

ABSTRACT
A key component of microbiome research is understanding the role of host genetic influence on gut microbial composition. However, it can be difficult to link host genetics with gut microbial composition because host genetic similarity and environmental similarity are often correlated. Longitudinal microbiome data can supplement our understanding of the relative role of genetic processes in the microbiome. These data can reveal environmentally contingent host genetic effects, both in terms of controlling for environmental differences and in comparing how genetic effects differ by environment. Here, we explore four research areas where longitudinal data could lend new insights into host genetic effects on the microbiome: microbial heritability, microbial plasticity, microbial stability, and host and microbiome population genetics. We conclude with a discussion of methodological considerations for future studies.
KEYWORDS: Gut microbiome, host genetics, time series, host-microbiome interactions, heritability, plasticity
Plain Language Summary
For humans and animals, host genes play a role in shaping the gut microbiome. However, measuring these effects is difficult because host genetic and environmental similarities are often correlated. For instance, relatives often live together and share similar diets and lifestyles—forces that can also change gut microbial communities. Watching the microbiome over time, through longitudinal sampling, can help solve this problem by breaking gene-environment correlations. Here, we review this idea and several other research areas where longitudinal data will be helpful for understanding host genetic effects on the microbiome. We believe this approach will shed new light on the evolution of host–microbe relationships and can inform new microbiome therapies.
Introduction
Gut microbial taxa – including those under host genetic influence – have widespread effects on host health and physical functioning, as reviewed in recent studies.1–3 They help to digest food,4,5 influence host behavior,6,7 and regulate host gene expression,8,9 metabolites,10,11 and immune mechanisms.12 On evolutionary scales, relationships between gut microbes and host genes may facilitate local adaptation to novel environments in humans (as reviewed in 2), shape adaptive phenotypic plasticity across mammalian hosts (as reviewed in 13), affect host genetic divergence between populations,14 and even, some argue, lead to selection of a host and its microbes as a single evolutionary unit.15 Given these evolutionary implications, it may seem surprising that only a minority of microbiome taxa are associated with host genetic variation. For instance, few genetic variants in hosts are consistently identified as having gut microbial correlations across population-specific studies (reviewed in 3). Further, in humans, very few microbial taxa are heritable (an estimated 3–13%, on average; 16–19), and those that are most frequently heritable have widely varying heritability estimates across studies (e.g., Christensenellaceae, one of the most consistently heritable taxa in humans, exhibits heritability ranging from 0.31 to 0.64 20).
A major challenge with estimating microbiome heritability is the difficulty in isolating host genetic effects from other drivers of microbiome variation–especially variation in the environment. Host environments play a strong role in shaping gut microbiome composition,21,22 and genetic similarity between hosts is often confounded by shared environments (e.g., diet and lifestyle 23). As such, even studies with large sample sizes struggle to detect host genetic effects in the face of environmental correlations.3 For example, Gacesa et al. 24 found that across 8,208 individuals from 2,756 family units, only 6.6% of microbes were heritable. In contrast, 48.6% of microbial abundances were affected by cohabitation, but because relatives often cohabitate, the correlation between shared genes and shared housing may have obscured the researchers’ ability to detect heritable microbes.
This complication is exacerbated by the dynamic nature of microbiome community composition within a host over time. For example, a study in which 20 individual hosts were sampled daily for 6 weeks found that variations in the abundances of most microbes were greater within individual hosts than between hosts.25 An additional complication is that genetic effects on dynamic host traits can be themselves dynamic. For example, the heritability of dynamic host traits, such as body mass index (BMI), changes over an individual’s lifetime as the relative importance of environmental and behavioral factors increase compared to genetic effects.26,27 Similar age-related changes in heritability have also been found in the gut microbiome.21
To date, most research on host genetic effects on the gut microbiome relies on cross-sectional data, but time-series data can supplement our understanding of the relative role of genetic processes in the microbiome in ways that cross-sectional data cannot. Broadly, sampling the same individual over time can help to reveal environmentally contingent host genetic effects, both in terms of controlling for environmental differences and in comparing how genetic effects differ by environment. Longitudinal sampling can also minimize cohort effects. For example, 20 note that in a study of age and microbiome composition, the association of Christensenellaceae abundance with age is based on one data point per host, such that if individuals in a certain age class have a similar diet or share other life history traits, the Christensenellaceae result may reflect between-cohort differences rather than a true age-related increase in Christensenellaceae abundance. Sampling the same host multiple times can reveal age-related changes in microbial abundances within individual hosts. Repeated sampling may also help to ameliorate the sample size and power limitations discussed in the past work3.
In this review, we highlight four areas where time-series data are particularly valuable for understanding host genetic effects on the microbiome and the implications of these patterns for evolution and host health. These four areas are (1) heritability of microbial phenotypes, (2) local adaptation and phenotypic plasticity of the microbiome, (3) lifetime stability and dynamism of microbial communities, and (4) microbiome population genetics. Additionally, we provide a roadmap for other researchers by summarizing methods for visualizing and modeling longitudinal microbiome data with associated host genetic information and proposing future directions for the field.
How time series data can inform host genetics effects on the gut microbiome
Microbiome phenotype heritability
Quantifying the heritability of complex, dynamic phenotypes requires parsing environmental effects from genetic effects. In the case of the gut microbiome, it is common to define phenotype heritability as the proportion of variation in either microbiome community composition or the abundances of individual microbial taxa that can be explained by host genetic variance.4,21,24 Larger sample sizes, which are often advocated for in studies of host genetic effects on microbiome composition,24 cannot necessarily overcome the limitation that host genetic similarity is often correlated with environmental similarity.
One approach that can help disentangle gene/environment correlations is to use study designs that leverage longitudinal microbiome samples from many individual hosts. For instance, a study using 16,234 samples from 585 wild baboons found that using multiple samples per host led to a striking increase in the number of heritable microbial taxa detected versus using one sample per host.21 Likewise, including multiple samples per subject over extended time scales can also help break the genetic relatedness/environmental similarity correlation that can affect heritability estimates in cross-sectional designs, as relatives may share environments at a given time point, but undergo individualized changes in their physical and social environments over time. Further, repeated sampling from the same individual removes cohort effects when testing how environments or host traits affect heritability estimates. For example, the relationship between host age and microbiome heritability can be better teased out when, for instance, all four-year-old hosts in the data set were not born in the same calendar year, and thus have dissimilar early life environmental effects. Comparing heritability estimates across environments is particularly important because heritability estimates of individual microbes are environmentally dependent.1 This effect has been demonstrated experimentally in a study of the corn rhizosphere across field sites 28 and in the observational study of baboon gut microbes described above.21 By repeated sampling of individual hosts across changing environments, it is possible to paint a broader picture of the context of the changing relative contributions of additive genetic versus environmental effects on microbial composition (as shown in Figure 1). Similar to sampling across multiple physical environments, sampling a host at multiple daily timepoints could provide an interesting contribution to our understanding of heritability, as several gut microbial taxa demonstrate a marked circadian rhythm in their relative abundances 29 (reviewed in 30).
Figure 1.
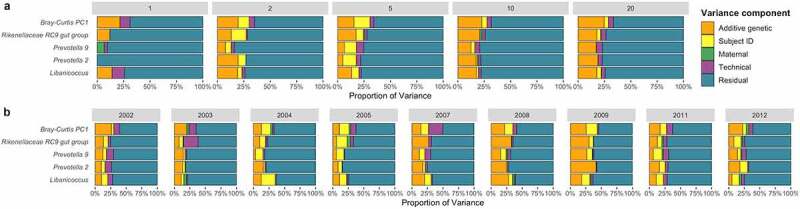
Genetic and non-genetic variance components of the microbiome for four highly heritable taxa and one metric of overall microbiome composition from up to 585 baboon subjects measured (A) at one, two, five, ten, and twenty timepoints per subject; and (B) with timepoints grouped by hydrological year, with varying numbers of timepoints per subject across a mean of 220 subjects per year. These data were generated from a data set published as part of.21 Figure 1A emphasizes that increasing the number of samples per subject affects our ability to detect host genetic effects, while Figure 1B shows that the relative contribution of genetic and environmental components to overall microbiome variance can change over time.
Longitudinal microbiome data are already being used to understand heritability in some health and agricultural applications. For example, questions about the heritability of early life gut colonization and community assembly in humans have been addressed by repeated sampling of multiple sets of triplet infants.31 In agricultural and livestock research, understanding the relationship between host genotype, microbes, and phenotype allows for targeted breeding of desired traits,1 such that parsing host gene-by-environment interactions on host phenotypes, as well as gene-environment-microbiome interactions, is a high priority.32 One such example is a longitudinal study of piglets that determined heritable microbes are associated with growth rates.33 Agricultural and livestock systems also provide great experimental setups for quantifying microbial heritability in changing environmental contexts. For example, planting inbred maize lines in different fields over 2 years revealed host genotype effects on microbiome rhizosphere colonization throughout differing environments.34
Phenotypic plasticity of the gut microbiome
Gut microbiome community composition is a plastic phenotype, but the dynamics of this plasticity are not well understood.35,36 Longitudinal data can reveal if gut microbial dynamics are individualized. Combining this information with data on host genetics could further reveal if host genotype predicts plasticity in the microbiome phenotype. For example, past studies have shown that when hosts undergo a change in their environment by moving to a new area, their microbiomes likewise change as they pick up local microbes.37–39 Further, the rate of local microbial acquisition varies between hosts. Six humans sampled pre-immigration and monthly for up to 9 months post-immigration to the US varied in the length of time; it took them to acquire local microbes, but by 9 months their guts had the same dominant taxa as long-term US residents.37 Similarly, a study of dispersing baboons found that males who had lived in a social group for less than a year varied considerably in how similar their gut microbiota were to other group members, but this variation decreased over time.39 Time-series samples from dispersing primates and other animals that periodically change environments, such as migratory birds, can provide natural experiments to test which environmental characteristics predict if hosts acquire local microbes, the length of time over which such shifts happen, and if the rate of acquisition has a genetic component. Quantifying the rate of change also provides insight into the mechanisms by which hosts acquire local microbes. An immediate change could suggest diet is the primary driver, whereas gradual acquisition of local microbes implicates horizontal transmission from conspecifics or other drivers.
Longitudinal studies could also further our understanding of when a plastic microbiome is adaptive and to what degree host genetics affects microbial plasticity. The ability to acquire the local microbiome may provide key fitness advantages to the host; indeed, studies have shown that bean bugs acquire bacteria from the soil that makes them resistant to pesticides,40 and woodrats acquire microbes that allow them to degrade dietary plant toxins.41 Longitudinal studies on these and other systems could further test local adaptation by experimentally swapping hosts between different environments and testing if fitness improves over time as they acquire the microbes of their new environment. More broadly, longitudinal data could reveal if host-associated microbes shift the mean or variance of a host phenotype. A shift in the mean could lead to a host phenotype better adapted to the current environment, but with less flexibility to adapt to a changing future environment. In contrast, a microbiome that increased variance in a phenotype could lead to increased capacity to adapt to environmental change.42
An additional aspect of microbial plasticity that could be considered in longitudinal designs is the plasticity of individual microbial taxa. Changes in microbial gene expression under different environmental conditions have been well studied in pathogenic bacteria (e.g. Salmonella typhi alters virulence factor expression in response to temperature 43), and temperature-dependent and other environmentally dependent changes in gut microbial gene expression could be a key component of temporal variation in host–microbiome interactions.44
The capacity to adapt to the local environment could start at birth; if there is selection for imperfect vertical transmission of microbes, it could lead to greater microbial variation between offspring, as proposed by.45 Microbial variation between offspring could increase fitness in variable environments, thus increasing the chances that one offspring will be adapted to the environment.45 These ideas could be tested by comparing temporal microbial trajectories between, for example, nestlings from similar hatching dates or pups from the same litter. Finally, longitudinal studies have the potential to provide insight into which taxa fluctuate and which ones are maintained at more constant levels, illuminating the tradeoff between plasticity and stability.
Lifetime stability of gut microbiome phenotypes
Measuring microbiome stability, or the degree of microbiome change within a host over time, requires time-series data by definition. Although individual host signatures on overall microbiome community composition can be detected in samples collected years apart,46 individual taxa components of the microbiome are temporally dynamic, with most microbes demonstrating large fluctuations over days or weeks (as shown in Figure 2).25 There are two main ways that host genetic information can inform our understanding of microbiome stability. First, microbial stability itself could be under host genetic influence; i.e., the magnitude of change in individual microbial taxa over time could be quantified as a phenotype, and heritability could be estimated. Second, incorporating host genetic information into time-series models of microbial stability can provide insight into the mechanisms regulating microbial stability.
Figure 2.
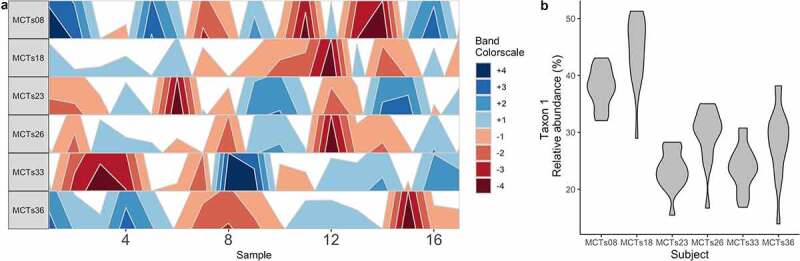
Stability and reproducibility of the microbiome. (A) A horizon plot shows how a taxon’s abundance does not change in consistent ways across time points between individual subjects. Band colors represent quartiles relative to the median. (B) A violin plot depicts the mean ± SD relative abundance of a single taxon in each individual subject, highlighting that stability is personalized. These data are from the demonstration data set from the BiomeHorizon package.47
It is possible that the stability of some microbiome phenotypes has a host genetic component; i.e., stability could be heritable. In other words, host genetics could control the day-to-day change in the microbiome. Although, to the best of our knowledge, this hypothesis has not been tested with real-world data, there are several reasons why maintaining a more stable microbiome can be beneficial for the host. For example, temporal stability of ecosystem functions makes ecosystem services likewise more stable, and therefore reliable;48 a stable microbiome has greater resistance against colonization by invading microbes, including pathogens;49 and major shifts in human microbiome composition are associated with illness.50 However, the microbiome can also settle into an unhealthy stable state, which may contribute to chronic diseases (e.g., insulin resistance and recurrent Clostridium dificile infections) with resistance to medical interventions.49 Longitudinal data sets could be used to test if taxa that demonstrate stability over a host’s lifetime are those with abundances that are already linked to host genetics, i.e., heritable. In support of this, microbiome taxa that have abundances that are highly personalized to individual hosts tend to be more heritable than taxa that exhibit similar abundance patterns across hosts.51 Individual taxon stability could also be measured as genetic stability; 46 studied SNP (single nucleotide polymorphism) haplotypes of microbes and SVs (genomic structural variants) and found smaller differences within than between hosts and that the degree of microbial genetic stability differed between microbes. A recent study3 states that at least one group of bacterial species that are genetically stable over time, Bifidobacterium, also has high heritability. Additionally, there may be selection at the level of gene functions or pathways, rather than at the level of individual microbial taxa, and selection could also occur on pathways or functional roles filled by multiple taxa.52 In support of the idea that hosts and their microbiomes experience selection for some functions to be stable over time, there is a phylogenetic signature for heritability such that taxa that are more closely related–and therefore may have similar functions and occupy similar niches – may exhibit similar host genetic effects.1 Future studies could further explore the link between host genetics and functional stability.
Combining host genetic information with microbiome time-series data and associated environmental metadata can reveal under what circumstances microbes are stable and provide insight into stability mechanisms. Even highly heritable microbial taxa demonstrate considerable variation in abundance over a host’s lifetime,21 and studies have found that the stability of microbiome community composition changes with age, sickness, and other traits.49,53 How individual hosts vary in their degree of microbial stability, which microbes are most affected, and quantifying the degree of environmental versus genetic drivers of this variation at different temporal time scales are key questions for understanding host–microbiome interactions.54 A likely mechanism for host genetic control of microbial stability is changes in host gene expression, as host gene expression differences have been linked to host genetic control of the microbiome.55 Future work could explore if the same changes in gene expression that control microbial taxa abundances at individual time points also regulate the stability of taxa or overall community composition.
Additionally, microbiome time-series data sets that include hormone information, such as those that measure metabolites of steroid hormones in fecal samples,56,57 will be particularly valuable for parsing out a second potential mechanism of lifetime microbial stability. Glucocorticoids have a pervasive genetic component,58–60 and a study on primate microbial endocrinology61 proposes that host–microbiome signaling via the endocrine system may promote microbiome stability. This suggests that one potential mechanism for the host genetic influence on microbial stability is via glucocorticoid levels.
Host and microbe population genetics
Genetic differences between host populations are also often correlated with environmental, social, and spatial characteristics, making it difficult to determine the relative contributions of these factors in shaping local microbiomes.62 Further, due to rapid generation times in bacteria, ecological and evolutionary processes operate at a similar temporal scale and may interact to shape microbial populations.63 Longitudinal data may be useful in teasing apart host population-level drivers of microbial differences by breaking these correlations and to address several unique questions: Is it possible to detect evidence of phylosymbiosis (i.e., when microbial community relationships parallel host species’ evolutionary relationships 64,65) or patterns of co-diversification (i.e., when the taxonomy of specific microbes parallels that of the hosts 66) after controlling for ecological differences? Do host populations demonstrate microbial change at different rates, and how is this rate affected by local landscape characteristics? Similarly, does a population always look like itself, or do environmental shifts cause host-associated microbial communities from temporally and geographically separated host populations to resemble each other?
Time-series data can help place evidence of phylosymbiosis or co-diversification in an ecological context. Past work has shown that the genetic structure of hosts may parallel (or not) the genetic structure of members of their microbial communities. For example, genetically similar stickleback fish populations have more similar microbes,67 but in humans, few microbes show biogeographical patterns that match host biogeography (reviewed in 63). Time-series data are useful because even among microbes whose phylogenies parallel host genetic relationships, environmental traits may modify these signatures. For example, microbiota from oysters in environmentally disturbed sites no longer exhibit genetic relationships that parallel their hosts’ population genetic structure.68 This phenomenon has also been shown experimentally; the microbiotas of woodrats brought into captivity and placed on uniform diets showed stronger host phylogenetic structuring than when they were in the wild, suggesting that signatures of phylosymbiosis on microbial composition may be obscured by strong environmental effects in wild populations.69 Time-series data can also reveal if landscape modification is reflected in microbiome changes. A recent study by Couch et al.62 advocates incorporating spatial elements to model how the landscape interacts with host ecology to influence population-level microbial variation. Past work has shown that geographic and environmental traits structure microbiomes between host populations of mice, tortoises, and baboons.70–72 By sampling host populations over time, as they shift their landscape use, it will be possible to test if changes in home range use parallel changes in microbial communities. Observational studies of long-term wild study systems will be of particular importance in answering these questions, as microbial patterns observed in controlled lab systems (or even captive animals) do not necessarily transfer to what is observed in their wild counterparts.1,73 These questions are also timely, as climate change causes shifts in local environments and in animal ranges.
Methodological considerations: incorporating host genetics into longitudinal microbiome statistics
Incorporating host genetic information into longitudinal microbiome models presents a unique statistical challenge, as the wide range of longitudinal study designs that can test different aspects of host genetic effects (e.g., twin studies, family-based designs, case–control, and prospective cohort studies) have a similarly wide range of statistical approaches. Even within the microbiome-diet subfield, there are no standardized best practices for longitudinal study designs.74 A second complication is that some studies are constrained to irregular sampling intervals, yielding datasets with uneven time between samples. Further, the unit of analysis also varies across studies; in this review, we have focused on repeated sampling of the same individual host, but some longitudinal studies may use cage, inbred line, social group, or population as their level of analysis.
An additional set of complications that especially affect longitudinal microbiome studies are those associated with data collection and processing, which broadly fall under the issue of reproducibility in microbiome studies.75–77 Some common methods of sample preservation do not yield consistent microbiome profiles when subjected to freeze-thaw cycles and other stresses common to fieldwork,75,76 although this can somewhat be ameliorated by treating data as compositional.78 Batch effects must also be controlled for, as samples collected over extended time periods are often not processed at the same time.21,79–81 Approaches to account for batch effects include running technical replicates across multiple plates and including sequencing plates in statistical models,21 and using decontam 82 or other software to identify and remove contaminants between batches.81
Once sequence data are processed, the first step in any longitudinal analysis, whether it includes host genetic information or not, is visualizing which taxa are changing over time. Stream or line graphs are a common approach for visualizing temporal microbial changes for a handful of microbes in a limited number of hosts.83–86 As an alternative, horizon plots can show the temporal dynamics of more taxa in a more condensed visual space. Several R packages provide horizon plot functionality, including CNEr and TSFEL.87,88 BiomeHorizon is the first R package designed to apply horizon plots to microbiome data.47
There are several other software packages in R and other statistical environments that are specifically designed to analyze longitudinal microbiome data, and future studies could explore integrating host genetic information as well. TIME, a web-based interface, guides users through multiple workflows to visualize microbial abundances and co-occurrences over time, and to predict causality (dynamic time warping, Granger causality, and Dickey–Fuller tests)86. Ridenhour et al. propose an autoregressive integrated moving average (ARIMA) time-series model modified to handle microbiome dynamics89. This model uses untransformed count data as the input, and incorporates environmental differences. A study by Wanger et al.90 focus on time-varying analyses of overall changes in microbial diversity. Chen and Li91ʹproposed ZIBR (two-part zero-inflated beta regression model with random effects) model accounts for the high number of zeros in longitudinal data. SynTracker is designed for the level of microbial strains92,93 and provides a user-friendly walkthrough of microbiome time-series analyses, as well as R and matlab tutorials. For a more detailed discussion of statistical challenges and approaches in analyzing longitudinal microbiome data, see the thorough review on the topic by Kodikara et al.94
Host relatedness data may also be integrated into other common models for measuring temporal variation in ecological systems, including generalized Lotka–Volterra equations,95 multispecies time-series data using first-order multivariate autoregressive (MAR(1)) models 96), generalized additive models (GAMs),51 and the stochastic logistic model with environmental noise (SLM).97
To incorporate longitudinal data into heritability models, the Animal Model includes options for multiple samples per individual.21,98,99 The ACE model, which is often used in twin studies, can be modified for repeated measures.100 Quantitative genetics models can also be extended to include genetic effects on the host and genetics effects on the microbiome.42 Bayesian models could also be used, including R packages for calculating heritability estimates from generalized linear mixed-effects models using a Markov chain Monte Carlo approach (MCMCglmm and QCglmm; 101,102). To address the issue of compositionality that is inherent to microbiome studies and has the potential to affect heritability estimates, we recommend permuting host identity in any heritability models (e.g., Fig. S10 in a study by Grieneisen et al.21).
Conclusion
The incorporation of host genetic data into longitudinal microbiome studies opens up many exciting directions in the microbiome field. The future work will leverage a combination of existing collections, natural history studies of human and wild animal populations, and controlled experimental approaches.103–105 Further, technical advances will improve our ability to characterize temporal changes in host gene–microbiome interactions. For example, database improvements with more GWAS studies will allow for more functional gene annotations.63 Although this review focused on the gut microbiome, incorporation of host genetic effects into longitudinal studies of other bodysite microbiomes, such as the oral microbiome,106,107 skin,108,109 and vagina,110 can provide similarly exciting insights into links between microbial dynamics and host health.
In addition to the four topic areas we focused on in this review, several recent articles highlight crucial areas where longitudinal microbiome data can contribute.63,104,111–113 We point out that many of these areas also have host genetic components. These include quantifying disease risk;113 understanding intra-individual variation;112 linking early life effects with lifetime microbiome consequences;104,111 matching microbial and host aging, modeling social dynamics and transmission, uncovering which microbial taxa are heritable, and linking microbial dynamics to host fitness;104 and modeling gene recombination rates and characteristics of microbes under selection.63
The results from such studies have broad real-world applications to human health. Detailing the relationship between the gut microbiome and host genetics could be important for translational medicine applications 114 and personalized therapeutics.103 Understanding temporal dynamics and host genotype effects could help contribute to our understanding of the stable colonization of therapeutic microbes.63 Further, they could contribute to the expanding field of microbiome breeding, defined as conducting artificial selection on microbiomes that “seeks to change the genetic composition of microbiomes in order to benefit plant or animal hosts”115.
Funding Statement
This work was supported by the Grand Challenges in Biology Postdoctoral Fellowship to LG; ASPIRE Program Fund to LG; National Institute on Aging under Grant R01 AG071684 to EA; National Science Foundation under Grant DEB 1840223 to EA; and by the National Institute of General Medical Sciences under Grant R35 GM128716 to RB.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.EP R, Davenport ER.. Host genetic determinants of the microbiome across animals: from Caenorhabditis elegans to cattle. Annu Rev Anim Biosci. 2022;10:203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki TA, Ley RE. The role of the microbiota in human genetic adaptation. Science. 2020;370:6521. doi: 10.1126/science.aaz6827. [DOI] [PubMed] [Google Scholar]
- 3.Sanna S, Kurilshikov A, van der Graaf A, Fu J, Zhernakova A. Challenges and future directions for studying effects of host genetics on the gut microbiome. Nat Genet. 2022;54:100–106. [DOI] [PubMed] [Google Scholar]
- 4.Goodrich JK, Davenport ER, Clark AG, Ley RE. The relationship between the human genome and microbiome comes into view. Annu Rev Genet. 2017;51:413–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin Y, Havulinna AS, Liu Y, Jousilahti P, Ritchie SC, Tokolyi A, Sanders JG, Valsta L, Brożyńska M, Zhu Q, et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat Genet. 2022;54(2):134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–1412. [DOI] [PubMed] [Google Scholar]
- 7.Buffington SA, Dooling SW, Sgritta M, Noecker C, Murillo OD, Felice DF, Turnbaugh PJ, Costa-Mattioli M. Dissecting the contribution of host genetics and the microbiome in complex behaviors. Cell. 2021;184:1740–1756.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grieneisen L, Muehlbauer AL, Blekhman R. Microbial control of host gene regulation and the evolution of host-microbiome interactions in primates. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards AL, Muehlbauer AL, Alazizi A, Burns MB, Findley A, Messina F, Gould TJ, Cascardo C, Pique-Regi R, Blekhman R, et al. Gut microbiota has a widespread and modifiable effect on host gene regulation. mSystems. 2019;4:5. doi: 10.1128/mSystems.00323-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kemis JH, Linke V, Barrett KL, Boehm FJ, Traeger LL, Keller MP, Rabaglia ME, Schueler KL, Stapleton DS, Gatti DM, et al. Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genet. 2019;15(8):e1008073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar N, Korem T, Weissbrod O, Zeevi D, Rothschild D, Leviatan S, Kosower N, Lotan-Pompan M, Weinberger A, Le Roy CI, et al. A reference map of potential determinants for the human serum metabolome. Nature. 2020;588(7836):135–140. [DOI] [PubMed] [Google Scholar]
- 12.Khan AA, Yurkovetskiy L, O’Grady K, Pickard JM, de Pooter R, Antonopoulos DA, Golovkina T, Chervonsky A. Polymorphic immune mechanisms regulate commensal repertoire. Cell Rep. 2019;29:541–550.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moeller AH, Sanders JG. Roles of the gut microbiota in the adaptive evolution of mammalian species. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudman SM, Greenblum S, Hughes RC, Rajpurohit S, Kiratli O, Lowder DB, Lemmon SG, Petrov DA, Chaston JM, Schmidt P. Microbiome composition shapes rapid genomic adaptation of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2019;116:20025–20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zilber-Rosenberg I, Rosenberg E. Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev. 2008;32:723–735. [DOI] [PubMed] [Google Scholar]
- 16.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, Ley RE. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y. Genome-Wide association studies of the human gut microbiota. PLoS One. 2015;10:e0140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48(11):1413–1417. [DOI] [PubMed] [Google Scholar]
- 20.Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grieneisen L, Dasari M, Gould TJ, Björk JR, Grenier J-C, Yotova V, Jansen D, Gottel N, Gordon JB, Learn NH, et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science. 2021;373(6551):181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 23.Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gacesa R, Kurilshikov A, Vich Vila A, Sinha T, Klaassen MAY, Bolte LA, Andreu-Sánchez S, Chen L, Collij V, Hu S, et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature. 2022. 1–8 [DOI] [PubMed] [Google Scholar]
- 25.Vandeputte D, De Commer L, Tito RY, Kathagen G, Sabino J, Vermeire S, Faust K, Raes J. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat Commun. 2021;12:6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet. 2008;9:255. [DOI] [PubMed] [Google Scholar]
- 27.Ge T, Chen C-Y, Neale BM, Sabuncu MR, Smoller JW. Phenome-wide heritability analysis of the UK Biobank. PLoS Genet. 2017;13:e1006711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions PNAS. 2013 110(16):6548. doi: 10.1073/pnas.1302837110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, Abramson L, Katz MN, Korem T, Zmora N, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell. 2014;159(3):514–529. [DOI] [PubMed] [Google Scholar]
- 30.Voigt RM, Forsyth CB, Green SJ, Engen PA, Keshavarzian A. Chapter Nine - circadian rhythm and the gut microbiome. In: Cryan JF, Clarke G editors. International review of neurobiology. Vol. 131. : Academic Press; 2016. p. 193–205. [DOI] [PubMed] [Google Scholar]
- 31.Palmeira O, Matos LRB, Naslavsky MS, Bueno HMS, Soler JP, Setubal JC, Zatz M. Longitudinal 16S rRNA gut microbiota data of infant triplets show partial susceptibility to host genetics. iScience. 2022;25:103861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oyserman BO, Cordovez V, Flores SS, Leite MFA, Nijveen H, Medema MH, Raaijmakers JM. Extracting the GEMs: genotype, environment, and microbiome interactions shaping host phenotypes. Front Microbiol. 2020;11:574053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergamaschi M, Maltecca C, Schillebeeckx C, McNulty NP, Schwab C, Shull C, Fix J, Tiezzi F. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Sci Rep. 2020;10:10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walters WA, Jin Z, Youngblut N, Wallace JG, Sutter J, Zhang W, González-Peña A, Peiffer J, Koren O, Shi Q, et al. 2018. Large-scale replicated field study of maize rhizosphere identifies heritable microbes PNAS . 2018;115(28):7368–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolodny O, Schulenburg H. Microbiome-mediated plasticity directs host evolution along several distinct time scales. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alberdi A, Aizpurua O, Bohmann K, Zepeda-Mendoza ML, Gilbert MTP. Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol Evol. 2016;31:689–699. [DOI] [PubMed] [Google Scholar]
- 37.Vangay P, Johnson AJ, Ward TL, Al-Ghalith GA, Shields-Cutler RR, Hillmann BM, Lucas SK, Beura LK, Thompson EA, Till LM. US immigration westernizes the human gut microbiome. Cell. 2018; 175(4):962–972.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomez A, Sharma AK, Mallott EK, Petrzelkova KJ, Jost Robinson CA, Yeoman CJ, Carbonero F, Pafco B, Rothman JM, Ulanov A, et al. Plasticity in the human gut microbiome defies evolutionary constraints. mSphere. 2019;4(4):e00271–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grieneisen LE, Livermore J, Alberts S, Tung J, Archie EA. Group living and male dispersal predict the core gut microbiome in wild baboons. Integr Comp Biol. 2017;57:770–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itoh H, Hori T, Sato Y, Nagayama A, Tago K, Hayatsu M, Kikuchi Y. Infection dynamics of insecticide-degrading symbionts from soil to insects in response to insecticide spraying. ISME J. 2018;12:909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohl KD, Dearing MD. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front Microbiol. 2016:7. doi: 10.3389/fmicb.2016.01165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henry LP, Bruijning M, Forsberg SKG, Ayroles JF. The microbiome extends host evolutionary potential. Nat Commun. 2021;12:5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brewer SM, Twittenhoff C, Kortmann J, Brubaker SW, Honeycutt J, Massis LM, Pham THM, Narberhaus F, Monack DM. A Salmonella Typhi RNA thermosensor regulates virulence factors and innate immune evasion in response to host temperature. PLoS Pathog. 2021;17:e1009345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hör J, Gorski SA, Vogel J. Bacterial RNA Biology on a Genome Scale. Mol Cell. 2018;70:785–799. [DOI] [PubMed] [Google Scholar]
- 45.Bruijning M, Henry LP, Forsberg SKG, Metcalf CJE, Ayroles JF. Natural selection for imprecise vertical transmission in host–microbiota systems. Nature Ecology & Evolution. 2021;6:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Wang D, Garmaeva S, Kurilshikov A, Vich Vila A, Gacesa R, Sinha T, Study LC, Segal E, Weersma RK, et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell. 2021. doi: 10.1016/j.cell.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 47.Fink I, Abdill RJ, Blekhman R, Grieneisen L. BiomeHorizon: visualizing microbiome time series data in R. mSystems. 2022. doi: 10.1128/msystems.01380-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilcox KR, Tredennick AT, Koerner SE, Grman E, Hallett LM, Avolio ML, La Pierre KJ, Houseman GR, Isbell F, Johnson DS, et al. Asynchrony among local communities stabilises ecosystem function of metacommunities. Ecol Lett. 2017;20(12):1534–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fassarella M, Blaak EE, Penders J, Nauta A, Smidt H, Zoetendal EG. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2020. doi: 10.1136/gutjnl-2020-321747. [DOI] [PubMed] [Google Scholar]
- 50.Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663. [DOI] [PubMed] [Google Scholar]
- 51.Björk JR, Dasari MR, Roche K, Grieneisen L, Gould TJ, Grenier J-C, Yotova V, Gottel N, Jansen D, Gesquiere LR, et al. Synchrony and idiosyncrasy in the gut microbiome of wild baboons. Nature Ecology & Evolution 6 . 2022;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doolittle WF, Booth A. It’s the song, not the singer: an exploration of holobiosis and evolutionary theory. Biol Philos. 2017;32:5–24. [Google Scholar]
- 53.Chen DW, Garud NR. Rapid evolution and strain turnover in the infant gut microbiome. bioRxiv. 2021; accessed 2021 Oct 4. doi: 10.1101/2021.09.26.461856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bubier JA, Chesler EJ, Weinstock GM. Host genetic control of gut microbiome composition. Mamm Genome. 2021;32:263–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gesquiere LR, Pugh M, Alberts SC, Markham AC. Estimation of energetic condition in wild baboons using fecal thyroid hormone determination. Gen Comp Endocrinol. 2018;260:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foley CAH, Papageorge S, Wasser SK. Noninvasive stress and reproductive measures of social and ecological pressures in free-ranging African elephants. Conserv Biol. 2001;15:1134–1142. [Google Scholar]
- 58.Stedman JM, Hallinger KK, Winkler DW, Vitousek MN. Heritable variation in circulating glucocorticoids and endocrine flexibility in a free-living songbird. J Evol Biol. 2017;30:1724–1735. [DOI] [PubMed] [Google Scholar]
- 59.Tucker-Drob EM, Grotzinger AD, Briley DA, Engelhardt LE, Mann FD, Patterson M, Kirschbaum C, Adam EK, Church JA, Tackett JL, et al. Genetic influences on hormonal markers of chronic hypothalamic-pituitary-adrenal function in human hair. Psychol Med. 2017;47:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jenkins BR, Vitousek MN, Hubbard JK, Safran RJ. 2014. An experimental analysis of the heritability of variation in glucocorticoid concentrations in a wild avian population Proceedings of the Royal Society B. 281(1790):20141302. doi: 10.1098/rspb.2014.1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benavidez KM, Iruri-Tucker A, Steiniche T, Wasserman MD. Primate microbial endocrinology: an uncharted frontier. Am J Primatol. 2019;81(10-11:e23053. [DOI] [PubMed] [Google Scholar]
- 62.Couch CE, Epps CW. Host, microbiome, and complex space: applying population and landscape genetic approaches to gut microbiome research in wild populations. J Hered. 2022. doi: 10.1093/jhered/esab078. [DOI] [PubMed] [Google Scholar]
- 63.Garud NR, Pollard KS. Population Genetics in the Human Microbiome. Trends Genet. 2020;36:53–67. [DOI] [PubMed] [Google Scholar]
- 64.Lim SJ, Bordenstein SR. An introduction to phylosymbiosis. Proc Biol Sci. 2020;287:20192900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theis KR, Dheilly NM, Klassen JL, Brucker RM, Baines JF, Bosch TCG, Cryan JF, Gilbert SF, Goodnight CJ, Lloyd EA, et al. Getting the hologenome concept right: an eco-evolutionary framework for hosts and their microbiomes. mSystems [Internet]. 2016;1:2. doi: 10.1128/mSystems.00028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kohl KD. Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith CCR, Snowberg LK, Gregory Caporaso J, Knight R, Bolnick DI. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015. doi: 10.1038/ismej.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wegner KM, Volkenborn N, Peter H, Eiler A. Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 2013;13:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinstein, SB , Martinez-Mota, R , Stapleton, TE , Klure, DM , Greenhalgh, R , Orr, TJ , Dale, C , Kohl, K , Dearing, MD. 2022. Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp.). PNAS 118;47: e2108787118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linnenbrink M, Wang J, Hardouin EA, Kunzel S, Metzler D, Baines JF. The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol Ecol. 2013;22:1904–1916. [DOI] [PubMed] [Google Scholar]
- 71.Yuan ML, Dean SH, Longo AV, Rothermel BB, Tuberville TD, Zamudio KR. Kinship, inbreeding and fine-scale spatial structure influence gut microbiota in a hindgut-fermenting tortoise. Mol Ecol. 2015;24:2521–2536. [DOI] [PubMed] [Google Scholar]
- 72.Grieneisen LE, Charpentier MJE, Alberts SC, Blekhman R, Bradburd G, Tung J, Archie EA. 2019. Genes, geology and germs: gut microbiota across a primate hybrid zone are explained by site soil properties, not host species Proceedings of the Royal Society B . . 286(1901):20190431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hird SM. Evolutionary biology needs wild microbiomes. Front Microbiol. 2017;8:725. doi: 10.3389/fmicb.2017.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson AJ, Zheng JJ, Kang JW, Saboe A, Knights D, Zivkovic AM. A guide to diet-microbiome study design. Front Nutr. 2020;7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schloss PD. Identifying and overcoming threats to reproducibility, replicability, robustness, and generalizability in microbiome research. MBio. 2018;9:3. doi: 10.1128/mBio.00525-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song SJ, Amir A, Metcalf JL, Amato KR, Xu ZZ, Humphrey G, Knight R. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems. 2016;1(3). http://msystems.asm.org/content/1/3/e00021-16.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berlow M, Kohl KD, Derryberry EP. Evaluation of non‐lethal gut microbiome sampling methods in a passerine bird. Ibis. 2020;162:911–923. [Google Scholar]
- 78.Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ. It’s all relative: analyzing microbiome data as compositions. Ann Epidemiol. 2016;26:322–329. [DOI] [PubMed] [Google Scholar]
- 79.Gibbons SM, Duvallet C, Alm EJ. Correcting for batch effects in case-control microbiome studies. PLoS Comput Biol. 2017;14:e1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goh WWB, Wang W, Wong L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol. 2017;35:498–507. [DOI] [PubMed] [Google Scholar]
- 81.Moossavi S, Fehr K, Khafipour E, Azad MB. Repeatability and reproducibility assessment in a large-scale population-based microbiota study: case study on human milk microbiota. Microbiome. 2021;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Personalized Microbiome Class Students, et al. Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe. 2019;25(6):789–802.e5. [DOI] [PubMed] [Google Scholar]
- 84.David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, Alm EJ. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014;15:R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Turroni S, Rampelli S, Biagi E, Consolandi C, Severgnini M, Peano C, Quercia S, Soverini M, Carbonero FG, Bianconi G, et al. Temporal dynamics of the gut microbiota in people sharing a confined environment, a 520-day ground-based space simulation, MARS500. Microbiome. 2017;5(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baksi KD, Kuntal BK, Mande SS. “TIME”: a web application for obtaining insights into microbial ecology using longitudinal microbiome data. Front Microbiol. 2018;9:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barandas M, Folgado D, Fernandes L, Santos S, Abreu M, Bota P, Liu H, Schultz T, Gamboa H. TSFEL: Time Series Feature Extraction Library. SoftwareX. 2020;11:100456. [Google Scholar]
- 88.Tan G, Polychronopoulos D, Lenhard B. CNEr: a toolkit for exploring extreme noncoding conservation. PLoS Comput Biol. 2019;15:e1006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ridenhour BJ, Brooker SL, Williams JE, Van Leuven JT, Miller AW, Dearing MD, Remien CH. Modeling time-series data from microbial communities. ISME J. 2017;11:2526–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wagner BD, Grunwald GK, Zerbe GO, Mikulich-Gilbertson SK, Robertson CE, Zemanick ET, Harris JK. On the use of diversity measures in longitudinal sequencing studies of microbial communities. Front Microbiol. 2018;9:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen EZ, Li H. A two-part mixed-effects model for analyzing longitudinal microbiome compositional data. Bioinformatics. 2016;32:2611–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Enav H, Ley RE. SynTracker: a synteny based tool for tracking microbial strains. bioRxiv. 2021; accessed 2021 Oct 11. doi: 10.1101/2021.10.06.463341. [DOI] [Google Scholar]
- 93.Coenen AR, Hu SK, Luo E, Muratore D, Weitz JS. A primer for microbiome time-series analysis. Front Genet. 2020;11:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kodikara S, Ellul S, Lê Cao K-A. Statistical challenges in longitudinal microbiome data analysis. Brief Bioinform. 2022;23:4. doi: 10.1093/bib/bbac273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stein RR, Bucci V, Toussaint NC, Buffie CG, Rätsch G, Pamer EG, Sander C, Xavier JB. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput Biol. 2013;9:e1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ives AR, Dennis B, Cottingham KL, Carpenter SR. Estimating community stability and ecological interactions from time-series data. Ecol Monogr. 2003;73:301–330. [Google Scholar]
- 97.Zaoli S, Grilli J. A macroecological description of alternative stable states reproduces intra- and inter-host variability of gut microbiome. Cold Spring Harbor Laboratory. 2021; accessed 2021 Feb 18. doi: 10.1101/2021.02.12.430897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.AJ Wilson, Réale D, MN Clements, MM Morrissey, Postma E, CA Walling, LEB Kruuk, DH Nussey. An ecologist’s guide to the animal model. J Anim Ecol. 2010;79(1):13–26. [DOI] [PubMed] [Google Scholar]
- 99.Thomson CE, Winney IS, Salles O, Pujol B. A guide to using a multiple-matrix animal model to disentangle genetic and nongenetic causes of phenotypic variance. PLoS One. 2018;13:e0197720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Llewellyn CH, Trzaskowski M, Plomin R, Wardle J. From modeling to measurement: developmental trends in genetic influence on adiposity in childhood. Obesity. 2014;22:1756–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.de Villemereuil P, Schielzeth H, Nakagawa S, Morrissey M. General methods for evolutionary quantitative genetic inference from generalized mixed models. Genetics. 2016;204:1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Villemereuil P. Quantitative genetic methods depending on the nature of the phenotypic trait. Ann N Y Acad Sci. 2018;1422:29–47. [DOI] [PubMed] [Google Scholar]
- 103.Yassour M, Vatanen T, Siljander H, A-M H, Härkönen T, Ryhänen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8:343ra81–343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Björk JR, Dasari M, Grieneisen L, Archie EA. Primate microbiomes over time: longitudinal answers to standing questions in microbiome research. Am J Primatol. 81 2019;e22970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trosvik P, de Muinck EJ. Ecology of bacteria in the human gastrointestinal tract—identification of keystone and foundation taxa. Microbiome. 2015;3:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Freire M, Moustafa A, Harkins DM, Torralba MG, Zhang Y, Leong P, Saffery R, Bockmann M, Kuelbs C, Hughes T, et al. Longitudinal study of oral microbiome variation in twins. Sci Rep. 2020;10(1):7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Demmitt BA, Corley RP, Huibregtse BM, Keller MC, Hewitt JK, McQueen MB, Knight R, McDermott I, Krauter KS. Genetic influences on the human oral microbiome. BMC Genomics. 2017;18:659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Si J, Lee S, Park JM, Sung J, Ko G. Genetic associations and shared environmental effects on the skin microbiome of Korean twins. BMC Genomics. 2015;16:992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moitinho-Silva L, Degenhardt F, Rodriguez E, Emmert H, Juzenas S, Möbus L, Uellendahl-Werth F, Sander N, Baurecht H, Tittmann L, et al. Host genetic factors related to innate immunity, environmental sensing and cellular functions are associated with human skin microbiota. Nat Commun. 2022;13(1):6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wright ML, Fettweis JM, Eaves LJ, Silberg JL, Neale MC, Serrano MG, Jimenez NR, Prom-Wormley E, Girerd PH, Borzelleca JF Jr, et al. Vaginal microbiome Lactobacillus crispatus is heritable among European American women. Commun Biol. 2021;4(1):872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martin R, Makino H, Cetinyurek Yavuz A, Ben-Amor K, Roelofs M, Ishikawa E, Kubota H, Swinkels S, Sakai T, Oishi K, et al. Early-Life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS One. 2016;11(6):e0158498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Olsson LM, Boulund F, Nilsson S, Khan MT, Gummesson A, Fagerberg L, Engstrand L, Perkins R, Uhlén M, Bergström G, et al. Dynamics of the normal gut microbiota: a longitudinal one-year population study in Sweden. Cell Host Microbe. 2022. doi: 10.1016/j.chom.2022.03.002. [DOI] [PubMed] [Google Scholar]
- 113.Mai V, Morris JG Jr. Need for prospective cohort studies to establish human gut microbiome contributions to disease risk. J Natl Cancer Inst. 2013;105:1850–1851. [DOI] [PubMed] [Google Scholar]
- 114.Luca F, Kupfer SS, Knights D, Khoruts A, Blekhman R. Functional genomics of host-microbiome interactions in humans. Trends Genet. 2018;34:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mueller UG, Linksvayer TA. Microbiome breeding: conceptual and practical issues. Trends Microbiol. 2022. doi: 10.1016/j.tim.2022.04.003. [DOI] [PubMed] [Google Scholar]