

Abstract
Extracellular vesicles (EVs) can carry pathological cargo and play an active role in disease progression. Neutral Sphingomyelinase-2 (nSMase2) is a critical regulator of EV biogenesis, and its inhibition has shown protective effects in multiple disease states. 2,6-Dimethoxy-4-(5-phenyl-4-thiophen-2-yl-1H-imidazol-2-yl)-phenol (DPTIP) is one of the most potent (IC50=30 nM) inhibitor of nSMase2 discovered to-date. However, DPTIP exhibits poor oral pharmacokinetics (PK), limiting its clinical development. To overcome DPTIP’s PK limitations, we synthesized a series of prodrugs by masking its phenolic hydroxyl group. When administered orally, the best prodrug (P18) with a 2’,6’-diethyl-1,4’-bipiperidinyl-promoiety exhibited >4-fold higher plasma (AUC0-t=1047 pmol.h/mL) and brain exposures (AUC0-t=247 pmol.h/g) versus DPTIP; and a significant enhancement of DPTIP half-life (2 h vs. ~0.5 h). In a mouse model of acute brain injury, DPTIP released from P18 significantly inhibited IL-1β-induced EV release into plasma and attenuated nSMase2 activity. These studies report the discovery of a DPTIP-prodrug with potential for clinical translation.
Keywords: Extracellular vesicles, neutral sphingomyelinase 2, prodrugs, brain penetration
Graphical Abstract

INTRODUCTION
Neutral sphingomyelinase2 (nSMase2; SMPD3), a member of the sphingomyelinase (SMase) enzyme family, catalyzes the hydrolysis of sphingomyelin to phosphorylcholine, and ceramide1. nSMase2 is largely localized to the inner leaflet of the plasma membrane and Golgi apparatus; and is prominently expressed in the brain2. Many published reports have identified roles for nSMase2 generated ceramide in regulating glial activation, myelination, neuronal plasticity and cell survival3–7. nSMase2-generated ceramide regulates cellular signaling largely through modulation of the biophysical properties of cellular membranes. Ceramides pack tightly together and are critical components of highly ordered membrane microdomains8, 9. These domains, also known as lipid rafts, are highly dynamic structures that serve to regulate signaling by controlling the localization and activation of membrane receptors and their association with secondary signal transduction machinery10, 11. nSMase2-generated ceramide also regulates cellular signaling by regulating the biogenesis of at-least one population of extracellular vesicles (EVs)12, 13.
EVs are small vesicular carriers shed from cells that contain a variety of cargo including proteins, nucleic acids, miRNAs, and bioactive lipids. The cargo of EVs is modified by the stimulus used to evoke their release. For example, EVs that are shed from astrocytes in response to ATP have trophic effects on neurons, while EVs shed from astrocytes in response to TNF downregulate neuronal activity and neural network connectivity based on modifications in the EV cargo14. EVs play an important role in neurodevelopmental processes15 including neural cell proliferation, differentiation, synaptic formation, and myelination process and thus can directly participate in embryonic neurogenesis and gliogenesis16.
Under normal conditions genetic deletion or inhibition of nSMase2 may lead to detrimental effects on growth and CNS functions17–22, considering the important role of nSMase2 as well as EVs in several physiological functions. For example, deficiency of nSMase2 was shown to perturb neuronal proteostasis leading to progressive cognitive impairment17. Similarly, inhibition of nSMase2 in normal wild-type mice exhibited worsening of the cognitive function22. Further, nSMase2-KO mice displayed disruption of hypothalamic-pituitary-adrenal axes, leading to chondrodysplasia and dwarfism, defects in bone formation, and overall hypoplasia18–21.
Conversely, under diseased conditions, inhibition of nSMase2 is beneficial, particularly where ceramide-mediated EVs play a key pathogenic role23–30. In-fact, chronic increase of nSMase2 activity has been implicated in multiple pathological disorders including brain inflammation31, cancer metastasis32, amyloid deposition33–35, tau protein propagation36, and HIV infection37–40. Moreover, pharmacological inhibition and/or normalization of nSMase2 activity has been reported to improve disease pathology in numerous experimental model systems for neurodegenerative diseases41, suggesting its utility for neuroprotection. As such, there is a rising interest in developing clinically viable inhibitors for nSMase2.
Unfortunately, there are no clinically useful nSMase2 inhibitors41. Current inhibitors are weak (μM-mM) with poor physicochemical properties and/or limited brain penetration. Our laboratory previously reported DPTIP (2,6-dimethoxy-4-(5-phenyl-4-thiophen-2-yl-1H-imidazol-2-yl)-phenol), as one of the most potent inhibitors of nSMase2 identified to date (IC50 = 30 nM)6, 42. Its IC50 is 10- to 160-fold lower than the known inhibitors1, 4, 43. DPTIP was found to be selective, and capable of dose-dependently inhibiting EV release in vitro and in vivo (when delivered systemically). However, DPTIP exhibits poor pharmacokinetics (PK) and rapid clearance (t1/2<0.5 h). Structural modifications on the DPTIP scaffold did not lead to substantial improvements42.
Given the excellent potency, selectivity, and stability of DPTIP, we aimed to advance DPTIP by optimizing its PK profile using prodrug strategies44–46. Notably, prodrug approaches are common in drug development; about 10–12 % of the approved drugs are prodrugs, and of these, most are designed to enhance permeation of the parent drug47 through biological membranes. Our group has successfully modulated PK properties of drug candidates by employing prodrug strategies including improving solubility48, enhancing permeability across biological membranes49, brain penetration50, and tumor targeting51.
Herein, we report synthesis, stability, in vivo PK evaluation, and target engagement of DPTIP prodrugs. These were designed based on previous successes in improving the PK properties of poorly permeable molecules. Amongst the series of DPTIP-prodrugs synthesized and characterized, we identified P18, a prodrug with a 2’,6’-diethyl-1,4’-bipiperidinyl-based promoiety on the phenolic site of DPTIP. P18 exhibited an excellent PK profile compared to DPTIP and significantly inhibited IL-1β-induced EV release by inhibition of brain nSMase2 activity. Thus, P18 is a novel DPTIP prodrug that can aid in clinical translation of this class of inhibitors.
RESULTS AND DISCUSSION
CHEMISTRY
We have previously evaluated PK of DPTIP analogs in rodents and reported O-glucuronidation at the phenolic hydroxyl group as their primary path of metabolism. This glucuronidation makes DPTIP susceptible to rapid clearance leading to poor half-life. One of our first strategies, therefore, was to deter glucuronidation by derivatization of the phenolic hydroxyl site of DPTIP; thus, improving its half-life. We further strategized that the promoiety architecture could be modulated to improve oral absorption and / or brain penetration of DPTIP. Pursuant to this strategy, we initially synthesized DPTIP prodrugs supporting alkyl chain promoieties coupled either by ester or carbamate linkages (P1 – P13, and P18). These also included the various piperidine promoieties as well as the piperidinopiperidine promoiety that has previously been employed to mask the phenolic site of SN-38, an FDA approved drug for the treatment of colorectal cancers (CPT-11; irinotecan hydrochloride)52. With phenolic site masked, the in vivo metabolism of the prodrugs was primarily dominated by carboxylesterase enzymes which cleave both the ester or carbamate linkage of the inactive prodrug to release DPTIP. The rate of metabolic release of DPTIP is thus controlled by the stability of the linker which can be tuned via ester / carbamate linkage in addition to the steric hindrance imparted by the promoiety.
DPTIP is lipophilic (cLogP = 4.72) which limits its solubility in formulations required for oral dosing. To overcome this physicochemical limitation, our second strategy included designing phosphate esters (P14-P16) to increase hydrophilicity as well as polar surface of DPTIP. We aimed to achieve a hydrophilic/lipophilic balance and enhance oral exposures by improving dissolution rate of DPTIP. This strategy has previously been reported to improve solubility of miproxifene via its phosphorylation53. We also have previously reported a mebendazole prodrug where its phosphate ester displayed a 2.2-fold higher plasma and 1.7-fold higher brain exposures when tested in rodents. Similarly, in dogs, phosphate ester of mebendazole resulted in 3.8-fold higher plasma exposures. A similar strategy was used to improve the absorption of propofol using a water-soluble phosphate ester prodrug fospropofol, which demonstrated 20–70% improvement (dose dependent) of oral bioavailability in rats54. P15 and P16 were designed to further modulate the stability of linkages of phosphate prodrugs in addition to optimizing their physicochemical properties. Overall, these strategies were anticipated to improve solubility of DPTIP-prodrugs and enhance their absorption, following activation in intestinal brush border or liver where resident phosphatase enzymes would trigger DPTIP release. Notably, these phosphate ester-based prodrugs were not designed to improve the brain penetration of DPTIP, but to impart an increased solubility due to the phosphate moieties that would increase overall plasma and brain exposures of DPTIP versus DPTIP administration.
Finally, we also synthesized a N-methyl-1,4-dihydronicotinic ester prodrug of DPTIP (P17) designed to passively absorb through GI tract and eventually through the blood brain barrier (BBB). The prodrug would then acquire a positive charge in the brain due to aromatization, forming a pyridinium salt impermeable to BBB55, 56. This ‘trapped’ prodrug accumulating in the brain then is expected to release DPTIP. We describe the synthesis and characterization of the 18 DPTIP prodrugs below.
Synthesis
DPTIP was synthesized using our previously reported methods6, 42. Prodrugs P1-P13 were synthesized in a single step reaction using DPTIP and respective amines as the starting material in the presence of triphosgene and Hünig’s base (DIPEA) to form either esters or carbamates (Scheme-1). Briefly, DPTIP was treated with triphosgene in the presence of DIPEA in dichloroethane at 0 °C and stirred for 1 h. The amine was then added to this mixture and the reaction was periodically monitored via thin layer chromatography (TLC) for completion (4–20 h). If carbonyl chlorides of the promoieties were available, the reaction was carried out under similar conditions as described above but in the absence of triphosgene. Detailed reaction conditions for synthesis of all prodrugs are provided in the experimental section.
Scheme-1: Synthesis of Prodrugs P1-P13.

(a) DIPEA, triphosgene, DCE, 0 °C – rt, 4 – 20 h; (b) DIPEA, DCE, 0 °C – rt, 4 – 20 h; (c) DIPEA, DCE, rt, 8 h.
The phosphoester of DPTIP P14 was synthesized by addition of phosphoryl chloride and DPTIP to DIPEA in DCE followed by potassium carbonate treatment (Scheme-2). The reaction was stirred until completion and then crude mixture purified using preparative column chromatography. P15 and P16 were obtained via the intermediate 4, which was synthesized by reaction of DPTIP with t-butyl-protected chloromethyl phosphate in presence of cesium carbonate and sodium iodide (Scheme-2). Intermediate 4 was treated with trifluoroacetic acid (TFA) to isolate P15. To obtain P16, intermediate 4 was first treated with silica and catalytic amount of conc. HCl for 24 h at room temperature, when partially deprotected intermediate 5 was obtained. This was then treated with chloromethyl isopropyl carbonate in presence of cesium carbonate and sodium iodide, followed by deprotection of the t-butyl ester using trifluoroacetic acid to reveal P16.
Scheme-2: Synthesis of Prodrugs P14-P16.

(a) (i) POCl3, DIPEA, 0 °C, 2 h, (ii) K2CO3, H2O, rt, 18 h; (b) Cs2CO3, NaI, DMF, rt, 24 h; (c) TFA, DCM, rt, 12 h; (d) silica, methanol, conc. HCl (cat.), rt, 24 h.
P17 was obtained in three steps via Steglich esterification of DPTIP with nicotinic acid followed by N-alkylation with methyl iodide (Scheme-3). In the final step the resulting pyridinium salt is treated with sodium dithionite to reveal P17, containing N-methyl-1,4-dihydropyridine moiety.
Scheme-3: Synthesis of Prodrug P17.

(a) EDC, DMAP, DMF, rt, 12 h; (b) CH3I, acetone, rt, 24 h; (c) Na2S2O4, NaHCO3, H2O, 0 °C, 3 h.
P18 was synthesized via reaction of DPTIP and intermediate 8 using the conditions described earlier (P1-P13). 2’,6’-Diethyl-1,4’-bipiperidine (8) was prepared in a 3-step synthetic procedure starting with reaction of 3-oxopentanedioic acid, propionaldehyde and benzylamine in water to form cyclized ketone 7. This intermediate 7 was then reacted with piperidine to form enamine, which underwent the reductive amination in the presence of sodium cyanoborohydride. In the third step, removing the benzyl group by palladium-mediated catalytic hydrogenation revealed intermediate 8 (Scheme-4).
Scheme-4: Synthesis of Prodrug P18.

(a) propionaldehyde, benzylamine, H2O, 0 °C – rt, 48 h; (b) piperidine, NaCNBH3, methanol, acetic acid, rt, 16 h; (c) 10% Pd/C, H2 (5 atm), 50 °C, methanol, acetic acid, 16 h; (d) triphosgene, DIPEA, DCE, 0 °C – rt, 16 h.
Prodrugs exhibited broad range of lipophilicity and metabolic stability
Eighteen prodrugs of DPTIP were designed, synthesized, and assessed with the goal to enhance metabolic stability as well as to the facilitate oral absorption and brain penetration. First, cLogP values were obtained in silico using ChemDraw Professional 20.1 software (Table 1). The prodrugs showed varying degrees of lipophilicity (cLogP from 2.99 to 7.36) compared to DPTIP (cLogP = 4.72). As expected, a majority of the prodrugs containing alkyl, piperidine, and piperidinopiperidine promoieties exhibited an increase in the cLogP (from 4.84 to 7.36) mirroring the lipophilic character of the promoieties. In contrast, compounds containing phosphate ester promoieties (P14 and P15) exhibited a decrease in lipophilicity (cLogP ranged from 2.99 to 3.18). The decrease in cLogP for these compounds was attributed to the charged nature of the promoieties for enhanced dissolution. P16 containing a phospho-ester with an isopropyloxy carbonyloxy methyl (POC) group depicted a higher cLogP due to masking of one of the charges. Owing to the low solubility and permeability of DPTIP, the 18 prodrugs were designed with a broad range of chemical groups to provide improvement in either solubility or permeability with the ultimate goal of enhancing oral absorption.
Table 1.
Lipophilicity and metabolic stabilities of DPTIP prodrugs P1 - P18

| |||||
|---|---|---|---|---|---|
| Compound | R | cLogP | Stability in mouse (% remaining at 1 h) Mean ± Std Error (n = 3) | ||
| Plasma | Liver | Brain | |||
| DPTIP | −H | 4.72 | 103 ± 2 | 98 ± 6 | 89 ± 4 |
| P1 |
|
5.20 | 90 ± 2 | 85 ± 3 | 48 ± 2 |
| P2 |

|
6.52 | 13 ± 1 | 1 ± 0 | 63 ± 1 |
| P3 |

|
4.41 | 28 ± 2 | 0 ± 0 | 6 ± 0 |
| P4 |

|
5.65 | 1 ± 0 | 0 ± 0 | 93 ± 1 |
| P5 |

|
4.06 | 96 ± 3 | 100 ± 1 | 100 ± 2 |
| P6 |

|
6.92 | 83 ± 3 | 80 ± 4 | 69 ± 2 |
| P7 |

|
6.19 | 50 ± 2 | 85 ± 3 | 86 ± 1 |
| P8 |

|
5.97 | 60 ± 6 | 83 ± 2 | 68 ±1 |
| P9 |
|
7.02 | 60 ± 4 | 97 ± 4 | 84 ± 4 |
| P10 |

|
7.36 | 78 ± 5 | 51 ± 1 | 90 ± 1 |
| P11 |
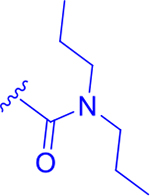
|
6.19 | 99 ± 2 | 62 ± 4 | 95 ± 4 |
| P12 |

|
5.58 | 85 ± 9 | 95 ± 3 | 76 ± 3 |
| P13 |

|
5.54 | 87 ± 2 | 78 ± 4 | 84 ± 1 |
| P14 |

|
3.18 | 91 ± 4 | 55 ± 1 | 102 ± 2 |
| P15 |

|
2.99 | 96 ± 6 | 21 ± 0 | 1 ± 0 |
| P16 |
|
5.97 | 100 ± 3 | 11 ± 0 | 79 ± 2 |
| P17 |

|
4.84 | 52 ± 2 | 22 ± 1 | 46 ± 0 |
| P18 |

|
5.55 | 92 ± 7 | 100 ± 3 | 100 ± 1 |
All synthesized prodrugs were next screened for in vitro stability in carboxylesterase 1 knockout (CES1−/−) mice plasma and tissues that have undetectable carboxylesterase activity in plasma but have normal carboxylesterase activity in tissues57 and thus recapitulate human prodrug metabolism51 (Table 1). Given the broad structural modifications, all prodrugs were screened for stability in plasma, liver and brain homogenates from CES1−/− mice as this mouse model was also used for subsequent in vivo PK studies. The objective was to assess post absorption conversion of prodrugs to DPTIP at these highly metabolically-active sites by ubiquitous metabolic enzymes. The assays were not tailored to evaluate specific enzymes involved in conversion of some prodrugs e.g. compounds P14 - P16 that would be expected to be metabolized by alkaline phosphatases. The results from these in vitro assays were used for the selection of prodrugs with low susceptibility to systemic metabolism thereby enabling biodistribution of the intact prodrugs to the target site, specifically the CNS. DPTIP can then be released from these prodrugs by the activity of local, endogenous enzymes. The most desirable prodrugs showed considerable metabolic stability in plasma and liver homogenates but modest metabolism and conversion in the brain homogenates. P1, P5, P6, and P10–16, P18 were found to be stable in CES1−/− mouse plasma with >75% intact prodrug remaining after 1 h of incubation at 37 °C. P7, P8, P9, and P17 were moderately stable (~50–60% remaining at 1 h) and P2, P3, and P4 were unstable (<30% remaining at 1 h). In mouse liver homogenates, P1, P5, P6, P12, P13, and P18 were metabolically stable with >75% intact prodrug remaining after 1 h of incubation at 37 °C. P10, P11, and P14 were moderately stable (>50% remaining at 1 h) and P2, P3, P4, P15, P16, and P17 were highly metabolized (<25% remaining at 1 h) in liver homogenates. In mouse brain homogenates, P4, P5, P14, P16, and P18 were found to be metabolically stable with >75% intact prodrug remaining after 1 h of incubation at 37 °C. P1, P2, P6, and P17 were moderately stable (~50% or higher remaining at 1 h); and P3 and P15 were completely metabolized in brain homogenates.
In general, compounds containing ester promoieties showed highest metabolism in plasma and liver as these are likely cleaved by the carboxylesterase enzymes highly abundant at these sites. One exception to this was P6, which showed modest metabolic stability against the esterase cleavage, likely due to the steric hinderance of the phenyl and pivalate promoieties. This is more evident when directly compared with P4 (with pivalate promoieties only) which converted rapidly. In contrast, prodrugs comprising the carbamate linkage (either alkyl carbamate or piperidinopiperidine promoiety) demonstrated higher metabolic stability in plasma and liver homogenates. Interestingly, a subset of the carbamate prodrugs (P1, P7, P8, P12) showed considerable metabolism in the brain homogenates. Lastly, the compounds containing the phospho-esters (P15, P16) were stable in plasma. However, P15 was completely metabolized and P16 (phospho-ester with POC group) was moderately metabolized in brain homogenates.
Given the broad stability profile for prodrugs P1-P18, we subsequently evaluated them in a single time point PK in plasma and brain; and selected the best of those for time-dependent PK evaluation.
Prodrugs improved oral absorption and brain penetration compared to DPTIP
As an initial in vivo screen, CES1−/− mice dosed orally (10 mg/kg, DPTIP equivalent; n = 3) were sacrificed 2 h after dose; plasma and brain were collected to measure the levels of intact prodrug and released DPTIP in these matrices. 2 h time point was selected because DPTIP was expected to show low levels due to its rapid clearance and only prodrugs which showed improvement were advanced for time-dependent evaluation. The results from the PK analysis of DPTIP prodrugs are shown in Figure 1 (A–C) and Figure S1. Amongst all the prodrugs tested, P2 with the hexanoate ester showed an increase in DPTIP release (Figure 1A and S1) in plasma (355 nM) and brain (36.8 nM); and although the brain levels were higher than DPTIP (36.8 nM vs. 4.9 nM), brain-to-plasma ratio remain unchanged (~0.1). The higher level of DPTIP released in plasma was attributed to high lipophilicity imparted by the hexanoate ester as well as instability of the ester in plasma and liver due to the presence of an ester linkage. Consistent with the stability results (Table-1) P2 showed poor levels of the intact compound in plasma and brain (25.2 nM and 13.3 nM respectively, Figure S1). In contrast, P5, a prodrug containing dimethyl carbamate promoiety, showed a notable improvement in oral absorption giving 4.3 μM intact levels (Figure S1); unfortunately, the released DPTIP levels (540.9 nM) were only 12% of the intact. This improvement of the plasma levels for the intact P5 was attributable to its high stability, relatively smaller promoiety, and lower clogP leading to improved dissolution, and high permeation. However, due to its poor conversion to DPTIP (~12%) and poor brain penetration index of released DPTIP (~0.1), it was not further advanced. P1, a piperidinopiperidine conjugate of DPTIP, released 72.3 nM DPTIP in brain at 2 h post-dose, thus showing a significant improvement versus DPTIP at same dose level (4.9 nM) (Figure 1A–B). Notably, P1 also showed a remarkable improvement in the brain-to-plasma ratio of released DPTIP (~0.7) as compared to DPTIP (~0.1). Intact P1 levels in the plasma and brain were 195.1 nM and 86.5 nM respectively, indicating good oral absorption of the prodrug. P17, a prodrug designed to be ‘trapped’ in brain with the N-methyl-1,4-dihydronicotinic ester promoiety did not show any improvements in the plasma (28.3 nM) or brain (2.2 nM) DPTIP levels. P17 also showed very low levels of the intact compound in plasma and brain (3.9 nM and 3.0 nM respectively, Figure S1). Further characterization using metabolite identification studies of P17 using high resolution mass spectrometry showed negligible quantities of the putative oxidized pyridinium metabolite in both brain and plasma (Fig S2). These data indicated poor oral absorption as well as poor bioactivation of P17. Interestingly, P18, that was designed combining the chemical attributes of P1 and P2, with a diethyl-piperidinopiperidine promoiety, exhibited desirable PK with higher plasma (189.3 nM vs. 23 nM), and brain (61.5 nM vs. 4.9 nM) levels as well as higher brain-to-plasma ratio of 0.33. Similar to P1, P18 also showed good intact levels in plasma and brain (282.9 nM and 35.2 nM respectively). Overall, In the single time point screening evaluation, almost all the prodrugs with carbamate linkages showed considerable levels of the intact prodrugs in plasma (~195 nM - 4.3 μM) and brain (~10 nM - 251 nM) (Figure S1) due to the higher stability of the carbamates. Predictably, the ester based prodrugs which present higher liabilities for chemical and enzymatic conversion showed low levels of intact prodrugs (maximum 43.2 nM and 13.4 nM in plasma and brain respectively) (Figure S1). From the screening study, P1 and P18 that showed high release of DPTIP in both plasma (>3-fold) and brain (>10 fold) versus DPTIP following oral administration as well as improvement in its brain/plasma ratio (e.g. P1) were further advanced for time-dependent PK evaluation. Moreover, prodrugs (P2-P17) that were unsuccessful in improving the brain and plasma levels of DPTIP and/or its brain penetration index were not further evaluated.
Figure 1: Single time-point pharmacokinetic screening of all prodrugs (P1 - P18) and DPTIP in CES1−/− mice dosed orally at 10 mg/kg DPTIP equivalent.

(A) Plasma levels of released DPTIP from the prodrugs. (B) Brain levels of released DPTIP from the prodrugs. (C) Brain to plasma ratio of released DPTIP at 2 h. Data expressed as mean ± SEM, n = 3.
P18 exhibited ~5-fold higher exposures in brain and plasma and improved brain penetration index following PO administration as compared to DPTIP
As described earlier, we identified P1 and P18 as the most promising candidates for time dependent PK. Detailed PK parameters of P1, P18 and DPTIP are given in the Figure 2 and Figure S3. Following oral administration in CES1−/− mice (n = 3), P1 exhibited 3.6-fold higher exposure of released DPTIP in brain (AUC0-t = 187.1 vs. 52.8 pmol.h/g) and a 3.7-fold improvement in brain penetration index (AUCbrain/plasma ratio 0.70 vs. 0.19) with a substantial improvement in apparent DPTIP half-life (t1/2 ~3 h vs. <30 min) when compared to equimolar DPTIP (Figure 2A). Pharmacokinetics of intact P1, indicated good plasma and brain exposures (AUC0-t = 829.9 pmol.h/mL and AUC0-t = 494.6 pmol.h/g, respectively) depicting about 40% conversion of P1 to DPTIP. P18 which showed best profile in single time point PK exhibited a remarkable 4.7-fold higher exposure in brain (AUC0-t = 247 vs. 52.8 pmol.h/g), and a 4-fold higher DPTIP exposures in plasma (AUC0-t =1047 vs. 270 pmol.h/mL), with a substantially improved DPTIP apparent half-life (t1/2 ~2 h vs. <30 min) when compared to equimolar DPTIP. P18 also showed a 1.3-fold improvement in brain penetration index (AUCbrain/plasma ratio 0.24 vs. 0.19). Intact P18 PK revealed good plasma and brain exposures (AUC0-t = 969 pmol.h/mL and AUC0-t = 179 pmol.h/g respectively) with >50% conversion to DPTIP in the brain in vivo, exhibiting desirable PK. Overall, P18 demonstrated best PK profile and was further evaluated in a mouse model of brain injury for inhibition of EV release and target engagement.
Figure 2: Time-dependent in vivo PK analysis of DPTIP (A) P1 (B) P18 (C) and their PK parameters (D) in CES1−/− mice.

DPTIP, P1, and P18 were dosed orally at 10 mg/kg DPTIP equivalent dose. Data expressed as mean ± SEM, n = 3.
P18 inhibited EV release and showed target engagement in a mouse model of brain injury
We have previously reported that nSMase2 regulates the secretion of EVs from astrocytes in response to a focal inflammatory brain lesion. Striatal injection of IL-1β in mice expressing GFAP-EGFP in astroglia evokes the release of GFP-labelled EVs that rapidly enter into plasma and target multiple organ systems31. We used this mouse model to evaluate the ability of P18 to inhibit EV release by the inhibition of nSMase2 activity in vivo and did a head-to-head comparison to DPTIP. The PK results (Figure 2) showed that the released DPTIP concentrations from P18 (10 mg/kg DPTIP equivalent; PO) were above the IC50 (30 nM) of DPTIP until at-least 4 hours and also provided ~ 4-fold higher exposures compared to equimolar DPTIP. Consequently, we used both 3 and 10 mg/kg (DPTIP equivalent) dose PO to evaluate its dose-response effects. Interleukin-1β-injected mice (n = 4), orally pre-dosed with either DPTIP (10 mg/kg) or P18 (30 min prior) were euthanized 4 h post-IL-1β injection. GFP labeled (GFP+) EVs were quantified in plasma and nSMase2 activity was measured in the brain striata. DPTIP levels were also measured in mouse plasma and brain to confirm brain penetration in this mouse model. Intrastriatal administration of IL-1β significantly increased the number of GFP+ EVs in plasma, and P18 significantly inhibited the release of brain derived GFP+ EVs into blood at both 3 and 10 mg/kg (DPTIP equivalent dose) (Figure 3Bi), while DPTIP did not show a significant inhibition even at the higher dose of 10 mg/kg. Similarly, P18 treatment normalized the heightened nSMase2 activity caused by IL-1β in a dose dependent manner (Figure 3Bii). The levels of released DPTIP and PK parameters in plasma and brain samples from IL-1β treated mice were similar in CES1−/− mice and mice administered intrastriatal IL-1β (Figure 3Biii). To confirm the free fraction levels of DPTIP, protein binding studies of DPTIP were conducted using ultra-filtration method58 in mouse plasma. DPTIP showed 90% protein binding giving free fractions, expected to provide 10–30% inhibition of the enzyme activity, similar to that observed in Figure 3B (ii).
Figure 3: Effects of orally administered P18 in a mouse model of acute brain injury.

Quantification of GFP labeled EVs in plasma when treated with (A) DPTIP or (Bi) P18. ###p < 0.001 (compared to saline + vehicle group); ***p <0.001 (compared to IL1-β + vehicle group). (Bii) nSMase2 activity in mouse striata under different treatments following IL-1β injection. n = 4/group, bars represent mean ± SEM. ####p < 0.0001 (compared to saline + vehicle group); ****p < 0.0001, ***p <0.001 and *p < 0.05 (compared to IL-1β + vehicle group). B(iii) Plasma and brain levels of released DPTIP from P18 in the same samples. n = 4/group, bars represent mean ± SEM
CONCLUSION
EVs are crucial for various normal physiological functions including neurodevelopmental processes. nSMase2 regulates the biogenesis and release of EVs through ceramides. Genetic deletion or inhibition of nSMase2 under normal conditions has been implicated in multiple neurological, skeletal, and developmental disorders. However, in pathological states, chronic increase of nSMase2 activity can be debilitating as EVs can carry pathological cargo and cause propagation of various diseases including neurological disorders, cancer, and HIV (see Tallon et. al.)41. Therefore, development of small molecule inhibitors of nSMase2 is highly desirable. DPTIP represents the most potent nSMase2 inhibitor identified to date. However, DPTIP exhibits poor PK in mice with low exposures in plasma and brain. Our previous SAR studies on the DPTIP scaffold revealed key pharmacophores essential for potent inhibition as well as structural modifications that can be tolerated by the enzyme. However, these structural analogs of DPTIP failed to improve upon PK properties of these nSMase2 inhibitors.
In an effort to address poor PK properties exhibited by DPTIP and enhance its delivery to the brain we employed a prodrug strategy. We strategized to mask the polar hydroxyl group of DPTIP by various promoieties such as simple alkyl esters or carbamates, piperidinyl carbamates, or phosphate esters in order to improve oral bioavailability, as well as plasma and brain exposures. We designed and synthesized DPTIP prodrugs P1 - P18, and evaluated them using in vitro stability, in vivo PK, EV release inhibition, and target engagement assays. Prodrugs containing the carbamate promoieties P1 and P18 demonstrated improved PK properties (i.e. oral absorption, brain penetration, and half-life) and were identified as the two most promising candidates for further evaluation. P18 showed excellent improvement of plasma exposures (~4.0-fold) and half-life (2–3 h) over DPTIP as well as the highest improvement in brain exposure (4.7-fold) following PO administration in mice as compared to DPTIP. P18 also demonstrated significant inhibition of nSMase2 activity and IL-1β-induced EV release in mice. Using a prodrug approach, we achieved a significant enhancement in the oral availability, brain penetration, and apparent half-life of DPTIP and these outcomes could aid in its clinical translation.
EXPERIMENTAL SECTION
General information:
1H NMR was recorded on a Bruker 400 MHz spectrometer, using residual signal of deuterated solvent as internal reference. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane. 1H NMR data are reported as follows: chemical shift (multiplicity, coupling constants, and number of hydrogens). Multiplicity is abbreviated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). The electrospray ionization7 high-resolution mass spectrometry (HRMS) was performed on a Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham MA) coupled with a Dionex ultra-high-performance LC system.
Purity of Final Prodrugs:
HPLC analysis was performed on a C8 reversed-phase column. The purity for all key compounds was >95%. Description of the purity analysis has been included in the Experimental section. Detailed HPLC information of key compounds (traces, retention times, and purity) are included in the Supporting Information. From our 1H NMR analysis, we found that there are two stable and inseparable tautomers for DPTIP and all our synthesized prodrugs. We confirmed our hypothesis by 1H NMR experiment with one of our prodrugs at different temperatures (see NMR data section in Supporting Information). While at 25 °C we observed 2 NH signals corresponding to 2 tautomers (slow exchange of hydrogen atom on imidazole ring), at 80 °C we detected only one broad signal for NH (fast exchange).
General synthetic procedure for prodrugs P1, P7–12:
To a stirred solution of DPTIP (800 mg, 2.11 mmol, 1.0 equiv.) in 1,2-dichloroethane (25 mL) cooled to 0 °C was added DIPEA (1.80 mL, 10.6 mmol, 5.0 equiv.) and triphosgene (692 mg, 2.32 mmol, 1.1 equiv.) and the resulting mixture was stirred for 1 h at 0 °C. Primary or secondary amine (3.17 mmol, 1.5 equiv.) was added to the above reaction mixture and the mixture was stirred at room temperature for further 3 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with DCM (50 mL), washed with water (2×25 mL) and brine solution (25 mL). Organic phase was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The residue was purified by Biotage isolera or preparative HPLC to give resulting products P1, P7-P12 in 25–40 % yields.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl [1,4’-bipiperidine]-1’-carboxylate (P1):
1,4’-bipiperidine (533 mg); mobile phase: 5–10% MeOH in DCM; P1 was obtained as an off white solid (483 mg) in 40 % yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.77 (s, 1H), 8.17 (t, J = 8.0 Hz, 2H), 7.61 (t, J = 8.0 Hz, 1H), 7.54 – 7.32 (m, 5H), 7.06 (s, 1H), 6.98 (s, 1H), 4.17 – 4.02 (m, 1H), 4.01 – 3.98 (m, 1H), 3.84 (s, 6H), 2.86 – 2.68 (m, 2H), 2.51 – 2.50 (m, 4H), 2.33 – 2.17 (m, 2H), 1.76 – 1.40 (m, 9H). HRMS 7: [M + H]+ (C32H37N4O4S) calculated 573.2535, found 573.2527.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl methyl (pentyl) carbamate (P7):
N-methylpentan-1-amine (321 mg); preparative HPLC; P7 was obtained as an off white solid (277 mg) in 26% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.77 (s, 1H), 9.93 (d, J = 8.0 Hz, 2H), 7.62 – 7.30 (m, 6H), 7.06 – 6.98 (m, 2H), 3.91 (s, 6H), 3.40 – 3.38 (m, 2H), 3.28 – 3.26 (m, 1H), 3.02 (s, 3H), 1.65 – 1.52 (m, 1H), 1.33 – 1.17 (m, 4H), 0.91 – 0.86 (m, 3H). HRMS 7: [M + H] + (C28H32N3O4S) calculated 506.2113, found 506.2114.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl pentylcarbamate (P8):
pentan-1-amine (276 mg); prep-HPLC; P8 was obtained as an off white solid (270 mg) in 26% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.75 (s, 1H), 7.64 – 7.60 (m, 3H), 7.54 – 7.38 (m, 5H), 7.37 – 7.34 (m, 1H), 7.06 – 6.97 (m, 2H), 3.84 (s, 6H), 3.04 (t, J = 4.0 Hz, 2H), 1.47 – 1.45 (m, 2H), 1.31 – 1.29 (m, 4H), 0.90 (t, J = 12.0 Hz, 3H). HRMS 7: [M + H]+ (C27H30N3O4S) calculated 492.1957, found 492.1960.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl heptylcarbamate (P9):
heptan-1-amine (365 mg); preparative HPLC; P9 was obtained as an off white solid (296 mg) in 27% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.75 (s, 1H), 7.65 – 7.34 (m, 9H), 7.05 – 6.98 (m, 2H), 3.85 (s, 6H), 3.06 – 3.02 (m, 2H), 1.47 – 1.44 (m, 2H), 1.29 – 1.24 (m, 8H), 0.89 (t, J = 8.0 Hz, 3H). HRMS 7: [M + H]+ (C29H34N3O4S) calculated 520.2270, found 520.2277.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl 2,6-diethylpiperidine-1-carboxylate (P10):
2,6-diethylpiperidine (448 mg); preparative HPLC; P10 was obtained as a pale brown solid (288 mg) in 25% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.76 (s, 1H), 7.63 – 7.60 (m, 2H), 7.54 – 7.34 (m, 6H), 7.06 – 6.97 (m, 2H), 4.03 – 4.02 (m, 1H), 3.86 (s, 6H), 2.68 – 2.67 (m, 3H), 1.70 – 1.54 (m, 8H), 0.90 (t, J = 8.0 Hz, 6H). HRMS 7: [M + H]+ (C31H36N3O4S) calculated 546.2426, found 546.2432.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl dipropylcarbamate (P11):
diisopropylamine (321 mg); preparative HPLC; P11 was obtained as an off white solid (288 mg) in 27% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.80 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.48 – 7.12 (m, 6H), 7.07 – 6.99 (m, 2H), 3.84 (s, 6H), 3.22 – 3.21 (m, 2H), 3.19 – 3.17 (m, 2H), 1.70 – 1.53 (m, 4H), 0.92 (t, J = 8.0 Hz, 6H). HRMS 7: [M + H]+ (C28H32N3O4S) calculated 506.2113, found 506.2118.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl 8-azabicyclo[3.2.1]octane-8-carboxylate (P12):
8-azabicyclo[3.2.1]octane hydrochloride (468 mg); preparative HPLC; P12 was obtained as an off white solid (272 mg) in 25% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.76 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.54 – 7.51 (m, 2H), 7.47 – 7.32 (m, 4H), 7.06 – 6.97 (m, 2H), 4.30 – 4.28 (m, 1H), 4.09 – 4.03 (m, 1H), 3.87 (s, 6H), 1.99 – 1.80 (m, 3H), 1.77 – 1.73 (m, 4H), 1.60 – 1.43 (m, 3H). HRMS 7: [M + H]+ (C29H30N3O4S) calculated 516.1957, found 516.1961.
Synthesis of 2,6-diethylpiperidine:
To a stirred solution of 2,6-diethylpyridine (1.50 g, 11.1 mmol, 1.0 equiv.) in methanol (25 mL) was added 5 % rhodium on carbon (300 mg) and few drops of acetic acid. The mixture was stirred at 60 °C for 16 h in an autoclave at 20 atm hydrogen pressure. The completion of the reaction was monitored by TLC. The reaction mixture was filtered through pad of celite and methanol was evaporated under reduced pressure to give 2,6-diethylpiperidine as a colorless liquid (830 mg) in 53% yield.
General synthetic procedure for prodrugs P2-P5:
To a stirred solution of DPTIP (200 mg, 0.528 mmol, 1.0 equiv.) in 1,2-dichloroethane (5 mL) was added DIPEA (P2: 0.46 mL, 2.64 mmol, 5.0 equiv.; P3-P5: 0.27 mL, 1.58 mmol, 3.0 equiv.) and acyl chloride (1.06 mmol, 2.0 equiv.) at room temperature. The mixture was stirred at room temperature for 8 h (P2), 12 h (P3-P4) or 20 h (P5). The completion of the reaction was monitored by TLC. The reaction mixture was diluted with DCM (10 mL), washed with water (2×10 mL) and brine solution (10 mL). Organic layer was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The residue was purified by Biotage isolera using 60–70% (P2) or 70–80% (P3-P5) ethyl acetate in petroleum ether as eluents to give off white solids P2-P5 in 15–60% yields.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl hexanoate (P2):
hexanoyl chloride (142 mg); P2 (93.1 mg), 37% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.82 (s, 1H), 7.62 – 7.21 (m, 8H), 7.06 (s, 1H), 6.99 (s, 1H), 3.85 (s, 6H), 3.32 – 3.26 (m, 2H), 1.68 – 1.62 (m, 2H), 1.37 – 1.32 (m, 4H), 0.92 (t, J = 8.0 Hz, 3H). HRMS 7: [M + H]+ (C27H29N2O4S) calculated 477.1848, found 477.1841.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl acetate (P3):
acetyl chloride (83.4 mg); P3 (55.5 mg), 25% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.79 (s, 1H), 7.62 – 7.34 (m, 8H), 7.06 (s, 1H), 6.98 (s, 1H), 3.86 (s, 6H), 2.28 (s, 3H). HRMS 7: [M + H]+ (C23H21N2O4S) calculated 421.1222, found 421.1223.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl pivalate (P4):
pivaloyl chloride (127 mg); P4 (147 mg), 60% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.82 (s, 1H), 7.68 – 7.20 (m, 8H), 7.06 (s, 1H), 6.98 (s, 1H), 3.84 (s, 6H), 1.32 (s, 9H). HRMS 7: [M + H]+ (C26H27N2O4S) calculated 463.1691, found 463.1691.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl dimethylcarbamate (P5):
dimethylcarbamic chloride (114 mg). P5 (35.6 mg), 15% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.76 (s, 1H), 7.67 – 7.60 (m, 2H), 7.56 – 7.24 (m, 6H), 6.99 (s, 1H), 6.97 (s, 1H), 3.85 (s, 6H), 3.05 (s, 3H), 2.90 (s, 3H). HRMS 7: [M + H]+ (C24H24N3O4S) calculated 450.1487, found 450.1488.
Synthesis of Prodrug P6:
Chloro(phenyl)methyl (2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl) carbonate (1):
To a stirred solution of DPTIP (400 mg, 1.06 mmol, 1.0 equiv.) in 1,2-dichloroethane (15 mL) cooled to 0 °C was added DIPEA (0.92 mL, 5.28 mmol, 5.0 equiv.) and chloro(phenyl)methyl carbonochloridate (325 mg, 1.59 mmol, 1.5 equiv.). The resulting mixture was stirred at room temperature for 8 h. The completion of the reaction was monitored by LC-MS. Then the reaction mixture was diluted with DCM (20 mL), washed with water (2×20 mL) and brine solution (20 mL). Organic layer was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The crude product 1 (320 mg) was used in the subsequent step without further purification.
(((2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy) carbonyl)oxy)(phenyl)methyl pivalate (P6):
To a stirred solution of intermediate 1 (320 mg, 0.584 mmol, 1.0 equiv.) in acetone (10 mL) was added triethylamine (0.41 mL, 2.92 mmol, 5.0 equiv.) and pivalic acid (179 mg, 1.75 mmol, 3.0 equiv.) at room temperature. The mixture was stirred at room temperature for 48 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with ethyl acetate (20 mL), washed with water (2×10 mL) and brine solution (10 mL). Organic layer was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The residue was purified by reverse phase prep-HPLC to give P6 as an off white solid (35.8 mg) in 10% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.82 (s, 1H), 7.63 – 7.60 (m, 4H), 7.58 – 7.51 (m, 5H), 7.47 – 7.44 (m, 3H), 7.39 – 7.34 (m, 2H), 7.06 (s, 1H), 6.97 (s, 1H), 3.86 (s, 6H), 1.24 (s, 9H). HRMS 7: [M + H]+ (C34H33N2O7S) calculated 613.2008, found 613.1998.
Synthesis of Prodrug P13:
tert-Butyl 3-(piperidin-1-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate:
To a stirred solution of tert-butyl 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylate (2.00 g, 8.87 mmol, 1.0 equiv.) in methanol (20 mL) was added piperidine (1.30 g, 13.3 mmol, 1.5 equiv.), few drops of acetic acid and sodium cyanoborohydride (836 mg, 13.3 mmol, 1.5 equiv.) at 0 °C. The mixture was stirred at room temperature for 18 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with DCM (50 mL), washed with water (25 mL) and brine solution (20 mL). Organic phase was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The residue was purified by silica gel column chromatography to give tert-butyl 3-(piperidin-1-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate as a colorless liquid (2.09 g) in 80% yield.
3-(Piperidin-1-yl)-8-azabicyclo[3.2.1]octane hydrochloride52:
To a stirred solution of tert-butyl 3-(piperidin-1-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate (1.20 g, 5.08 mmol, 1.0 equiv.) in 1,4-dioxane (10 mL) was added 4M solution of HCl in 1,4-dioxane (6 mL) at 0 °C. The mixture was stirred at room temperature for 5 h. The completion of the reaction was monitored by TLC. The volatiles were removed under reduced pressure to give 3-(piperidin-1-yl)-8-azabicyclo[3.2.1]octane hydrochloride as a colorless liquid (879 mg) in 75% yield. The crude product was used to the following step without further purification.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl 3-(piperidin-1-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate (P13):
To a stirred solution of DPTIP (800 mg, 2.11 mmol, 1.0 equiv.) in 1,2-dichloroethane (25 mL) was added DIPEA (1.80 mL, 10.6 mmol, 5.0 equiv.) and triphosgene (692 mg, 2.32 mmol, 1.1 equiv.) at 0 °C. The mixture was stirred at 0 °C for 1 h. 3-(Piperidin-1-yl)-8-azabicyclo[3.2.1]octane hydrochloride (700 mg, 3.17 mmol, 1.5 equiv.) was added to the above reaction mixture and the mixture was stirred at room temperature for 3 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with DCM (50 mL), washed with water (2×25mL) and brine solution (25mL). Organic phase was separated, dried over anhydrous sodium sulphate and volatiles were removed under reduced pressure. The residue was purified by prep-HPLC to give P13 as an off white solid (316 mg) in 25% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.83 (s, 1H), 7.68 – 7.60 (m, 4H), 7.55 – 7.40 (m, 4H), 7.06 – 6.97 (m, 2H), 4.35 – 4.33 (m, 1H), 4.14 – 4.12 (m, 1H), 3.84 (s, 6H), 2.97 – 2.96 (m, 1H), 2.67 – 2.51 (m, 4H), 2.01 – 1.94 (m, 3H), 1.75 – 1.72 (m, 5H), 1.69 – 1.63 (m, 4H), 1.41 – 1.40 (m, 2H). HRMS 7: [M + H]+ (C34H39N4O4S) calcd 599.2692, found 599.2697.
Synthesis of Prodrug P14:
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl dihydrogen phosphate (P14):
To a stirred solution of DPTIP (200 mg, 0.528 mmol, 1.0 equiv.) in 1,2-dichloroethane (5 mL) was added DIPEA (0.46 mL, 2.64 mmol, 5.0 equiv.) and POCl3 (122 mg, 0.793 mmol, 1.5 equiv.) at 0 °C. The mixture was stirred at 0 °C for 2 h. To the resulting mixture was added potassium carbonate (365 mg, 2.64 mmol, 5.0 equiv.) and water (5 mL) and the mixture was stirred at room temperature for further 18 h. The completion of the reaction was monitored by TLC. The reaction mixture was completely concentrated under reduced pressure. The residue was purified by preparative HPLC to give P14 as an off white solid (53.3 mg) in 22% yield. 1H-NMR (400 MHz, DMSO-d6): δ 13.39 (s, 1H), 7.63 (t, J = 4.0 Hz, 2H), 7.57 – 7.44 (m, 6H), 7.28 (s, 1H), 7.17 (s, 1H), 3.86 (s, 6H). HRMS 7: [M + H]+ (C21H20N2O6PS) calculated 459.0779, found 459.0780.
Synthesis of Prodrug P15:
Di-tert-butyl ((2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy)methyl) phosphate (4):
To a stirred solution of DPTIP (1.50 g, 3.96 mmol, 1.0 equiv.) in DMF (20 mL) was added cesium carbonate (2.58 g, 7.92 mmol, 2.0 equiv), di-tert-butyl (chloromethyl) phosphate (2.05 g, 7.92 mmol, 2.0 equiv) and sodium iodide (594 mg, 3.96 mmol, 1.0 equiv.) at room temperature. The mixture was stirred at room temperature for 24 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with ethyl acetate (50 mL), washed with water (2×25 mL) and brine solution (25mL). Organic layer was separated, dried over anhydrous sodium sulphate and volatiles were evaporated under reduced pressure. The residue was purified by biotage isolera using 60–70% ethyl acetate in petroleum ether as eluent to give intermediate 4 as a pale-yellow solid (1.14 g) in 48% yield.
(2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy) methyl dihydrogen phosphate (P15):
To a stirred solution of intermediate 4 (200 mg, 0.333 mmol, 1.0 equiv.) in DCM (5 mL) was dropwise added trifluoroacetic acid (2 mL) in DCM (18 mL). The mixture was stirred at room temperature for 12 h. The completion of the reaction was monitored by TLC. The volatiles were evaporated under reduced pressure. The residue was purified by reverse phase prep-HPLC to give P15 as off white solid (48.8 mg) in 30% yield. 1H-NMR (400 MHz, DMSO-d6): δ 7.62 (t, J = 4.0 Hz, 2H), 7.45 – 7.03 (m, 8H), 5.31 (s, 2H), 3.86 (s, 6H). HRMS 7: [M + H]+ (C22H22N2O7PS) calculated 489.0885, found 489.0877.
Synthesis of Prodrug P16:
tert-Butyl ((2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy)methyl) hydrogen phosphate (5):
To a stirred solution of intermediate 4 (520 mg, 0.866 mmol, 1.0 equiv.) in methanol (10 mL) was added silica gel (5 g, mess size 60–120 μm) and few drops of conc. HCl (37%). The mixture was stirred at room temperature for 24 h. The completion of the reaction was monitored by LC-MS. The reaction mixture was filtered and the volatiles were removed under reduced pressure. The crude intermediate 5 (470 mg, quantitative yield) was used to the following step without further purification.
((tert-Butoxy((2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy)methoxy)phosphoryl)oxy)methyl isopropyl carbonate:
To a stirred solution of intermediate 5 (470 mg, 0.866 mmol, 1.0 equiv.) in DMF (10 mL) was added cesium carbonate (564 mg, 1.73 mmol, 2.0 equiv.), chloromethyl isopropyl carbonate (264 mg, 1.73 mmol, 2.0 equiv.) and sodium iodide (130 mg, 0.866 mmol, 1.0 equiv.) at room temperature. The mixture was stirred at room temperature for 24 h. The completion of reaction was monitored by LC-MS. The reaction mixture was filtered and volatiles were removed under reduced pressure. The crude product was obtained in 48% yield (275 mg) and was used in the subsequent step without further purification.
((((2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy)methoxy)(hydroxy)phosphoryl)oxy)methyl isopropyl carbonate (P16):
To a stirred solution of ((tert-butoxy((2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy)methoxy)phosphoryl)oxy)methyl isopropyl carbonate (320 mg, 0.484 mmol, 1.0 equiv.) in DCM (5 mL) was slowly added trifluoroacetic acid (2 mL) in DCM (18 mL). The mixture was stirred at room temperature for 12 h. The completion of reaction was monitored by LC-MS. Volatiles were removed under reduced pressure and the residue was purified by reverse phase preparative HPLC to give P16 as an off white solid (32.2 mg) in 11% yield. 1H-NMR (400 MHz, DMSO-d6): δ 7.61 (t, J = 8.0 Hz, 2H), 7.48 (t, J = 8.0 Hz, 2H), 7.41 – 7.28 (m, 3H), 7.18 – 7.13 (m, 1H), 7.05 (s, 1H), 7.00 (s, 1H), 5.37 – 5.31 (m, 4H), 4.81 – 4.75 (m, 1H), 3.85 (s, 6H), 1.23 (d, J = 4.0 Hz, 6H). HRMS 7: [M + H]+ (C27H30N2O10PS) calculated 605.1358, found 605.1348.
Synthesis of Prodrug P17:
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl nicotinate:
To the stirred solution of DPTIP (1.00 g, 2.64 mmol, 1.0 equiv.) in DMF (15 mL) was added nicotinic acid (488 mg, 3.96 mmol, 1.5 equiv.), EDC*HCl (760 mg, 3.96 mmol, 1.5 equiv.), HOBt (536 mg, 3.96 mmol, 1.5 equiv.) and triethyalmine (1.10 mL, 7.92 mmol, 3.0 equiv.). The mixture was stirred at room temperature for 12 h until the completion of reaction (monitored by TLC). The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (2×10 mL) and brine solution (10 mL). Organic layer was separated, dried over anhydrous sodium sulphate and solvents were evaporated under reduced pressure. The residue was purified by biotage isolera using 0–80% ethyl acetate in petroleum ether as an eluent to give 2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl) phenyl nicotinate as an off white solid (894 mg) in 70% yield.
3-((2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenoxy) carbonyl)-1-methylpyridin-1-ium iodide (6):
To the stirred solution of 2,6-dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl nicotinate (700 mg, 1.44 mmol, 1.0 equiv.) in acetone (20 mL) was added iodomethane (0.18 mL, 2.89 mmol, 2.0 equiv.). The mixture was stirred at room temperature for 24 h. The completion of the reaction was monitored by TLC. The volatiles were removed under reduced pressure to give the title compound 6 as a pale-yellow solid (802 mg) in 89% yield. The crude product was used to the following step without further purification.
2,6-Dimethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl 1-methyl-1,4-dihydropyridine-3-carboxylate (P17):
To a stirred solution of intermediate 6 (780 mg, 1.56 mmol, 1.0 equiv.) in water (20 mL) at 0 °C was added sodium bicarbonate (657 mg, 7.82 mmol, 5.0 equiv.) and sodium dithionite (817 mg, 4.69 mmol, 3.0 equiv.). The mixture was stirred at 0 °C for 3 h. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (10 mL) and brine solution (10 mL). Organic layer was separated, dried over anhydrous sodium sulphate and ethyl acetate was evaporated under reduced pressure. The residue was purified by reverse phase prep-HPLC to afford P17 as a pale-yellow solid (39.0 mg) in 5% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.80 (s, 1H), 7.62 – 7.31 (m, 9H), 7.10 (s, 1H), 7.09 (s, 1H), 5.92 (s, 1H), 4.83 – 4.79 (m, 1H), 3.87 (s, 6H), 3.32 – 3.04 (m, 5H). HRMS 7: [M + H]+ (C28H26N3O4S) calculated 500.1644, found 500.1642.
Synthesis of Prodrug P18:
1-Benzyl-2,6-diethylpiperidin-4-one (7):
To a stirred solution of 3-oxopentanedioic acid (10.0 g, 68.4 mmol, 1.0 equiv.) in water (50 mL) was added propionaldehyde (9.8 mL, 137 mmol, 2.0 equiv.) and the resulting mixture was stirred at room temperature for 15 min. The reaction mixture was cooled to 0 °C, benzylamine (7.4 mL, 68.4 mmol, 1.0 equiv.) was added and mixture was stirred at room temperature for 48 h. The progress of the reaction was monitored by TLC. The reaction mixture was cooled to room temperature, acidified with 1N HCl up to pH=2, later neutralized with saturated NaHCO3 solution (to pH=7) and extracted with ethyl acetate (2×200 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography eluting with 0–50% ethyl acetate in hexane to afford the intermediate 7 as a brown oil (6.5 g) in 65% yield. 1H-NMR (400 MHz, DMSO-d6): δ 7.39 (t, J = 8.0 Hz, 2H), 7.34 (t, J = 8.0 Hz, 2H), 7.22 (t, J = 8.0 Hz, 1H), 3.92 (d, J = 12.0 Hz, 1H), 3.66 (d, J = 12.0 Hz, 1H), 2.93 – 2.90 (m, 2H), 2.42 – 2.41 (m, 2H), 2.11 – 2.10 (m, 2H), 1.55 – 1.53 (m, 2H), 1.35 – 1.33 (m, 2H), 0.85 (t, J = 8.0 Hz, 6H).
1’-Benzyl-2’,6’-diethyl-1,4’-bipiperidine:
To a stirred solution of intermediate 7 (5.00 g, 15.9 mmol, 1.0 equiv.) in methanol (50 mL) was added piperidine (2.00 g, 23.8 mmol, 1.5 equiv.), acetic acid (catalytic) and sodium cyanoborohydride (1.49 g, 23.8 mmol, 1.5 equiv.) at 0 °C. The mixture was stirred at room temperature for 16 h. Completion of the reaction was monitored by TLC. The reaction mixture was diluted with ethyl acetate (2×100 mL) and washed with water (50 mL). The combined organic layers were separated, and dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography eluting with 0–80% ethyl acetate in hexane to afford the 1’-benzyl-2’,6’-diethyl-1,4’-bipiperidine as a brown gum (3.1 g) in 62 % yield.
2’,6’-Diethyl-1,4’-bipiperidine (8):
To a degassed solution of 1’-benzyl-2’,6’-diethyl-1,4’-bipiperidine (3.10 g, 9.86 mmol, 1.0 equiv.) in methanol (40 mL) was added 10% of palladium on carbon (600 mg, 0.564 mmol, 0.06 equiv.) and few drops of acetic acid. The mixture was stirred in an autoclave under hydrogen pressure (5 atm) at 50 °C for 16 h. Completion of the reaction was monitored by TLC. The reaction mixture was filtered through a pad of celite and methanol was evaporated under reduced pressure to give intermediate 8 (1.28 g) in 58% yield.
2,6-Dimethoxy-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl 2’,6’-diethyl-[1,4’-bipiperidine]-1’-carboxylate (P18):
To a stirred solution of DPTIP (800 mg, 2.11 mmol, 1.0 equiv.) in 1,2-dichloroethane (25 mL) was added DIPEA (1.80 mL, 10.6 mmol, 5.0 equiv.) and triphosgene (692 mg, 2.32 mmol, 1.1 equiv.) at 0 °C. The mixture was stirred at 0 °C for 1 h. 2’,6’-Diethyl-1,4’-bipiperidine 8 (711 mg, 3.17 mmol, 1.5 equiv.) was added to the above reaction mixture and the resulting mixture was stirred at room temperature for 3 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with DCM (50 mL), washed with water (2×25mL) and brine solution (25mL). The organic layer was separated, dried over anhydrous sodium sulphate and volatiles were removed under reduced pressure. The residue was purified by prep-HPLC to give P18 as an off white solid (332 mg) in 25% yield. 1H-NMR (400 MHz, DMSO-d6): δ 12.96 (s, 1H), 7.61 (t, J = 8.0 Hz, 2H), 7.47 – 7.41 (m, 8H), 4.04 – 4.01 (m, 1H), 3.85 (s, 6H), 2.69 (t, J = 8.0 Hz, 1H), 2.51 – 2.50 (m, 3H), 1.96 – 1.92 (m, 5H), 1.75 – 1.73 (m, 2H), 1.53 – 1.50 (m, 7H), 1.40 – 1.39 (m, 2H), 0.92 (t, J = 8.0 Hz, 6H). HRMS 7: [M + H]+ (C36H45N4O4S) calculated 629.3161, found 629.3149.
Metabolic Stability Studies.
The in vitro metabolic stability analysis of DPTIP prodrugs were performed in CES1−/− mouse plasma, liver, and brain homogenates as previously described48. For the tissue homogenates, washed tissues were diluted 10-fold in 0.1 M potassium phosphate buffer and homogenized using a probe sonicator. To evaluate the stability of the intact prodrug over time, each of the crude homogenates and plasma were aliquoted to 1 mL and then spiked with a final assay concentration of 10 μM of each prodrug followed by incubation in an orbital shaker at 37 °C for 1 h (in triplicate). Sample from each incubation at predetermined time points (0 min, and 1h) was quenched with three volumes of acetonitrile containing the internal standard (IS; losartan: 0.5 μm). Samples were vortex-mixed for 30 s and centrifuged at 10,000 × g for 10 min at 4 °C. Disappearance of intact prodrugs from these samples were performed on a Dionex ultra-high-performance LC system coupled with Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham MA). The separation of analytes was achieved using the Agilent Eclipse Plus column (100 × 2.1 mm i.d.; maintained at 35 °C) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Pumps were operated at a flow rate of 0.4 mL/min for 9 min using gradient elution. The mass spectrometer controlled by Xcalibur software 4.0.27.13 (Thermo Scientific) was operated with a heated electrospray ionization (HESI) ion source in positive ionization mode. Quantification of the prodrugs were performed in the full-scan mode (from m/z 50 to 1600) by comparing t = 0 samples with t = 60 min samples.
Pharmacokinetic Study in CES1−/− Mice.
All PK studies in CES1−/− mice were conducted according to protocols approved by the Animal Care and Use Committee at Johns Hopkins University. The C57BL/6 CES1−/− mice were obtained as a generous gift from the United States Army Medical Research Institute of Chemical Defense, Maryland, and maintained on a 12 h light–dark cycle with ad libitum access to food and water. DPTIP was administered orally (PO) at a dose of 10 mg/kg (5% DMSO, 10% Tween-80 in saline). Prodrugs were dose at a dose of 10 mg/kg DPTIP equivalent (5% DMSO, 10% Tween-80 in saline) and administered as a single peroral (PO) dose. For example, 15.12 mg/kg dose of P1 and 16.62 mg/kg dose of P18 would be molar equivalent to 10 mg/kg dose of DPTIP. All of the formulations were freshly prepared prior to the dosing. The mice were sacrificed at specified time points (either 2 h or 0.25, 0.5, 1, 2, 4, 6, and 8 h) post drug administration. For the collection of plasma and brain tissue, animals were euthanized with CO2, and blood samples were collected in heparinized microtubes by cardiac puncture. Brains were dissected and immediately flash frozen (−80 °C). Blood samples were spun at 2000g for 15 min at 4°C, and plasma was removed and stored at −80 °C until LC– MS/MS analysis. Prior to extraction, frozen samples were thawed on ice. To demonstrate that ex-vivo metabolism/conversion of the prodrugs does not occur during sample handling and bioanalysis, we performed additional stability analysis using P1, P2, P3 and P18. These prodrugs were analyzed for their stability in CES1−/− mouse plasma and incubated over ice for 30 min which represents the samples handling condition and time. The stability data is presented in the supporting information (Table S2)
Bioanalysis.
Quantitation of the prodrugs and the DPTIP released from prodrugs were performed using our published DPTIP bioanalytical LC/MS/MS method6, 43, with necessary minor modifications. Briefly, calibration standards were prepared using respective tissue (naïve plasma and brain) with additions of respective DPTIP prodrugs and DPTIP. For quantifying the intact prodrugs and released DPTIP in the PK samples, plasma samples (20 μL) were processed using a single liquid extraction method by addition of 100 μL of acetonitrile containing internal standard (losartan: 0.5 μM), followed by vortex-mixing for 30 s and then centrifugation at 10,000 × g for 10 min at 4 °C. Brain tissues were diluted 1:5 w/v with acetonitrile containing losartan (0.5 μm) and homogenized, followed by vortex-mixing and centrifugation at 10,000 g for 10 min at 4 °C. A 50 μL aliquot of the supernatant were diluted with 50 μL of water and transferred to 250 μL polypropylene autosampler vials sealed with teflon caps. Then, 2 μL of the sample were injected into the LC/MS/MS system for analysis. Chromatographic analysis was performed using an Accela ultra-high-performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer. Separation of analyte was achieved at ambient temperature using Agilent Eclipse Plus column (100 ×2.1 mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution were used. The [M+H]+ ion transition of DPTIP (m/z 378.956→363.073, 200.055) and losartan (IS) (m/z 423.200 → 207.107, 180.880) were used. The ion transitions for the respective prodrugs are given the supplementary information (Table S1). Plasma concentrations (nmol/ml) as well as brain tissue concentrations (nmol/g) were determined and plots of mean plasma concentration versus time were constructed. Non-compartmental analysis modules in Phoenix WinNonlin version 7.0 (Certara USA, Inc., Princeton, NJ) were used to quantify exposures (AUC0–t). The brain vasculature accounts for about ~3–4% of the total brain exposure 18 in PK bioanalysis59. For our prodrugs, the brain AUCs were ranging from 20–70% of the plasma AUCs and therefore, considering that a very small contribution came from the blood vasculature, we did not correct for this.
Measurement of EV release in vivo in mice.
Striatal injections and EV measurements were performed as previously described by our group31, 60. Briefly, male (2–3 month), GFAP–EGFP transgenic mice that allow for the evaluation of fluorescent EVs in plasma that are generated in the brain (Jackson Laboratories, n = 4) were anaesthetized with 3% isoflurane (Baxter) in oxygen (Airgas) and placed in a stereotaxic frame (Stoelting Co). We chose male mice to avoid effect of female specific hormones (i.e., estrogen) in inflammation that is known to be anti-inflammatory61. A small burr hole was drilled in the skull over the left striatum using a dental drill (Fine Scientific Tools). IL-1β (0.1 ng/3 μl) was injected (total volume of 3 μl) at the rate 0.5 μl·min−1 via a pulled glass capillary (tip diameter < 50 μm60). The stereotaxic coordinates based on bregma as reference point were A/P + 1; M/L −2; and −3 D/V31, 62. Saline was used as a control. P18 (3 and 10 mg/kg DPTIP equivalent, 5% DMSO, 10% Tween-80 in saline) was given orally 30 min before IL-1β injection. Mice subjected to intrastriatal injection of IL-1β were also injected with the NSAID carprofen (rimadyl, 5 mg/kg, i.p.) and closely monitored during recovery; no adverse reactions were observed. Following infusion, the capillary was held in place for 5 min to allow for the solution to diffuse into the tissue. Fifteen mice (n=4 for treatments and vehicle; n=3 for saline+vehicle group) were utilized to perform EVs counts. Mice were euthanized at 4 hr post-IL-1β treatment with overdose of anaesthetic (isoflurane). Blood samples were taken at death by cardiac puncture with heparin (Sigma) coated syringes and EDTA tubes (BD). Blood was immediately centrifuged at 2,700×g for 15 min (20 °C) to obtain plasma. Plasma was further centrifuged at 10,000 g for 15 min (4 °C) to generate platelet- free plasma. This procedure removes large particles such as apoptotic bodies. Quantitation of Plasma EVs: Dynabeads M-450 Epoxy (Invitrogen) were coupled with anti-GFP antibody (Thermo Fisher) at a ratio of 200-μg antibody per 4 × 108 beads. Plasma from GFAP–GFP mouse (50 μl) was incubated with 2 × 107 anti-GFP antibody-coupled Dynabeads at 4 °C overnight. The beads were washed and placed on a magnet to separate EVs bound to anti-GFP antibody-coupled Dynabeads. The precipitated EVs were eluted using 0.1-M glycine (pH 3.0). The concentration of immunoprecipitated GFP + EVs was quantified using ZetaView nanoparticle tracking analysis (Particle Metrix) and corresponding ZetaView software (8.05.14.SP7). The instrument was calibrated with 100 nm diameter beads (Thermo Scientific) prior to use. Instrument preacqusition were set to following parameters; temperature 23 °C, sensitivity 85, frame rate of 30 frames per second (fps), shutter speed of 100, and a laser pulse duration equal to that of shutter duration. Post-acquisition parameters were set to a minimum brightness of 25, maximum size of 200 pixels, and a minimum size of 10 pixels. For each sample, 1 ml of diluted EVs were injected into the sample-carrier cell and the particle count was measured at five positions, with two cycles of reading per position. The sample-carrier cell was washed with PBS after every sample. The data were collected by an investigator blinded to experimental conditions.
Measurement of nSMase2 activity in vivo in mice.
Interleukin-1β-injected mice, orally pre-dosed with either vehicle or P18 (3 and 10 mg/kg DPTIP equivalent, 30 min before IL-1β), were euthanized after four hours of IL-1β injection, the striata dissected and analyzed for nSMase2 activity using a modification of previously published protocols4, 6. Briefly, mice striata were homogenized in ice-cold Tris-HCl buffer (0.1 M, pH 7.5) containing 250 mM sucrose, 10 mM EGTA (Research Products International, Prospect, IL), 100 μM sodium molybdate and protease inhibitors (Cell Signaling, Danvers, MA) using Biomasher II and then sonicated using Kontes’ Micro Ultrasonic Cell Disrupter (three pulses of 15 s duration on ice with 30 s between pulses). The resulting lysates were collected for both nSMase2 activity measurements and total protein analysis. nSMase2 activity measurements were initiated upon the addition of Sphingomyelin (SM) and coupling enzymes in the Amplex Red system (25 μl) 4, 6 and SM hydrolysis carried out in total reaction volumes of 50 μl in 384-well microplates for 3 h at 37 °C. At the end of the reaction period, the relative fluorescence units were measured at Ex 530nm, Em 590nm. Finally, total protein measurements were carried as per manufacturer’s instructions using BioRad’s Detergent Compatible Protein Assay kit and data presented as RFU/mg/h.
Measurement of protein binding of DPTIP using ultra-filtration.
Centrifugal filter units (Amicon Ultra 0.5 mL filters, MWCO 3K) were obtained from Millipore (Billerica, MA). Compounds were added to 400 μL of mouse plasma at 10 M. One 50 μL aliquots was removed before placing in a Amicon device. This 50 μL aliquot was immediately processed for measurement of initial compound concentration (C1). The top and bottom chambers of the Amicon Ultra device were weighed before compound addition and then again after spinning to obtain the approximate volume of the top plasma (V2) and bottom ultrafiltrate (V3) samples, assuming a density of one for both samples. The device was spun at 14000 × g for 5 min at RT. A 50 μL aliquot of the top plasma (C2) and bottom ultrafiltrate (C3) were then removed for detection. For traditional measurement of nonspecific binding, plasma was replaced with PBS (pH 7.4), and samples were processed as above. 50 μL of the spiked PBS was used for initial concentration detection (Cpbs), and 50 μL of the ultrafiltrate was used for detection in ultrafiltrate (Cpbs-u). An equal volume of blank plasma was added to all the samples to create analytically identical matrices for LC–MS/MS analysis.
Statistical Analysis.
All results are expressed as mean ± SEM. Statistical analysis was performed with GraphPad PRISM 9 software. The significance of the differences was measured by one-way ANOVA followed by Tukey’s multiple comparison test (∗ p ≤ 0.05; ∗∗ p ≤ 0.01; ### or ∗∗∗ p ≤ 0.001; #### or ∗∗∗∗p ≤ 0.0001).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by R01 AG063831 to B.S.S. and R.R. and R01 AG059799 to B.S.S. and T.T. from the NIH, and a Tau Pipeline Enabling Program (T-PEP)-Alzheimer’s Disease Foundation grant to B.S.S.
ABBREVIATIONS:
- AUC
Area under the curve
- ATP
adenosine tri-phosphate
- CES1
Carboxylesterase-1
- CES1−/−
carboxylesterase-1 knockout
- DCE
Dichloroethane
- DCM
Dicholoromethane
- DIPEA
N,N-Diisopropylethylamine
- DPTIP
2,6-Dimethoxy-4-(5-phenyl-4-thiophen-2-yl-1H-imidazol-2-yl)-phenol
- EDCI
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EV
Extracellular vesicle
- GI
Gastrointestinal
- HOBt
Hydroxybenzotriazole
- HPLC
High performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- IL-β
Interleukin-1 beta
- nSMase2
Neutral Sphingomyelinase 2
- PK
Pharmacokinetics
- SMase
Sphingomyelinase
- SMPD
Sphingomyelin phosphor-diesterases
- TLC
Thin layer chromatography
- TEA
Triethylamine
- TFA
Trifluoroacetic acid
- TNF
tumor necrosis factor
Footnotes
The authors declare the following financial interests which may be considered as potential competing interests: A.P., N.H., B.S.S., and R.R. are listed as inventors in patent applications filed by Johns Hopkins Technology Ventures covering novel compositions of nSMase2 inhibitors and their utility.
ASSOCIATED CONTENT
Supporting Information: Details of 1H NMR and HPLC analysis, 1H NMR spectra, HPLC chromatograms; table containing mass transitions for LC–MS/MS analysis (Table S1); Supplementary Figures S1, S2 and S3; Additional stability data (Table S2).
References
- 1.Luberto C; Hassler DF; Signorelli P; Okamoto Y; Sawai H; Boros E; Hazen-Martin DJ; Obeid LM; Hannun YA; Smith GK, Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J Biol Chem 2002, 277 (43), 41128–41139. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann K; Tomiuk S; Wolff G; Stoffel W, Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc Natl Acad Sci U S A 2000, 97 (11), 5895–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shamseddine AA; Airola MV; Hannun YA, Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul 2015, 57, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Figuera-Losada M; Stathis M; Dorskind JM; Thomas AG; Bandaru VV; Yoo SW; Westwood NJ; Rogers GW; McArthur JC; Haughey NJ; Slusher BS; Rojas C, Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS One 2015, 10 (5), e0124481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu L; Huang B; Shen W; Gao L; Ding Z; Wu H; Guo J, Early activation of nSMase2/ceramide pathway in astrocytes is involved in ischemia-associated neuronal damage via inflammation in rat hippocampi. J Neuroinflammation 2013, 10, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rojas C; Barnaeva E; Thomas AG; Hu X; Southall N; Marugan J; Chaudhuri AD; Yoo S-W; Hin N; Stepanek O; Wu Y; Zimmermann SC; Gadiano AG; Tsukamoto T; Rais R; Haughey N; Ferrer M; Slusher BS, DPTIP, a newly identified potent brain penetrant neutral sphingomyelinase 2 inhibitor, regulates astrocyte-peripheral immune communication following brain inflammation. Scientific Reports 2018, 8 (1), 17715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo SW; Agarwal A; Smith MD; Khuder SS; Baxi EG; Thomas AG; Rojas C; Moniruzzaman M; Slusher BS; Bergles DE; Calabresi PA; Haughey NJ, Inhibition of neutral sphingomyelinase 2 promotes remyelination. Sci Adv 2020, 6 (40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiPasquale M; Deering TG; Desai D; Sharma AK; Amin S; Fox TE; Kester M; Katsaras J; Marquardt D; Heberle FA, Influence of ceramide on lipid domain stability studied with small-angle neutron scattering: The role of acyl chain length and unsaturation. Chem Phys Lipids 2022, 245, 105205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ventura AE; Varela ARP; Dingjan T; Santos TCB; Fedorov A; Futerman AH; Prieto M; Silva LC, Lipid domain formation and membrane shaping by C24-ceramide. Biochim Biophys Acta Biomembr 2020, 1862 (10), 183400. [DOI] [PubMed] [Google Scholar]
- 10.Lingwood D; Simons K, Lipid rafts as a membrane-organizing principle. Science 2010, 327 (5961), 46–50. [DOI] [PubMed] [Google Scholar]
- 11.Simons K; Toomre D, Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000, 1 (1), 31–39. [DOI] [PubMed] [Google Scholar]
- 12.Trajkovic K; Hsu C; Chiantia S; Rajendran L; Wenzel D; Wieland F; Schwille P; Brugger B; Simons M, Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319 (5867), 1244–1247. [DOI] [PubMed] [Google Scholar]
- 13.Tallon C; Picciolini S; Yoo SW; Thomas AG; Pal A; Alt J; Carlomagno C; Gualerzi A; Rais R; Haughey NJ; Bedoni M; Slusher BS, Inhibition of neutral sphingomyelinase 2 reduces extracellular vesicle release from neurons, oligodendrocytes, and activated microglial cells following acute brain injury. Biochem Pharmacol 2021, 194, 114796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datta Chaudhuri A; Dasgheyb RM; DeVine LR; Bi H; Cole RN; Haughey NJ, Stimulus-dependent modifications in astrocyte-derived extracellular vesicle cargo regulate neuronal excitability. Glia 2020, 68 (1), 128–144. [DOI] [PubMed] [Google Scholar]
- 15.You Y; Borgmann K; Edara VV; Stacy S; Ghorpade A; Ikezu T, Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J Extracell Vesicles 2020, 9 (1), 1706801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bahram Sangani N; Gomes AR; Curfs LMG; Reutelingsperger CP, The role of Extracellular Vesicles during CNS development. Prog Neurobiol 2021, 205, 102124. [DOI] [PubMed] [Google Scholar]
- 17.Stoffel W; Jenke B; Schmidt-Soltau I; Binczek E; Brodesser S; Hammels I, SMPD3 deficiency perturbs neuronal proteostasis and causes progressive cognitive impairment. Cell Death Dis 2018, 9 (5), 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoffel W; Hammels I; Jenke B; Binczek E; Schmidt-Soltau I; Brodesser S; Schauss A; Etich J; Heilig J; Zaucke F, Neutral sphingomyelinase (SMPD3) deficiency disrupts the Golgi secretory pathway and causes growth inhibition. Cell Death Dis 2016, 7 (11), e2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoffel W; Hammels I; Jenke B; Schmidt-Soltau I; Niehoff A, Neutral Sphingomyelinase 2 (SMPD3) Deficiency in mice causes chondrodysplasia with unimpaired skeletal mineralization. Am J Pathol 2019, 189 (9), 1831–1845. [DOI] [PubMed] [Google Scholar]
- 20.Stoffel W; Jenke B; Holz B; Binczek E; Gunter RH; Knifka J; Koebke J; Niehoff A, Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am J Pathol 2007, 171 (1), 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoffel W; Jenke B; Block B; Zumbansen M; Koebke J, Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc Natl Acad Sci U S A 2005, 102 (12), 4554–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabatadze N; Savonenko A; Song H; Bandaru VV; Chu M; Haughey NJ, Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J Neurosci Res 2010, 88 (13), 2940–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes AR; Sangani NB; Fernandes TG; Diogo MM; Curfs LMG; Reutelingsperger CP, Extracellular vesicles in CNS developmental disorders. Int J Mol Sci 2020, 21 (24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma P; Mesci P; Carromeu C; McClatchy DR; Schiapparelli L; Yates JR 3rd; Muotri AR; Cline HT, Exosomes regulate neurogenesis and circuit assembly. Proc Natl Acad Sci U S A 2019, 116 (32), 16086–16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamlett ED; Ledreux A; Potter H; Chial HJ; Patterson D; Espinosa JM; Bettcher BM; Granholm AC, Exosomal biomarkers in Down syndrome and Alzheimer’s disease. Free Radic Biol Med 2018, 114, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milenkovic I; Jarc J; Dassler E; Aronica E; Iyer A; Adle-Biassette H; Scharrer A; Reischer T; Hainfellner JA; Kovacs GG, The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down syndrome. Neuropathol Appl Neurobiol 2018, 44 (3), 314–327. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Gonzalez R; Gauthier SA; Sharma A; Miller C; Pawlik M; Kaur G; Kim Y; Levy E, A pleiotropic role for exosomes loaded with the amyloid beta precursor protein carboxyl-terminal fragments in the brain of Down syndrome patients. Neurobiol Aging 2019, 84, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsilioni I; Theoharides TC, Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1beta. J Neuroinflammation 2018, 15 (1), 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burrello J; Biemmi V; Dei Cas M; Amongero M; Bolis S; Lazzarini E; Bollini S; Vassalli G; Paroni R; Barile L, Sphingolipid composition of circulating extracellular vesicles after myocardial ischemia. Sci Rep 2020, 10 (1), 16182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caobi A; Nair M; Raymond AD, Extracellular vesicles in the pathogenesis of viral infections in humans. Viruses 2020, 12 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickens AM; Tovar YRLB; Yoo SW; Trout AL; Bae M; Kanmogne M; Megra B; Williams DW; Witwer KW; Gacias M; Tabatadze N; Cole RN; Casaccia P; Berman JW; Anthony DC; Haughey NJ, Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal 2017, 10 (473). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosaka N; Iguchi H; Hagiwara K; Yoshioka Y; Takeshita F; Ochiya T, Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem 2013, 288 (15), 10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinkins MB; Enasko J; Hernandez C; Wang G; Kong J; Helwa I; Liu Y; Terry AV Jr.; Bieberich E, Neutral sphingomyelinase-2 deficiency ameliorates Alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci 2016, 36 (33), 8653–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinkins MB; Dasgupta S; Wang G; Zhu G; Bieberich E, Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging 2014, 35 (8), 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sardar Sinha M; Ansell-Schultz A; Civitelli L; Hildesjo C; Larsson M; Lannfelt L; Ingelsson M; Hallbeck M, Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 2018, 136 (1), 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asai H; Ikezu S; Tsunoda S; Medalla M; Luebke J; Haydar T; Wolozin B; Butovsky O; Kugler S; Ikezu T, Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015, 18 (11), 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barclay RA; Schwab A; DeMarino C; Akpamagbo Y; Lepene B; Kassaye S; Iordanskiy S; Kashanchi F, Exosomes from uninfected cells activate transcription of latent HIV-1. J Biol Chem 2017, 292 (28), 11682–11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalvi P; Sun B; Tang N; Pulliam L, Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Sci Rep 2017, 7 (1), 9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shukla K; Ferraris DV; Thomas AG; Stathis M; Duvall B; Delahanty G; Alt J; Rais R; Rojas C; Gao P; Xiang Y; Dang CV; Slusher BS; Tsukamoto T, Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem. 2012, 55 (23), 10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun B; Dalvi P; Abadjian L; Tang N; Pulliam L, Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 2017, 31 (14), F9–F17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tallon C; Hollinger KR; Pal A; Bell BJ; Rais R; Tsukamoto T; Witwer KW; Haughey NJ; Slusher BS, Nipping disease in the bud: nSMase2 inhibitors as therapeutics in extracellular vesicle-mediated diseases. Drug Discov Today 2021, 26 (7), 1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepanek O; Hin N; Thomas AG; Dash RP; Alt J; Rais R; Rojas C; Slusher BS; Tsukamoto T, Neutral sphingomyelinase 2 inhibitors based on the 4-(1H-imidazol-2-yl)-2,6-dialkoxyphenol scaffold. European Journal of Medicinal Chemistry 2019, 170, 276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rojas C; Sala M; Thomas AG; Datta Chaudhuri A; Yoo S-W; Li Z; Dash RP; Rais R; Haughey NJ; Nencka R; Slusher B, A novel and potent brain penetrant inhibitor of extracellular vesicle release. British Journal of Pharmacology 2019, 176 (19), 3857–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aungst BJ; Hussain MA, Use of prodrugs of 3-hydroxymorphinans to prevent bitter taste upon buccal, nasal or sublingual administration. Google Patents: 1987.
- 45.Hussain AA; AL-Bayatti AA; Dakkuri A; Okochi K; Hussain MA, Testosterone 17β-N, N-dimethylglycinate hydrochloride: A prodrug with a potential for nasal delivery of testosterone. J. Pharm. Sci. 2002, 91 (3), 785–789. [DOI] [PubMed] [Google Scholar]
- 46.Kao HD; Traboulsi A; Itoh S; Dittert L; Hussain A, Enhancement of the systemic and CNS specific delivery of L-dopa by the nasal administration of its water soluble prodrugs. Pharm. Res. 2000, 17 (8), 978–984. [DOI] [PubMed] [Google Scholar]
- 47.Rautio J; Meanwell NA; Di L; Hageman MJ, The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018. [DOI] [PubMed] [Google Scholar]
- 48.Zimmermann SC; Tichy T; Vavra J; Dash RP; Slusher CE; Gadiano AJ; Wu Y; Jancarik A; Tenora L; Monincova L; Prchalova E; Riggins GJ; Majer P; Slusher BS; Rais R, N-substituted prodrugs of mebendazole provide improved aqueous solubility and oral bioavailability in mice and dogs. J Med Chem 2018, 61 (9), 3918–3929. [DOI] [PubMed] [Google Scholar]
- 49.Dash RP; Tichý T; Veeravalli V; Lam J; Alt J; Wu Y; Tenora L; Majer P; Slusher BS; Rais R, Enhanced oral bioavailability of 2-(phosphonomethyl)-pentanedioic acid (2-PMPA) from its (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-based prodrugs. Molecular Pharmaceutics 2019, 16 (10), 4292–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rais R; Jancarik A; Tenora L; Nedelcovych M; Alt J; Englert J; Rojas C; Le A; Elgogary A; Tan J; Monincova L; Pate K; Adams R; Ferraris D; Powell J; Majer P; Slusher BS, Discovery of 6-diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: A potential treatment for glioblastoma. J Med Chem 2016, 59 (18), 8621–8633. [DOI] [PubMed] [Google Scholar]
- 51.Tenora L; Alt J; Dash RP; Gadiano AJ; Novotná K; Veeravalli V; Lam J; Kirkpatrick QR; Lemberg KM; Majer P; Rais R; Slusher BS, Tumor-targeted delivery of 6-diazo-5-oxo-l-norleucine (DON) using substituted acetylated lysine prodrugs. J Med Chem 2019, 62 (7), 3524–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slatter JG; Schaaf LJ; Sams JP; Feenstra KL; Johnson MG; Bombardt PA; Cathcart KS; Verburg MT; Pearson LK; Compton LD; Miller LL; Baker DS; Pesheck CV; Lord RS 3rd, Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos 2000, 28 (4), 423–433. [PubMed] [Google Scholar]
- 53.Heimbach T; Oh DM; Li LY; Forsberg M; Savolainen J; Leppänen J; Matsunaga Y; Flynn G; Fleisher D, Absorption rate limit considerations for oral phosphate prodrugs. Pharm Res 2003, 20 (6), 848–856. [DOI] [PubMed] [Google Scholar]
- 54.Wozniak KM; Vornov JJ; Mistry BM; Wu Y; Rais R; Slusher BS, Gastrointestinal delivery of propofol from fospropofol: its bioavailability and activity in rodents and human volunteers. J Transl Med 2015, 13, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bodor N; Farag HH, Improved delivery through biological membranes. 11. A redox chemical drug-delivery system and its use for brain-specific delivery of phenylethylamine. J Med Chem 1983, 26 (3), 313–318. [DOI] [PubMed] [Google Scholar]
- 56.Buchwald P; Bodor N, Brain-targeting chemical delivery systems and their cyclodextrin-based formulations in light of the contributions of Marcus E. Brewster. J Pharm Sci 2016, 105 (9), 2589–2600. [DOI] [PubMed] [Google Scholar]
- 57.Duysen EG; Koentgen F; Williams GR; Timperley CM; Schopfer LM; Cerasoli DM; Lockridge O, Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chem Res Toxicol 2011, 24 (11), 1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang C; Williams NS, A mass balance approach for calculation of recovery and binding enables the use of ultrafiltration as a rapid method for measurement of plasma protein binding for even highly lipophilic compounds. J Pharm Biomed Anal 2013, 75, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chugh BP; Lerch JP; Yu LX; Pienkowski M; Harrison RV; Henkelman RM; Sled JG, Measurement of cerebral blood volume in mouse brain regions using micro-computed tomography. Neuroimage 2009, 47 (4), 1312–1318. [DOI] [PubMed] [Google Scholar]
- 60.McCluskey L; Campbell S; Anthony D; Allan SM, Inflammatory responses in the rat brain in response to different methods of intra-cerebral administration. J Neuroimmunol 2008, 194 (1–2), 27–33. [DOI] [PubMed] [Google Scholar]
- 61.Zhang QG; Wang R; Tang H; Dong Y; Chan A; Sareddy GR; Vadlamudi RK; Brann DW, Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol Cell Endocrinol 2014, 389 (1–2), 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paxinos G, & Franklin KBJ, The mouse brain in stereotaxic coordinates. Second ed.; Academic Press: 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.