

ABSTRACT
Many anticancer agents exert cytotoxicity and trigger apoptosis through the induction of mitochondrial dysfunction. Mitophagy, as a key mitochondrial quality control mechanism, can remove damaged mitochondria in an effective and timely manner, which may result in drug resistance. Although the implication of mitophagy in neurodegenerative diseases has been extensively studied, the role and mechanism of mitophagy in tumorigenesis and cancer therapy are largely unknown. In a recent study, we found that the inhibition of PINK1-PRKN-mediated mitophagy can significantly enhance the anticancer efficacy of magnolol, a natural product with potential anticancer properties. On the one hand, magnolol can induce severe mitochondrial dysfunction, including mitochondrial depolarization, excessive mitochondrial fragmentation and the generation of mitochondrial ROS, leading to apoptosis. On the other hand, magnolol induces PINK1-PRKN-dependent mitophagy via activation of two rounds of feedforward amplification loops. The blockage of mitophagy through genetic or pharmacological approaches promotes rather than attenuates magnolol-induced cell death. Furthermore, inhibition of mitophagy by using distinct inhibitors targeting different mitophagic stages effectively enhances magnolol’s anticancer efficacy in vivo. Taken together, our findings strongly indicate that manipulation of mitophagy in cancer treatment will be a promising therapeutic strategy for overcoming cancer drug resistance and improving the therapeutic efficacy of anticancer agents.
Mitochondria are well known as the “powerhouse” of cells to provide ATP through aerobic respiration. Paradoxically, mitochondria play a central role in cell death through the regulation of the intrinsic apoptosis pathway. In addition, mitochondria participate in many other fundamental biological processes, such as fatty acid oxidation, ROS production, and calcium balance maintenance. Thus, it is conceivable that dysfunctional mitochondria are implicated in many human diseases, including cancer, neurodegenerative disorders, and heart failure, etc. As a key mitochondrial quality control mechanism, mitophagy can remove dysfunctional mitochondria through the autophagy-lysosome pathway to keep a healthy mitochondrial network. At present, while the implication of mitophagy in neurodegenerative diseases has been extensively studied, the role and mechanism of mitophagy in tumorigenesis and cancer therapy remain a mystery. It is well known that many anticancer agents exert cytotoxicity through induction of mitochondrial damage and the intrinsic apoptosis pathway. Thus, it is possible that cancer cells possibly employ mitophagy to eliminate damaged mitochondria and evade apoptosis, which can lead to drug resistance and therapeutic failure. Therefore, targeting mitophagy and the development of novel mitophagy-based therapeutic intervention may be a promising strategy for overcoming drug resistance and for better cancer therapy.
The anticancer properties of natural products derived from various natural sources have drawn substantial attention in recent years due to their few side effects and reduced toxicity to normal cells and tissues. However, for most conditions, the lack of enough scientific evidence to show their exact underlying molecular mechanism limits the application of such natural products in clinical applications. Magnolol, isolated from the root and stem bark of Magnolia officinalis, has been used as a gentle herb with a long history in traditional medicine. Magnolol exhibits many beneficial properties in clinics, including anti-inflammation, antioxidant activity, anti-neurodegeneration, cardiovascular protection, and hormone regulation. Nowadays, it has even been used as a dietary supplement and ingredient in a variety of health-care products. More noticeably, numerous preclinical studies have shown that magnolol displays potent anticancer activities for various types of cancer, including skin cancer, glioblastoma, non-small cell lung cancer, and breast cancer. However, it is worth noting that magnolol also exhibits cytotoxicity, such as inhibition of cell viability and cell proliferation. Therefore, it is important to explore the underlying molecular mechanism and function of magnolol.
In our study [1], through immunostaining and transmission electron microscopy, we observed that magnolol disrupts the mitochondrial network evidenced by mitochondrial membrane rupture and fragmentation. We also observed that some damaged mitochondria are sequestered within autophagosome-like (double membrane) and autolysosome-like (single membrane) structures. It is well known that the unique compartmentalized structures of mitochondria offer the optimal microenvironment for their energy harvesting and other essential functions. Also, damaged mitochondria are the main source of cellular ROS. Expectedly, our results demonstrated that magnolol markedly reduces mitochondrial membrane potential and increases the production of mitochondrial ROS evidenced by the good colocalization of MitoSOX dye with mitochondria.
To explore the possible regulatory role of magnolol in mitophagy, we first confirmed that magnolol indeed induces autophagy in different types of cancer cells. Interestingly, we found that magnolol treatment results in perinuclear clusters of mitochondria and promotes the distribution of LC3 onto damaged mitochondria. Those effects are similar with those caused by well-known mitophagy inducers, such as CCCP. Next, we utilized two methods, immunoblotting and immunostaining, to demonstrate that magnolol is a novel mitophagy inducer evidenced by the significant degradation of multiple mitochondrial proteins and mitochondrial DNA. Mechanistically, we provided convincing evidence that magnolol induces mitophagy by activating two rounds of feedforward amplification loops: (i) a PINK1-pUb-PRKN positive feedback loop; and (ii) an LC3-OPTN-CALCOCO2/NDP52 positive feedback loop. It remains to be further studied whether magnolol can affect negative regulators of mitophagy, such as USP30 and PTEN-L.
In our study, we noticed that magnolol treatment can cause apoptotic cell death, based on the evidence that such cell death can be attenuated by the caspase inhibitor ZVAD but not by the ferroptosis inhibitor ferrostatin-1 or the necroptosis inhibitor necrostatin-1. Consistently, magnolol treatment causes clear cleavage of CASP9, CASP3 and PARP, the hallmark proteins of apoptosis. Next, we utilized a mouse model by using a physiologically relevant human neuroblastoma SH-SY5Y cell line, and found that magnolol remarkedly inhibits tumor growth and prolongs the survival time of tumor-bearing mice with few side effects. These results are generally consistent with many previous studies. For instance, it has been reported that magnolol inhibits tumor cells via release of AIFM1 (apoptosis inducing factor mitochondria associated 1) or CYCS (cytochrome c, somatic) from the mitochondria to the cytosol. However, there is also evidence suggesting that magnolol induces autophagic cell death rather than apoptotic cell death. Noticeably, we also found that magnolol induces mitophagy in the tumor tissues, which impelled us to explore the function of mitophagy during magnolol’s anticancer process.
In the mitophagy field, due to the lack of specific inhibitors of PINK1-PRKN-mediated mitophagy, the most widely used methods to inhibit mitophagy are either to block lysosomal acidification (bafilomycin A1 or chloroquine) or block autophagosome formation (wortmannin). However, using these inhibitors, we cannot rule out the possibilities that these inhibitors affect general autophagy or other molecular pathways. Thus, we tried to use a relatively specific inhibitor of PINK1 or PRKN in our study. We and others found that Ac220 is possibly a powerful inhibitor for PINK1 and/or PRKN. We found that Ac220 effectively blocks PINK1 accumulation and completely prevents PRKN mitochondrial translocation, finally inhibiting mitophagy. To provide more convincing evidence, we utilized three distinct inhibitors to inhibit mitophagy at different stages, including: (i) wortmannin, a PtdIns3K inhibitor, to block the formation of mitophagosomes; (ii) hydroxychloroquine to inhibit the formation of mitolysosomes; (iii) Ac220 to prevent PINK1 accumulation and PRKN recruitment. In the in vivo animal model, we found that all combined treatments (magnolol-wortmannin, magnolol-hydroxychloroquine or magnolol-Ac220) effectively further reduce tumor growth more than a single agent, which is consistent with our in vitro cell death results.
Our study thus demonstrates that inhibition of mitophagy can significantly enhance the anticancer efficacy of magnolol (Figure 1). As drug resistance has become a major obstacle for cancer treatment, the development of an effective treatment strategy represents one of the most urgent unmet clinical needs for cancer therapy. Our study thus supports the notion that a combination of mitophagy inhibitors with anticancer agents to overcome drug resistance in cancer therapy is an effective approach.
Figure 1.
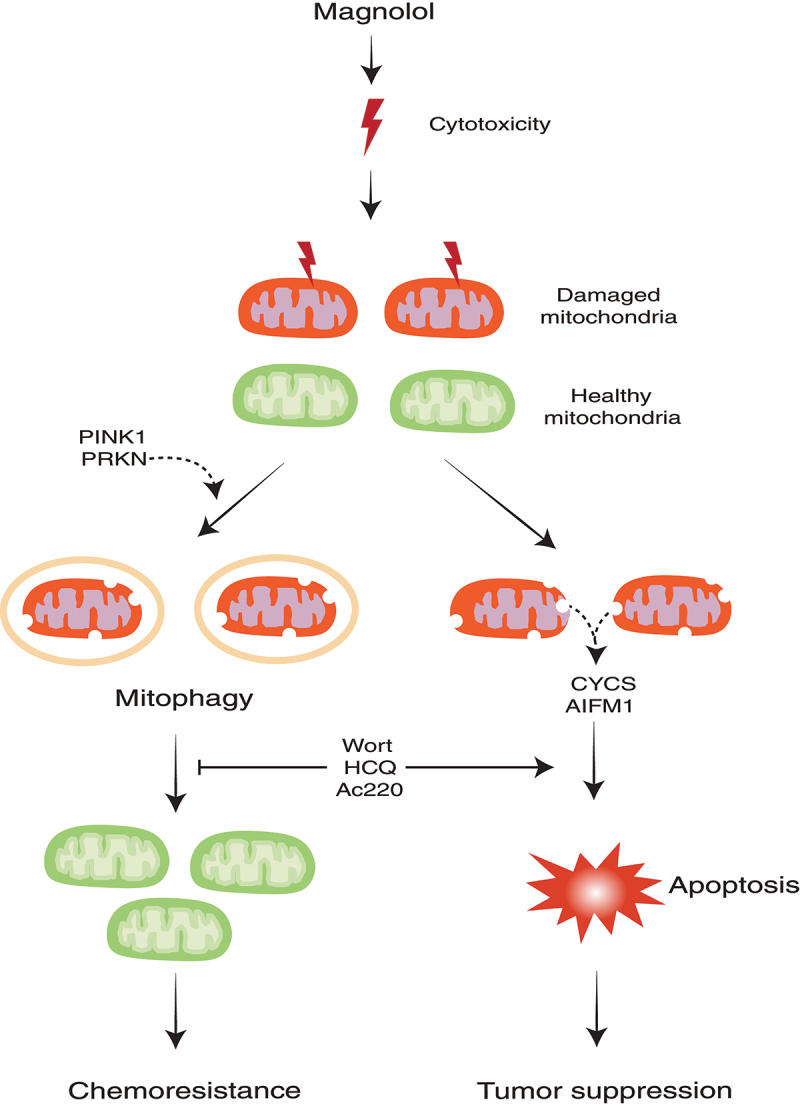
Illustration of how targeting mitophagy promotes apoptosis and improves cancer therapy. HCQ, hydroxychloroquine; Wort, wortmannin.
Funding Statement
This work was supported by research grants from the National Natural Science Foundation of China (82071441 to LMW; 82074123 to HBC), Innovation and Technology Fund (PRP/036/20FX and MHP/023/20) and Health and Medical Research Fund (MHRF-16170251) of Hong Kong to HBC.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Tang Y, Wang L, Yi T, et al. Synergistic effects of autophagy/mitophagy inhibitors and magnolol promote apoptosis and antitumor efficacy. Acta Pharm Sin B. 2021;11(12):3966–3982. DOI: 10.1016/j.apsb.2021.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]