

ABSTRACT
Innovative approaches in the design of T cell-engaging (TCE) molecules are ushering in a new wave of promising immunotherapies for the treatment of cancer. Their mechanism of action, which generates an in trans interaction to create a synthetic immune synapse, leads to complex and interconnected relationships between the exposure, efficacy, and toxicity of these drugs. Challenges thus arise when designing optimal clinical dose regimens for TCEs with narrow therapeutic windows, with a variety of dosing strategies being evaluated to mitigate key side effects such as cytokine release syndrome, neurotoxicity, and on-target off-tumor toxicities. This review evaluates the current approaches to dose optimization throughout the preclinical and clinical development of TCEs, along with perspectives for improvement of these strategies. Quantitative approaches used to aid the understanding of dose-exposure-response relationships are highlighted, along with opportunities to guide the rational design of next-generation TCE molecules, and optimize their dose regimens in patients.
KEYWORDS: T-cell engager, immune oncology, bispecific antibody, dose optimization, quantitative clinical pharmacology, translational PK/PD modeling, quantitative systems pharmacology
Introduction
Using the body’s immune system to recognize and kill cancer cells is an enticing prospect within oncology, showing great promise with several different therapeutic approaches. Among these are bispecific T-cell engagers (TCE), which can overcome some of the limitations of the body’s natural immune system by recruiting T cells to the tumor site and stimulating their activation and proliferation to help override the immune escape mechanisms developed by cancer cells.1 The potential of TCE therapy in oncology is beginning to be realized, with recent regulatory approvals in the US and EU for tebentafusp in uveal melanoma, for teclistamab in multiple myeloma, and for mosunetuzumab in follicular lymphoma, adding to the worldwide approval of blinatumomab in acute lymphoblastic leukemia, first accepted by the FDA in 2014. Emerging data from clinical trials of TCEs continue to provide insights on the efficacy and safety profiles of this class of therapy, and recent efforts have focused on optimizing clinical dose regimens to mitigate side effects related to overstimulation of the immune system, with particular emphasis on cytokine release syndrome (CRS) and neurotoxicity.2
For the currently approved TCEs, a key discovery was that by incrementally increasing the dose administered to a patient before reaching the target dose level, the body’s immune system could be primed in a more gradual manner, thereby modulating the balance between T cell activation and expansion, and cytokine-mediated efficacy and toxicity. This is known as step-up dosing, and various forms of this approach have been used to optimize TCE dose regimens to reduce the incidence or severity of CRS during clinical trials.3,4 However, care must be taken in considering this as a “one-size-fits-all” approach, since other mechanisms of immune-mediated toxicity may have different temporal exposure-response relationships. For example, localized inflammatory responses and subsequent damage to healthy tissues which also express the target antigen may not be merely “first-dose” phenomena, and thus may not be effectively mitigated by step-up dosing. Alternative strategies such as subcutaneous dosing have been shown to provide both slower and lower peak drug concentrations (Cmax), although it is still unclear whether some on-target off-tumor toxicities may be mediated by cumulative rather than acute drug exposure. Moreover, phase I studies for TCEs are often very protracted compared to a conventional biologic, stymying progress for the field and for patients. Therefore, a thorough quantitative characterization of the relationships between exposure, efficacy, and toxicity is essential to understand their relative contributions to the overall benefit-risk profile and enable more rapid progress to clinical proof of concept.
Retrospective analysis of the clinical dose-exposure-response relationships across different TCE molecules is complicated by their wide range of targets and molecular formats, which have differing impacts on the various facets of the exposure-response relationships for both efficacy and toxicity. Innovations in molecular design are continually emerging with the goal of improving the therapeutic index (TI) of TCEs and providing more convenient dose regimens for patients, but major challenges remain in solid tumor indications or for TCEs where the clinically evaluated dose regimens have not yielded an acceptable balance between efficacy and toxicity.5–8 A quantitative understanding of the impact of novel molecular design characteristics on the exposure-response relationships of TCEs, in combination with the underlying biological mechanisms of variability in response, is vital in guiding the development of future generations of these molecules.9 Quantitative modeling approaches can also help enable the rational evaluation of different dosing regimens during clinical studies, and better anticipate the efficacy and safety of TCEs in more diverse patient populations.
This review first examines the impact of molecular design characteristics on the pharmacokinetics (PK) and biodistribution of TCEs, along with the subsequent impact on exposure-response relationships. Current approaches to select optimal dose regimens within clinical studies of TCEs are then evaluated, and alternative strategies are considered. Throughout, it is highlighted where quantitative modeling and simulation approaches have been used at various stages of preclinical and clinical development to increase the understanding of exposure-response relationships, and to influence clinical dosing strategies. Finally, perspectives and challenges for the next generation of TCEs are outlined, with the intention of providing more effective and convenient therapies and dose regimens to improve the lives of patients with cancer.
Impact of molecular design on the pharmacokinetic and pharmacodynamic characteristics of TCEs
The first generation of bispecific T-cell engager (BiTE) molecules, including the anti-CD19 TCE blinatumomab, were composed of dual single-chain variable antibody fragments (scFv) targeting the CD3 component of the T-cell receptor (TCR), and a specific tumor associated antigen (TAA). By forming a molecular synapse between CD3 and the TAA, BiTE molecules were designed to mimic the naturally-occurring interactions between the TCR and specific tumor-derived peptides presented on the surface of tumor cells, leading to a cascade of T cell-mediated functions ultimately resulting in cancer cell death.10 As a consequence of their small size, BiTEs typically have systemic half-lives of a few hours, requiring frequent administration or even continuous intravenous (IV) infusion regimens in order to provide sufficient exposure for efficacy while minimizing acute peaks in concentration which potentially lead to toxicity. The desire to provide more convenient dose regimens for patients subsequently led to efforts focused on designing TCE molecules with longer systemic half-lives, thereby allowing less frequent administration. Additional protein engineering strategies were also carried out with the goal of improving the pharmacodynamic (PD) characteristics of TCE molecules, but the PK and biodistribution properties of TCEs may also potentially be modified as a consequence. This in turn can have an impact on the exposure of the TCE at the site of action and may thereby influence the appropriate dose regimen for patients. Careful consideration of all of these aspects must be taken when designing TCEs.
Molecular structure
The wealth of possibilities within the molecular design of bispecific antibodies has led to the development of TCEs ranging in size and structure, from antibody fragment-derived constructs of around 50 kDa to immunoglobulin-like molecules of 150 kDa and higher.11,12 Interestingly, it has been reported that molecular size and structural conformation of the CD3 and TAA binding domains can affect the cytotoxic potency of the synapse formed between tumor cells and T cells, depending on the location of the TAA binding epitope.13 Since the pharmacological activity of a drug depends on both the potency and the amount of drug which reaches the target site, it is important to understand the impact of molecular structure on both the PK and the biodistribution of TCEs to tumor cells, in order to fully elucidate their exposure-response relationships.14 Although smaller-sized molecules may have more rapid or extensive tumor penetration, this could be offset by a higher systemic clearance, and therefore the frequency of administration required to provide an optimal magnitude and duration of exposure, while minimizing resistance through T cell exhaustion, requires thorough characterization.
A variety of half-life extension strategies have been developed, including the incorporation of albumin-binding domains or fragment crystallizable regions (Fc) into the original scFv-derived construct,15 as well as the utilization of immunoglobulin (Ig)-like molecular formats. The particular Ig subtype (e.g., IgG1, IgG2, IgG4, IgM) selected to form the structural backbone may influence its PK through several potential mechanisms,16 including via interaction with the neonatal Fc receptor (FcRn). The impact of FcRn-mediated recycling on the systemic exposure of Fc-containing molecules is influenced by the affinity and pH dependency of binding of their Fc domain to FcRn within cellular endosomes. The binding affinity of TCEs to FcRn can be modified by engineering within the Fc domain, such as during heavy or light-chain heterodimerization, and by the introduction of amino acid mutations for humanization or for attenuation of Fc-mediated effector functions in an attempt to reduce off-target toxicity. Therefore, in addition to carrying out functional assays to evaluate the molecular stability and effector function as a result of antibody engineering, it is also important to determine whether modifications to the Fc region alter its FcRn binding kinetics, either by using in vitro binding assays, or in vivo using appropriate preclinical species such as transgenic FcRn mice. Transgenic FcRn mice have a similar abundance and distribution of FcRn compared to human, and could potentially enable a better prediction of human clearance via allometric scaling compared to non-transgenic mice, potentially allowing prediction of human PK prior to studies in nonhuman primates.17 In silico physiologically based PK (PBPK) models have been developed which include FcRn within tissue compartments,18 and, as such, could be used to predict the influence of modified FcRn binding on the time course of TCE exposure in both blood and tissues.
Modifications to the native IgG structure can also result in changes in the physicochemical properties of the engineered antibody in terms of overall thermal stability, hydrophobicity, charge, aggregation, cell-membrane interactions and nonspecific binding to cell-surface receptors.19 To evaluate the net effect of these processes on the PK and biodistribution of antibody-derived molecules, data from in vitro assays used to evaluate the physicochemical and nonspecific binding properties of the molecule can be integrated within a PBPK modeling framework, in order to quantitatively predict the time course of drug concentrations in blood and tissues.20
CD3-targeting domain
The discovery that different signaling thresholds are required for triggering cytotoxicity versus cytokine production by T cells in vitro led to the hypothesis that cytokine release may be “dispensible” for cytotoxic T cell activity of TCEs.21,22 Moreover, the natural pMHC/TCR interaction affinity is measured in the order of 1–100 µM as opposed to the low nM range for the early CD3-TCEs.23 Therefore, focus has shifted toward the development of novel anti-CD3 antibodies for TCEs, with optimized affinity for cytotoxic activity while aiming to reduce cytokine release in patients.24,25 Several molecules with lower affinity to CD3 than the first-generation TCEs are currently being evaluated in clinical studies, such as the anti-BCMA TCEs CC-93269 and ABBV-383/TNB-383B, the anti-CD19 TCE AZD0486/TNB-486, the anti-STEAP1 TCE AMG 509, and the anti-PSMA TCE AMG 340/TNB-585. The feasibility of this approach to reduce cytokine release and improve the tolerability of TCEs in patients, while still yielding robust clinical efficacy will be eagerly awaited.
Aside from the ultimate goal of reducing the incidence and severity of cytokine-mediated toxicity, modulation of CD3 binding affinity has also been found to modify the PK and biodistribution characteristics of some TCEs in preclinical species, affecting their relative distribution to the tumor versus to CD3+ T cell-rich lymphoid tissues in mouse models.26 Furthermore, a two- to three-fold higher serum exposure was observed for a weak CD3-affinity TCE compared to moderate CD3-affinity version of the same molecule, for both CD20- and BCMA-targeting TCEs in cynomolgus monkeys.27 The clinical impact of differing CD3-affinities on the exposure, efficacy and toxicity of TCEs will be challenging to evaluate via meta-analysis, not only due to the complex physiological interplay between cytokines, T cell regulation and tumor cell death, but also due to the variety in TAAs being targeted by the current crop of TCEs, as well as their differing binding affinities to each of their target antigens. Interestingly, two BCMA-targeting TCEs of the same structural format are being evaluated concurrently in clinical trials; one with high affinity to CD3 (REGN5458), and one with a 37-fold lower affinity to CD3 (REGN5459),28 which could allow differences in their clinical PK as a result of CD3 affinity, as well as their efficacy and toxicity profiles to be determined.
Tumor-targeting domain
The selection of an appropriate TAA and the affinity of the associated target binding domain(s) within the TCE can have an impact on the PK and biodistribution of the molecule, particularly at doses which result in low drug concentrations in comparison with the expression levels of the target. It is also important to quantify the contribution of the target-mediated components of distribution and clearance, since heterogeneity in target expression across species and patients could significantly influence interspecies translation, as well as the inter-individual variability in systemic exposure of the TCE within a patient population. For immune agonists such as TCEs, pharmacological activity can occur at lower receptor occupancy compared to receptor antagonists, and consequently low doses of drug may be administered relative to the levels of both target antigens. Therefore, the physiological distribution and expression levels of the TAA and CD3 within the body, along with the binding affinity of the drug to each of these targets may have a vital impact on its PK and distribution.29 The influence of target-mediated disposition on the PK of the molecule can be quantitatively characterized using model-based approaches, provided that sufficient data concerning the tumor target abundance, turnover rate, and binding kinetics to the TCE are available.30 If the target antigen is also expressed in healthy tissues, this can further impact the biodistribution and PK of the TCE, as well as potentially resulting in on-target off-tumor toxicity.31 Furthermore, target shedding from either tumor cells or healthy cells may also act as a soluble sink for the TCE, and therefore could have a detrimental impact on drug exposure in blood and at the tumor site.32,33 One strategy which may increase the selectivity of TCEs to tumor cells expressing high levels of TAA, as opposed to healthy tissues or soluble shed antigen, is to increase the valency of the TCE binding domains to the tumor antigen, thus prioritizing avidity-driven binding over monovalent binding.34 This could potentially result in higher potency along with greater efficacy at lower doses compared to monovalently binding TCEs, but could also increase the contribution of target-mediated clearance to the overall clearance of the molecule at these lower doses. Consequently, since target antigen loss has emerged as a potential resistance mechanism for some immunotherapies, depletion of the TAA-expressing tumor cells over time may also impact the time course of any target-mediated clearance of the TCE, and could thus give rise to time-dependent PK.35 The complex interplay between the molecule and its target antigens on T cells, tumor cells, healthy cells, and shed antigen can be quantified using in silico modeling approaches,36 which could be of value when predicting the extent and time course of systemic drug exposure and response of TCEs across the range of doses to be evaluated in clinical studies. This will be discussed in subsequent sections of this review.
Immunogenicity
Immunogenicity can affect the exposure of therapeutic proteins, which can potentially have a detrimental impact on their efficacy, as well as possible safety issues associated with antibody cross-linking. This can be a critical issue for TCEs as the doses achieved clinically are often too low to enable “dosing through” the anti-drug antibodies (ADA). The likelihood of immunogenicity is further exacerbated for TCEs as a result of direct CD4+ T cell activation supporting a humoral response.37 Additionally, the more highly engineered nature of bispecific antibodies compared to monoclonal antibodies can lead to a wide range of ADA incidence.38 The impact of modifications to the natural structure of antibody-derived molecules on the potential for immunogenicity should be evaluated prior to clinical studies, where possible. An integrated approach to characterize the ADA of bispecific antibodies has been previously reported using in silico, in vitro and in vivo studies to determine which binding epitopes or components of the molecule were the main contributors to the immunogenic response.39 Immunogenicity of TCEs is often apparent in preclinical in vivo PK studies as a reduction in free drug concentrations due to the formation of ADA-TCE complexes, and can thus pose challenges inunderstanding preclinical exposure-response relationships. This was addressed for the CD123-targeting Dual-Affinity Re-Targeting (DART) molecule flotetuzumab by using a PK/PD modeling approach to characterize the impact of ADA on flotetuzumab-mediated T cell trafficking and depletion of CD123-expressing cells in non-human primates, with the aim of proposing different clinical dosing strategies via model simulations.40 Translation of this type of PK/PD model to humans could allow the simulation of the potential impact of ADA on predicted exposure-response relationships in patients, although assumptions regarding different theoretical incidence of ADA in patients would be required, since this currently cannot be predicted accurately from preclinical species.
Overall, it is becoming increasingly clear that modifications to the molecular design of TCEs must be evaluated not just in terms of their impact on efficacy or toxicity, but also their impact on PK, biodistribution and immunogenicity of TCEs, which may in turn affect their exposure and thus clinical dose regimens. All of the above elements are interconnected within dose-exposure-response relationships in patients, and will be important to consider when optimizing dosing strategies throughout the clinical development of TCEs.
Improving dose selection and anticipating therapeutic index prior to clinical studies
The overproduction of cytokines is a common consequence of immune agonism by therapeutic agents such as TCEs, and can result in potentially severe clinical side effects such as CRS which may arise via the complex interactions between the body’s immune cells following their stimulation, proliferation, and activation by TCE molecules.41 Given the difficulty in predicting when and in which individuals CRS will occur, conservative approaches have generally been used for the selection of first-in-human (FIH) starting doses for TCEs.42 However, this has often resulted in lengthy dose escalation phases, with large differences between the starting dose and the recommended phase 2 dose (RP2D). For example, the approved dose of the anti-CD20 TCE mosunetuzumab was over 1000-fold higher than the starting dose.43 This is in contrast to the starting dose approach for chimeric antigen receptor (CAR)-T therapies which have a similar mechanism of action and toxicity profile, but generally require very few dose escalations to reach the clinically efficacious dose.44 To try to compensate for this discrepancy, single patient cohorts are commonly used in the early phase of dose escalation for TCEs, often with criteria based on emerging toxicity signals such as grade 2 CRS to inform the switch to multiple-patient cohorts. However, insufficient knowledge regarding predictors of the incidence and severity of CRS could mean that such criteria may trigger the switch from single- to multiple-patient cohorts either prematurely, or belatedly. CRS-based criteria also do not allow for the possibility that other on-target off-tumor toxicities may be more critical to tolerability than CRS, so other toxicity-guided criteria such as any grade 2 adverse event have sometimes been used as an alternative. Patients who are treated at sub-efficacious dose levels are often subsequently permitted to receive higher dose levels as the dose escalation phase progresses; however, the duration of time required to reach efficacious dose levels may be too long for individuals in late-line settings whose disease has progressed or who have dropped out of the trial. A better understanding of the interplay between dose, exposure, efficacy, and toxicity of TCEs in patients could thus allow efficacious doses to be achieved earlier in patients, while maximizing tolerability. This understanding can be aided by using a variety of mechanistic modeling approaches to characterize and predict exposure-response relationships for TCEs (Table 1).
Table 1.
Examples of quantitative PK/PD modeling approaches used to characterize dose-exposure-response relationships of TCEs.
| Area of focus | Modeling approach | Drug | Reference |
|---|---|---|---|
| Molecular structure and target binding | QSP model including multiple binding domains | Trispecific CD3-CD28-CD38 TCE | 114 |
| QSP model accounting for differences in molecular synapse properties and epitope binding on cytotoxic potency | BCMA-CD3 diabody-Fc and IgG TCEs | 13 | |
| PBPK model to predict biodistribution and immune synapse formation for a variety of molecular formats of TCEs in tumor and lymphoid tissues | PF-06863135 and AMG 420 | 45 | |
| Starting dose prediction | Model-based characterization of in vitro data used to inform starting dose selection | Cibisatamab | 52 |
| Starting dose prediction via in vitro to in vivo translation of trimer concentration | P-Cadherin LP DART | 51 | |
| Translational PK/PD modeling of in vivo cytokine release in monkey with in vitro potency correction, to predict starting dose | Glofitamab | 54 | |
| Exposure-efficacy relationships | Translational prediction of efficacious dose range using trimer concentration to drive preclinical and clinical efficacy | P-Cadherin LP DART Solitamab Cibisatamab Blinatumomab, AMG 330, anti-FLT3 TCE, anti-CD20 TCE |
64 65 62 48 |
| Model-based evaluation of impact of immunogenicity on preclinical exposure-response | Flotetuzumab | 40 | |
| Exposure-response model to evaluate appropriate clinical dose regimen and predict receptor occupancy | Mosunetuzumab Glofitamab |
46 56 |
|
| Semi-mechanistic PK/PD model to evaluate appropriate clinical dose regimen and predict saturation of trimer concentrations and clinical response | Epcoritamab | 65 | |
| QSP model to elucidate the dose-exposure-efficacy relationship and identify potential PD biomarkers of clinical response | Cibisatamab | 76 | |
| QSP model to mechanistically identify contribution of components to clinical response within combination studies | Cibisatamab + atezolizamab | 103 | |
| PK/PD of cytokine release | Semi-mechanistic PK/PD model to evaluate the impact of tumor target dynamics on cytokine release | Blinatumomab, P-Cadherin LP DART | 80 |
| QSP model to evaluate the impact of different dose regimens on cytokine release | Mosunetuzumab, Blinatumomab Tebentafusp |
79, 111 | |
| Minimal PBPK/PD model to characterize target cell depletion and cytokine release under various physiological conditions, such as E:T ratio | Blinatumomab | 67 | |
| PBPK model to predict cytokine-mediated DDI potential | Blinatumomab | 109 |
DDI: drug-drug interaction, E:T ratio: effector-to-tumor ratio, PBPK: physiologically based pharmacokinetic, PD: pharmacodynamic, QSP: quantitative systems pharmacology.
Starting dose selection
The minimum anticipated biological effect level (MABEL) approach has traditionally been used to select the starting dose in FIH studies of CD3-targeting immune agonists.42 This conservative approach to dose selection is considered to be more appropriate than a receptor occupancy (RO)-based method for drugs with this mechanism of action, since significant clinical activity of many TCEs can occur even at low RO. For example, clinical activity of mosunetuzumab occurs at dose levels corresponding to 0.2 to 10% receptor occupancy of CD20.46 Among TCEs using the MABEL approach to select the starting dose, a range of values for “minimal biological activity” have been reported, determined from a variety of different assays and biomarkers. Typically evaluated in vitro assays include cytotoxicity assays, T cell activation and proliferation assays, and cytokine release assays. For the early TCEs, the most sensitive assay was often used to select the FIH dose, with the concentration corresponding to 20% of the maximal effect (EC20) commonly considered to represent minimal biological activity. Based on the continually evolving clinical experience with TCEs, this approach could now be considered too conservative given the large orders of magnitude often encountered between starting dose levels and the recommended phase 2 dose (RP2D).
Using potency measured in in vitro assays as a benchmark for minimal biological activity of TCEs is prone to potentially high levels of uncertainty, since experimental assay conditions can differ substantially from conditions in the in vivo environment. This can in turn have an influence on the starting dose selected using in vitro drug potency within the MABEL approach. For example, the growth rate of tumor cells cultured in vitro is higher than observed clinically, and the ratio between the number of effector T cells and tumor cells (E:T ratio) used in in vitro cytotoxicity and cytokine release assays is often substantially higher than under physiological conditions in vivo, which could lead to an overestimation of biological activity. The interplay between the various factors leading to experimental uncertainty can also confound the interpretation of the data generated, as demonstrated by in vitro cytotoxicity data for blinatumomab determined at different E:T ratios appearing to result in differing levels of maximum cell killing after a 24-h incubation.47 A mechanistic model-based analysis of these data deduced that the major impact of E:T was on the rate of cell killing, rather than on the maximum cytotoxic effect (Emax) or the EC50, and thus the duration of the incubation could affect the apparent Emax value obtained.48 For other TCEs, it has been shown that varying the E:T ratio resulted in different experimentally determined values of EC50.49–51 However, if a single time point is used to estimate in vitro potency, the particular time point chosen can also highly influence the apparent EC50 value. This was shown for the anti-CEA TCE cibisatamab, where the apparent EC50 of a variety of biological endpoints varied substantially depending on the time point selected to measure the effect. It was then demonstrated that a more robust determination of EC50 could be obtained when taking into account multiple time points by using the area under the effect curve to derive a time-independent measure of potency.52 Other factors such as the inter-individual variability across peripheral blood mononuclear cell (PBMC) donors can also have an impact on the potency values determined using these assays. The above analysis for cibisatamab showed that the time-independent approach actually gave a similar EC50 value compared to single time point approach as long as the most appropriate time point was chosen, and the authors proposed a pragmatic workflow in which the time course of biological activity could initially be used to determine potency using relatively few PBMC donors, and then the most relevant time point could be selected and EC50 determined with a greater number of PBMC donors in order to evaluate inter-donor variability.
It has also been demonstrated that a more rational selection and interpretation of data could affect the choice of clinical starting dose. As for many of the early TCEs, the starting dose of cibisatamab in patients was selected using the EC20 in the most sensitive cytotoxicity assay as the minimal biological activity.53 However, this starting dose was substantially lower than the clinically active dose range, and 7700-fold lower than the maximum tolerated dose (MTD) of 400 mg. The authors considered that cytokine release measured in a more disease-relevant tumor cell line, rather than cytotoxicity measured in a hCEA-transfected cell line was the more appropriate surrogate biomarker for toxicity in patients at early dose levels. Additionally, a biological activity threshold of 30% rather than 20% was used, based on insights from Saber et al.42 According to this retrospective analysis, a starting dose of 450 μg could feasibly have been proposed for cibisatamab, which was ninefold higher than the actual starting dose of 52 µg used in the FIH study. This underlines the importance of a rational and robust approach to the determination of the relevant biological activity in vitro, and will be especially important for next-generation TCEs designed to improve TI by generating lower levels of cytokines, and for which the “most sensitive assay” may not be the most clinically relevant assay.
In addition to considering the relevant in vitro and in vivo pharmacodynamic data with which to select the starting dose, elucidation of the most appropriate exposure metric with which to drive the predicted biological activity in patients is also vital. It is generally not the concentrations of the TCE itself that drive pharmacological activity, since cytotoxicity should theoretically only occur upon dual binding of the drug to CD3 on T cells and to the tumor antigen to form a CD3-TAA-TCE ternary complex, or trimer. As shown in Figure 1, saturation of binding to one or both of the targets within the tumor environment can result in a nonlinear relationship between dose and trimer exposure, which can thereby give rise to a nonlinear dose-response curve, even when the serum exposure of TCE appears to be linear with respect to dose. This could ultimately result in a “bell-shaped” relationship for trimer formation where the concentration of TCE exceeds the concentration of available receptors, which favors monovalent binding and the formation of dimers between the TCE and its respective targets. A consequent decrease in response can frequently be observed in vitro since TCEs generally cannot elicit pharmacological activity in their dimeric forms, although this is less commonly observed in vivo since the TCE concentrations required to exceed maximal trimer formation are rarely reached due to limited tolerability at this dose range. For a given concentration of TCE, the extent of trimer formation in vitro may also exceed the level of trimer formed at the same concentration of TCE in vivo, due to differences in E:T ratio. Therefore, relating the trimer concentration, rather than the TCE concentration, to biological activity may provide a more appropriate means of scaling preclinical in vitro and in vivo data to human.36 This approach was carried out for P-Cadherin LP-DART using a mechanistic in vitro PK/PD model of trimer concentrations to drive tumor cell killing and T cell proliferation.51 Once characterized in vitro, simulation of trimer concentrations in human was achieved by predicting the human PK and distribution of the TCE to the tumor, and the subsequent formation of trimer in the tumor microenvironment as a function of the intra-tumoral TCE, CD3 and TAA levels. Thus, the human dose giving a tumor trimer exposure equal to the estimated EC20 of trimer in the chosen in vitro assay was determined as the MABEL.
Figure 1.
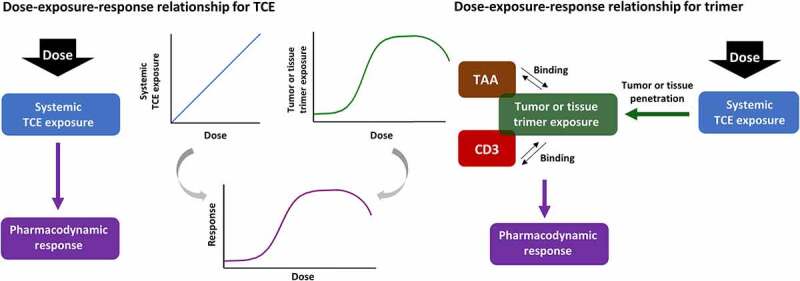
Schematic representation of the relationship between dose, exposure and pharmacodynamic response for the TCE, and for the trimeric complex (“trimer”) formed upon dual target engagement of TCE with CD3 and TAA.
While the above approach used predicted exposure of the trimer in tumor cells to drive the biological activity, it may be possible to evaluate some PD biomarkers such as cytokines in vivo using toxicology studies in species which express the target on healthy cells, even in the absence of tumor. As an alternative to an in vitro-based MABEL approach, PK/PD modeling of in vivo data was carried out to select the starting dose for the anti-CD20 TCE glofitamab.54 Cytokine release resulting from serum exposure of glofitamab and B cell levels in the blood was modeled in the monkey, and translation of the relevant model parameters allowed the prediction of systemic cytokine levels in patients for different doses of glofitamab, assuming a range of B cell counts in patients. Furthermore, this approach combined both in vivo and in vitro knowledge, by correcting for the 70-fold higher observed in vitro potency for cytokine release in human versus monkey whole blood. Such holistic translational modeling strategies, which integrate preclinical data from various sources, could provide a better overall prediction of safe starting dose ranges for TCEs by accounting for multiple underlying mechanisms of toxicity. However, cytokine release resulting in CRS may entail several components: binding of the TCE to CD3 in the absence of TAA cross-linking, and TAA-mediated cross-linking with CD3 which can occur in both tumor cells and healthy tissue cells expressing the TAA. Both the off-target and the on-target off-tumor toxicity can be evaluated in cross-reactive species which express the TAA in healthy tissues, but the on-target on-tumor toxicity is not accounted for in commonly used toxicology species such as non-human primates. Consequently, an additional 10-fold safety factor was applied to the model-determined starting dose of glofitamab to compensate for potential cytokine release from tumor which was not included in the translational PK/PD model. Nevertheless, the final starting dose was still ~30-fold higher than the dose that would have been selected using an in vitro-based MABEL approach. Although the PD was driven by glofitamab concentrations and B cells in peripheral blood, for other TCEs it is possible that trimer exposure within healthy tissues expressing the TAA could be more relevant to toxicity, thus the prediction of trimer concentrations in these tissues as well as in tumor could be a more appropriate approach for interspecies translation, albeit with the significant challenge of obtaining accurate values for TAA and CD3 levels within healthy tissues in both preclinical species and in patients. Humanized immunocompetent tumor-bearing mouse models may also be of value since they could allow the simultaneous evaluation of toxicity and efficacy, provided that the expression levels and function of both immune cells and TAA within tumor and healthy tissues is similar enough to that in patients, or can be scaled appropriately.
The above examples demonstrate that a more rational and mechanistic selection of starting dose for TCEs could be achieved using appropriate exposure metrics and relevant assays for biological activity, as well as by quantitatively accounting for differences in experimental conditions or preclinical species versus the human in vivo environment. This may ultimately provide confidence in selecting starting doses which are both tolerable and closer to the efficacious dose range in patients, allowing more patients to receive doses that are likely to provide clinical benefit. Another key component of this approach is therefore to predict the potentially efficacious dose range in patients, and to anticipate whether it can be achieved with acceptable levels of tolerability.
Anticipating the efficacious dose range and therapeutic index of TCEs in patients
For some immunotherapeutic molecules such as immune checkpoint inhibitors, their mechanism of action relies upon a sufficient extent of saturation of the target antigen, and thus the clinical activity of these molecules can often be correlated to receptor occupancy.55 However, for immune agonists such as TCEs, the complex pharmacological cascade downstream of dual target engagement means that RO alone cannot reliably be used to prospectively determine the clinically efficacious dose range. This was illustrated during an exposure-response analysis for glofitamab, for which an objective response rate (ORR) of 54% in the pivotal phase 2 study corresponded to an average CD20 RO of less than 1%.56 The correlation between RO and efficacy can vary substantially among different TCEs, depending on the potency of the immune synapse formed, the abundance and heterogeneity of receptor expression on tumor cells, and the nature and magnitude of the downstream mediators of cytotoxicity. Thus, determining the concentration of TCE required to obtain a desired level of pharmacological activity, as opposed to a certain receptor occupancy, may be a more appropriate approach to predict clinically active doses of TCEs. This approach was carried out for the anti-BCMA TCE teclistamab during dose escalation, by comparing the observed serum concentrations of teclistamab in patients with ex vivo potency measured in bone marrow mononuclear cells derived from MM patients, co-incubated with human PBMCs obtained from a range of donors.57 For the dose route and regimen selected as the RP2D, the mean steady-state serum concentrations in patients were shown to be maintained above the maximum ex vivo EC90 value across the different PBMC donors.58 Using a similar approach, the mean steady-state serum concentrations of the anti-GPRC5D TCE talquetamab were also shown to exceed the ex vivo EC90 in patient-derived multiple myeloma (MM) cells co-incubated with PBMCs.59 Since in vitro potency is generally a static value, i.e., measured in the presence of non-fluctuating drug concentrations after a pre-determined incubation duration, as well as using artificially high E:T ratios and with drug readily accessible to both target cells, these assays may not necessarily provide potency values that are reflective of the in vivo environment at the target site, particularly when compared with serum concentrations of drug in patients. Therefore, the in vivo exposure at efficacious doses in preclinical pharmacology models was used as an alternative approach to anticipate the clinically active dose range during the dose escalation of the anti-CD20 TCE odronextamab.60 Serum concentrations of odronextamab in patients were compared with the maximum and trough concentrations measured at the efficacious dose in a CD20-expressing mouse xenograft tumor model. A subsequent retrospective analysis of the translational value of these data found that the exposure corresponding to 100% inhibition of tumor growth in the mouse model was able to give a good agreement with the clinically efficacious exposure of odronextamab.61
Although these examples provided useful indicators of the therapeutic dose ranges for some TCEs, these approaches are prone to the same limitations and assumptions as those used to determine the starting dose, in terms of the differences in physiological conditions between the human tumor environment and the preclinical systems, so care must be taken when applying these approaches to other TCEs. The influence of parameters such as E:T ratio on the in vitro cytotoxic potency for a range of different TCE molecules was quantitatively evaluated using a mechanistic modeling framework, by characterizing the potency as a measure of the predicted trimer concentrations driving in vitro pharmacological activity across a range of E:T ratios and experimental conditions.48 The analysis also demonstrated that by using the trimer potency and by translating the relevant physiological model parameters to their relevant values in patients, the observed clinical exposure-response relationship of blinatumomab was able to be predicted by the model. The incorporation of additional biomarkers relevant to the pharmacological pathway within a mechanistic modeling framework could help to elucidate the key mechanisms driving PD under different experimental conditions, and thus further improve the prediction of clinical activity in human, while also guiding the selection of potential clinical biomarkers. For example, by including activated T cells as an intermediate biomarker between trimer formation and cell killing within the mechanistic in vitro model for cibisatamab, the variability in cytotoxicity across cell lines expressing different levels of the TAA was able to be accounted for via differences in T cell activation downstream of target engagement and trimer formation.62
For TCEs in poorly penetrable tumors, the correlation between in vitro potency and the clinically efficacious concentration range may be further complicated by differences in drug exposure within the tumor versus in the bloodstream, due to the complex interplay between extravasation, diffusion within the tumor interstitial fluid, and interactions with the target antigens within the tumor microenvironment.63 Therefore, physiologically based translational modeling approaches may provide a more accurate prediction of the PK/PD relationship of the drug-target complex at the site of action. An example of this type of physiologically based mechanistic modeling approach was reported for P-Cadherin LP-DART, in which the tumor concentrations of trimer were predicted at the efficacious dose range in mouse xenograft models, and the physiological model parameters were then scaled to their equivalent human values in order to predict the dose which would result in the same tumor trimer exposure in patients.64 Translational physiologically based models such as these could also potentially allow the simulation of various scenarios relating to heterogeneity in the human tumor environment, such as tumor burden and immune cell enrichment.
Predicting the efficacious dose range is only part of the challenge in anticipating the clinical therapeutic window. In a similar model-based analysis to those described above, the trimer concentration required for efficient in vitro cell killing by the anti-Epcam BiTE solitomab was determined to be very close to the predicted trimer exposure at the observed MTD in the FIH clinical study.65 As such, it is important to anticipate the clinical dose range which is expected to result in significant toxicity, in order to predict the TI. Preclinical-to-clinical translation of toxicity is a major challenge for TCEs,66 particularly given the apparent discrepancy between the maximum tolerated dose in human versus in animals reported by Saber et al, albeit across TCEs with a variety of targets and molecular formats.42 The steep and nonlinear dose-toxicity relationship often observed for TCEs is challenging to anticipate prior to clinical studies, due to interspecies differences in the relative expression levels and tissue distribution of the target antigen, as well as heterogeneity across patient populations due to disease-related factors or prior treatments. In order to characterize and translate preclinical exposure-toxicity relationships to patients, PBPK models may be of value since they allow the prediction of the time course of drug concentrations in different locations within the body, and can be customized to include target-mediated interactions via incorporation of the tissue-located target antigens within the relevant model compartments. However, it is currently challenging to quantitatively determine the abundance and turnover of the target antigen on the cell surface of these tissues in both preclinical species and in patients, which will be a key prerequisite for the prediction of on-target off-tumor toxicity of TCEs. Additionally, tissue toxicity may not necessarily be solely due to on-target off-tumor binding to the target antigen, and nonspecific binding to other receptors present within tissues could also trigger undesired T cell-mediated local inflammatory responses in the presence of T cells. Evaluating both on- and off-target toxicity prior to clinical studies will be essential to provide an overall assessment of the expected safety profile in patients. Since the mechanisms behind the pharmacological activity and toxicity of TCEs are inherently linked, obtaining the desired balance between efficacy and tolerability may be finely poised. Therefore, mechanistic modeling approaches that incorporate the impact of the pharmacological mediators of TCE activity on both efficacy and toxicity within the same model framework could be of value in predicting the therapeutic window in patients. For example, a quantitative systems pharmacology (QSP) model was developed in order to characterize the effect of blinatumomab on both target B cell depletion and cytokine release in patients, thus providing a holistic view of the overall exposure-response relationship with respect to both efficacy and toxicity.67 Models such as these could indicate whetherthe TI in human is predicted to be narrow, and thus help guide the design of appropriate dose regimens within the FIH study in order to maximize the benefit-risk relationship.
Optimization of efficacy and safety during the clinical development of TCEs
The complex interplay between the pharmacodynamic mediators of both the efficacy and toxicity of TCEs is a key challenge in the selection of an optimal dose regimen. As one of the key cytokine-induced side effects, CRS is generally managed in the clinical setting by the administration of corticosteroids such as dexamethasone, along with antihistamines and antipyretics, and prophylactic use of these agents has been incorporated into the trial design of many TCEs. Targeted CRS intervention strategies such as the interleukin (IL)-6 receptor antagonist tocilizumab have also been used to inhibit the activity of key individual cytokines which are involved in the inflammatory pathways of CRS.68 There is some debate as to whether suppression of the immune response via corticosteroid administration negatively impacts the efficacy of immune stimulatory therapies,69,70 therefore it is also desirable to evaluate alternative clinical dosing strategies for TCEs in order to find the optimal regimen to reduce the incidence and severity of cytokine-mediated toxicity.
Optimization of dose regimen to reduce CRS
During the clinical development of blinatumomab, it was discovered that an initial dose regimen of 5 µg/m2/day for the first 7 days followed by 15 µg/m2/day resulted in lower levels of cytokine secretion compared to in patients receiving a fixed dose regimen of 15 µg/m2/day.71 This gave rise to the hypothesis that step-up dosing could potentially be used to reduce the incidence and severity of CRS for other TCEs, a concept which was further supported by a 36% increase in the RP2D of the gp100-targeting TCE tebentafusp when using a 3-week step-up dosing regimen compared with the MTD obtained using a fixed weekly dosing schedule.72 A pharmacodynamic analysis for mosunetuzumab revealed that cytokine induction was primarily associated with the first administration, with no further increase in IL-6 levels at the higher doses subsequently administered within the treatment cycle.73 Therefore as depicted in Figure 2, initial doses which are lower than the target dose may be administered within the first treatment cycle in order to provide a stepwise increase in peak drug concentrations when compared to fixed dosing schedules, thus “priming” the immune system more gradually in order to prevent an early, uncontrolled inflammatory response which could lead to CRS or other immune-related toxicities. Fractionated step-up dosing (Figure 2c) may also be implemented to provide a similar overall first-cycle exposure of drug compared to the exposure at the target dose regimen, thus compensating for the lower exposure of each individual step-up dose.
Figure 2.
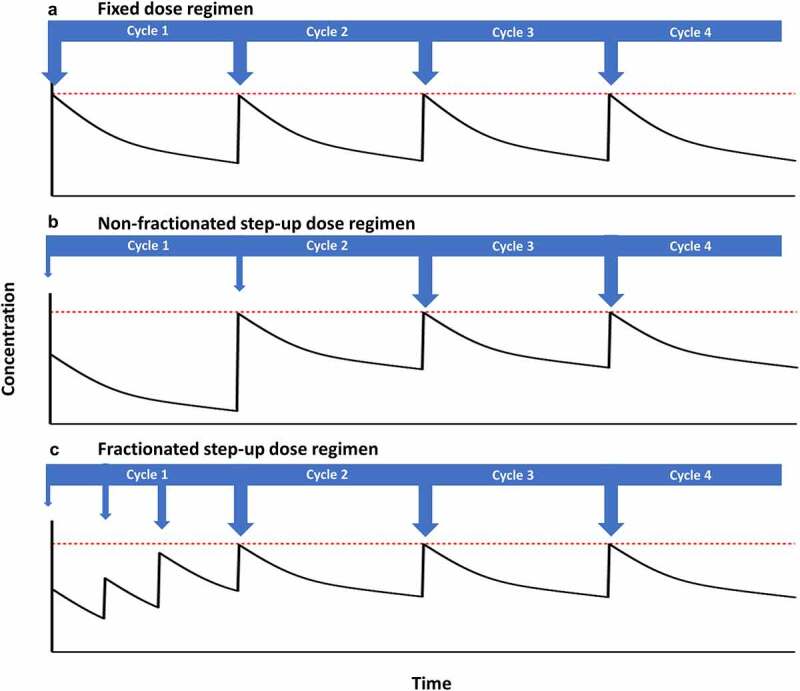
Schematic representation of the impact of fixed and step-up dose regimens on drug concentration versus time profiles (solid black lines). Dose level and frequency is indicated by arrows, and the maximum desired peak concentration is indicated by the dashed red lines.
The diverse array of step-up dose regimens that have been evaluated for TCEs raises the question of how to determine the appropriate dose intensity and frequency of the priming doses. For some molecules such as the anti-CD20 TCE epcoritamab and the anti-MUC16 TCE ubamatamab, step-up dose regimens were implemented from the initial dose escalation cohort, with the step dose being a “reasonable” multiple of the target starting dose (e.g. one-third), and the step dose for subsequent cohorts being equal to the target dose for the preceding cohort. As target dose levels increased during the escalation phase, the step dose was then fixed to a level with acceptable tolerability. To “bridge the widening gap” between the step dose and the escalating target dose, an additional intermediate step dose may then be introduced.74 The a priori selection of an appropriate frequency and intensity of step-up dosing is not trivial, and may depend upon a variety of factors relating to the molecule, the target, and the nature and severity of disease. In view of this uncertainty, four different step-up dose regimens along with a fixed dosing regimen were evaluated within the FIH trial of the anti-CD123 TCE vibecotamab,75 whereas the dose escalation phase for the anti-CD20 TCE plamotamab FIH trial included a three-part design, with the goal of establishing appropriate levels for the initial priming doses in subsequent parts of the study.76 However, the evaluation of different step-up dose regimens during dose escalation could pose significant and potentially unnecessary challenges in the interpretation of the dose-response relationship, particularly if dose levels early in the escalation phase are not expected to result in severe CRS, based on translational modeling analyses discussed in the previous section. Instead, step-up dosing regimens may be triggered based upon certain toxicity-related criteria, such as for the anti-BCMA TCE teclistamab where step-up dosing was initiated following the first incidence of grade ≥2 CRS.77
As clinical development progresses, population PK/PD and exposure-response modeling can help to determine appropriate step-up and target dose regimens for further evaluation in pivotal trials and in additional patient populations, as demonstrated for mounetuzumab, glofitinib, and blinatumomab.46,56,78 These empirical model analyses can provide key insights during clinical development, but since they rely on using observed data from patients, they can only be developed once sufficient clinical data is available. Translational PK/PD modeling could allow earlier insights into when and how to implement step-up dosing, using predicted exposure-response relationships based on preclinical data. For example, a semi-physiological PK/PD model was developed to characterize the trafficking of CD8 + T cells and CD19- and CD20-expressing B cells between the blood and lymphoid tissues in monkeys and humans, and to simulate the impact of the immune cell dynamics on the magnitude and time course of IL-6 dynamics (i.e. safety), and target cell depletion (i.e. efficacy).79 Simulations using models such as these can aid in determining optimal step-up dose regimens, which could provide the desired balance between safety and efficacy for TCEs. The time course of TCE-induced cytokine production and disposition may differ in solid tumors versus hematologic malignancies, due to differences in T cell trafficking and cancer cell dynamics within the tumor microenvironment. The impact of these differences on cytokine levels within the bloodstream can be evaluated using mechanistic modeling approaches which account for temporal changes in TAA levels due to differences in the dynamics of tumor burden. For example, a model developed to capture the observed clinical cytokine data for blinatumomab administered at fixed versus step-up dose regimens was then modified to characterize the cytokine release of P-cadherin LP DART in monkeys, via incorporation of the relevant dynamics of target antigen turnover in P-cadherin-expressing tissues as a “surrogate” for cytokine release subsequent to TCE binding in solid tumors.80 However, CRS may not be the only immune-related toxicity of concern for some TCEs, and tissue damage may occur due to localized cytokine release and cytolytic activity within healthy tissues which also express the target antigen. To account for this, the anti-PSMA TCE HPN424 and the anti-mesothelin TCE HPN536 incorporated the criteria of grade ≥2 drug-related toxicity, rather than CRS-related criteria, to trigger step-up dosing cohorts in parallel to fixed dosing cohorts during dose escalation. It must be noted that an assumption inherent to the step-up dosing approach is that the relevant toxicity is an initial-dose phenomenon, which may not always be the case. For example, the SSTR2-targeting TCE tidutamab included step-up dosing during the dose escalation phase of the FIH study, but gastrointestinal toxicity was nevertheless considered to be dose-limiting by the fourth dose level, and ultimately development of tidutamab was terminated.81 In such cases, it may be beneficial to evaluate alternative administration strategies instead of, or in addition to step-up dosing, in search of the optimal therapeutic window.
Route of administration
As well as providing a more convenient dosing regimen for patients, subcutaneous administration has been evaluated in the FIH studies of several TCEs with the aim of achieving higher drug exposure while minimizing Cmax-driven adverse events such as CRS and neurotoxicity. Subcutaneous administration generally results in a lower peak-to-trough ratio of drug concentrations, as well as a more gradual release of drug into the systemic circulation. This was shown to markedly reduce the incidence of grade 2 and grade 3 CRS for mosunetuzumab, while achieving lower Cmax and higher trough concentrations at the target subcutaneous dose regimen compared to the previously evaluated IV dose regimen.82 Subcutaneous administration has been incorporated within the FIH dose escalation phases of FIH trials following initial evaluation of IV administration for some TCEs such as teclistamab and talquetamab, resulting in higher dose levels being achieved compared to the maximum IV dose levels evaluated.83,84 Subcutaneous dosing may also provide the benefit of reducing the frequency of administration, since trough concentrations may be maintained over a longer duration compared to an IV dosing regimen. As such, model-based simulations of subcutaneous dose regimens based on clinical PK data obtained after IV administration for some TCEs such as plamotamab have suggested that subcutaneous administration with less frequent dosing compared to the IV regimen would allow trough concentrations to be maintained, while potentially reducing CRS via lower Cmax levels.76 It will be of interest to evaluate the impact that a lower peak and longer time course of TCE concentrations, as well as less frequent dosing may have on efficacy and on resistance mechanisms such as T cell exhaustion in both solid and hematological cancers.
The success of subcutaneous dose regimens for TCEs is also likely to depend on achieving an appropriately high bioavailability, in order to provide higher systemic exposures and limit localized sequestration of the molecule within the site of administration. For example, the reported bioavailability of mosunetuzumab in patients was over 80%.82 But since bioavailability can vary across therapeutic proteins, it cannot necessarily be assumed that subcutaneous administration will give a similar exposure to IV administration for a given dose. In the FIH study for teclistamab, the subcutaneous dosing rationale was informed by modeling the early clinical IV PK data, and simulating the expected PK assuming a bioavailability of 60% after subcutaneous administration.85 Simulations using this type of modeling approach can guide dose selection based on the expected reduction in peak-to-trough concentration ratio after subcutaneous compared to IV dosing, assuming “plausible” or “worst-case” scenarios for bioavailability. Alternatively, some TCEs such as epcoritamab have been administered subcutaneously in FIH studies without IV dosing ever being evaluated in patients. Although it is currently challenging to accurately predict the human bioavailability of biotherapeutics during preclinical development, initiatives to improve prediction tools such as translational PBPK models may ultimately allow the prospective simulation of the PK of TCEs in patients in order to guide the choice of dose route in the FIH study.86,87
A note of caution is often raised on whether subcutaneously administered therapeutic proteins carry a greater risk of immunogenicity.88 For TCEs, the impact of route of administration on ADA incidence can be difficult to elucidate when the dose and exposure levels differ substantially between the two routes of administration. For example, the ADA incidence for the anti-PSMA TCE JNJ-081 after IV administration was 17% compared to 63% after subcutaneous administration, but the differences in JNJ-081 exposure observed for the two routes of administration meant that it was considered inconclusive as to whether the higher ADA incidence was caused by subcutaneous administration.89 Further caution must be exercised when comparing immunogenicity data across dose routes for TCEs targeting tumor antigens expressed on B cells, since a low incidence of ADA is generally expected as a result of B cell depletion and subsequent suppression of immunoglobulin production. The impact of route-dependent immunogenicity on efficacy or safety will also depend on whether neutralizing or non-neutralizing ADA are generated, and the extent to which they impact the exposure of the TCE.
Selection of the optimal clinical dose
The complexity of the various dosing strategies used during the dose escalation phase of FIH trials for TCEs may lead to difficulties in deciding upon the optimal dose regimen for further study in patients. Increasing emphasis on improving the process of dose optimization within oncology will require a rigorous analysis of the exposure, efficacy and safety data across the evaluated dose range, as opposed to merely selecting the highest tolerated dose.90 As such, it may be necessary to select more than one dose level or regimen for further evaluation in the dose expansion phase of the FIH study or in subsequent studies, in order to more robustly characterize the dose-exposure-response relationship. For example, the anti-DLL3 TCE tarlatamab evaluated two different dose levels in a randomized phase 2 study in small cell lung cancer patients, following the FIH study.91 It may also be necessary to consider additional evaluation of different step-up dose regimens, as demonstrated by the inclusion of two separate expansion cohorts in the FIH study of the anti-FcRH5 TCE cevostamab, which aimed to further characterize the impact of single step-up versus double step-up dosing on tolerability.92
As well as obtaining additional data in clinical studies, quantitative characterization of the relevant PK/PD relationships for efficacy and safety can be utilized to guide the selection of the optimal dose regimen for TCEs. A model-informed approach was proposed to support the selection of RP2D of epcoritamab, for which a semi-mechanistic PK/PD model was developed during the preclinical stage of development and further refined using clinical PK, PD and response data from the FIH study, in order to thoroughly characterize the exposure-response relationship and to determine the clinical dose at which efficacy reached an optimal peak.93 Because the model included prediction of the PK of epcoritamab within the tumor cell environment, as well as T cell and target antigen abundance and dynamics, it was possible to predict the concentration of the trimer complex at each dose level administered during the study. Using model simulations, the clinical dose at which trimer formation reached a plateau was able to be determined, with higher doses not predicted to result in higher concentrations of trimer. The model was also linked to an in silico tumor growth model using the baseline tumor size in patients receiving epcoritamab, to enable the prediction of clinical outcome in terms of tumor growth inhibition and objective response rate. Since drug concentrations may increase proportionally with dose but trimer concentrations may not, depending on the saturation of CD3 and/or the tumor antigen, this highlights the importance of considering the appropriate driver of efficacious exposure, i.e., the trimer, as opposed to the TCE itself, during modeling and simulation of exposure-response relationships. By using a holistic, translational modeling approach, the value of this modeling framework can be evaluated as clinical development continues (“learn and confirm”) and refined for the development of subsequent TCE molecules.
Another key component in aiding the selection of optimal dose regimens for further clinical development involves the identification of dose- or exposure-dependent modulation of PD biomarkers, which may be predictive of clinical response. For glofitamab, it was demonstrated that the magnitude of T cell margination and expansion of effector memory cells was dose-dependent, and positively associated with clinical benefit.94 In some tumors with low baseline levels of T cells residing in the tumor microenvironment, redirection of cytotoxic T cells from peripheral locations to the tumor may be critical for efficacy. For tebentafusp, early drug-dependent changes in biomarkers related to the CXCL10-CXCR3 chemoattractant axis involved in T cell redirection were associated with greater tumor shrinkage and longer overall survival.95 Therefore, mechanistic PK/PD modeling approaches which incorporate the drug-dependent impacts on both the infiltration and proliferation of T cells contributing to tumor growth inhibition could further aid in evaluating the relevant pharmacodynamic predictors of clinical response. Additionally, the PD of the tumor target antigen may also provide a potentially predictive biomarker for correlation with clinical response. A QSP model developed based on the PK and PD characteristics of cibisatamab predicted that a higher tumor mutational burden and higher CEA expression on cancer cells corresponded to an increased reduction in tumor volume following simulation of cibisatamab administration in a virtual population of colorectal cancer patients, whereas CD3 expression on effector and regulatory T cells did not affect the anti-tumor response.96 This model analysis also deduced that while the amount of CEA influenced the simulated ORR, this was much reduced above a certain CEA threshold level, after which no further clinically relevant change in tumor response was predicted. Thus, models which include the influence of tumor antigen levels on response rates could potentially help to guide patient selection or stratification criteria, based on target antigen cutoff values. Furthermore, by including the time course of the PD of TAA in response to the drug, these models could also be used to characterize potential resistance mechanisms such as antigen loss, which has been observed for immunotherapies targeting CD19.97 As well as a progressive decrease in efficacy, the loss of antigen could also manifest as a time-dependent change in clearance of drug, as demonstrated by the population PK analysis included in the regulatory submission of mozenutuzumab,98 and it will be important to determine whether this may cause confounding effects on exposure-efficacy relationships. Additional investigation of T cell-related resistance mechanisms could be carried out using models such as the one developed for cibisatamab, which incorporated different subpopulations of T cells, including immune suppressive regulatory T cells (Treg). Although the model did not predict a substantial impact of Treg levels on tumor growth inhibition by cibisatamab, it has been shown that Treg levels in patients were predictive of clinical response to other TCEs such as blinatumomab, where responders had a lower percentage of Treg cells in peripheral blood.99 Therefore, care must be taken when extrapolating the interpretations from these types of models across different TCEs.
Beyond RP2D
Given the complex temporal relationship between the time course of drug exposure and T cell dynamics, phenotype, and function, additional consideration may be required regarding the refinement of the dosing regimen over a longer duration of treatment. It is common for patients with hematological cancers to receive fixed durations of treatment, and for many of the TCEs with once-weekly dose regimens in early cycles, less frequent dosing schedules have been implemented in subsequent cycles. While this can reduce the treatment burden for patients, it may also help to reduce T cell exhaustion resulting from chronic exposure of the T cells to antigen, which can be a major resistance mechanism to immunotherapy in oncology.100 Another approach to combat T cell exhaustion is to combine TCEs with immune checkpoint inhibitors targeting the PD-1/PD-L1 axis, since this has the potential to counteract the upregulation of PD-1 as a mediator of T cell dysfunction.101,102 Several clinical studies combining TCEs with PD-1 or PD-L1 inhibitors are currently ongoing, and quantitative modeling approaches have also been carried out to characterize the relative contributions of each drug to the T cell response.103 These types of mechanistic models could also help to rationally select combination drug partners with complementary mechanisms of action, and guide the selection of appropriate dose intensities and scheduling for evaluation in clinical trials. Translational modeling of in vivo data in preclinical species may also provide valuable insights into the mechanisms driving the combination effect, either in parallel or prior to clinical studies. This was recently demonstrated for a TCE targeting TYRP1 using an immune-humanized mouse model of melanoma, in which higher doses of TCE as monotherapy resulted in greater tumor cell killing, but also increasingly rapid tumor regrowth. Combining the TCE with anti-PD-L1 therapy reduced both the rate and time to tumor regrowth, and a translational PK/PD model was developed to quantify these processes and predict clinical response of the combination versus monotherapy.104
An alternative approach to boost the anti-tumor immune response is to combine TCEs with other immune stimulating agents such as 4–1BB and CD28 agonists.105,106 Interestingly, in one such clinical trial combining the bispecific immune engagers REGN4018 and REGN5668 which target CD3 and CD28 respectively, both molecules also bind to the same TAA (MUC16) on tumor cells.107 Mechanistic modeling could aid in characterizing the pharmacokinetic and pharmacodynamic interplay between these molecules, including competition for the TAA. In addition to pharmacodynamic drug interactions, potential pharmacokinetic drug-drug interactions (DDI) may sometimes require consideration when TCEs are combined with small molecules given as standard of care. It is known that elevated cytokine levels can influence the expression and function of drug-metabolizing enzymes such as cytochrome P450 (CYP450).108 Therefore, the potential for DDI between cytokine-modulating TCEs and CYP450-metabolized drugs may need to be evaluated, as exemplified by the PBPK modeling approach carried out to predict the impact of blinatumomab-mediated cytokine modulation on CYP450-mediated drug metabolism.109 The magnitude of DDI for blinatumomab was not predicted to be clinically relevant, so PBPK evaluation may be important to help determine whether or not clinical DDI studies are required. Some TCEs are also currently being evaluated in combination with antibody-drug conjugates,110 and as such it may be of particular importance to assess the potential for cytokine-mediated DDI on the PK of the small molecule drug released from the ADC, especially if its metabolism is mediated by CYP450 enzymes. Finally, it will also be important to consider whether the combination of TCEs with chemotherapeutic drugs which cause significant lymphodepletion may affect the PK and PD of TCEs via their impacts on T cell levels, which may potentially counterbalance their tumor debulking effects. This is currently an area which requires further investigation and may be critical to the success of TCEs as they move into earlier lines of therapy.
Perspectives for the next generation of TCEs
With significant progress being made in evolving clinical dosing paradigms (Figure 3), TCE therapies are demonstrating increasingly robust clinical activity in hematological cancers, and are beginning to show promise in some solid tumor indications. However, fulfilling the potential of TCE therapy will rely on addressing several key challenges which further expand the scope of the dose-exposure-response paradigm. A better understanding of the immune cell-related components of the PK/PD relationship could help to account for factors which may influence not only the variability in therapeutic outcomes, but also the disposition of the TCE itself. The complexity of the pharmacodynamic cascade elicited by TCE therapy requires mechanistic and time-dependent characterization of both the number of resident and tumor-infiltrating immune cells within the biophase, along with their phenotypic differences, all of which may vary over the duration of treatment and disease. Physiologically-based models which account for the production, turnover and trafficking of immune cells from lymphoid tissues to tumor tissue, as well as the dynamics of TAA-expressing cells and the distribution of the TCE have previously been developed to aid PD-focused molecular design of TCEs.45 Models such as these could also be vital in understanding the overall response of patients to TCE therapy in a manner which is not solely driven by “input” drug concentrations, but by each component of the resulting immune synapse. This could ultimately help to elucidate which factor – T cells, TAA-expressing cells, or drug levels – has the greatest impact on the clinical efficacy and safety profile for a given TCE and indication, and may thereby guide the implementation of the most appropriate strategy for patient selection based on either immune status or TAA levels. As demonstrated with a QSP model developed for tebentafusp using clinical data for model verification, simulation of different dosing scenarios could guide the selection of step-up dose regimens which provide an optimal balance between efficacy and safety, in order to improve the benefit/risk profile.111
Figure 3.
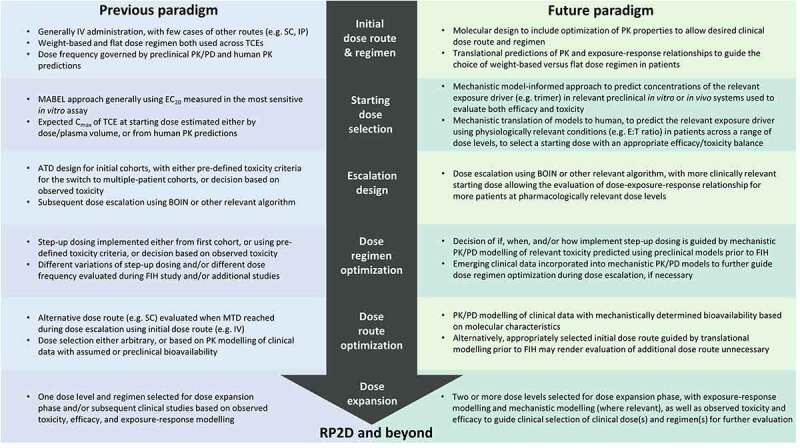
Comparison of different clinical dosing strategies for TCEs.
In cases where the micro-environment of the tumor is unfavorable to T cell infiltration or proliferation, combination therapy may be required to enhance the function of TCEs.112 As an alternative to combining TCEs with other immune-modulating therapies, efforts are underway to design next-generation TCE molecules with binding domains to other costimulatory immune targets, such as CD28.113 Multi-specific antibodies will present challenges for quantitative in silico modeling of exposure-response relationships, since the respective binding kinetics of each targeting domain will have an impact on both the PK and the PD of the molecule. A recently published QSP modeling analysis for a TCE targeting CD3, CD28, and CD38 was carried out to explore the hypothesis that co-stimulation of CD28, while not essential for the cytotoxic activity of CD3-targeting TCEs, could nevertheless be beneficial in increasing the functional capacity and durability of effector T cells that are already differentiated into an early exhausted state, while potentially also triggering the activation of naive T cells.114 Other TCEs have been designed to target both PD-1 and CD3, in addition to a specific TAA, thereby aiming to mimic the anti-PD-1 combination therapy approach to reduce T cell dysfunction or to overcome the immunosuppressive tumor microenvironment.115,116 When developing molecules with multiple target antigens on immune cells, particularly in the case of inhibitory versus agonistic mechanisms such as PD-1 and CD3, it will be important to combine data generated using a range of appropriate in vitro assays with modeling and simulation, in order to achieve the optimal receptor occupancy balance.
Another strategy to improve the therapeutic index of TCEs has been to reduce their off-tumor activity by targeting two separate TAAs which are co-expressed on tumor cells, but with limited or no co-expression on healthy cells.117 Tumor selectivity depends upon sufficiently high receptor density of both antigens on tumor cells to permit avidity-driven target engagement and CD3 cross-linking, while minimizing T cell activation upon monovalent binding to the individual TAAs in their respective locations within healthy tissues. Affinity optimization may not be straightforward, and model-guided approaches could provide valuable input into the design of appropriate assays to thoroughly characterize the relationship between antigen expression, avidity and activity, thus improving confidence in predicting PK/PD in patients. Alternatively, prodrug-like TCEs designed for reduced off-tumor activity via “masked” binding domains to either CD3 or the TAA have recently entered clinical trials.118,119 Tumor-specific activation is achieved by cleavage of the “mask” by proteases that are preferentially located or activated within the tumor microenvironment. For prodrug-like TCEs, the serum concentrations of TCE may not be an appropriate surrogate measure of the exposure which drives pharmacological activity, therefore early implementation of bioanalytical strategies to allow monitoring of the relevant components of the molecule with which to evaluate preclinical exposure-response relationships will be essential. Furthermore, a more complete mechanistic characterization of the processes governing the tumor distribution and cleavage of the prodrug, including accounting for potential inter-patient variability in protease levels, will be required in order to translate preclinical in vitro or in vivo efficacy and toxicity to patient populations.120 The implementation of these model-based approaches as early as possible during the development of these next-generation TCEs is therefore encouraged.
In conclusion, it is essential to develop rational, mechanistically guided approaches to dose selection and clinical study design, in order to facilitate the development of immune engaging molecules with a better therapeutic index in patients. Significant improvements have been made in characterizing dose-exposure-response relationships for efficacy and toxicity of TCEs throughout their development, and it will be important to continue optimizing clinical dosing paradigms in order to successfully deliver on the promise of TCE therapy in oncology.
Funding Statement
This work was funded by AstraZeneca.
Abbreviations
ADA: Anti-drug antibody; BCMA: B cell maturation antigen; BiTE: Bispecific T-cell engager; CAR: Chimeric antigen receptor; CD: Cluster of differentiation; CEA: Carcinoembryonic antigen; Cmax: Maximum concentration; CRS: Cytokine release syndrome; CXCL: CXC motif chemokine ligand; CXCR: CXC motif chemokine receptor; CYP450: Cytochrome P450; DART: Dual-Affinity Re-Targeting; DDI: Drug-drug interaction; DLL3: Delta-like ligand 3; EC: Effective concentration; Emax: Maximum effect; E:T: Effector:Tumor ratio; EU: European Union; Fc: Fragment crystallizable; FcRH5: Fc receptor-like protein 5; FcRn: Neonatal Fc receptor; FDA: Food and Drug Administration; FIH: First-in-human; GPRC5D: G Protein-Coupled Receptor Class C Group 5 Member D; Ig: Immunoglobulin; IL: Interleukin; IV: Intravenous; MABEL: Minimum anticipated biological effect level; MTD: Maximum tolerated dose; MM: Multiple myeloma; MUC: Mucin; ORR Objective response rate; PBMC: Peripheral blood mononuclear cell; PBPK: Physiologically based pharmacokinetic; PD: Pharmacodynamic; PD-1: Programmed cell death protein 1; PD-L1: Programmed cell death ligand 1; PK: Pharmacokinetic; pMHC: Peptide major histocompatibility complex; PSMA: Prostate-specific membrane antigen; QSP: Quantitative systems pharmacology; RO: Receptor occupancy; RP2D: Recommended phase 2 dose; scFv: Single-chain variable antibody fragment; SSTR2: Somatostatin receptor 2; STEAP1: Six-transmembrane epithelial antigen of prostate-1; TAA: Tumor associated antigen; TCE: T-cell engager; TCR: T-cell receptor; TI: Therapeutic index; Treg: regulatory T cell; TYRP1: Tyrosinase-related protein 1; US: United States.
Disclosure statement
The authors report there are no competing interests to declare.
References
- 1.Beatty GL, Gladney WL.. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–18. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022;22(2):85–96. doi: 10.1038/s41577-021-00547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salvaris R, Ong J, Gregory GP. Bispecific antibodies: a review of development, clinical efficacy and toxicity in B-cell lymphomas. J Pers Med. 2021;11(5). doi: 10.3390/jpm11050355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol. 2021;14(1):1–18. doi: 10.1186/s13045-021-01084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fucà G, Spagnoletti A, Ambrosini M, de Braud F, Di Nicola M. Immune cell engagers in solid tumors: promises and challenges of the next generation immunotherapy. ESMO Open. 2021;6(1):100046. doi: 10.1016/j.esmoop.2020.100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y, Yi M, Zhu S, Wang H, Wu K. Recent advances and challenges of bispecific antibodies in solid tumors. Exp Hematol Oncol. 2021;10(1):1–14. doi: 10.1186/s40164-021-00250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A, Dees S, Grewal IS. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br J Cancer. 2021;124(6):1037–48. doi: 10.1038/s41416-020-01225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arvedson T, Bailis JM, Britten CD, Klinger M, Nagorsen D, Coxon A, Egen JG, Martin F. Targeting solid tumors with bispecific T cell engager immune therapy. Annu Rev Cancer Biol. 2022;6:17–34. doi: 10.1146/annurev-cancerbio-070620-104325. [DOI] [Google Scholar]
- 9.Long M, Mims AS, Li Z. Factors affecting the cancer immunotherapeutic efficacy of T cell bispecific antibodies and strategies for improvement. Immunol Invest. 2022:1–39. doi: 10.1080/08820139.2022.2131569. [DOI] [PubMed] [Google Scholar]
- 10.Stieglmaier J, Benjamin J, Nagorsen D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin Biol Ther. 2015;15(8):1093–99. doi: 10.1517/14712598.2015.1041373. [DOI] [PubMed] [Google Scholar]
- 11.You G, Won J, Lee Y, Moon D, Park Y, Lee SH, Lee S-W. Bispecific antibodies: a smart arsenal for cancer immunotherapies. Vaccines. 2021;9(7):724. doi: 10.3390/vaccines9070724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arvedson T, Bailis JM, Urbig T, Stevens JL. Considerations for design, manufacture, and delivery for effective and safe T-cell engager therapies. Curr Opin Biotechnol. 2022;78:102799. doi: 10.1016/j.copbio.2022.102799. [DOI] [PubMed] [Google Scholar]
- 13.Chen W, Yang F, Wang C, Narula J, Pascua E, Ni I, Ding S, Deng X, Chu MLH, Pham A, et al. One size does not fit all: navigating the multi-dimensional space to optimize T-cell engaging protein therapeutics. mAbs. 2021;13(1). doi: 10.1080/19420862.2020.1871171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. 2019;201:103–19. doi: 10.1016/j.pharmthera.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, Kufer P, Iskander K, Kantarjian HM. The BiTE (bispecific T-cell engager) platform: development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer. 2020;126(14):3192–201. doi: 10.1002/cncr.32909. [DOI] [PubMed] [Google Scholar]
- 16.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. mAbs. 2018;10(5):751–64. doi: 10.1080/19420862.2018.1462429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 19.Boswell CA, Tesar DB, Mukhyala K, Theil F-P, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21(12):2153–63. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- 20.Hu S, Datta-Mannan A, D’Argenio DZ. Physiologically based modeling to predict monoclonal antibody pharmacokinetics in humans from in vitro physiochemical properties. MAbs. 2022;14(1):2056944. doi: 10.1080/19420862.2022.2056944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Müller S, Valitutti S. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci. 2003;100(24):14145–50. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Piskol R, Ybarra R, Chen YJJ, Li J, Slaga D, Hristopoulos M, Clark R, Modrusan Z, Totpal K, et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. 2019;11(508):1–13. doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 23.Germain RN, Stefanová I. THE DYNAMICS OF T CELL RECEPTOR SIGNALING: complex orchestration and the key roles of tempo and cooperation. Annu Rev Immunol. 1999;17(1):467–522. doi: 10.1146/annurev.immunol.17.1.467. [DOI] [PubMed] [Google Scholar]
- 24.Trinklein ND, Pham D, Schellenberger U, Buelow B, Boudreau A, Choudhry P, Clarke SC, Dang K, Harris KE, Iyer S, et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. mAbs. 2019;11(4):639–52. doi: 10.1080/19420862.2019.1574521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vafa O, Trinklein ND. Perspective: designing T-cell engagers with better therapeutic windows. Front Oncol. 2020;10(April):1–7. doi: 10.3389/fonc.2020.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, Ho J, Hristopoulos M, Clark R, Totpal K, et al. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17(4):776–85. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 27.Haber L, Olson K, Kelly MP, Crawford A, DiLillo DJ, Tavaré R, Ullman E, Mao S, Canova L, Sineshchekova O, et al. Generation of T-cell-redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci Rep. 2021;11(1):1–17. doi: 10.1038/s41598-021-93842-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith E, Olson K, Delfino F, DiLillo D, Kirshner J, Sineshchekova O, Zhang Q; Regeneron Pharmaceuticals, Inc . Bispecific anti-BCMA x anti-CD3 antibodies and uses thereof. United States patent US 20200024356A1. 2020 January 23.
- 29.Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154(October 2018):102–17. doi: 10.1016/j.ymeth.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 30.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 31.Grimm HP. Gaining insights into the consequences of target-mediated drug disposition of monoclonal antibodies using quasi-steady-state approximations. J Pharmacokinet Pharmacodyn. 2009;36(5):407–20. doi: 10.1007/s10928-009-9129-5. [DOI] [PubMed] [Google Scholar]
- 32.Gibiansky L, Gibiansky E. Target-mediated drug disposition model for drugs that bind to more than one target. J Pharmacokinet Pharmacodyn. 2010;37(4):323–46. doi: 10.1007/s10928-010-9163-3. [DOI] [PubMed] [Google Scholar]
- 33.Lee J-H, Kim H, Yao Z, Szajek LP, Grasso L, Kim I, Paik CH. Tumor-shed antigen affects antibody tumor targeting: comparison of two 89 Zr-labeled antibodies directed against shed or nonshed antigens. Contrast Media Mol Imaging. 2018;2018:1–12. doi: 10.1155/2018/2461257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bacac M, Klein C, Umana P. CEA TCB: a novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8):1–3. doi: 10.1080/2162402X.2016.1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petitcollin A, Bensalem A, Verdier M-C, Tron C, Lemaitre F, Paintaud G, Bellissant E, Ternant D. Modelling of the time-varying pharmacokinetics of therapeutic monoclonal antibodies: a literature review. Clin Pharmacokinet. 2020;59(1):37–49. doi: 10.1007/s40262-019-00816-7. [DOI] [PubMed] [Google Scholar]
- 36.Betts A, van der Graaf PH. Mechanistic quantitative pharmacology strategies for the early clinical development of bispecific antibodies in oncology. Clin Pharmacol Ther. 2020;108(3):528–41. doi: 10.1002/cpt.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, De Groot AS. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review 2020. Front Immunol. 2020:11. doi: 10.3389/fimmu.2020.01301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Penny HL, Kroenke MA, Bautista B, Hainline K, Chea LS, Parnes J, Mytych DT. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J Immunother Cancer. 2022;10(4):e004225. doi: 10.1136/jitc-2021-004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen S, Chung S, Spiess C, Lundin V, Stefanich E, Laing ST, Clark V, Brumm J, Zhou Y, Huang C, et al. An integrated approach for characterizing immunogenic responses toward a bispecific antibody. MAbs. 2021;13(1). doi: 10.1080/19420862.2021.1944017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campagne O, Delmas A, Fouliard S, Chenel M, Chichili GR, Li H, Alderson R, Scherrmann J-M, Mager DE. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. 2018;24(11):2631–41. doi: 10.1158/1078-0432.CCR-17-2265. [DOI] [PubMed] [Google Scholar]
- 41.Leclercq G, Servera LA, Danilin S, Challier J, Steinhoff N, Bossen C, Odermatt A, Nicolini V, Umaña P, Klein C, et al. Dissecting the mechanism of cytokine release induced by T-cell engagers highlights the contribution of neutrophils. Oncoimmunology. 2022;11(1):2039432. doi: 10.1080/2162402X.2022.2039432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saber H, Del Valle P, Ricks TK, Leighton JK. An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharmacol. 2017;90:144–52. doi: 10.1016/j.yrtph.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Budde LE, Assouline S, Sehn LH, Schuster SJ, Yoon SS, Yoon DH, Matasar MJ, Bosch F, Kim WS, Nastoupil LJ, et al. Single-agent mosunetuzumab shows durable complete responses in patients with relapsed or refractory B-cell lymphomas: phase I dose-escalation study. J Clin Oncol. 2022;40(5):481–91. doi: 10.1200/JCO.21.00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holstein SA, Venkatakrishnan K, van der Graaf PH. Quantitative clinical pharmacology of CAR T‐cell therapy. Clin Pharmacol Ther. 2022;112(1):11–15. doi: 10.1002/cpt.2631. [DOI] [PubMed] [Google Scholar]
- 45.Yoneyama T, Kim M-S, Piatkov K, Wang H, Zhu AZX, Rubinstein R. Leveraging a physiologically-based quantitative translational modeling platform for designing B cell maturation antigen-targeting bispecific T cell engagers for treatment of multiple myeloma. PLoS Comput Biol. 2022;18(7):e1009715. doi: 10.1371/journal.pcbi.1009715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li -C-C, Bender B, Yin S, Li Z, Zhang C, Hernandez G, Kwan A, Sun L, Adamkewicz JI, Wang H, et al. Exposure-response analyses indicate a promising benefit/risk profile of mosunetuzumab in relapsed and refractory non-hodgkin lymphoma. Blood. 2019;134(Supplement_1):1285–1285. doi: 10.1182/blood-2019-123961. [DOI] [Google Scholar]
- 47.Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, Kufer P, Riethmuller G, Bargou R, Baeuerle PA. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100(6):690–97. doi: 10.1002/ijc.10557. [DOI] [PubMed] [Google Scholar]
- 48.Jiang X, Chen X, Carpenter TJ, Wang J, Zhou R, Davis HM, Heald DL, Wang W. Development of a Target cell-Biologics-Effector cell (TBE) complex-based cell killing model to characterize target cell depletion by T cell redirecting bispecific agents. MAbs. 2018;10(6):876–89. doi: 10.1080/19420862.2018.1480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, Sinclair AM, Kischel R, Frankel SR, Walter RB. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554–61. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hipp S, Tai Y-T, Blanset D, Deegen P, Wahl J, Thomas O, Rattel B, Adam PJ, Anderson KC, Friedrich M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31(8):1743–51. doi: 10.1038/leu.2016.388. [DOI] [PubMed] [Google Scholar]
- 51.Chen X, Haddish-Berhane N, Moore P, Clark T, Yang Y, Li H, Xuan D, Barton HA, Betts AM, Barletta F. Mechanistic projection of first-in-human dose for bispecific immunomodulatory P-cadherin LP-DART: an integrated PK/PD modeling approach. Clin Pharmacol Ther. 2016;100(3):232–41. doi: 10.1002/cpt.393. [DOI] [PubMed] [Google Scholar]
- 52.Van De Vyver A, Eigenmann M, Ovacik M, Pohl C, Herter S, Weinzierl T, Fauti T, Klein C, Lehr T, Bacac M, et al. A novel approach for quantifying the pharmacological activity of T-cell engagers utilizing in vitro time course experiments and streamlined data analysis. AAPS J. 2022;24(1):1–13. doi: 10.1208/s12248-021-00637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dudal S, Hinton H, Giusti AM, Bacac M, Muller M, Fauti T, Colombetti S, Heckel T, Giroud N, Klein C, et al. Application of a MABEL approach for a T-cell-bispecific monoclonal antibody: CEA TCB. J Immunother. 2016;39(7):279–89. doi: 10.1097/CJI.0000000000000132. [DOI] [PubMed] [Google Scholar]
- 54.Frances N, Bacac M, Bray-French K, Christen F, Hinton H, Husar E, Quackenbush E, Schäfer M, Schick E, Van De Vyver A, et al. Novel in vivo and in vitro pharmacokinetic/pharmacodynamic-based human starting dose selection for glofitamab. J Pharm Sci. 2022;111(4):1208–18. doi: 10.1016/j.xphs.2021.12.019. [DOI] [PubMed] [Google Scholar]
- 55.Desnoyer A, Broutin S, Delahousse J, Maritaz C, Blondel L, Mir O, Chaput N, Paci A. Pharmacokinetic/pharmacodynamic relationship of therapeutic monoclonal antibodies used in oncology: part 2, immune checkpoint inhibitor antibodies. Eur J Cancer. 2020;128:119–28. doi: 10.1016/j.ejca.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Djebli N, Morcos PN, Jaminion F, Guerini E, Kratochwil NA, Justies N, Schick E, Kwan A, Humphrey K, Lundberg L, et al. Population pharmacokinetics and exposure-response analyses for glofitamab in relapsed/refractory B-cell non-hodgkin lymphoma (R/R NHL): confirmation of efficacy and CRS mitigation in patients with step-up dosing. Blood. 2020;136(Supplement 1):1–2. doi: 10.1182/blood-2020-136311.32430499 [DOI] [Google Scholar]
- 57.Girgis S, Lin SXW, Pillarisetti K, Banerjee A, Stephenson T, Ma X, Shetty S, Yang T-Y, Hilder BW, Jiao Q, et al. Translational modeling predicts efficacious therapeutic dosing range of teclistamab for multiple myeloma. Target Oncol. 2022;(123456789). doi: 10.1007/s11523-022-00893-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krishnan AY, Garfall AL, Mateos M-V, van de Donk NWCJ, Nahi H, San-Miguel F, Oriol J, Rosiñol A, Chari L, Bhutani A, et al. Updated phase 1 results of teclistamab, a B-cell maturation antigen (BCMA) × CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). J Clin Oncol. 2021;39(15_suppl):8007–8007. doi: 10.1200/JCO.2021.39.15_suppl.8007. [DOI] [Google Scholar]
- 59.Berdeja JG, Krishnan AY, Oriol A, van de Donk NWCJ, Rodríguez-Otero P, Askari E, Mateos M-V, Minnema MC, Costa LJ, Verona R, et al. Updated results of a phase 1, first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D (GPRC5D) × CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). J Clin Oncol. 2021;39(15_suppl):8008–8008. doi: 10.1200/JCO.2021.39.15_suppl.8008. [DOI] [Google Scholar]
- 60.Bannerji R, Allan JN, Arnason JE, Brown JR, Advani RH, Barnes JA, Ansell SM, O’Brien SM, Chavez J, Duell J, et al. Clinical activity of REGN1979, a bispecific human, anti-CD20 x anti-CD3 antibody, in patients with Relapsed/Refractory (R/R) B-cell non-hodgkin lymphoma (B-NHL). Blood. 2019;134(Supplement_1):762–762. doi: 10.1182/blood-2019-122451. [DOI] [Google Scholar]
- 61.Zhu M, Olson K, Kirshner JR, Khaksar Toroghi M, Yan H, Haber L, Meagher C, Flink DM, Ambati SR, Davis JD, et al. Translational findings for odronextamab: from preclinical research to a first-in-human study in patients with CD20+ B-cell malignancies. Clin Transl Sci. 2022;15(4):954–66. doi: 10.1111/cts.13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van de Vyver AJ, Weinzierl T, Eigenmann MJ, Frances N, Herter S, Buser RB, Somandin J, Diggelmann S, Limani F, Lehr T, et al. Predicting tumor killing and t-cell activation by t-cell bispecific antibodies as a function of target expression: combining in vitro experiments with systems modeling. Mol Cancer Ther. 2021;20(2):357–66. doi: 10.1158/1535-7163.MCT-20-0269. [DOI] [PubMed] [Google Scholar]
- 63.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29(2):57–61. doi: 10.1016/j.tips.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Betts A, Haddish-Berhane N, Shah DK, van der Graaf PH, Barletta F, King L, Clark T, Kamperschroer C, Root A, Hooper A, et al. A translational quantitative systems pharmacology model for CD3 bispecific molecules: application to quantify T cell-mediated tumor cell killing by P-cadherin LP DART®. AAPS J. 2019;21(4). doi: 10.1208/s12248-019-0332-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin L, Wille L, Betts A, Hua F, Hagen D, Park J, Apgar J, Burke J. Abstract A70: bridging nonclinical studies to clinical design using quantitative systems pharmacology model of T cell-engaging bispecifics. Cancer Immunol Res. 2020;8(3_Supplement):A70–A70. doi: 10.1158/2326-6074.TUMIMM19-A70. [DOI] [Google Scholar]
- 66.Kamperschroer C, Shenton J, Lebrec H, Leighton JK, Moore PA, Thomas O. Summary of a workshop on preclinical and translational safety assessment of CD3 bispecifics. J Immunotoxicol. 2020;17(1):67–85. doi: 10.1080/1547691X.2020.1729902. [DOI] [PubMed] [Google Scholar]
- 67.Jiang X, Chen X, Jaiprasart P, Carpenter TJ, Zhou R, Wang W. Development of a minimal physiologically-based pharmacokinetic/pharmacodynamic model to characterize target cell depletion and cytokine release for T cell-redirecting bispecific agents in humans. Eur J Pharm Sci. 2020;146:105260. doi: 10.1016/j.ejps.2020.105260. [DOI] [PubMed] [Google Scholar]
- 68.Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, Kochanek M, Böll B, von Bergwelt-Baildon MS. Cytokine release syndrome. J Immunother Cancer. 2018;6(1). doi: 10.1186/s40425-018-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, Wu T, Chen D, Chang AH, Gao Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):20–23. doi: 10.1038/s41408-020-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun Z, De Xun R, Liu MS, Wu XQ, Qu HT. The association between glucocorticoid administration and the risk of impaired efficacy of axicabtagene ciloleucel treatment: a systematic review. Front Immunol. 2021;12(April):1–8. doi: 10.3389/fimmu.2021.646450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nägele V, Kratzer A, Zugmaier G, Holland C, Hijazi Y, Topp MS, Gökbuget N, Baeuerle PA, Kufer P, Wolf A, et al. Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp Hematol Oncol. 2017;6(1):1–14. doi: 10.1186/s40164-017-0074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carvajal RD, Nathan P, Sacco JJ, Orloff M, Hernandez-Aya LF, Yang J, Luke JJ, Butler MO, Stanhope S, Collins L, et al. Phase I study of safety, tolerability, and efficacy of tebentafusp using a step-up dosing regimen and expansion in patients with metastatic uveal melanoma. J Clin Oncol. 2022;40(17):1939–48. doi: 10.1200/JCO.21.01805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bartlett NL, Sehn LH, Assouline SE, Bosch F, Magid Diefenbach CS, Flinn I, Hong J, Kim WS, Matasar MJ, Nastoupil LJ, et al. Managing cytokine release syndrome (CRS) and neurotoxicity with step-fractionated dosing of mosunetuzumab in relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL). J Clin Oncol. 2019;37(15_suppl):7518–7518. doi: 10.1200/JCO.2019.37.15_suppl.7518. [DOI] [Google Scholar]
- 74.Liu JF, O’Malley DM. Phase 1/2 study of REGN4018, a MUC16 x CD3 bispecific antibody, as monotherapy or in combination with cemiplimab in patients with recurrent ovarian cancer. SGO 2021; Virtual meeting. Poster 10818. [Google Scholar]
- 75.Ravandi F, Bashey A, Stock W, Foran JM, Mawad R, Egan D, Blum W, Yang A, Pastore A, Johnson C, et al. Complete responses in relapsed/refractory Acute Myeloid Leukemia (AML) patients on a weekly dosing schedule of vibecotamab (XmAb14045), a CD123 x CD3 T cell-engaging bispecific antibody; initial results of a phase 1 study. Blood. 2020;136(Supplement 1):4–5. doi: 10.1182/blood-2020-134746.32614961 [DOI] [Google Scholar]
- 76.Patel K, Michot J-M, Chanan-Khan A, Ghesquieres H, Bouabdallah K, Byrd JC, Cartron G, Portell CA, Solh M, Tilly H, et al. Safety and anti-tumor activity of plamotamab (XmAb13676), an anti-CD20 x anti-CD3 bispecific antibody, in subjects with relapsed/refractory non-hodgkin’s lymphoma. Blood. 2021;138(Supplement 1):2494–2494. doi: 10.1182/blood-2021-144350. [DOI] [Google Scholar]
- 77.Usmani SZ, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, Rosinol L, Chari A, Bhutani M, Karlin L, et al. Teclistamab, a B-cell maturation antigen × CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): a multicentre, open-label, single-arm, phase 1 study. Lancet. 2021;398(10301):665–74. doi: 10.1016/S0140-6736(21)01338-6. [DOI] [PubMed] [Google Scholar]
- 78.Hijazi Y, Klinger M, Kratzer A, Wu B, Baeuerle PA, Kufer P, Wolf A, Nagorsen D, Zhu M. Pharmacokinetic and pharmacodynamic relationship of blinatumomab in patients with non-hodgkin lymphoma. Curr Clin Pharmacol. 2018;13(1):55–64. doi: 10.2174/1574884713666180518102514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hosseini I, Gadkar K, Stefanich E, Li CC, Sun LL, Chu YW, Ramanujan S. Mitigating the risk of cytokine release syndrome in a Phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. Npj Syst Biol Appl. 2020;6(1). doi: 10.1038/s41540-020-00145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen X, Kamperschroer C, Wong G, Xuan D. A modeling framework to characterize cytokine release upon T-cell–engaging bispecific antibody treatment: methodology and opportunities. Clin Transl Sci. 2019;12(6):600–08. doi: 10.1111/cts.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.El-Rayes B, Hendifar AE, Pant S, Wilky BA, Reilley M, Benson AB, Chow WA, Konda B, Starr J, Ahn DH, et al. Preliminary safety, pharmacodynamic, and antitumor activity of tidutamab, an SSTR2 x CD3 bispecific antibody, in subjects with advanced neuroendocrine tumors. Presented at: 2021 NANETS Annual Symposium; November 3–6, 2021; Virtual meeting. Abstract 109. [Google Scholar]
- 82.Bartlett NL, Giri P, Budde LE, Schuster SJ, Assouline S, Matasar MJ, Yoon -S-S, Canales M, Gutierrez NC, Fay K, et al. Subcutaneous (SC) administration of mosunetuzumab with cycle 1 step-up dosing is tolerable and active in patients with relapsed/refractory B-Cell Non-Hodgkin Lymphomas (R/R B-NHL): initial results from a phase I/II study. Blood. 2021;138(Supplement 1):3573–3573. doi: 10.1182/blood-2021-147937. [DOI] [Google Scholar]
- 83.Nooka AK, Moreau P, Usmani SZ, Garfall AL, van de Donk NWCJ, San-Miguel JF, Oriol Rocafiguera A, Chari A, Karlin L, Mateos M-V, et al. Teclistamab, a B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM): updated efficacy and safety results from MajesTEC-1. J Clin Oncol. 2022;40(16_suppl):8007–8007. doi: 10.1200/JCO.2022.40.16_suppl.8007. [DOI] [Google Scholar]
- 84.Minnema MC, Krishnan AY, Berdeja JG, Oriol Rocafiguera A, van de Donk NWCJ, Rodríguez-Otero P, Morillo D, Mateos M-V, Costa LJ, Caers J, et al. Efficacy and safety of talquetamab, a G protein-coupled receptor family C group 5 member D x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM): updated results from MonumenTAL-1. J Clin Oncol. 2022;40(16_suppl):8015–8015. doi: 10.1200/JCO.2022.40.16_suppl.8015. [DOI] [Google Scholar]
- 85.Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, Nooka AK, Martin T, Rosinol L, Chari A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;1–11. doi: 10.1056/nejmoa2203478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Z, Yu X, Li Y, Verma A, Chang HP, Shah DK. A two-pore physiologically based pharmacokinetic model to predict subcutaneously administered different-size antibody/antibody fragments. AAPS J. 2021;23(3):62. doi: 10.1208/s12248-021-00588-8. [DOI] [PubMed] [Google Scholar]
- 87.Sánchez-Félix M, Burke M, Chen HH, Patterson C, Mittal S. Predicting bioavailability of monoclonal antibodies after subcutaneous administration: open innovation challenge. Adv Drug Deliv Rev. 2020;167:66–77. doi: 10.1016/j.addr.2020.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Hamuro L, Kijanka G, Kinderman F, Kropshofer H, Bu D, Zepeda M, Jawa V. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106(10):2946–54. doi: 10.1016/j.xphs.2017.05.030. [DOI] [PubMed] [Google Scholar]
- 89.Lim EA, Schweizer MT, Chi KN, Aggarwal RR, Agarwal N, Gulley JL, Attiyeh EF, Greger J, Wu S, Jaiprasart P, et al. Safety and preliminary clinical activity of JNJ-63898081 (JNJ-081), a PSMA and CD3 bispecific antibody, for the treatment of metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. 2022;40(6_suppl):279–279. doi: 10.1200/JCO.2022.40.6_suppl.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shah M, Rahman A, Theoret MR, Pazdur R. The drug-dosing conundrum in oncology — when less is more. N Engl J Med. 2021;385(16):1445–47. doi: 10.1056/NEJMp2109826. [DOI] [PubMed] [Google Scholar]
- 91.Ramalingam SS, Ahn M-J, Akamatsu H, Blackhall FH, Borghaei H, Hummel H-D, Johnson ML, Reck M, Zhang Y, Jandial D, et al. Phase 2 study of tarlatamab, a DLL3-targeting, half life–extended, bispecific T-cell engager (HLE BiTE)immuno-oncology therapy, in relapsed/refractory small cell lung cancer (SCLC). J Clin Oncol. 2022;40(16_suppl):8603–8603. doi: 10.1200/JCO.2022.40.16_suppl.TPS8603. [DOI] [Google Scholar]
- 92.Trudel S, Cohen AD, Krishnan AY, Fonseca R, Spencer A, Berdeja JG, Lesokhin A, Forsberg PA, Laubach JP, Costa LJ, et al. Cevostamab monotherapy continues to show clinically meaningful activity and manageable safety in patients with heavily pre-treated Relapsed/Refractory Multiple Myeloma (RRMM): updated results from an ongoing phase I study. Blood. 2021;138(Supplement 1):157–157. doi: 10.1182/blood-2021-147983. [DOI] [Google Scholar]
- 93.Li T, Hiemstra IH, Chiu C, Oliveri RS, Elliott B, DeMarco D, Salcedo T, Egerod FL, Sasser K, Ahmadi T, et al. Semimechanistic physiologically‐based pharmacokinetic/pharmacodynamic model informing epcoritamab dose selection for patients with <scp>B‐Cell</scp> lymphomas. Clin Pharmacol Ther. 2022;112(5):1108–19. doi: 10.1002/cpt.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bröske A-ME, Korfi K, Belousov A, Wilson S, Ooi C-H, Bolen CR, Canamero M, Alcaide EG, James I, Piccione EC, et al. Pharmacodynamics and molecular correlates of response to glofitamab in relapsed/refractory non-hodgkin lymphoma. Blood Adv. 2022;6(3):1025–37. doi: 10.1182/bloodadvances.2021005954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Middleton MR, McAlpine C, Woodcock VK, Corrie P, Infante JR, Steven NM, Evans TRJ, Anthoney A, Shoushtari AN, Hamid O, et al. Tebentafusp, A TCR/Anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res. 2020;26(22):5869–78. doi: 10.1158/1078-0432.CCR-20-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma H, Wang H, Sove RJ, Jafarnejad M, Tsai CH, Wang J, Giragossian C, Popel AS. A quantitative systems pharmacology model of T cell engager applied to solid tumor. AAPS J. 2020;22(4):1–16. doi: 10.1208/s12248-020-00450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357–62. doi: 10.1016/j.csbj.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kang C. Mosunetuzumab: first approval. Drugs. 2022;0123456789:1–6. doi: 10.1007/s40265-022-01749-5. [DOI] [PubMed] [Google Scholar]
- 99.Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, Rasche L, Hartmann E, Dandekar T, Einsele H, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia. 2017;31(10):2181–90. doi: 10.1038/leu.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Catakovic K, Klieser E, Neureiter D, Geisberger R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal. 2017;15(1):1. doi: 10.1186/s12964-016-0160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kobold S, Pantelyushin S, Rataj F, Vom Berg J. Rationale for combining bispecific T cell activating antibodies with checkpoint blockade for cancer therapy. Front Oncol. 2018:8. doi: 10.3389/fonc.2018.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sam J, Colombetti S, Fauti T, Roller A, Biehl M, Fahrni L, Nicolini V, Perro M, Nayak T, Bommer E, et al. Combination of T-cell bispecific antibodies with PD-L1 checkpoint inhibition elicits superior anti-tumor activity. Front Oncol. 2020;10. doi: 10.3389/fonc.2020.575737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ma H, Wang H, Sové RJ, Wang J, Giragossian C, Popel AS. Combination therapy with T cell engager and PD-L1 blockade enhances the antitumor potency of T cells as predicted by a QSP model. J Immunother Cancer. 2020;8(2):1–11. doi: 10.1136/jitc-2020-001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sánchez J, Nicolini V, Fahrni L, Waldhauer I, Walz A-C, Jamois C, Fowler S, Simon S, Klein C, Umaña P, et al. Preclinical InVivo data integrated in a modeling network informs a refined clinical strategy for a CD3 T-cell bispecific in combination with anti-PD-L1. AAPS J. 2022;24(6):106. doi: 10.1208/s12248-022-00755-5. [DOI] [PubMed] [Google Scholar]
- 105.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019;11(496). doi: 10.1126/scitranslmed.aav5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Herter S, Sam J, Ferrara Koller C, Diggelmann S, Bommer E, Schönle A, Claus C, Bacac M, Klein C, Umana P. RG6076 (CD19-4-1BBL): CD19-targeted 4-1BB ligand combination with glofitamab as an off-the-shelf, enhanced T-cell redirection therapy for B-cell malignancies. Blood. 2020;136(Supplement 1):40–40. doi: 10.1182/blood-2020-134782. [DOI] [Google Scholar]
- 107.Winer IS, Shields AF, Yeku OO, Liu JF, Peterman MJ, Yoo SY, Lowy I, Yama-Dang NA, Goncalves PH, Kroog G. A phase I/II, multicenter, open-label study of REGN5668 (mucin [MUC]16 x CD28 bispecific antibody [bsAb]) with cemiplimab (programmed death [PD]-1 Ab) or REGN4018 (MUC16 x CD3 bsAb) in recurrent ovarian cancer (rOVCA). J Clin Oncol. 2021;39(15_suppl):5602–5602. doi: 10.1200/JCO.2021.39.15_suppl.TPS5602. [DOI] [Google Scholar]
- 108.Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease–drug–drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89(5):735–40. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- 109.Xu Y, Hijazi Y, Wolf A, Wu B, Sun Y, Zhu M. Physiologically based pharmacokinetic model to assess the influence of blinatumomab-mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharmacometrics Syst Pharmacol. 2015;4(9):507–15. doi: 10.1002/psp4.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Budde LE, Ghosh N, Chavez JC, Lossos IS, Mehta A, Dorritie KA, Kamdar MK, Negricea R, Pham S, Hristopoulos M, et al. Promising tolerability and efficacy results from dose-escalation in an ongoing phase Ib/II study of mosunetuzumab (M) with polatuzumab vedotin (Pola) in patients (pts) with relapsed/refractory (R/R) B-cell non-Hodgkin’s lymphoma (B-NHL). J Clin Oncol. 2021;39(15_suppl):7520–7520. doi: 10.1200/JCO.2021.39.15_suppl.7520. [DOI] [Google Scholar]
- 111.Weddell J. Mechanistically modeling peripheral cytokine dynamics following bispecific dosing in solid tumors. CPT Pharmacom & Syst Pharma. 2023; Early View. doi: 10.1002/psp4.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662–80. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 113.Wu L, Seung E, Xu L, Rao E, Lord DM, Wei RR, Cortez-Retamozo V, Ospina B, Posternak V, Ulinski G, et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat Cancer. 2020;1(1):86–98. doi: 10.1038/s43018-019-0004-z. [DOI] [PubMed] [Google Scholar]
- 114.Abrams RE, Pierre K, Murr NE, Seung E, Wu L, Luna E, Mehta R, Li J, et al. Quantitative systems pharmacology modeling sheds light into the dose response relationship of a trispecific T cell engager in multiple myeloma. Sci Rep. 2022;12(1):10976. doi: 10.1038/s41598-022-14726-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A, Rataj F, Mocikat R, Metzeler KH, Spiekermann K, et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood. 2018;132(23):2484–94. doi: 10.1182/blood-2018-05-849802. [DOI] [PubMed] [Google Scholar]
- 116.Passariello M, Yoshioka A, Takahashi K, Hashimoto S, Rapuano Lembo R, Manna L, Nakamura K, De Lorenzo C. Novel bi-specific immuno-modulatory tribodies potentiate T cell activation and increase anti-tumor efficacy. Int J Mol Sci. 2022;23(7):3466. doi: 10.3390/ijms23073466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dicara DM, Bhakta S, Go MA, Ziai J, Firestein R, Forrest B, Gu C, Leong SR, Lee G, Yu S-F, et al. Development of T-cell engagers selective for cells co-expressing two antigens. MAbs. 2022;14(1). doi: 10.1080/19420862.2022.2115213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Panchal A, Seto P, Wall R, Hillier BJ, Zhu Y, Krakow J, Datt A, Pongo E, Bagheri A, Chen THT, et al. COBRA™: a highly potent conditionally active T cell engager engineered for the treaTMent of solid tumors. MAbs. 2020;12(1):1792130. doi: 10.1080/19420862.2020.1792130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boustany LM, LaPorte SL, Wong L, White C, Vinod V, Shen J, Yu W, Koditek D, Winter MB, Moore S, et al. A Probody T Cell–engaging bispecific antibody targeting EGFR and CD3 inhibits colon cancer growth with limited toxicity. Cancer Res. 2022;82(22):4288–98. doi: 10.1158/0008-5472.CAN-21-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stroh M, Sagert J, Burke JM, Apgar JF, Lin L, Millard BL, Kavanaugh WM. Quantitative systems pharmacology model of a masked, tumor-activated antibody. CPT Pharmacometrics Syst Pharmacol. 2019;8(9):676–84. doi: 10.1002/psp4.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]