archival paraffin samples of human tumors. Biol Open. 2012;1(4):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Linsley PS, Greenbaum CJ, Rosasco M, et al. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new-onset type 1 diabetes. Genes Immun. 2019;20(4):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Linsley PS, Greenbaum CJ, Speake C, et al. B lymphocyte alterations accompany abatacept resistance in new-onset type 1 diabetes. JCI Insight. 2019;4(4). DOI: 10.1172/jci.insight.126136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Linsley PS, Speake C, Whalen E, et al. Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS One. 2014;9(10):e109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy. 2008;4(3):354–356. [DOI] [PubMed] [Google Scholar]
- [34].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Koeneke E, Witt O, Oehme I. HDAC family members intertwined in the regulation of autophagy: a druggable vulnerability in aggressive tumor entities. Cells. 2015;4(2):135–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Phelan JD, Young RM, Webster DE, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560(7718):387–391. DOI: 10.1038/s41586-018-0290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barnes BJ, Richards J, Mancl M, et al. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. 2004;279(43):45194–45207. [DOI] [PubMed] [Google Scholar]
- [39].Akkaya M, Traba J, Roesler AS, et al. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat Immunol. 2018;19(8):871–884. DOI: 10.1038/s41590-018-0156-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Weisel FJ, Mullett SJ, Elsner RA, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol. 2020;21(3):331–342. DOI: 10.1038/s41590-020-0598-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Viglianti GA, Lau CM, Hanley TM, et al. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19(6):837–847. [DOI] [PubMed] [Google Scholar]
- [42].Faridi MH, Khan SQ, Zhao W, et al. Cd11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest. 2017;127(4):1271–1283. DOI: 10.1172/JCI88442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].O’Gorman WE, Hsieh EW, Savig ES, et al. Single-Cell systems-level analysis of human Toll-like receptor activation defines a chemokine signature in patients with systemic lupus erythematosus. J Allergy Clin Immunol. 2015;136(5):1326–1336. DOI: 10.1016/j.jaci.2015.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S a. 2008;105(36):13520–13525. DOI: 10.1073/pnas.0804295105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Young RM, Wu T, Schmitz R, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S a. 2015;112(44):13447–13454. DOI: 10.1073/pnas.1514944112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ciccacci C, Perricone C, Alessandri C, et al. Evaluation of ATG5 polymorphisms in Italian patients with systemic lupus erythematosus: contribution to disease susceptibility and clinical phenotypes. Lupus. 2018;27(9):1464–1469. DOI: 10.1177/0961203318776108 [DOI] [PubMed] [Google Scholar]
- [47].Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol. 2017;176:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–920. DOI: 10.1136/annrheumdis-2013-204343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tam JM, Reedy JL, Lukason DP, et al. Tetraspanin CD82 organizes Dectin-1 into signaling domains to mediate cellular responses to Candida albicans. J Immunol. 2019;202(11):3256–3266. DOI: 10.4049/jimmunol.1801384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bates M, Jones SA, Zhuang X. Stochastic optical reconstruction microscopy (STORM): a method for superresolution fluorescence imaging. Cold Spring Harb Protoc. 2013;2013(6):498–520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data associated with the paper is deposited in GEO (gene expression omnibus) database with accession number GSE209755.