

ABSTRACT
Gerstmann–Sträussler–Scheinker (GSS) disease is an autosomal dominant neurodegenerative disease, and it is characterized by progressive cerebellar ataxia. Up to now, GSS cases with the p.P102L mutation have mainly been reported in Caucasian, but rarely in Asian populations. A 54-year-old female patient presented with an unstable gait in the hospital. Last year, she was unable to walk steadily and occasionally choked, could not even walk independently gradually. After taking her medical history, we found that she was misdiagnosed with schizophrenia before the gait problems. The patient’s father showed similar symptoms and was diagnosed with brain atrophy at the age of 56, but her daughter showed no similar symptoms at present. On arrival at the Neurology Department, the patient’s vital signs and laboratory examinations showed no abnormality. As the proband presented with cerebellar ataxia and had an obvious family history, we were sure that she had hereditary cerebellar ataxia. Then, patient’s brain MRI showed an abnormal signal in the right parietal cortex and bilateral small ischaemic lesions in the frontal lobe. A gene panel (including 142 ataxia-related genes) was performed, and a heterozygous mutation PRNP Exon2 c.305C>T p. (Pro102Leu) was identified. Her daughter had the same heterozygous mutation. The patient was diagnosed with GSS with mental disorders as initial symptoms. After 2 months of TCM treatment, the patient’s walking instability decreased, and her emotional fluctuations were less than before. In conclusion, we have reported a rare case of GSS in Sichuan, China, and the family with mental disorder as the first symptom was finally confirmed with GSS PRNP P102L mutation.
KEYWORDS: Gerstmann–Sträussler–Scheinker disease, PRNP, P102L mutation, mental disorder
1. Introduction
Gerstmann–Sträussler–Scheinker (GSS) disease is an autosomal dominant neurodegenerative disease, which is characterized by progressive cerebellar ataxia, dementia, Parkinsonism, and pyramidal signs [1]. GSS results from mutations in the prion protein (PRNP) gene, of which P102L mutation is the commonest mutation in GSS cases worldwide [2,3]. Patients with P102L mutation usually exhibit a classic onset of cerebellar ataxia in midlife, while it is difficult to distinguish GSS mental symptoms from other diseases clinically at the early stage of the disease [4].
GSS is a rare progressive fatal condition with an incidence rate of 1–100 per 100 million [5]. Moreover, epidemiological data has indicated that the onset age of GSS patients varies from 30 to 60 years, and the course of the disease ranges from 3.5 to 9.5 years [6]. Unfortunately, there is no effective treatment for GSS currently, thus prompt detection and early diagnosis are crucial to ameliorate the later life of GSS patients.
Herein, we reported a rare family GSS case from Sichuan, China. The patient was originally showed mental disorders as the initial symptoms, and she was finally diagnosed with GSS PRNP P102L mutation via an ataxia-related gene panel test. Our study is expected to give more reference information for the detection and diagnosis of the rare disease in the clinical work.
2. Case presentation
A 54-year-old female patient from Sichuan Province presented with an unstable gait. Last year, she was unable to walk steadily and occasionally choked. About a half year ago, she was diagnosed with chronic ischaemia after brain computerized tomography (CT) and an electroencephalography (EEG) at a local hospital, whereas her symptoms were not alleviated after treatment with conventional Western medicine, such as mecobalamin. Subsequently, she was unable to walk independently.
During taking her medical history, according to her daughter’s telling, the patient’s personality had already changed significantly before the onset of gait instability, for instance reduced speech and a lack of expression. Subsequently, she was misdiagnosed with schizophrenia by a psychiatrist. She did not complain of memory loss. She had free history of frequent diarrhoea, the common cold, or drug/food allergies. She was not an alcohol drinker or smoker. Exposure to toxic agents was also excluded. In addition, she had not taken any medication for a long time.
The patient’s parents were non-consanguineous. Her father had similar symptoms and had been diagnosed with brain atrophy at another hospital at the age of 56, but the cause of these symptoms was unknown. He had died when he was 60 years old. Her daughter had no similar symptoms at present, but we gave her a genetic test due to her family history (Figure 1, Table 1).
Figure 1.
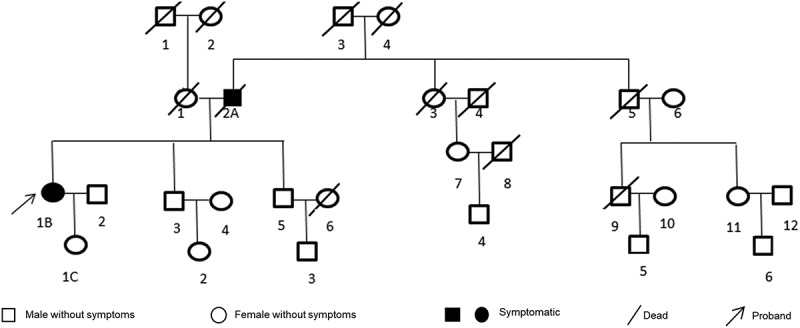
Family tree of the patient. The proband in this family was the present patient (1b). Her father (2a) used to have similar symptoms and dead at 60 years old. Her daughter (1c) have no similar symptom up to now.
Table 1.
Clinical data of the patient’s family.
| Patient | Age of onset (years) | Course of disease (years) | First symptom | Main clinical manifestations | Related inspection | Pathology | Genetic analysis |
|---|---|---|---|---|---|---|---|
| 2A (father) | 56 | 4 | Psychiatric symptoms (depression, hallucinations) | Unstable gait, dysarthria, dysphagia, muscle atrophy of both lower extremities, advanced severe dementia, and two obstacles | Unknown | Not done | Not done |
| 1B (proband) | 54 | ≥1 | Psychiatric symptoms (depression, emotional fluctuations) | Unstable gait, psychiatric symptoms, dementia, and dysfunction in recent follow-up | See case data for details | Not done | P102L mutation in PRNP gene |
| 1C (daughter) | 32 (Currently without symptoms) | – | – | – | – | – | P102L mutation in PRNP gene |
On arrival at the Neurology Department, the patient’s vital signs showed no abnormality. After a comprehensive neurological examination, her abnormal signs included a slight decrease in muscle tone in the inferior, a wide-based unsteady gait, and difficulty with the left finger–nose test and bilateral heel–knee–shin test. Rapid alternating movements of the hands were clumsy. Romberg’s test indicated her abnormality when walking with the eyes open and closed. Deep tendon reflexes were exaggerated in the upper limbs. Babinski’s sign was positive for the left foot, and other pathological reflexes were negative.
No obvious abnormalities were observed in laboratory examinations, including routine blood tests, blood biochemistry, coagulation indices, routine urine tests, stool tests, glycated haemoglobin, thyroid function, and tumour markers. Antibodies for common autoimmune diseases, including autoimmune encephalitis and paraneoplastic syndromes, were also negative. In addition, an electrocardiogram and cerebrospinal fluid tests were both normal. No abnormalities in muscles and nerves were detected by electromyography. Blood lipids were listed as below: cholesterol: 5.85 mmol/L and low-density lipoprotein cholesterol: 3.67 mmol/L. The patient had a total score of 21 points on the Mini-Mental State Examination (MMSE) and 20 points on the Montreal Cognitive Assessment (MOCA). The Hamilton Anxiety Scale and Hamilton Depression Scale assessments showed moderate anxiety and depression. There was no abnormality in the head CT scan performed outside the hospital half a year ago.
As the proband presented with cerebellar ataxia and had an obvious family history, we confirmed that she had hereditary cerebellar ataxia. Her symptoms were not alleviated after administration of a routine neurotrophic drug (methylcobalamin) and a circulation-improving drug (butylphthalide). On the seventh day after admission, patient’s brain magnetic resonance imaging (MRI) showed an abnormal signal in the right parietal cortex and bilateral small ischaemic lesions in the frontal lobe (Figure 2A, B, C). Based on the abnormal manifestations of brain MRI, we further improved the EEG examination, and diffuse slow waves with sharp waves and three-phase waves were observed (Figure 3). Then, the patient insisted to be discharged. However, in order to further clarify the specific classification of the disease in combination with the brain MRI and EEG results, we analysed a gene panel (Kingmed Diagnostics, Sichuan, China), comprising direct sequencing of 142 genes relating to common and rare hereditary forms of ataxia. We identified a heterozygous mutation, namely PRNP Exon2 c.305C>T p. (Pro102Leu) (Figure 4), which presented in GSS. Hence, the patient was eventually diagnosed with GSS. Considering the patient’s genetic mutation, a genetic analysis was also performed on her daughter and her daughter also had the same heterozygous mutation. The patient and her family refused to undergo a brain biopsy. Her two brothers and their children had not shown symptoms of cerebellar ataxia up to now, but they refused to undergo a PRNP gene analysis.
Figure 2.
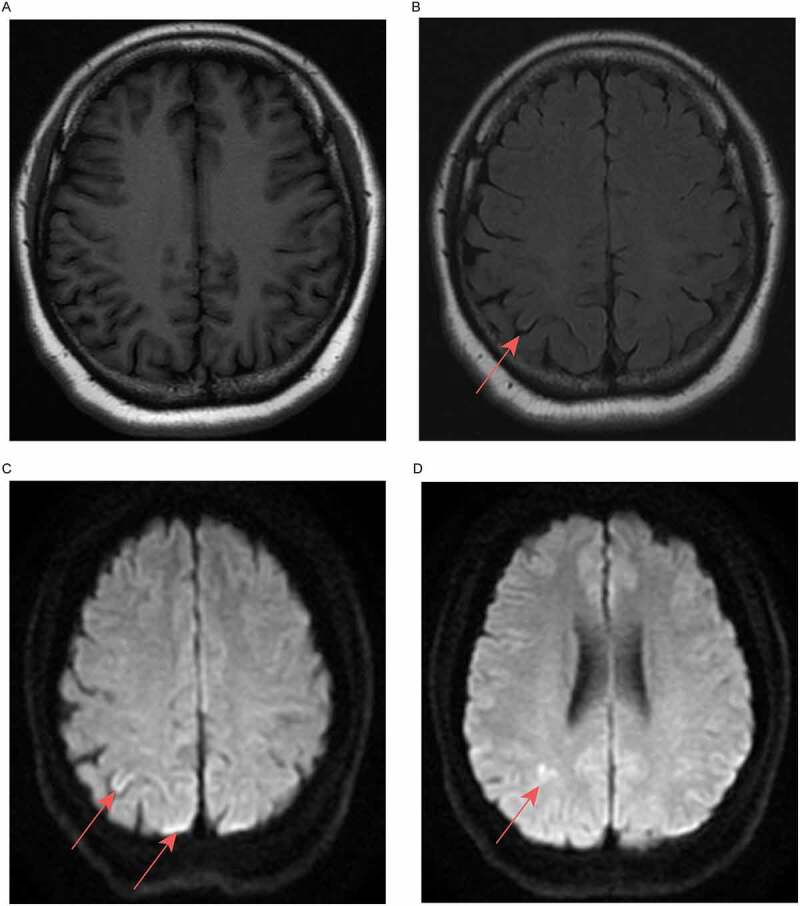
Brain MRI images of the patient (acquired 11-04-2018). (a) Isointensity on T1-weighted fluid-attenuated inversion recovery. (b) Right side of the parietal cortex, T2-weighted fluid-attenuated inversion recovery, slightly high signal. (c, d) Right side of the parietal cortex, diffusion-weighted imaging, high signal.
Figure 3.
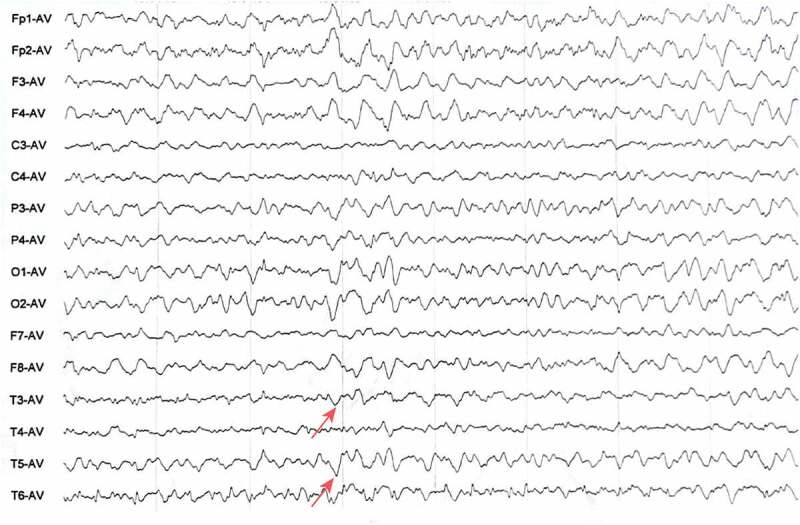
EEG results of the patient. The results showed diffuse slow waves with sharp waves and three-phase waves.
Figure 4.

Gene mapping of the patient (performed on 17-05-2018). A missense mutation (C to T) was identified at nt 305 in one allele of PRNP, which led to a change from proline (Pro) to leucine (Leu) at codon 102.
Owing to the limitations of Western medicine, we employed traditional Chinese medicine (TCM) to treat the patient. After 2 months of TCM treatment, including Radix Rehmanniae Praeparata, Corni Fructus, Cuscutae Semen, Euryales Semen, Alpiniae Oxyphyllae Fructus, Coicis Semen, Alismatis Rhizoma, Poria, Lycii Fructus, and Radix Codonopsis, the patient’s walking instability decreased, and her emotional fluctuations were less than before (More detailed treatment information was summarized in Table S1). We therefore reviewed her brain MRI images, and the results showed no significant changes. However, an EEG showed that epileptic waves disappeared and there were mainly slow waves (Figure 5). The total score of the MMSE increased from 21 to 25, and the MOCA score increased from 20 to 24. Then the patient was prescribed the formula in pill form for long-term use. During the following 6 months of telephone follow-up, she was able to walk dependently, but only for a short time, and she had dysarthria and memory loss. We recommended that she should undergo re-examination via MRI and EEG, but she and her family refused. One year later, the patient died of multiple infections.
Figure 5.
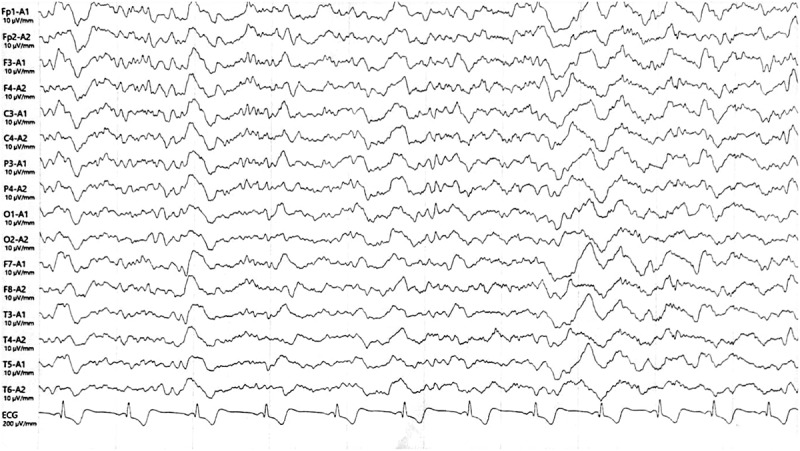
EEG results of the patient after 2 months of TCM treatments. EEG showed diffuse slow waves without sharp waves.
3. Discussion
GSS is a kind of rare disease around world, and there were only a few cases in China, to the best of our knowledge. Clinical manifestations of GSS could vary between the family members and generations. Common symptoms comprise cerebellar ataxia, dementia, dysarthria, blindness, deafness, myoclonus, spastic paraparesis, parkinsonian signs, and so on. In 2017, Long et al. have reported a GSS patient in southern China with PRNP p.P102L mutation exhibited cerebellar ataxia initially [7]. However, the GSS patient in our present case with PRNP p.P102L mutation showed mental disorders initially. As far as we know, she was the first GSS case that showed mental changes before ataxia symptoms, especially in western China.
To date, more than 60 variants in PRNP have been identified worldwide, approximately one quarter of which are correlated with GSS [8–11]. A total of 15 PRNP mutations have been reported in more than 50 families with GSS, including P102L, P105L, A117V, Q160X, F198S, Q217R, Y218N, Y226X, and Q227X, of which p.P102L is the commonest in GSS cases worldwide [4,6]. However, GSS cases with the p.P102L mutation have mainly been found in populations of Caucasian origin but have been extremely rarely recorded in Asian populations [12,13]. Since 2000, few GSS cases with the p.P102L mutation in Asia have been reported, especially in China. Among the cases in China, five GSS families have been reported by Li et al. [14]. All of these lineages have been confirmed with the PRNP p.P102L mutation, which may be the largest series of GSS PRNP p.P102L mutation-related reports in China. They provided the detailed clinical features of five probands with GSS, and all these patients exhibited progressive ataxia at the onset age, ranging from 37 to 59 years (48.44 ± 6.31). Moreover, cognitive impairment was found in 4/5 probands. The patient in our present case started GSS with mental disorder and gradually developed to cerebellar ataxia. However, she was misdiagnosed in some previous hospitals, and was finally diagnosed with GSS via genetic analysis. Currently, the diagnosis of GSS mainly depends on the genetic family history, clinical manifestations, genetic testing, and neuropathology, of which neuropathology is the gold standard for diagnosis. The most sensitive and specific method of diagnosing GSS mainly comprised the detection of mutations in PRNP gene. Owing to the low incidence of GSS, the diagnosis would remain ambiguous without analysing the gene panel containing a broad range of hereditary diseases.
Furthermore, GSS patients with the p.P102L mutation often showed a classic onset of midlife cerebellar ataxia with or without early dementia, such as apathy, depression, or other mental disorders, whereas the present patient’s personality has changed a lot before the ataxia, exhibiting reduced speech and a lack of expression. Accordingly, she was previously misdiagnosed as having schizophrenia. In our clinical cases, it is indeed a challenge to make accurate diagnosis promptly, facing the situation that the mental disorders manifest several months earlier than other symptoms of GSS. On the other hand, it has been widely documented that at the early stage of GSS syndrome, there are no mental disorder-related manifestations, and it is clinically difficult to distinguish GSS from other types of hereditary ataxia, especially spinocerebellar ataxia (SCA), as their genetic model and clinical manifestations often exhibit great similarity. Thus, appropriate gene test was probably a powerful tool to make differential diagnosis.
In terms of auxiliary examinations, positive changes were observed both in the brain MRI images and EEG of the patient. Most reports of midbrain MRI mainly reveal extensive cerebellar atrophy, whereas some typical manifestations, such as the hockey-stick sign in the subcortical nuclei, have been rarely reported [15]. Moreover, the EEG is helpful to detect functional changes in the brain via voltage fluctuations associating with ion flow in neurons, besides, EEG is able to reflect the functional status of the brain sensitively. Accordingly, the EEG results could help to evaluate the brain dysfunction and to assess/predict the prognosis of patients with dementia or cognitive impairment. Accordingly, we also paid special attention to the results of EEG. We found that the patient exhibited epileptic waves (sharp waves and three-phase waves). It has been demonstrated that about only 10% of prion disease patients have periodic synchronous three-phase waves on their EEGs [16]. Additionally, no similar epileptic waves were reported in the GSS cases in China as far as we know. After TCM treatment, the epileptic waves had disappeared, implying that patient’s symptoms were indeed attenuated. However, whether the epileptic waves were helpful in the differential diagnosis of GSS could not be concluded basing on one case.
4. Conclusions
In conclusion, we have reported an extremely rare case of GSS in Sichuan, China. The proband exhibited unusual mental disorders as the initial symptoms, and she was finally confirmed with GSS PRNP P102L mutation. Herein, patient’s mental disorders began before the common symptoms of ataxia, which provided more reference information regarding the differential diagnosis of schizophrenia and GSS. Meanwhile, for families with dominant hereditary ataxia, especially with early mental disorders, particular attention should be paid to PRNP gene screening to consider the possibility of GSS syndrome. Necessary genetic analysis would be conducive to the prompt accurate diagnosis.
Supplementary Material
Funding Statement
This work was supported by the Hospital of Chengdu University of Traditional Chinese Medicine (Approval No. 202YTS01), and the Academic Credit Foundation of the Hospital of Chengdu University of Traditional Chinese Medicine (Approval No. 20JF216).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19336896.2023.2180255
References
- [1].Katherine Young H, Clark B, Pedro Piccardo SR, et al. Gerstmann–Sträussler–Scheinker disease with the PRNP P102L mutation and valine at codon129. Mol Brain Res. 1997;Vol. 44:147–150. PMID: 33879752. [DOI] [PubMed] [Google Scholar]
- [2].Riudavets MA, Sraka MA, Schultz M, et al. Gerstmann–Sträussler–Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102l mutation. Brain Pathol. 2014;24:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].AR G, GD F, Aresi A, et al. Atypical frontotemporal dementia a new clinical phenotype of Gerstmann–Straussler–Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol Sci. 2008;29:405–410. [DOI] [PubMed] [Google Scholar]
- [5].Araujo AQ. Prionic diseases. Arq Neuropsiquiatr. 2013;71:731–737. [DOI] [PubMed] [Google Scholar]
- [6].Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ling L, Cai X, Shu Y, et al . A family with hereditary cerebellar ataxia finally confirmed as Gerstmann–Sträussler–Scheinker syndrome with P102L mutation in PRNP gene. Neuroences. 2017;22:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones M, Odunsi S, du Plessis D, et al. Gerstmann–Sträussler–Scheinker disease: novel PRNP mutation and VGKC-complex antibodies. Neurology. 2014;82:2107–2111. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liberski PP. Gerstmann–Sträussler–Scheinker disease. Adv Exp Med Biol. 2012;724:128–137. [DOI] [PubMed] [Google Scholar]
- [10].Peoc’h K, Levavasseur E, Delmont E, et al. Substitutions at residue 211 in the prion protein drive a switch between CJD and GSS syndrome, a new mechanism governing inherited neurodegenerative disorders. Hum Mol Genet. 2012;21(26):5417–5428. [DOI] [PubMed] [Google Scholar]
- [11].Popova SN, Tarvainen I, Capellari S, et al. Divergent clinical and neuropathological phenotype in a Gerstmann–Sträussler–Scheinker P102L family. Acta Neurol Scand. 2012;126:315–323. [DOI] [PubMed] [Google Scholar]
- [12].Takazawa T, Ikeda K, Ito H, et al. A distinct phenotype of leg hyperreflexia in a Japanese family with Gerstmann–Sträussler–Scheinker syndrome (P102L). Intern Med. 2010;49:339–342. [DOI] [PubMed] [Google Scholar]
- [13].Webb TE, Poulter M, Beck J, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131:2632–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hong-Fu L, Liu Z-J, Dong H-L, et al. Clinical features of Chinese patients with Gerstmann–Sträussler–Scheinker identified by targeted next-generation sequencing[J]. Neuro Agi. 2016;49:216.e1–216.e5. [DOI] [PubMed] [Google Scholar]
- [15].Kim MO, Takada LT, Wong K, et al. Genetic PrPrion Diseases. Cold Spring Harb Perspect Biol. 2018;10:a033134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Franko E, Wehner T, Joly O, et al. Quantitative EEG parameters correlate with the progression of human prion diseases. J Neurol Neurosurg. 2016;87:1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.