

ABSTRACT
Beyond potency, a good developability profile is a key attribute of a biological drug. Selecting and screening for such attributes early in the drug development process can save resources and avoid costly late-stage failures. Here, we review some of the most important developability properties that can be assessed early on for biologics. These include the influence of the source of the biologic, its biophysical and pharmacokinetic properties, and how well it can be expressed recombinantly. We furthermore present in silico, in vitro, and in vivo methods and techniques that can be exploited at different stages of the discovery process to identify molecules with liabilities and thereby facilitate the selection of the most optimal drug leads. Finally, we reflect on the most relevant developability parameters for injectable versus orally delivered biologics and provide an outlook toward what general trends are expected to rise in the development of biologics.
KEYWORDS: Biologics, developability, antibodies, drug development, biotherapeutics, drug discovery, drug properties, half-life, immunogenicity, manufacturability
Introduction
Bringing a biologic from early discovery to a marketed product is an immensely expensive endeavor, with the average investment being further compounded by the high attrition rates in clinical development.1 While the clinical success of a biologic ultimately depends on its safety and efficacy in human patients, the underlying properties of these endpoints include everything from drug potency, specificity, immunogenicity, and pharmacokinetics to biophysical behavior in vivo and during storage.2,3 Moreover, with more advanced biologics such as bispecific antibodies and antibody-drug conjugates in the pipeline, chemistry, manufacturing, and control (CMC) and regulatory aspects can be key to commercial success, when the biologic must be repeatedly produced at consistently high quality.4 Combined, these performance measures are often referred to as a biologics’ “developability”.5 In this review, the different developability parameters for biologics will be presented together with a discussion on how to assess and optimize these using both in silico, in vitro, and in vivo methods (Figure 1). When optimally employed, the assessment and improvement of developability properties can lead to lower attrition rates, as well as improved manufacturability, enabling the production of higher quality, lower cost biologics for the benefit of patients worldwide.
Figure 1.
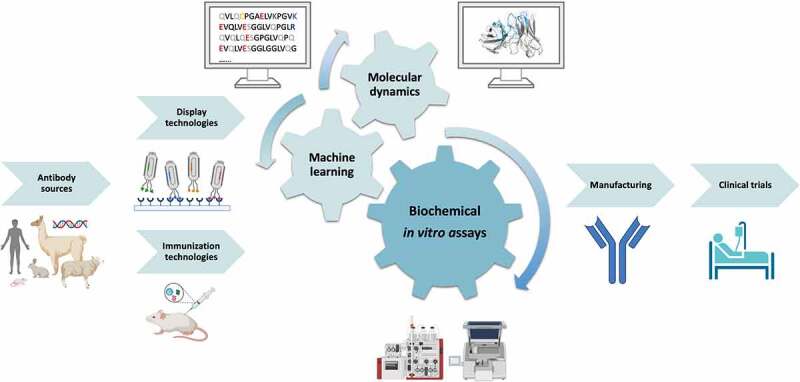
Schematic overview of antibody sources, discovery strategies, in vitro assays, and in silico methods used to assess developability properties during the development of biologics. Antibodies can be of either natural or synthetic origin and are typically discovered using various display technologies or immunization strategies, or a combination of these. In addition, antibodies can be derived from patient populations. During the development of antibodies, various in vitro assays in combination with in silico methods for machine learning and molecular dynamics can be applied to assess the antibodies’ developability properties and select candidates with the best developability profile.
Antibody sources
Antibodies and antibody fragments comprise the largest group of biologics.6 Antibodies are typically sourced from blood plasma,7 hybridomas,8 or recombinant cell lines,9 and originate from natural (animal or human) or synthetic sequences. Both the source and the origin may affect the antibodies’ developability profile. Natural antibodies, which are derived from natural, non-engineered sequences and typically sourced from (immunized) animals or patient populations, are to date the most commonly used source of antibodies.7 Such antibodies inherently carry the benefit of having been derived from a living creature and have gone through affinity maturation and self-tolerance in vivo, which often results in a good developability profile.7 However, in the context of clinical use, there is a stark difference between human and animal-derived antibodies. Whilst the former constitutes antibodies that have a low risk of immunogenicity, antibodies of non-human origin may elicit severe immune responses in patients, depending on the animal from which they were derived. The risk of immunogenicity can be reduced in several ways. For example, antibodies can be humanized or animals transgenic for human immunoglobulins (e.g., rabbits10 or mice11–14) can be used for immunization. A potential limitation of antibodies derived from immunization campaigns is that they will primarily target the most immunogenic epitopes of an antigen. This can limit epitope diversity and potentially prevent the discovery of binders to important, but low- or non-immunogenic antigen epitopes. Furthermore, inadequate immunization strategies can prevent antibodies from being discovered toward targets that are highly homologous to targets expressed within the immunized host due to self-tolerance.
To avoid these limitations, other types of antibodies have increased in popularity, i.e., synthetic antibodies,15,16 as they allow the discovery of binders toward, in principle, any target. Such antibodies may originate from rationally designed recombinant antibody repertoires and are typically discovered via in vitro display technologies.17 Using such methodologies grants substantial control over the antibody libraries. For example, certain amino acid residues known to be detrimental to binding interactions can be removed, lengths of variable regions can be adjusted, new formats can be experimented with, and overall antibody libraries with variability beyond nature can be created.18 As a result, this allows for their use against a substantially larger target space. Synthetic antibodies, however, typically come with the drawback of not having undergone in vivo maturation and being derived from a natural origin. Thus, they may potentially carry unexpected developability issues, such as unwanted polyreactivity, immunogenicity, propensity for aggregation, and poor expression profiles. Nevertheless, in combination with sophisticated display technologies and advanced computational tools, these risks can be minimized.
Advances in display technologies
Over the past few decades, display technologies have permitted the creation of large and diverse populations of antibodies and other proteins and peptides, from which individual variants with desired target-binding properties can be isolated.19 The requirements for generating an antibody drug, however, do not solely revolve around the binding specificity or affinity of the antibody. Other factors can affect the likelihood that a lead candidate antibody can be developed into an efficacious, manufacturable, safe, and stable drug. For example, the propensity of the antibody to aggregate upon formulation at higher concentration, the extent to which non-specific interactions occur, and the pharmacokinetics of the antibody can lead to the failure of a lead molecule during the development process.3,5,20
In applying display technologies, populations of antibodies may be derived from re-arranged antibody genes from B cells of human and non-human donors, constructed from synthetic antibody genes where variation has been introduced during oligonucleotide synthesis or a combination of both.21 Developability problems can arise irrespective of the starting source and even “natural”, immune-derived antibodies may suffer from biophysical liabilities. It has also been shown that engineering of lead antibodies with a sole focus on improving affinity can generate affinity-enhanced variants with poorer biophysical properties.22,23 For example, Buchanan et al.24 identified an unpaired cysteine, and Dobson et al.22 used hydrogen deuterium exchange to identify hydrophobic residues (W30 and F31 in VH complementary-determining region (CDR)1 and L56 in VH CDR2), which were protected in the initial dimerization preceding aggregation. Alternatively, structures (or structural models) could be used to identify hydrophobic or positively charged patches as used by Bethea et al.25 (F99, H100 and W100a in VH CDR3) and Dyson et al.23 (S52, F54, and R57 in VH CDR2). Based on these approaches, individual variants can be produced and assessed for improved biophysical characteristics. In some cases, the developability issues of clones can be solved by identifying problematic regions (e.g., hydrophobic patches), producing individual variants, and assessing these for improved biophysical characteristics.22,24,25
Historically, the consideration of developability issues has been addressed after the initial discovery, affinity optimization, and selection of an antibody drug lead for preclinical development. Increasingly, drug developers are assessing biophysical characteristics earlier in the discovery process, recognizing that this will avoid losses in time and money later. Rather than waiting to screen tens to hundreds of output clones for biophysical characteristics, the ability to create large display libraries of variants and select directly for good biophysical characteristics would greatly facilitate the search for antibody variants with optimal properties.
The power of selection technology, best exemplified by display on bacteriophage (phage display),26 has been applied to selecting clones for greater thermostability27 by exposing libraries of clones to elevated temperatures and selecting for retained binding to protein A or protein L, which requires correct folding.28 Others have subjected libraries of antibodies to acidic conditions to identify again clones that combined thermodynamic stability and aggregation-resistant unfolded states.28
While selection of clones with low thermostability will help identify aggregation propensity, there may be clones with normal melting temperature where biophysical problems only emerge when the antibody is concentrated (e.g., to 10–100 mg/mL as required for subcutaneous administration). Therefore, additional selection methods are needed. The principle underlying the power of phage display (coupling an encoding gene to its displayed product) has also been applied to other display systems using baculovirus,29 ribosomes,30 yeast,31 and higher eukaryotes, such as mammalian cells.29,31 Mammalian display, in particular, appears to offer benefits over the other systems. Using a mammalian display system23 showed that closely related clones, with differing biophysical properties, can be distinguished based simply on display levels. The underlying mammalian display system, first described by Parthiban et al.,32 achieves transcriptional normalization by integrating antibody genes into a single genome locus. Thus, display levels are determined by the properties of the antibody itself, rather than variable transcriptional activity. In contrast to secretion-based systems, the expressed antibodies are retained on the cell surface via a transmembrane domain and achieve high local concentrations in the endoplasmic reticulum or cell surface. Antibodies are therefore exposed to high concentrations and clones with poor biophysical properties are likely to aggregate at such high surface concentrations, with aggregates presumably removed by the quality control machinery of the cell. In turn, this results in low presentation levels of such aggregation-prone or “sticky” clones.
This ability to detect biophysical properties by mammalian display then permitted selection of clones with improved developability profiles from libraries of variants.23 In practice, libraries were created, and the clones with highest levels of presentation were selected. Multiparametric flow cytometry allows simultaneous screening for optimal biophysical properties while retaining antigen binding, and thereby allows paratopic residues involved in target binding to be addressed for improved biophysical properties while retaining target binding. The power of such a system was exemplified by creating variants of an anti-PCSK9 antibody with superior biophysical properties and reduced immunogenicity compared to the parental antibody (bococizumab). These and other experiments thereby demonstrate how in vitro display technologies can be fine-tuned for the discovery of antibodies with improved developability profiles ab initio.
Biophysical characterization
Irrespective of which strategy is used for antibody discovery, there typically are several early-stage candidates, ranging from tens to thousands, that must be reduced to only one candidate for cell line development and manufacturing. To select the optimal candidate, an increasing amount of attention is being paid to biophysical characterization of therapeutic antibodies in addition to their functional properties, which enables the deselection of antibodies with poor developability properties early in the drug development process. There are multiple developability assays to facilitate this selection, which have been reported to correlate to different extents with attrition rates of clinical-stage antibodies.2,3
One key feature of antibody drugs is their high specificity, defined by high on-target binding and low off-target and non-specific binding, which is important to reduce the risks of abnormal pharmacokinetics and fast antibody clearance.33,34 To evaluate non-specific binding, enzyme-linked immunosorbent assays (ELISAs), which typically involve binding to multiple non-targets, such as single-stranded DNA, double-stranded DNA, lipopolysaccharide (LPS), insulin, and keyhole limpet hemocyanin (KLH), have been most commonly used.3,35–37 For increased sensitivity, assays such as the polyspecificity particle (PSP) assay38 can be used, which involves detecting the binding of either complex antigen mixtures (e.g., soluble membrane proteins from Chinese hamster ovary (CHO) cells) or defined protein reagents (e.g., ovalbumin) to immobilized antibodies on Protein A coated beads via flow cytometry. Similar to the PSP assay, other assays also use complex antigen mixtures, including baculovirus particles,39 whole cells,38 and cell lysates (polyspecificity reagent, PSR).40 PSR has been shown to correlate quite well with cross-interaction chromatography (CIC).41 Thus, another way to study non-specific interactions is chromatography. In CIC, non-specific protein interactions, such as monoclonal antibodies interacting with immobilized polyclonal antibodies, are detected via their relative retention times.42–44 A related chromatography method, namely standup monolayer chromatography (SMAC), instead detects non-specific interactions between monoclonal antibodies and the column.45 For example, heparin chromatography has been described for identifying antibodies with abnormal pharmacokinetics via increased cell-surface interactions, leading to excessive pinocytosis.46 In addition to specificity, the SMAC measurements identify antibodies with increased likelihood of precipitation and aggregation. Non-specific binding can also be induced by surface hydrophobicity, which is often quantified using hydrophobic interaction chromatography (HIC).47,48
Another key feature of antibody drugs is their high colloidal stability and low propensity to self-association and aggregation, which is especially important for concentrated liquid formulations used for subcutaneous delivery.49 Several assays have been reported for evaluating antibody self-association, including static and dynamic light scattering.50,51 While these assays have proven to be valuable, they are not compatible with early-stage development due to their requirement for high antibody concentrations and purity. Therefore, alternative assays have been developed, including self-interaction chromatography (SIC),52,53 CIC,41,43 and clone self-interaction by biolayer interferometry,44 the latter of which has been shown to correlate with SIC and CIC. To enable even higher throughput and the use of low antibody concentrations, two nanoparticle-based assays have been reported, affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS)54–57 and charge-stabilized self-interaction nanoparticle spectroscopy (CS-SINS).50 AC-SINS is most commonly performed in a solution mimicking physiological conditions (pH 7.4, phosphate-buffered saline),3 and its measurements have most commonly been linked to pharmacokinetic properties,33,58 although it has also been used for formulation applications.59 CS-SINS is performed in a common formulation condition (pH 6, 10 mM histidine) and has been reported to identify antibodies with low viscosity and opalescence in concentrated antibody formulations.50
A third key feature of therapeutic antibodies is their high folding stability. Given the goal that formulated antibodies have a shelf-life of several years, obtaining real-time stability data are extremely time-consuming. To accelerate stability analysis, various stress conditions are commonly used. Thermal stability is typically measured using differential scanning calorimetry or differential scanning fluorimetry.60 In addition to temperature, surface-mediated stress can be used to evaluate antibody stability. For example, the recently described hydrophobic nanoparticles surface-stress assay was used with 14 antibody variants spanning a range of solubility values to identify variants characterized by high instability against agitation in the presence of air–water interfaces. Furthermore, aggregation assessment by this surface-mediated stress assay correlated well with other approaches to assess biophysical properties, such as temperature-induced aggregation and AC-SINS.61 Taken together, these and other in vitro assays allow the drug developer to select antibodies with combinations of preferable developability features. However, all in vitro assays require the expression of antibodies, sometimes including purification and formulation, followed by experiments to be carried out in the laboratory. To enable screening of an even higher number of antibodies, reduce cost, manual labor, and the requirement for antibody proteins, in silico methodologies are now gaining increased attention and multiple computational strategies to address developability exist, and additional ones are being rapidly developed.
Big data, machine learning, and computational assessments of developability
Computational assessments, which play an important role in assessing developability of biologics, are particularly useful in the early stages of biotherapeutic drug discovery where usually little to no experimental data is available.62–66 For example, an ideal stage for applying the computational assessments is immediately after sequencing the fragment variable regions (Fvs) of the antibody binders obtained from immunizations or display experiments, where there is a need to select a subset of binders for further experiments (Figure 2). Typical strategies for this selection include clustering of antibodies with respect to diversity of VH-VL germline pairs and epitope/paratope diversity. Inclusion of developability assessments at this stage is important to ensure focused use of available experimental resources, given that immunization campaigns often yield several thousands of potential hit sequences. Large portions of the encoded antibodies may either not bind in the subsequent confirmatory experiments or show multiple developability challenges. Flagging such antibodies via computational analyses helps prioritize antibodies for experimental testing (Figure 2). The developability assessments at this stage can include both sequence and structure-based methods or a combination thereof.63,64,67,68
Figure 2.
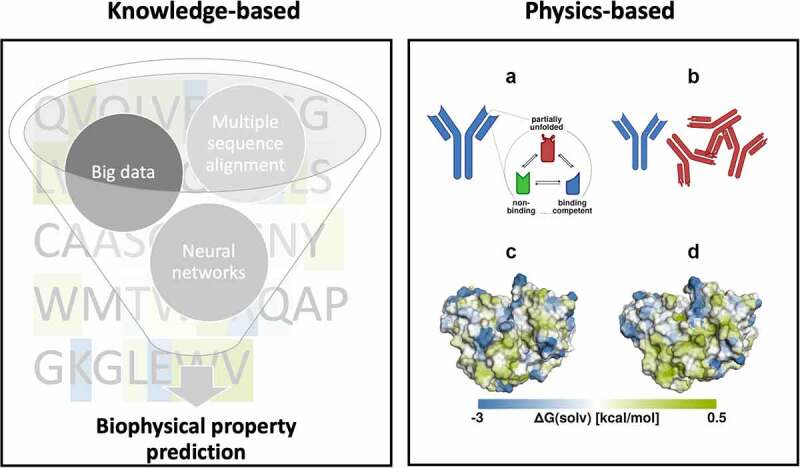
Knowledge and physics-based approaches for characterizing biophysical properties of antibodies. Left panel, knowledge-based: Overview of critical steps for sequence-based in silico prediction of biophysical properties from several thousands of potential hit sequences. Right panel, physics-based: A) The antibody binding interface exists as an ensemble of conformations, which includes binding competent as well as non-binding states. Partially unfolded conformations also exist with a lower probability. B) Different conformations exhibit different properties, where partially unfolded conformations may aggregate which leads to further unfolding. In C) and D), the hydrophobicity profile of two different conformations of the TNF-α binding antibody golimumab is mapped on its molecular surface using localized free energy of hydration. The two conformations show a significantly altered hydrophobicity profile and will therefore most likely interact differently with other hydrophobic molecules.
Typical components of developability assessments should include detection of motifs for chemical degradation, such as oxidation, deamidation, Asp isomerization, and along with those, motifs for glycosylation and presence of non-canonical Cys residues. Percent humanness (or “naturalness”),69 potential aggregation-prone regions and MHC class II binding immune epitopes can also be predicted using sequence-based methods.64,70,71 Advances in structural prediction of antibody variable regions in recent years have enabled fast and reliable high-throughput modeling of the antibody variable regions on a regular basis.72–76 Nevertheless, recent contributions on understanding the conformational behavior of CDR-H3 and its prediction indicate that the need for better models still exist.72,77,78 It is now feasible to model hundreds or even thousands of antibody structures with moderate computational resources. These Fv models can be used to compute several physicochemical descriptors, such as isoelectric point (pI), charge, hydrophobic imbalance, surface areas buried at the VH-VL interface along with molecular surface patches.64,79,80 These descriptors describe electrostatic as well as hydrophobic properties of the antibodies along with their conformational stabilities.
The pI of an antibody is an important property that potentially impacts the developability in multiple ways. It can affect various aspects, including those related to antibody purification, formulation (e.g., stability and viscosity), and pharmacokinetic properties. Generally, the isoelectric point of therapeutic antibodies is between 6 and 9. However, various developability challenges have been reported for some antibodies with relatively low (pI <6.5-7) or high (pI >8.5-9) isoelectric points.2,36,81–84 For example, Bailly et al. revealed that increasing the initially low pIs (pIs 6.3-6.7) of humanized antibodies to higher values (pIs >7) improved their purification yields and reduced their tendency to aggregate.2 Furthermore, the antibody pI affects pharmacokinetics, tissue distribution,81 and binding to FcRn and thereby the antibody half-life.82 By comparing the computed properties of the newly discovered confirmed binders (hits) with those of the antibodies in clinic or those currently available in the market,3,63,79,80 one can profile drug-likeness of the hits.
Among the antibodies chosen for experimental testing, the computational developability assessments can help identify potential lead molecules that possess an optimal combination of functional as well as physicochemical attributes. However, depending on peculiarities of individual drug discovery projects, these lead molecules may need to be affinity matured,85,86 humanized,87–89 formatted in novel molecular constructs,90 and further optimized for fitness with development platforms. This calls for the use of computational protein engineering tools to optimize the lead molecules. If the final molecular format of the lead molecule is a monoclonal IgG antibody, then computational engineering of the variable regions for improved developability attributes is more likely to translate into experimentally verifiable results and, surprisingly, only a few mutations can make substantial improvements in the CMC properties of the drug candidates.68,91–93
Once one or a few optimal lead molecule(s) have been selected, the discovery project then moves into the early development phase. In this phase, the optimized lead candidates are assessed for their fitness with the development platform(s). Computational developability assessments should now involve modeling the full-length antibody structure for a more accurate description of the computed properties and their agreement with the standard development experiments that involve larger amounts of materials and larger sample volumes. Use of multi-scale molecular simulations and machine learning models at these stages can help with identifying early formulation process challenges, such as aggregation, diffusion interaction, viscosity and solubility,94–104 physicochemical degradation,105–107 and immunogenicity.108,109 Specifically, use of explicit solvent molecular dynamics (MD) simulations can potentially provide a molecular-level understanding of molecular response to thermal and other stresses.110 Expanding the scope of such simulations to include the considerations of formulation buffers, salt, pH, and excipients will pave the way toward in silico formulation development for biologics. Finally, while most sequence and developability reports are based on biophysical models, machine learning methods for predicting developability parameters are being developed that infer developability parameters from the antibody sequence without the need for structural modeling.111,112
While the above-discussed machine learning approaches are discrimination tasks (e.g., prediction of variable X), deep learning also allows for the possibility of “generative machine learning”, which consists in learning the underlying features of a training dataset of antibody sequences with a specific property and then generating new antibody sequences, different from the training dataset, but with similar properties (also called features) as in the training dataset, where features may consist of developability parameters,113 binding parameters, or both.69,114–116 Interestingly, using a simulation framework, it was recently shown that generative learning can explore new binding and developability spaces.114 However, it remains to be determined to what extent out-of-distribution (generation of antibody sequences with features that were not included in the training dataset) learning is feasible and how much training data is necessary for achieving this task.64 In addition, so far, no antibody-focused generative approaches exist that would allow the generation of antibodies with multiple pre-specified features.117,118 Large-scale simulations may help in understanding the minimal data complexity necessary for such tasks.114,119–122 In summary, computation plays an important role in assessing the developability of biologics.
Biophysical properties of biologics from structure and molecular dynamics
Structural and dynamic characterization of antibodies is a prerequisite for engineering properties, such as chemical modifications, antigen recognition and receptor binding.123,124 The three-dimensional structure of proteins, in particular antibodies, is not static, but fluctuates constantly.125,126 These fluctuations can occur on different timescales, ranging from the low nanosecond timescale up to seconds.127 Even rare conformations can be relevant if they lead to a modification that is irreversible or part of a one-sided equilibrium, for example in aggregation or chemical modifications (Figure 2A).128–130 Furthermore, several studies have used molecular dynamics simulations to estimate the thermal stabilities of antibodies. For instance, the fraction of native contacts computed from simulations at high temperature has been shown to correlate with experimental melting temperatures.131
To elucidate the function and properties of antibodies, single-static structures are not always sufficient and thus, the antibody paratope may rather be characterized as conformational ensembles in solution.125,127,132 These paratope ensembles have been described by correlated CDR loop movements and interdomain and elbow angle rearrangements.125,133,134 This high flexibility and conformational diversity of the antigen-binding site, in particular of the CDR-H3 loop, challenges antibody structure prediction.132,135 Despite the substantial advances in antibody structure prediction,72–76 it is critical to carefully evaluate the structure model before further processing, as some of the models can contain structural inaccuracies, substantially deteriorating biophysical surface property predictions.136–138 Special care has to be taken when predicting antibody structures based on apo X-ray structures, which can be distorted by crystal packing effects and consequently do not correspond to the dominant solution-structure.125,132,139
Accounting for the high conformational diversity of antibodies by considering them as ensembles in solution can facilitate not only structure prediction but also guide the engineering workflow, facilitate identification of developability liabilities and optimize biophysical properties.136,140 For instance, it has been shown that aggregation of antibodies is accelerated by low-population states (Figure 2A-B) that become more frequent near hydrophobic surfaces and at phase boundaries.141 The hydrophobic interaction with the surface leads to a conformational shift toward more hydrophobic conformations, which in turn are more prone to aggregate (Figure 2A-B). In the same way, elevated temperatures shift the ensemble toward aggregation-prone conformations. This effect is experimentally seen as an irreversible aggregation.128,142,143 Therefore, it is necessary to describe antibody properties as an ensemble of structures. Molecular dynamics simulations127,144 provide such an ensemble in solution, thereby increasing the probability that the conformations responsible for hydrophobic or aggregation behavior are included (Figure 2C-D). Several works have studied the effect of conformational ensembles on hydrophobicity. While more coarse methods based on hydrophobicity scales are in general less sensitive to structural differences,145,146 a study based on explicit solvent thermodynamics found a critical influence of input structures and side chain orientations.136
It has been shown that both binding and biophysical properties46 of antibodies can be charge-dependent. However, the pKa of amino acids in proteins remains highly challenging to predict. In contrast to empirical methods such as protein pKa calculation,147 which assign protonation states for a single structure, constant pH molecular dynamics allows for the incorporation of protonation changes in structural sampling.148,149 Capturing the changes in protonation is particularly important for designing pH responsive antibodies, for example targeting acidified tumor microenvironments.
Thus, we strongly suggest considering antibodies as conformational ensembles in solution, which can improve structure prediction and allow assessment of biophysical properties that facilitate the development of antibody therapeutics.
Cellular assays for developability assessment
To a patient’s immune system, therapeutic biologics are foreign molecules, and therefore they may be immunogenic. Immunogenicity arising from structural traits and formulations can trigger both acute and long-term issues, such as innate and adaptive immune activation,150 acute cytokine storms,151 or a rise of anti-drug antibodies (ADA) causing neutralization of the drug and loss of therapeutic efficacy.152,153 Complex formation between ADA and the biologic can have detrimental effects to patients,154 which may be tied to an innate immune response leading to antigen presentation, secretion of inflammatory cytokines, and activation of T and B cells.155 Depending on the type of immune cells involved, immune activation may or may not lead to ADAs.155–157 A driver for antibody immunogenicity is T cell activation and subsequent cytokine release.158,159 This may be addressed in vitro by the use of immune cell activation assays, where pooled peripheral blood mononuclear cells are exposed to candidate biologics to reveal the presence of activating T cell epitopes.158 The general principle of such assays is illustrated in Figure 3. This type of assay has been shown to correlate with clinical immunogenicity of monoclonal IgG antibodies by revealing increased T cell proliferation and release of pro-inflammatory cytokines such as IL-2 and IFN-γ,160 and have been highlighted as important tools by both the European Medicines Agency and the US Food and Drug Administration.161,162 While such tools are often used to de-risk pre-clinical development, the in-patient immunogenicity of biologics remains difficult to predict, likely due to variations in drug delivery, the therapeutic context in which a given biologic is used, as well as the complexity of the human immune repertoire, including individual haplotype of human leukocyte antigens.
Figure 3.
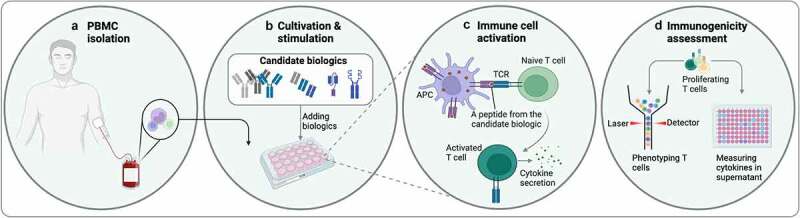
Schematic illustration of a Peripheral Blood Mononuclear Cell (PBMC) immunogenicity assay. A) PBMCs are isolated from healthy donors. B) Isolated cells are cultured in cell media with added candidate biologics. C) Candidate biologics are taken up by antigen presenting cells (APCs) and presented to T cells in culture. If the biologic is immunogenic, this may lead to T cell activation and concurrent cytokine secretion. D) Immunogenicity assessment can then be performed by phenotyping the T cells following stimulation with candidate biologics and measuring cytokine levels in culture supernatant.
Both IgG antibodies and albumin-based biologics have favorable transport properties within and across barriers, providing them with a long plasma half-life of three weeks on average in humans. This arises from their ability to bind to the neonatal Fc receptor (FcRn), which resides predominantly in acidified endosomes in a multitude of both non-hematopoietic and hematopoietic cell types.163,164 Here, FcRn encounters its ligands following their cellular entry by fluid-phase pinocytosis and binds to them in a pH-dependent fashion, where binding occurs at mildly acidic pH, and no binding or release occurs at neutral pH. This directs cellular recycling or transcytosis, and as such, FcRn engagement rescues the ligands from intracellular degradation, which results in high blood concentrations and long plasma half-life. The efficiency by which different biologics undergo this process has an enormous impact on their pharmacokinetic properties and biodistribution. This has spurred establishment of biophysical methods to determine pH-dependent binding kinetics toward FcRn by, for instance, the use of surface plasmon resonance, microscale thermophoresis, and affinity chromatography techniques, with the aim to predict the efficiency of FcRn-mediated transport.165–168 While these accessible and high-throughput biophysical methods can provide information regarding binding at a given pH condition, they typically do not unravel how the molecules bind FcRn throughout a pH gradient, and therefore do not directly mimic a cellular setting. However, the pH gradient can be mimicked by analytical FcRn affinity chromatography, where a gradual increase in pH is used to address dissociation of FcRn-targeted molecules from the receptor coupled to the matrix.166,169 While such studies can reveal valuable information, caution should be taken, as they do not account for the fact that FcRn is embedded in a negatively charged cell membrane and follows transport pathways involving different endosomal structures.170–172 Furthermore, they do not account for the diversity by which different cell types use FcRn for both ligand transport and antigen presentation and processing of immune complexes in concert with Fcγ receptors.164,173–178 In addition, the stoichiometry of FcRn in complex with both IgG and albumin in a cellular context is complex and far from fully understood.179–181 Thus, a need remains for reliable assays that can be used to dissect the determinants of cellular handling of IgG and albumin-based formats in both a FcRn-independent and dependent manner. This type of insights may guide lead selection by identifying so-called “red flags”, including, for example, polyreactivity, stability, and unfavorable pharmacokinetics early in the discovery process.3,79,80,182 These traits arise from biophysical properties such as surface charge, charge patches, isoelectric point, and hydrophobicity, and affect both FcRn binding, cellular transport, and in vivo performance.80,168,169,183 Therefore, cellular assays mimicking these traits are vital, and may provide necessary data input for computational analysis.3,80
Indeed, cellular assays addressing both FcRn-mediated cellular recycling and transcytosis exist and have been used to reveal insights on how FcRn-engaging formats are taken up and sorted.84,165,173,174,183–188 These assays have further been used to predict in vivo characteristics, exemplified by correlations between cellular transport properties and plasma half-life or clearance.84,165 The assays can include advanced live cell imaging to track both the receptor and its ligands and yield valuable mechanistic insights.171,189,190 However, while useful and yielding high resolution, imaging relies on protein labeling, which may affect overall stability as well as FcRn binding and transport.184,191 While many reports focus on measuring the amounts of molecules transported out of, or across cells, the rate of intracellular accumulation and degradation should also be accounted for. The importance of considering these parameters is exemplified by the use of a human endothelial cell-based recycling assay (HERA), where the cells overexpress human FcRn.165 HERA can be used for screening of the ability of FcRn-targeted molecules to be taken up, followed by their rescue from intracellular accumulation and/or degradation (Figure 4A). Notably, HERA only requires small amounts of proteins, and there is no need for labeling, as the parameters can be measured by ELISA setups on collected samples. Furthermore, the assay allows for manipulation of pH and blocking of the ligand binding sites, as well as modulation of the receptor expression level,165,168,183 which enables tailoring of the assay to specific needs and questions (Figure 4B). This provides a broad utility, and the cellular readout correlates with in vivo data. For example, HERA screening of IgG1 Fc-engineered variants with distinct FcRn binding kinetics revealed a predictive correlation with their plasma half-life in human FcRn transgenic mice (Figure 4C).165 In a recent study, HERA was used to address the efficiency by which different IgG1 Fc-containing biologics undergo cellular recycling, which, in combination with studies in transgenic mouse models, hints at the underlying reasons for the observably short half-life of IgG1 Fc-fusions compared to monoclonal IgG1 antibodies with corresponding specifities.168 Importantly, the study also addresses the discrepancy between recycling and transcytosis, which may be revealed by combining recycling assays with transwell studies.84,168,186 The latter allows for quantification of FcRn-mediated transcytotic transport, which, as opposed to recycling, correlates with increased clearance.84
Figure 4.
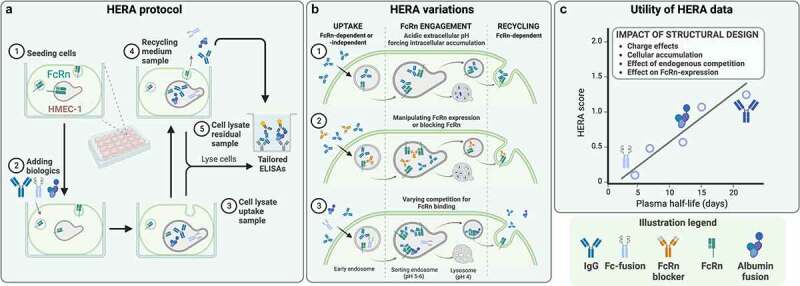
Human Endothelial Recycling Assay (HERA) as a tool for in vitro pharmacokinetic assessment and addressing FcRn-targeting strategies. A) Generalized HERA protocol. (1) Stably FcRn-transfected human microvascular endothelial cells (HMEC)-1 are seeded, prior to (2) adding FcRn-binding candidate biologics to two parallel cell plates. Following an incubation period, (3) cells from one plate are lysed to obtain an uptake sample. For the other plate, the media is exchanged to recycling medium, and after another incubation period, (4) the medium is harvested as a recycling sample, and (5) the cells are lysed to obtain a residual sample. Candidate biologics in all samples are quantified by an ELISA tailored for specific detection of the assessed biologic. B) Variations of the HERA protocol, enabling analysis of both FcRn-dependent and -independent uptake, cellular accumulation, and FcRn-dependent recycling. Variations include (1) performing the uptake step at mildly acidic extracellular pH, effectively forcing intracellular accumulation of biologics by preventing FcRn-mediated recycling, (2) manipulating FcRn-expression or blocking binding to FcRn to analyze FcRn-dependent and -independent cellular accumulation, and (3) introducing competition for FcRn binding to mirror endogenous competition on the ligand-binding sites of FcRn and its effects on the cellular transport for FcRn-binding biologics. C) HERA data can be used to address the impact of structural design of candidate biologics on FcRn-mediated cellular transport and unspecific cellular accumulation. For some candidate biologics, HERA data may allow for calculation of a score that correlates with plasma half-life in human FcRn-transgenic mice.
To realize the full utility of the cellular assays described above, the biophysical properties of biologics need to be considered. Factors such as the surface charge of Fc-fused structures may affect the cellular handling of biologics in both FcRn-dependent and independent manners. For example, the antibodies briakinumab and ustekinumab bind to the same target, the p40 subunit of IL12/23, but their varying charge profiles confer vastly different half-lives in humans.169,183,192,193 Specifically, the charge of the briakinumab Fv alters its interaction with FcRn.169,183,194 Cellular studies have revealed that this causes FcRn-independent cellular accumulation of briakinumab, which explains its shorter half-life.183 Interestingly, the same study found that the half-life of briakinumab can be improved by Fc-engineering, exemplifying that modulating the core binding to FcRn may adjust for unfavorable biophysical traits of Fc-fused structures. It also exemplifies the complexity tailored cellular assays may reveal. These are just a few of many existing examples of how distal structural traits may affect Fc binding to FcRn.164,194,195 Furthermore, HERA and other cellular assays can also be used to address transport of albumin-based molecules, which reflect their in vivo pharmacokinetic properties and ability to cross polarized mucosal epithelial cell surfaces.165,167,184,196
Preclinical pharmacokinetic assessment in mice
While cellular assays can guide selection and design of a limited set of lead biologic candidates, the in vivo behavior of drug candidates must subsequently be evaluated in reliable animal models. While non-human primates closely mimic human biology, their use for early development is limited by cost, accessibility, and ethical concerns. Hence, use of smaller animals, like mice, are preferred. However, such studies should be carefully planned and account for cross-species binding and expression differences between mice and humans, such as target recognition of antibody-based biologics. For example, while being a potent vascular endothelial growth factor (VEGF)-blocker in humans, the widely used anti-VEGF human IgG1 bevacizumab is unable to block mouse VEGF, implying that mice could not have been used in its development.197,198 Additionally, our understanding of FcRn biology has revealed major differences that must be taken into consideration when conventional mice are used. This is due to large differences in ligand binding to mouse and human FcRn, where mouse IgG binds very weakly to the human form, and human IgG binds stronger to mouse FcRn than to the human counterpart.199,200 Despite reports of a correlation between the pharmacokinetics of wild-type human IgG1 variants in conventional mice and humans,199 IgG1 variants that have been Fc engineered for enhanced human FcRn engagement and half-life extension fail to engage mouse FcRn efficiently due to loss of pH-dependent binding, resulting in short plasma half-life.165 These cross-species differences have motivated the development of mouse strains transgenic for human FcRn and lacking mouse FcRn. These transgenic mice are today the gold standard for pharmacokinetic evaluation of human IgG-based biologics,201–204 and importantly, data generated in them correlates with data from both non-human primates and clinical observations.205,206
An important, largely overlooked aspect of pharmacokinetic studies of antibodies in mice is their unusually low levels of endogenous IgG, which arises from the pathogen-free housing needed for experimental animal facilities.201,207,208 Naturally, injected IgG and albumin-based therapeutics will compete for binding to FcRn in the presence of large amounts of IgG and albumin, which have concentrations of approximately 12 and 40 mg/mL, respectively.164,209 This places competitive pressure on the ligand binding sites of FcRn, which modulates the plasma half-life of injected IgG and albumin.196,201,210,211 The absence of this pressure may mask relevant differences between candidate biologics,196 which should thus be accounted for in order to accurately predict pharmacokinetic properties. This can be achieved by pre-injection of high concentrations of intravenous IgG (IVIg).201,208 In addition, advances have recently been made in offering a relevant competitive setting in mice by creating human FcRn transgenic mice that also express human IgG1-Fc.201,202 Similarly, there are large differences regarding albumin binding across species. For instance, mouse FcRn binds poorly to human albumin,200,212 which effectively prevents human albumin-based formats from being rescued from intracellular degradation. Under these conditions, the plasma half-life of albumin drops to levels similar to irrelevant proteins of a size above the renal clearance threshold.213 Using human FcRn transgenic mice may correct for this issue. Additionally, such mice constitutively produce large amounts of endogenous albumin, effectively introducing a competitive environment for human albumin.196 In fact, human FcRn binds more efficiently to mouse albumin than the human counterpart,200,212 which increases the competitive pressure thathuman albumin-based biologics face in these mice. Alternatives offering a more biologically relevant setting include using human FcRn-expressing mice lacking endogenous albumin, and preloading them with human albumin, much like introducing IVIg when studying IgG,167,196,204 or using transgenic mice where mouse albumin has been replaced with the human counterpart.214,215
Cell line development and manufacturing of biologics
Most biologics are expensive therapeutic agents administered directly into the body of patients. Therefore, it is of the utmost importance that they are produced in an efficient, safe, and reproducible manner. The use of mammalian cells for production of marketed antibody therapeutics is most common (www.antibodysociety.org/antibody-therapeutics-product-data), but biologics may also be made in bacteria, yeast, and cell free expression systems.216 Production via mammalian cell cultivation most often involves billions of living cells, and it is challenging to control and reliably reproduce the complex biological processes involved at large scales. Therefore, substantial efforts go into the development of ideal cell lines for manufacturing biologics that stably express the product for more than 60 generations at high yields with consistent product quality in a highly reproducible process. As a result of these efforts, the development of production cell lines has been improved over the past decades,217 going from random integration of protein-expressing gene(s) followed by extensive screening for high-producing clones218 to more targeted approaches, including targeted gene integration for reproducible growth and yield219,220 and insertion of larger genetic elements221 to obtain robust high producing cell lines, and incorporating numerous cell engineering strategies to gain consistent product quality.222–226
Even though high-yielding and robust cell line development approaches have substantially advanced over the past decades, efficient production of biologics remains a continuous challenge. One contributing issue is the increasing complexity of biologics, such as heterologous proteins that can cause added cellular stresses and potentially cell death. Another challenge is the change in expression systems for biologics that often occurs between the early research stage and the later cell line development stage. Transient human expression systems are often preferred early on due to the ease and speed of production of workable product quantities,227 while stable CHO cell lines are preferred for commercial production.9 This change can cause profound differences in the product yield and quality (e.g., glycosylation, aggregation, protein folding), which means the product needs to be thoroughly re-characterized in the new expression system prior to commercial manufacture. Issues such as low product yield and changes in product quality that may compromise efficacy, quality, and patient safety are frequently encountered during cell line development.
The earlier the cell line development is considered during drug development, the higher the chances are for successful manufacturing. Numerous studies have shown that poor biophysical properties, such as aggregation propensity, are often linked to inefficient production in stable cell lines, underlining the importance of using prediction tools for biophysical properties as an early manufacturability indicator.93,228 The chances of successful manufacturing can also be increased by using a robust and flexible cell line development platform that combines early product assessment and stable cell line generation. A targeted integration platform with predictable high yield has strong potential in this regard, where the biologic can be produced in the same cell line from discovery to commercial manufacturing.219,229–231 Flexibility can be added by for example having a library of different glycoengineered cell lines,226 from where the desired glycosylation profile that determines the biologic’s stability, plasma half-life, and immunogenicity can be investigated and chosen. The cell line that provides the desired glycosylation profile of the product could then be used directly for large-scale manufacturing.
Antibodies for oral application
To date, most approved biologics are delivered as injectables, and the molecules, therefore, enter the circulatory system and exert their activity there or in tissues. However, alternative approaches are being explored, particularly for combating gastrointestinal (GI) infections.232–234 For such biologics, developability aspects are equally important as for injectable molecules. However, while properties such as plasma half-life and immunogenicity are critical factors for intravenously administered biologics, other aspects, such as stability in the GI tract and shelf-life are more important for orally available biologics that exert their function in the GI tract. Moreover, different quality parameters, such as product purity and presence of other proteins, may potentially be of less concern for orally administered biologics, as the GI tract normally encounters a wide range of macromolecules. As an example of how biologics can be optimized for the oral route, Fiil et al.235 engineered a highly biophysically stable homodivalent VHH construct for feed applications and demonstrated its functionality in inhibiting proliferation of enterotoxigenic E. coli (ETEC) in piglets.235 A key factor in their early design was the choice of a linker that was surprisingly more stable under GI conditions compared to the more natural hinge region of IgG3, which had previously been used as a linker for other orally delivered proteins.235,236 Similarly, Virdi et al. experimented with other formats, such as IgA-like molecules, which were also demonstrated to be effective in inhibiting ETEC proliferation in the GI tract of piglets.237 In this case, it was speculated that the incorporation of an Fc region would increase the retention time in the GI tract, which is conceptually similar to extending half-life.
In some cases, the biophysical stability of a biologic can be further optimized during the discovery process or via subsequent protein engineering efforts. In the case of nanobodies, one approach involves intracellular selection, which has been shown to select for nanobodies with higher stability.238 This can be followed with experiments in which nanobodies are subjected to elevated temperatures before the screening process, enabling the identification of nanobodies with higher refolding capacity,239 as well as protein engineering efforts where nanobodies are further stabilized against high temperature and proteases via the introduction of additional disulfide bonds between opposing beta strands.240–242
Finally, when a biologic is to be delivered orally, the use of alternative expression systems, such as microbial fermentation, the use of algae or transgenic plants, or even in situ production by engineered cells, may be considered.243–246 Such systems can potentially reduce both cost and time for production of the biologics, which could expand the range of applications for such molecules.
Concluding remarks
Recently, a shift has occurred within the discovery and development of biologics, from mainly focusing on high affinity and specificity to the target, hitting the right epitope, and conveying the desired function, to now also taking developability aspects into consideration. This broadened discovery and multidimensional engineering mindset is likely to yield better drug candidates, as well as reducing the number of late-stage failures during drug development. However, in many cases, especially with completely novel types of biologics, it is not always clear what constitutes a good developability profile, although some examples of such profiles (e.g., marketed drug-likeness79 and the Therapeutic Antibody Profiler80) are beginning to emerge. With more sequence information and biophysical data becoming publicly available, the task of establishing guiding principles on developability is becoming more approachable. Within this field, we expect that in silico predictions will play an increasingly larger role early in the discovery process, as they allow for very high-throughput analysis at low cost (when established). However, using in silico models may come with some inherent uncertainties due to biases in existing datasets, and re-evaluating algorithms and both expanding existing and building new datasets will continue to be important. During generation of these datasets, it is likely beneficial to include and explore molecules that may not be predicted to have superior developability profiles.
Another complication is the fact that some biophysical properties are inherently dynamic, such as aggregation and partial unfolding, and it can be important that in silico methods are able to incorporate molecular behavior in their prediction. As an example, as structure models are not always exact, simulations that take this into account are likely needed as a complement to steady-state models to improve the accuracy and reliability of biophysical predictions. To further improve the in silico approaches for the development of optimal biologics, both knowledge-based and physics-based methods are needed. Another hurdle for development of reliable in silico models is the lack of self-consistent and reproducible data obtained from experiments performed on a large number of molecules performed under similar conditions using similar protocols and instruments. The ongoing digital transformation of the biopharmaceutical industry is expected to ameliorate this difficulty and facilitate the generation of improved artificial intelligence and machine learning methods. Finally, the formation of collaborative consortia between industry and academia in a pre-competitive space to make self-consistent and reproducible data available for machine learning will be another great step forward in the improvement of in silico approaches and models.
In addition to in silico methods, we foresee that biophysical methods will continue to play a role to assess developability measures. Automation, miniaturization, and digitalization are key trends within drug development. Combined with novel in vitro assays, this may enable much more powerful and intelligent screening and characterization early in the discovery process for new biologics. However, a challenge in this area remains. While having a powerful discovery engine can be a major advantage for developing new biologics, it is both complicated and expensive to build up such capacity and educate personnel in the use of advanced systems. Moreover, as medicines are becoming increasingly more tailored and personalized, versatility and modularity of large discovery platforms need to be improved, so that they are not only optimized to discover modalities against a single type of indication.
Another avenue that will aid the development of biologics is the generation and use of animal models that better reflect the clinical setting and how the biologic performs in humans. In this regard, some important aspects include pharmacokinetics, immunogenicity, efficacy, and engagement of effector functions.
As biologics are often complex molecules to manufacture, it is of high importance that expression systems and purification methods are optimized to enable repeated, reproducible production of high-quality material. This involves optimization of yields, folding, post-translational modifications, such as glycosylation, and reduction of host cell proteins. Here, it is expected that glycoengineering will continue to play an important role, and we foresee that the consideration of glycosylation patterns (and other post-translational modifications) early in the discovery process might improve success rates for many protein-based biologics. In this area, however, much remains unknown, and it will be important to better establish knowledge and guidelines for how not only to engineer biologics to have human-like post-translational modifications, but also to have modifications that are even better than the corresponding human ones.
As a final remark, it is worth mentioning that most biologics to date are administered as injectables. In the future, it is expected that more biologics will be delivered orally, by the pulmonary route, or by other routes. This will undoubtedly have an influence on how new biologics should be developed and formulated, and may allow use of entirely new production systems.
Funding Statement
A.H.L. is supported by a grant from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program [850974], by a grant from the Villum Foundation [00025302], and a grant from Wellcome [221702/Z/20/Z]. V.G. is supported by The Helmsley Charitable Trust [2019PG-T1D011], UiO World-Leading Research Community, UiO:LifeSciences Convergence Environment Immunolingo, EU Horizon 2020 iReceptorplus [825821], a Research Council of Norway FRIPRO project [300740], a Research Council of Norway IKTPLUSS project [311341], and a Norwegian Cancer Society grant [215817]. T.P.J. has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie program [713683] (COFUNDfellowsDTU). J.T.A. received funding from the Research Council of Norway (grants: 287927, 314909). M.F.Q received funding the postdoctoral fellowship APART-MINT from the Austrian Academy of Sciences. K.R.L, F.W. and M.F.Q received funding from the Austrian Science Fund grant: P34518. B.V. and L.M.G. are supported by the Novo Nordisk Foundation [NNF20SA0066621]. P.M.T. is supported by the National Institutes of Health RF1AG059723, R35GM136300 (RF1AG059723 and R35GM136300), National Science Foundation CBET1813963, CBET1605266, CBET1804313 (CBET 1813963, 1605266 and 1804313), and Albert M. Mattocks Chair;
Abbreviations
- AC-SINS
Affinity-capture self-interaction nanoparticle spectroscopy
- ADA
Anti-drug antibodies
- APC
Antigen presenting cell
- CDR
Complementary-determining region
- CHO
Chinese hamster ovary
- CIC
Cross-interaction chromatography
- CMC
Chemistry, manufacturing, and control
- CS-SINS
Charge-stabilized self-interaction nanoparticle spectroscopy
- ETEC
Enterotoxigenic E. coli
- FcRn
Neonatal Fc receptor
- Fvs
Fragment variable regions
- GI
Gastrointestinal
- HERA
Human endothelial cell-based recycling assay
- HIC
Hydrophobic interaction chromatography
- KLH
Keyhole limpet hemocyanin
- LPS
Lipopolysaccharide
- MD
Molecular dynamics
- PBMC
Peripheral blood mononuclear cell
- PSP
Polyspecificity particle assay
- PSR
Polyspecificity Reagent
- SIC
Self-interaction chromatography
- SMAC
Standup monolayer chromatography
- VH
Variable heavy
- VL
Variable light
Conflict of Interest
J.T.A. is a co-founder, shareholder, and chairman of the board in the antibody-related company Authera AS. T.T.G. is a co-founder, shareholder and executive in Authera AS. A.H.L is a co-founder, shareholder, and board member in the antibody-related companies Bactolife A/S and VenomAid Diagnostics ApS. G.G. and H.K. are Roche employees and own Roche stocks; Roche has an interest in developing antibody-based therapeutics. S.K. is an employee of Boehringer-Ingelheim Pharmaceutical Inc. USA. V.G. declares advisory board positions in aiNET GmbH, Enpicom B.V, Specifica Inc, Adaptyv Biosystems, EVQLV, Omniscope, Diagonal Therapeutics, and Absci. V.G. is a consultant for Roche/Genentech, immunai, and Proteinea. J.M.C. is a co-founder, shareholder, and employee at Maxion Therapeutics and a shareholder of IONTAS Ltd.
References
- 1.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL.. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9(3):203–19. [DOI] [PubMed] [Google Scholar]
- 2.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. MAbs. 2020;12(1):1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5):944–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutierrez L, Cauchon NS, Christian TR, Giffin MJ, Abernathy MJ. The Confluence of Innovation in Therapeutics and Regulation: Recent CMC Considerations. J Pharm Sci. 2020;109(12):3524–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–98. [DOI] [PubMed] [Google Scholar]
- 6.Strohl WR, Strohl LM. 1-Introduction to biologics and monoclonal antibodies. Therapeutic Antibody Engineering. Woodhead Publishing Series in Biomedicine. Sawston, Cambridge: Woodhead Publishing; 2012. p. 1–595. [Google Scholar]
- 7.Pucca MB, Cerni FA, Janke R, Bermúdez-Méndez E, Ledsgaard L, Barbosa JE, Laustsen AH. History of Envenoming Therapy and Current Perspectives. Front Immunol. 2019;10:1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–97. [DOI] [PubMed] [Google Scholar]
- 9.Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018;36(12):1136–45. [DOI] [PubMed] [Google Scholar]
- 10.Ros F, Offner S, Klostermann S, Thorey I, Niersbach H, Breuer S, Zarnt G, Lorenz S, Puels J, Siewe B, et al. Rabbits transgenic for human IgG genes recapitulating rabbit B-cell biology to generate human antibodies of high specificity and affinity. MAbs. 2020;12(1):1846900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol. 2005;23(9):1117–25. [DOI] [PubMed] [Google Scholar]
- 12.Lee E-C, Liang Q, Ali H, Bayliss L, Beasley A, Bloomfield-Gerdes T, Bonoli L, Brown R, Campbell J, Carpenter A, et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol. 2014;32(4):356–63. [DOI] [PubMed] [Google Scholar]
- 13.Drabek D, Janssens R, van Haperen R, Grosveld F, Transgenic Heavy A. Chain IgG Mouse Platform as a Source of High Affinity Fully Human Single-Domain Antibodies for Therapeutic Applications. In: Hussack G, Henry KA, editors. Single-Domain Antibodies: Methods and Protocols. New York, NY: Springer US; 2022. p. 121–41. [DOI] [PubMed] [Google Scholar]
- 14.Lee E-C OM. The Application of Transgenic Mice for Therapeutic Antibody Discovery. In: Proetzel G, Ebersbach H, editors. Antibody Methods and Protocols. Totowa, NJ: Humana Press; 2012. p. 137–48. [DOI] [PubMed] [Google Scholar]
- 15.Gray AC, Bradbury A, Dübel S, Knappik A, Plückthun A, Borrebaeck CAK. Reproducibility: bypass animals for antibody production. Nature. 2020; 581:262. [DOI] [PubMed] [Google Scholar]
- 16.Bradbury ARM, Dübel S, Knappik A, Plückthun A. Animal- versus in vitro -derived antibodies: avoiding the extremes. MAbs. 2021;13(1):1950265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray A, Bradbury ARM, Knappik A, Plückthun A, Borrebaeck CAK, Dübel S. Animal-free alternatives and the antibody iceberg. Nat Biotechnol. 2020;38(11):1234–39. [DOI] [PubMed] [Google Scholar]
- 18.Borrebaeck CAK, Ohlin M. Antibody evolution beyond Nature. Nat Biotechnol. 2002;20(12):1189–90. [DOI] [PubMed] [Google Scholar]
- 19.Ledsgaard L, Ljungars A, Rimbault C, Sørensen CV, Tulika T, Wade J, Wouters Y, McCafferty J, Laustsen AH. Advances in antibody phage display technology. Drug Discov Today. 2022;27(8):2151–69. [DOI] [PubMed] [Google Scholar]
- 20.Starr CG, Tessier PM. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotechnol. 2019;60:119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.AH Laustsen, Greiff V, Karatt-Vellatt A, Muyldermans S, TP Jenkins. Animal Immunization in Vitro Display Technologies, and Machine Learning for Antibody Discovery. Trends Biotechnol. 2021;39(12):1263–73. [DOI] [PubMed] [Google Scholar]
- 22.Dobson CL, Devine PWA, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6(1):38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyson MR, Masters E, Pazeraitis D, Perera RL, Syrjanen JL, Surade S, Thorsteinson N, Parthiban K, Jones PC, Sattar M, et al. Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. MAbs. 2020;12(1):1829335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchanan A, Clementel V, Woods R, Harn N, Bowen MA, Mo W, Popovic B, Bishop SM, Dall’Acqua W, Minter R, et al. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. MAbs. 2013;5(2):255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bethea D, Wu S-J, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 2012;25(10):531–37. [DOI] [PubMed] [Google Scholar]
- 26.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348(6301):552–54. [DOI] [PubMed] [Google Scholar]
- 27.Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012;109(27):10879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376(4):926–31. [DOI] [PubMed] [Google Scholar]
- 29.Boublik Y, Di Bonito P, Jones IM. Eukaryotic virus display: engineering the major surface glycoprotein of the Autographa californica nuclear polyhedrosis virus (AcNPV) for the presentation of foreign proteins on the virus surface. Biotechnology. 1995;13(10):1079–84. [DOI] [PubMed] [Google Scholar]
- 30.Plückthun A. Ribosome display: a perspective. Methods Mol Biol. 2012;805:3–28. [DOI] [PubMed] [Google Scholar]
- 31.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–57. [DOI] [PubMed] [Google Scholar]
- 32.Parthiban K, Perera RL, Sattar M, Huang Y, Mayle S, Masters E, Griffiths D, Surade S, Leah R, Dyson MR, et al. A comprehensive search of functional sequence space using large mammalian display libraries created by gene editing. MAbs. 2019;11(5):884–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LB Avery, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, MS Kavosi, Penn S, Cirelli D, JC Kurz, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunningham O, Scott M, Zhou ZS, Finlay WJJ. Polyreactivity and polyspecificity in therapeutic antibody development: risk factors for failure in preclinical and clinical development campaigns. MAbs. 2021;13(1):1999195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–77. [DOI] [PubMed] [Google Scholar]
- 36.Rabia LA, Zhang Y, Ludwig SD, Julian MC, Tessier PM, Daggett V. Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng Des Sel. 2018;31(11):409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mouquet H, Scheid JF, Zoller MJ, Krogsgaard M, Ott RG, Shukair S, Artyomov MN, Pietzsch J, Connors M, Pereyra F, et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature. 2010;467(7315):591–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makowski EK, Wu L, Desai AA, Tessier PM. Highly sensitive detection of antibody nonspecific interactions using flow cytometry. MAbs. 2021;13(1):1951426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hötzel I, Theil F-P, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4(6):753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26(10):663–70. [DOI] [PubMed] [Google Scholar]
- 41.Tessier PM, Sandler SI, Lenhoff AM. Direct measurement of protein osmotic second virial cross coefficients by cross-interaction chromatography. Protein Sci. 2004;13(5):1379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.A-M WP, Sormanni P, Andersen JS, Sakhnini LI, Rodriguez-Leon I, Bjelke JR, Gajhede AJ, De Maria L, Otzen DE, Vendruscolo M, et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs. 2019;11(2):388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs SA, Wu S-J, Feng Y, Bethea D, O’Neil KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates. Pharm Res. 2010;27(1):65–71. [DOI] [PubMed] [Google Scholar]
- 44.Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Vásquez M, Wittrup KD, et al. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. MAbs. 2015;7(4):770–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7(4):752–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kraft TE, Richter WF, Emrich T, Knaupp A, Schuster M, Wolfert A, Kettenberger H. Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis. MAbs. 2020;12(1):1683432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. MAbs. 2014;6(4):852–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roettger BF, Ladisch MR. Hydrophobic interaction chromatography. Biotechnol Adv. 1989;7(1):15–29. [DOI] [PubMed] [Google Scholar]
- 49.Tessier PM, Wu J, Dickinson CD. Emerging methods for identifying monoclonal antibodies with low propensity to self-associate during the early discovery process. Expert Opin Drug Deliv. 2014;11(4):461–65. [DOI] [PubMed] [Google Scholar]
- 50.Starr CG, Makowski EK, Wu L, Berg B, Kingsbury JS, Gokarn YR, Tessier PM. Ultradilute Measurements of Self-Association for the Identification of Antibodies with Favorable High-Concentration Solution Properties. Mol Pharm. 2021;18(7):2744–53. [DOI] [PubMed] [Google Scholar]
- 51.Shahfar H, Du Q, Parupudi A, Shan L, Esfandiary R, Roberts CJ. Electrostatically Driven Protein-Protein Interactions: Quantitative Prediction of Second Osmotic Virial Coefficients to Aid Antibody Design. J Phys Chem Lett. 2022;13(5):1366–72. [DOI] [PubMed] [Google Scholar]
- 52.Tessier PM, Lenhoff AM, Sandler SI. Rapid measurement of protein osmotic second virial coefficients by self-interaction chromatography. Biophys J. 2002;82(3):1620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patro SY, Przybycien TM. Self-interaction chromatography: a tool for the study of protein-protein interactions in bioprocessing environments. Biotechnol Bioeng. 1996;52(2):193–203. [DOI] [PubMed] [Google Scholar]
- 54.Wu J, Schultz JS, Weldon CL, Sule SV, Chai Q, Geng SB, Dickinson CD, Tessier PM. Discovery of highly soluble antibodies prior to purification using affinity-capture self-interaction nanoparticle spectroscopy. Protein Eng Des Sel. 2015;28(10):403–14. [DOI] [PubMed] [Google Scholar]
- 55.Estep P, Caffry I, Yu Y, Sun T, Cao Y, Lynaugh H, Jain T, Vásquez M, Tessier PM, Xu Y. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. MAbs. 2015;7(3):553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 2014;6:483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sule SV, Sukumar M, Weiss WF. 4th, Marcelino-Cruz AM, Sample T, Tessier PM. High-throughput analysis of concentration-dependent antibody self-association. Biophys J. 2011;101:1749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu S, Datta-Mannan A, D’Argenio DZ. Physiologically Based Modeling to Predict Monoclonal Antibody Pharmacokinetics in Humans from in vitro Physiochemical Properties. MAbs. 2022;14(1):2056944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phan S, Walmer A, Shaw EW, Chai Q. High-throughput profiling of antibody self-association in multiple formulation conditions by PEG stabilized self-interaction nanoparticle spectroscopy. MAbs. 2022;14(1):2094750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kopp MRG, A-M WP, Zucca MV, Capasso Palmiero U, Friedrichsen B, Lorenzen N, Arosio P. An accelerated surface-mediated stress assay of antibody instability for developability studies. MAbs. 2020;12(1):1815995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilman W, Wróbel S, Bielska W, Deszynski P, Dudzic P, Jaszczyszyn I, Kaniewski J, Młokosiewicz J, Rouyan A, Satława T, et al. Machine-designed biotherapeutics: opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief Bioinform 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khetan R, Curtis R, Deane CM, Hadsund JT, Kar U, Krawczyk K, Kuroda D, Robinson SA, Sormanni P, Tsumoto K, et al. Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. MAbs. 2022;14(1):2020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akbar R, Bashour H, Rawat P, Robert PA, Smorodina E, Cotet T-S, Flem-Karlsen K, Frank R, Mehta BB, Vu MH, et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. MAbs. 2022;14(1):2008790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21(5):1549–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang B, Gallolu Kankanamalage S, Dong J, Liu Y. Optimization of therapeutic antibodies. Antib Ther. 2021;4(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaa R, Menang-Ndi E, Pratapa S, Nguyen C, Kumar S, Doerner A. Versatile and rapid microfluidics-assisted antibody discovery. MAbs. 2021;13(1):1978130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narayanan H, Dingfelder F, Butté A, Lorenzen N, Sokolov M, Arosio P. Machine Learning for Biologics: Opportunities for Protein Engineering, Developability, and Formulation. Trends Pharmacol Sci. 2021;42(3):151–65. [DOI] [PubMed] [Google Scholar]
- 69.Bachas S, Rakocevic G, Spencer D, Sastry AV, Haile R, Sutton JM, Kasun G, Stachyra A, Gutierrez JM, Yassine E, et al. Antibody optimization enabled by artificial intelligence predictions of binding affinity and naturalness. bioRxiv. 2022;1–39. [Google Scholar]
- 70.Kumar S, Singh SK. Developability of Biotherapeutics: Computational Approaches. Boca Raton: CRC Press; 2015. [Google Scholar]
- 71.Kumar S, Plotnikov NV, Rouse JC, Singh SK. Biopharmaceutical Informatics: supporting biologic drug development via molecular modelling and informatics. J Pharm Pharmacol. 2018;70(5):595–608. [DOI] [PubMed] [Google Scholar]
- 72.Ruffolo JA, Sulam J, Gray JJ. Antibody structure prediction using interpretable deep learning. Patterns (N Y). 2022;3(2):100406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leem J, Dunbar J, Georges G, Shi J, Deane CM. ABodyBuilder: Automated antibody structure prediction with data-driven accuracy estimation. MAbs. 2016;8(7):1259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruffolo JA, Chu L-S, Mahajan SP, Gray JJ. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. bioRxiv. 2022. Aug15. doi: 10.1101/2022.04.20.488972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marks C, Deane CM. How repertoire data are changing antibody science. J Biol Chem. 2020;295(29):9823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Almagro JC, Beavers MP, Hernandez-Guzman F, Maier J, Shaulsky J, Butenhof K, Labute P, Thorsteinson N, Kelly K, Teplyakov A, et al. Antibody modeling assessment. Proteins. 2011;79(11):3050–66. [DOI] [PubMed] [Google Scholar]
- 77.Abanades B, Georges G, Bujotzek A, Deane CM, Xu J. ABlooper: Fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics. 2022;38(7): 1877–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruffolo JA, Guerra C, Mahajan SP, Sulam J, Gray JJ. Geometric potentials from deep learning improve prediction of CDR H3 loop structures. Bioinformatics. 2020;36(Supplement_1):i268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmed L, Gupta P, Martin KP, Scheer JM, Nixon AE, Kumar S. Intrinsic physicochemical profile of marketed antibody-based biotherapeutics. Proc Natl Acad Sci U S A.2021; 118(37): e2020577118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U S A. 2019;116(10):4025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boswell CA, Tesar DB, Mukhyala K, Theil F-P, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21(12):2153–63. [DOI] [PubMed] [Google Scholar]
- 82.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169(9):5171–80. [DOI] [PubMed] [Google Scholar]
- 83.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111(52):18601–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chung S, Nguyen V, Lin YL, Lafrance-Vanasse J, Scales SJ, Lin K, Deng R, Williams K, Sperinde G, Li JJ, et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs. 2019;11:942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Conti S, Lau EY, Ovchinnikov V. On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations. Antibodies. 2022;11(3):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahmad B, Batool M, Kim M-S CS, Choi S. Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies. Int J Mol Sci. 2021; 22. (11): 5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goulet DR, Chatterjee S, Lee W-P, Waight AB, Zhu Y, Mak AN-S. Engineering an Enhanced EGFR Engager: Humanization of Cetuximab for Improved Developability. Antibodies (Basel). 2022;11(1): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marks C, Hummer AM, Chin M, Deane CM. Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinformatics. 2021;37(22): 4041–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prihoda D, Maamary J, Waight A, Juan V, Fayadat-Dilman L, Svozil D, Bitton DA. BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. MAbs. 2022;14(1):2020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wade J, Rimbault C, Ali H, Ledsgaard L, Rivera-de-Torre E, Abou Hachem M, Boddum K, Mirza N, Bohn M-F, Sakya SA, et al. Generation of Multivalent Nanobody-Based Proteins with Improved Neutralization of Long α-Neurotoxins from Elapid Snakes. Bioconjug Chem. 2022;33(8):1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumar S, Roffi K, Tomar DS, Cirelli D, Luksha N, Meyer D, Mitchell J, Allen MJ, Li L. Rational optimization of a monoclonal antibody for simultaneous improvements in its solution properties and biological activity. Protein Eng Des Sel. 2018;31(7–8):313–25. [DOI] [PubMed] [Google Scholar]
- 92.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7(1):212–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bauer J, Mathias S, Kube S, Otte K, Garidel P, Gamer M, Blech M, Fischer S, Karow-Zwick AR. Rational optimization of a monoclonal antibody improves the aggregation propensity and enhances the CMC properties along the entire pharmaceutical process chain. MAbs. 2020;12(1):1787121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prabakaran R, Rawat P, Yasuo N, Sekijima M, Kumar S, Gromiha MM. Effect of charged mutation on aggregation of a pentapeptide: Insights from molecular dynamics simulations. Proteins. 2022;90(2):405–17. [DOI] [PubMed] [Google Scholar]
- 95.Prabakaran R, Rawat P, Kumar S, Michael Gromiha M. ANuPP: A Versatile Tool to Predict Aggregation Nucleating Regions in Peptides and Proteins. J Mol Biol. 2021;433(11):166707. [DOI] [PubMed] [Google Scholar]
- 96.Tomar DS, Li L, Broulidakis MP, Luksha NG, Burns CT, Singh SK, Kumar S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs. 2017;9(3):476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tomar DS, Kumar S, Singh SK, Goswami S, Li L. Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. MAbs. 2016;8(2):216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tomar DS, Singh SK, Li L, Broulidakis MP, Kumar S. In Silico Prediction of Diffusion Interaction Parameter (kD), a Key Indicator of Antibody Solution Behaviors. Pharm Res. 2018;35(10):193. [DOI] [PubMed] [Google Scholar]
- 99.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8(1):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buck PM, Chaudhri A, Kumar S, Singh SK. Highly viscous antibody solutions are a consequence of network formation caused by domain-domain electrostatic complementarities: insights from coarse-grained simulations. Mol Pharm. 2015;12(1):127–39. [DOI] [PubMed] [Google Scholar]
- 101.Hashemi A, Basafa M, Behravan A. Machine learning modeling for solubility prediction of recombinant antibody fragment in four different E. coli strains Sci Rep. 2022;12(1):5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khurana S, Rawi R, Kunji K, Chuang G-Y, Bensmail H, Mall R, Valencia A. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics. 2018;34(15):2605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lai P-K, Fernando A, Cloutier TK, Kingsbury JS, Gokarn Y, Halloran KT, Calero-Rubio C, Trout BL. Machine Learning Feature Selection for Predicting High Concentration Therapeutic Antibody Aggregation. J Pharm Sci. 2021;110(4):1583–91. [DOI] [PubMed] [Google Scholar]
- 104.Rawat P, Prabakaran R, Kumar S, Gromiha MM. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci Rep. 2021;11(1):13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Plotnikov NV, Singh SK, Rouse JC, Kumar S. Quantifying the Risks of Asparagine Deamidation and Aspartate Isomerization in Biopharmaceuticals by Computing Reaction Free-Energy Surfaces. J Phys Chem B. 2017;121(4):719–30. [DOI] [PubMed] [Google Scholar]
- 106.Kaliman I, Nemukhin A, Varfolomeev S. Free Energy Barriers for the N-Terminal Asparagine to Succinimide Conversion: Quantum Molecular Dynamics Simulations for the Fully Solvated Model. J Chem Theory Comput. 2010;6(1):184–89. [DOI] [PubMed] [Google Scholar]
- 107.Salsbury Jr FR Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010;10(6):738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hashemi N, Hao B, Ignatov M, Paschalidis I, Vakili P, Vajda S, Kozakov D. Improved Predictions of MHC-Peptide Binding using Protein Language Models. bioRxiv. 2022. doi: 10.1101/2022.02.11.479844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tomar DS, Licari G, Bauer J, Singh SK, Li L, Kumar KS. Stress-dependent Flexibility of a Full-length Human Monoclonal Antibody: Insights from Molecular Dynamics to Support Biopharmaceutical Development. J Pharm Sci. 2022;111(3):628–37. [DOI] [PubMed] [Google Scholar]
- 111.Chen X, Dougherty T, Hong C, Schibler R, Zhao YC, Sadeghi R, Matasci N, Wu Y-C KI. Predicting Antibody Developability from Sequence using Machine Learning. bioRxiv. 2020. doi: 10.1101/2020.06.18.159798 [DOI] [Google Scholar]
- 112.Lai P-K, Fernando A, Cloutier TK, Gokarn Y, Zhang J, Schwenger W, Chari R, Calero-Rubio C, Trout BL. Machine Learning Applied to Determine the Molecular Descriptors Responsible for the Viscosity Behavior of Concentrated Therapeutic Antibodies. Mol Pharm. 2021;18(3):1167–75. [DOI] [PubMed] [Google Scholar]
- 113.Amimeur T, Shaver JM, Ketchem RR, Alex Taylor J, Clark RH, Smith J, Van Citters D, Siska CC, Smidt P, Sprague M, et al. Designing Feature-Controlled Humanoid Antibody Discovery Libraries Using Generative Adversarial Networks. bioRxiv. 2020. doi: 10.1101/2020.04.12.024844 [DOI] [Google Scholar]
- 114.Akbar R, Robert PA, Weber CR, Widrich M, Frank R, Pavlović M, Scheffer L, Chernigovskaya M, Snapkov I, Slabodkin A, et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. MAbs. 2022;14(1):2031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Khan A, Cowen-Rivers AI, Deik D-G-X, Grosnit A, Dreczkowski K, Robert PA, Greiff V, Tutunov R, Bou-Ammar D, Wang J, et al. AntBO: Towards Real-World Automated Antibody Design with Combinatorial Bayesian Optimisation. Cell Reports Methods. 2023;3: 100374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Makowski EK, Kinnunen PC, Huang J, Wu L, Smith MD, Wang T, Desai AA, Streu CN, Zhang Y, Zupancic JM, et al. Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat Commun. 2022;13(1):3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Freschlin CR, Fahlberg SA, Romero PA. Machine learning to navigate fitness landscapes for protein engineering. Curr Opin Biotechnol. 2022;75:102713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Das P, Sercu T, Wadhawan K, Padhi I, Gehrmann S, Cipcigan F, Chenthamarakshan V, Strobelt H, Dos Santos C, Chen P-Y, et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat Biomed Eng. 2021;5(6):613–23. [DOI] [PubMed] [Google Scholar]
- 119.Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, Chernigovskaya M, Scheffer L, Slabodkin A, Mehta BB , et al. One billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction Nature Computational Science. 2022;2:845-865. [DOI] [PubMed] [Google Scholar]
- 120.Sandve GK, Greiff V. Access to ground truth at unconstrained size makes simulated data as indispensable as experimental data for bioinformatics methods development and benchmarking. Bioinformatics. 2022. [DOI] [PMC free article] [PubMed]
- 121.Weber CR, Akbar R, Yermanos A, Pavlović M, Snapkov I, Sandve GK, Reddy ST, Greiff V, Schwartz R. immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking. Bioinformatics. 2020;36(11):3594–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jafari M, Guan Y, Wedge DC, Ansari-Pour N. Re-evaluating experimental validation in the Big Data Era: a conceptual argument. Genome Biol. 2021;22(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kralj S, Hodošček M, Podobnik B, Kunej T, Bren U, Janežič D, Konc J. Molecular Dynamics Simulations Reveal Interactions of an IgG1 Antibody With Selected Fc Receptors. Front Chem. 2021;9:705931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ieong P, Amaro RE, Li WW. Molecular dynamics analysis of antibody recognition and escape by human H1N1 influenza hemagglutinin. Biophys J. 2015;108(11):2704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fernández-Quintero ML, Pomarici ND, Math BA, Kroell KB, Waibl F, Bujotzek A, Georges G, Liedl KR. Antibodies exhibit multiple paratope states influencing VH-VL domain orientations. Commun Biol. 2020;3(1):589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450(7172):964–72. [DOI] [PubMed] [Google Scholar]
- 127.Fernández-Quintero ML, Georges G, Varga JM, Liedl KR. Ensembles in solution as a new paradigm for antibody structure prediction and design. MAbs. 2021;13(1):1923122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schrag JD, M-è P, Gaudreault F, Gagnon L-P, Baardsnes J, Manenda MS, Sheff J, Deprez C, Baptista C, Hogues H, et al. Binding symmetry and surface flexibility mediate antibody self-association. MAbs. 2019;11(7):1300–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Samantray S, Schumann W, Illig A-M, Carballo-Pacheco M, Paul A, Barz B, Strodel B. Molecular Dynamics Simulations of Protein Aggregation: Protocols for Simulation Setup and Analysis with Markov State Models and Transition Networks. In: Li MS, Kloczkowski A, Cieplak M, Kouza M, editors. Computer Simulations of Aggregation of Proteins and Peptides. New York, NY: Springer US; 2022. p. 235–79. [DOI] [PubMed] [Google Scholar]
- 130.Schmid I, Bonnington L, Gerl M, Bomans K, Thaller AL, Wagner K, Schlothauer T, Falkenstein R, Zimmermann B, Kopitz J, et al. Assessment of susceptible chemical modification sites of trastuzumab and endogenous human immunoglobulins at physiological conditions. Commun Biol. 2018;1(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bekker G-J, Ma B, Kamiya N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. 2019;28(2):429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fernández-Quintero ML, Kraml J, Georges G, Liedl KR. CDR-H3 loop ensemble in solution - conformational selection upon antibody binding. MAbs. 2019;11(6):1077–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fernández-Quintero ML, Kroell KB, Heiss MC, Loeffler JR, Quoika PK, Waibl F, Bujotzek A, Moessner E, Georges G, Liedl KR. Surprisingly Fast Interface and Elbow Angle Dynamics of Antigen-Binding Fragments. Front Mol Biosci. 2020;7:609088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fernández-Quintero ML, Hoerschinger VJ, Lamp LM, Bujotzek A, Georges G, Liedl KR. V H -V L interdomain dynamics observed by computer simulations and NMR. Proteins. 2020;88(7):830–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Regep C, Georges G, Shi J, Popovic B, Deane CM. The H3 loop of antibodies shows unique structural characteristics. Proteins. 2017;85(7):1311–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Waibl F, Fernández-Quintero ML, Kamenik AS, Kraml J, Hofer F, Kettenberger H, Georges G, Liedl KR. Conformational Ensembles of Antibodies Determine Their Hydrophobicity. Biophys J. 2021;120(1):143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Waibl F, ML F-Q, FS W, Kettenberger H, Georges G, KR L. Comparison of hydrophobicity scales for predicting biophysical properties of antibodies. Frontiers in Molecular Biosciences. 2022;9:960194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fernández-Quintero ML, Kokot J, Waibl F, Fischer A-LM, Quoika PK, Deane CM, Liedl KR. Challenges in antibody structure prediction. bioRxiv. 2022. doi: 10.1101/2022.11.09.515600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernández-Quintero ML, Heiss MC, Pomarici ND, Math BA, Liedl KR. Antibody CDR loops as ensembles in solution vs. canonical clusters from X-ray structures. MAbs. 2020;12:1744328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernández-Quintero ML, Vangone A, Loeffler JR, Seidler CA, Georges G, Liedl KR. Paratope states in solution improve structure prediction and docking. Structure. 2022;30(3):430–40.e3. [DOI] [PubMed] [Google Scholar]
- 141.Hauptmann A, Hoelzl G, Loerting T. Distribution of Protein Content and Number of Aggregates in Monoclonal Antibody Formulation After Large-Scale Freezing. AAPS PharmSciTech. 2019;20(2):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chakroun N, Hilton D, Ahmad SS, Platt GW, Dalby PA. Mapping the Aggregation Kinetics of a Therapeutic Antibody Fragment. Mol Pharm. 2016;13(2):307–19. [DOI] [PubMed] [Google Scholar]
- 143.Robinson MJ, Matejtschuk P, Bristow AF, Dalby PA. T m -Values and Unfolded Fraction Can Predict Aggregation Rates for Granulocyte Colony Stimulating Factor Variant Formulations but Not under Predominantly Native Conditions. Mol Pharm. 2018;15(1):256–67. [DOI] [PubMed] [Google Scholar]
- 144.Jo S, Xu A, Curtis JE, Somani S. AD MacKerell Jr. Computational Characterization of Antibody-Excipient Interactions for Rational Excipient Selection Using the Site Identification by Ligand Competitive Saturation-Biologics Approach. Mol Pharm. 2020;17:4323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012;101(1):102–15. [DOI] [PubMed] [Google Scholar]
- 146.Sankar K, Krystek SR, Carl SM, Day T, Maier JKX. AggScore: Prediction of aggregation-prone regions in proteins based on the distribution of surface patches. Proteins. 2018;86(11):1147–56. [DOI] [PubMed] [Google Scholar]
- 147.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7(2):525–37. [DOI] [PubMed] [Google Scholar]
- 148.Swails JM, York DM, Roitberg AE. Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J Chem Theory Comput. 2014;10(3):1341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Radak BK, Chipot C, Suh D, Jo S, Jiang W, Phillips JC, Schulten K, Constant-pH Molecular RB. Dynamics Simulations for Large Biomolecular Systems. J Chem Theory Comput. 2017;13(12):5933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thacker SG, Her C, Kelley-Baker L, Ireland DDC, Manangeeswaran M, Pang ES, Verthelyi D. Detection of innate immune response modulating impurities (IIRMI) in therapeutic peptides and proteins: Impact of excipients. Front Immunol. 2022;13:970499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Modernizing medical product development through collaboration . FDA Consum 2006; 40:5. [PubMed] [Google Scholar]
- 152.Vaisman-Mentesh A, Rosenstein S, Yavzori M, Dror Y, Fudim E, Ungar B, Kopylov U, Picard O, Kigel A, Ben-Horin S, et al. Molecular Landscape of Anti-Drug Antibodies Reveals the Mechanism of the Immune Response Following Treatment With TNFα Antagonists. Front Immunol. 2019;10:2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Garcês S, Demengeot J. The Immunogenicity of Biologic Therapies. Curr Probl Dermatol. 2018;53:37–48. [DOI] [PubMed] [Google Scholar]
- 154.Krishna M, Nadler SG. Immunogenicity to Biotherapeutics - The Role of Anti-drug Immune Complexes. Front Immunol. 2016;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pratt KP. Anti-Drug Antibodies: Emerging Approaches to Predict, Reduce or Reverse Biotherapeutic Immunogenicity. Antibodies (Basel). 2018;7(2):1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Horton C, Shanmugarajah K, Fairchild PJ. Harnessing the properties of dendritic cells in the pursuit of immunological tolerance. Biomed J. 2017;40(2):80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Blumberg RS,Lillicrap D, IgG Fc Immune Tolerance Group. Tolerogenic properties of the Fc portion of IgG and its relevance to the treatment and management of hemophilia. Blood. 2018;131(20):2205-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, De Groot AS. T-Cell Dependent Immunogenicity of Protein Therapeutics Pre-clinical Assessment and Mitigation-Updated Consensus and Review 2020. Front Immunol. 2020;11:1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, Scott DW, Martin W. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes.”. Blood. 2008;112(8):3303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lutfi RA. Informational processing of complex sound I: Intensity discrimination. J Acoust Soc Am. 1989;86:934–44. [DOI] [PubMed] [Google Scholar]
- 161.Committee for Medicinal Products for Human Use (CHMP) . Guideline on Immunogenicity assessment of therapeutic proteins. European Medicines Agency; 2017. [Google Scholar]
- 162.U.S . Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER). Guidance for Industry Immunogenicity Assessment for Therapeutic Protein Products. 2014.
- 163.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. [DOI] [PubMed] [Google Scholar]
- 164.Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The Neonatal Fc Receptor (FcRn): A Misnomer?. Front Immunol. 2019;10:1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Grevys A, Nilsen J, Sand KMK, Daba MB, Øynebråten I, Bern M, McAdam MB, Foss S, Schlothauer T, Michaelsen TE. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat Commun. 2018;9(1):621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs. 2013;5(4):576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nilsen J, Trabjerg E, Grevys A, Azevedo C, Brennan SO, Stensland M, Wilson J, Sand KMK, Bern M, Dalhus B, et al. An intact C-terminal end of albumin is required for its long half-life in humans. Commun Biol. 2020;3(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gjølberg TT, Frick R, Mester S, Foss S, Grevys A, Høydahl LS, Øk J, Schlothauer T, Sandlie I, Moe MC, et al. Biophysical differences in IgG1 Fc-based therapeutics relate to their cellular handling, interaction with FcRn and plasma half-life. Commun Biol. 2022;5(1):832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci USA. 2015;112(19):5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Acquiring PM. English at different ages: the English displacement effect and other findings. J Psycholinguist Res. 1990;19(1):57–70. [DOI] [PubMed] [Google Scholar]
- 171.Gan Z, Ram S, Ober RJ, Ward ES. Using multifocal plane microscopy to reveal novel trafficking processes in the recycling pathway. J Cell Sci. 2013;126(5):1176–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Louis FJ, Fargier JJ, Maubert B, Louis JP, Hounsinou C, Le Bras J, Touzé JE. Severe malaria attacks in adults in Cameroon: comparison of 2 therapeutic protocols using quinine via parenteral route. Ann Soc Belg Med Trop. 1992;72:179–88. [PubMed] [Google Scholar]
- 173.Foss S, Grevys A, Sand KMK, Bern M, Blundell P, Michaelsen TE, Pleass RJ, Sandlie I, Andersen JT. Enhanced FcRn-dependent transepithelial delivery of IgG by Fc-engineering and polymerization. J Control Release. 2016;223:42–52. [DOI] [PubMed] [Google Scholar]
- 174.Deissler HL, Lang GK, Lang GE. Neonatal Fc receptor FcRn is involved in intracellular transport of the Fc fusion protein aflibercept and its transition through retinal endothelial cells. Exp Eye Res. 2017;154:39–46. [DOI] [PubMed] [Google Scholar]
- 175.Hubbard JJ, Pyzik M, Rath T, Kozicky LK, Sand KMK, Gandhi AK, Grevys A, Foss S, Menzies SC, Glickman JN, et al. FcRn is a CD32a coreceptor that determines susceptibility to IgG immune complex-driven autoimmunity. J Exp Med 2020;217(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shimotsu H, Kuroda MI, Yanofsky C, Henner DJ. Novel form of transcription attenuation regulates expression the Bacillus subtilis tryptophan operon. J Bacteriol. 1986;166(2):461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Longo JM, Rodríguez-Cabello J, Bilbao JI, Aquerreta JD, Ruza M, Mansilla F. Popliteal vein thrombosis and popliteal artery compression complicating fibular osteochondroma: ultrasound diagnosis. J Clin Ultrasound. 1990;18(6):507–09. [DOI] [PubMed] [Google Scholar]
- 178.Baker K, Rath T, Lencer WI, Fiebiger E, Blumberg RS. Cross-presentation of IgG-containing immune complexes. Cell Mol Life Sci. 2013;70:1319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994;372(6504):379–83. [DOI] [PubMed] [Google Scholar]
- 180.Raghavan M, Wang Y, Bjorkman PJ. Effects of receptor dimerization on the interaction between the class I major histocompatibility complex-related Fc receptor and IgG. Proc Natl Acad Sci U S A. 1995;92(24):11200–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Berschneider HM, Martens H, Powell DW. Effect of BW 942C, an enkephalinlike pentapeptide, on sodium and chloride transport in the rabbit ileum. Gastroenterology. 1988;94(1):127–36. [DOI] [PubMed] [Google Scholar]
- 182.Shehata L, Maurer DP, Wec AZ, Lilov A, Champney E, Sun T, Archambault K, Burnina I, Lynaugh H, Zhi X, et al. Affinity Maturation Enhances Antibody Specificity but Compromises Conformational Stability. Cell Rep. 2019;28(13):3300–3308. [DOI] [PubMed] [Google Scholar]
- 183.Grevys A, Frick R, Mester S, Flem-Karlsen K, Nilsen J, Foss S, Sand KMK, Emrich T, Fischer JAA, Greiff V, et al. Antibody variable sequences have a pronounced effect on cellular transport and plasma half-life. iScience. 2022;25(2):103746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Schmidt EGW, Hvam ML, Antunes F, Cameron J, Viuff D, Andersen B, Kristensen NN, Howard KA. Direct demonstration of a neonatal Fc receptor (FcRn)-driven endosomal sorting pathway for cellular recycling of albumin. J Biol Chem. 2017;292(32):13312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sockolosky JT, Tiffany MR, Szoka FC. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci U S A. 2012;109(40):16095–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liu C, Kim YS, Lowe JH-N, Chung S. A cell-based FcRn-dependent recycling assay for predictive pharmacokinetic assessment of therapeutic antibodies. Bioanalysis. 2021;13(14):1135–44. [DOI] [PubMed] [Google Scholar]
- 187.Claypool SM, Dickinson BL, Yoshida M, Lencer WI, Blumberg RS. Functional reconstitution of human FcRn in Madin-Darby canine kidney cells requires co-expressed human beta 2-microglobulin. J Biol Chem. 2002;277(31):28038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ramalingam TS, Detmer SA, Martin WL, Bjorkman PJ. IgG transcytosis and recycling by FcRn expressed in MDCK cells reveals ligand-induced redistribution. EMBO J. 2002;21(4):590–601. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189.Weflen AW, Baier N, Tang Q-J, Van den Hof M, Blumberg RS, Lencer WI, Massol RH, Gruenberg JE. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol Biol Cell. 2013;24(15):2398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gan Z, Ram S, Vaccaro C, Ober RJ, Ward ES. Analyses of the recycling receptor, FcRn, in live cells reveal novel pathways for lysosomal delivery. Traffic. 2009;10(5):600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sato K, Howell JN, Greene MH, Maher VM, McCormick JJ. Relationship between sensitivity of cells from patients with hereditary cutaneous malignant melanoma to killing and mutations by 4-nitroquinoline 1-oxide and adduct formation. Cancer Res. 1988;48:5145–50. [PubMed] [Google Scholar]
- 192.Zhu Y, Hu C, Lu M, Liao S, Marini JC, Yohrling J, Yeilding N, Davis HM, Zhou H. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2009;49(2):162–75. [DOI] [PubMed] [Google Scholar]
- 193.Lima XT, Abuabara K, Kimball AB, Lima HC. Briakinumab. Expert Opin Biol Ther. 2009;9(8):1107–13. [DOI] [PubMed] [Google Scholar]
- 194.Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD. A Two-pronged Binding Mechanism of IgG to the Neonatal Fc Receptor Controls Complex Stability and IgG Serum Half-life. Mol Cell Proteomics. 2017;16(3):451–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’Hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. MAbs. 2018;10(1):81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bern M, Nilsen J, Ferrarese M, Sand KMK, Gjølberg TT, Lode HE, Davidson RJ, Camire RM, Bækkevold ES, Foss S, et al. An engineered human albumin enhances half-life and transmucosal delivery when fused to protein-based biologics. Sci Transl Med. 2020;12(565):1-13. [DOI] [PubMed] [Google Scholar]
- 197.Liang W-C, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J, Fu L, Malik AK, Gerber H-P, Ferrara N, et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281(2):951–61. [DOI] [PubMed] [Google Scholar]
- 198.Yu L, Wu X, Cheng Z, Lee CV, LeCouter J, Campa C, Fuh G, Lowman H, Ferrara N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci. 2008;49(2):522–27. [DOI] [PubMed] [Google Scholar]
- 199.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13(12):1551–59. [DOI] [PubMed] [Google Scholar]
- 200.Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem. 2010;285(7):4826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Low BE, Christianson GJ, Lowell E, Qin W, Wiles MV. Functional humanization of immunoglobulin heavy constant gamma 1 Fc domain human FCGRT transgenic mice. MAbs. 2020;12(1):1829334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lee C-H, Kang TH, Godon O, Watanabe M, Delidakis G, Gillis CM, Sterlin D, Hardy D, Cogné M, Macdonald LE, et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat Commun. 2019;10(1):5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mariani G, Abbeele AD, Venkateshan CN, Kaldany A, Ito S, Adelstein SJ, Kassis AI. Monoclonal antibody internalization by tumor cells: an experimental model for potential radioimmunotherapy applications. Int J Rad Appl Instrum B. 1989;16(2):147–50. [DOI] [PubMed] [Google Scholar]
- 204.Roopenian DC, Low BE, Christianson GJ, Proetzel G, Sproule TJ, Wiles MV. Albumin-deficient mouse models for studying metabolism of human albumin and pharmacokinetics of albumin-based drugs. MAbs. 2015;7(2):344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan -Y-Y, Dowty ME, Zhang M, Zhang Y, Cheng A, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8(6):1064–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs FcRn-transgenic mice reveal a rodent model with predictive capabilities . MAbs. 2013;5:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Huggins MA, Jameson SC, Hamilton SE. Embracing microbial exposure in mouse research. J Leukoc Biol. 2019;105(1):73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18(12):1759–69. [DOI] [PubMed] [Google Scholar]
- 209.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170:3528–33. [DOI] [PubMed] [Google Scholar]
- 210.Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49(4):673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, Zhao Y, Kleijer M, Sandlie I, de Haas M, et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun. 2011;2(1):599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Nilsen J, Bern M, Sand KMK, Grevys A, Dalhus B, Sandlie I, Andersen JT. Human and mouse albumin bind their respective neonatal Fc receptors differently. Sci Rep. 2018;8(1):14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S. Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J Biol Chem. 2014;289(19):13492–502. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Viuff D, Antunes F, Evans L, Cameron J, Dyrnesli H, Thue Ravn B, Stougaard M, Thiam K, Andersen B, Kjærulff S, et al. Generation of a double transgenic humanized neonatal Fc receptor (FcRn)/albumin mouse to study the pharmacokinetics of albumin-linked drugs. J Control Release. 2016;223:22–30. [DOI] [PubMed] [Google Scholar]
- 215.Mandrup OA, Ong SC, Lykkemark S, Dinesen A, Rudnik-Jansen I, Dagnæs-Hansen NF, Andersen JT, Alvarez-Vallina L, Howard KA. Programmable half-life and anti-tumour effects of bispecific T-cell engager-albumin fusions with tuned FcRn affinity. Commun Biol. 2021;4(1):310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Silverman AD, Karim AS, Jewett MC. Cell-free gene expression: an expanded repertoire of applications. Nat Rev Genet. 2020;21(3):151–70. [DOI] [PubMed] [Google Scholar]
- 217.Tihanyi B, Nyitray L. Recent advances in CHO cell line development for recombinant protein production. Drug Discov Today Technol. 2020;38:25–34. [DOI] [PubMed] [Google Scholar]
- 218.Yang W, Zhang J, Xiao Y, Li W, Wang T. Screening Strategies for High-Yield Chinese Hamster Ovary Cell Clones. Front Bioeng Biotechnol. 2022;10:858478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sergeeva D, Lee GM, Nielsen LK, Grav LM. Multicopy Targeted Integration for Accelerated Development of High-Producing Chinese Hamster Ovary Cells. ACS Synth Biol. 2020;9(9):2546–61. [DOI] [PubMed] [Google Scholar]
- 220.Lee JS, Kildegaard HF, Lewis NE, Lee GM. Mitigating Clonal Variation in Recombinant Mammalian Cell Lines. Trends Biotechnol. 2019;37(9):931–42. [DOI] [PubMed] [Google Scholar]
- 221.Rajendran S, Balasubramanian S, Webster L, Lee M, Vavilala D, Kulikov N, Choi J, Tang C, Hunter M, Wang R, et al. Accelerating and de-risking CMC development with transposon-derived manufacturing cell lines. Biotechnol Bioeng. 2021;118(6):2301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kol S, Ley D, Wulff T, Decker M, Arnsdorf J, Schoffelen S, Hansen AH, Jensen TL, Gutierrez JM, Chiang AWT, et al. Multiplex secretome engineering enhances recombinant protein production and purity. Nat Commun. 2020;11(1):1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Amann T, Hansen AH, Kol S, Hansen HG, Arnsdorf J, Nallapareddy S, Voldborg B, Lee GM, Andersen MR, Kildegaard HF. Glyco-engineered CHO cell lines producing alpha-1-antitrypsin and C1 esterase inhibitor with fully humanized N-glycosylation profiles. Metab Eng. 2019;52:143–52. [DOI] [PubMed] [Google Scholar]
- 224.Dovgan T, Golghalyani V, Zurlo F, Hatton D, Lindo V, Turner R, Harris C, Targeted CT. CHO cell engineering approaches can reduce HCP-related enzymatic degradation and improve mAb product quality. Biotechnol Bioeng. 2021;118(10):3821–31. [DOI] [PubMed] [Google Scholar]
- 225.Ha TK, Hansen AH, Kildegaard HF, Lee GM. Knockout of sialidase and pro-apoptotic genes in Chinese hamster ovary cells enables the production of recombinant human erythropoietin in fed-batch cultures. Metab Eng. 2020;57:182–92. [DOI] [PubMed] [Google Scholar]
- 226.Heffner KM, Wang Q, Hizal DB, Ö C, Betenbaugh MJ. Glycoengineering of Mammalian Expression Systems on a Cellular Level. Adv Biochem Eng Biotechnol. 2021;175:37–69. [DOI] [PubMed] [Google Scholar]
- 227.Sifniotis V, Cruz E, Eroglu B, Kayser V. Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies (Basel). 2019;8(2): 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.van der Kant R, Karow-Zwick AR, Van Durme J, Blech M, Gallardo R, Seeliger D, Aßfalg K, Baatsen P, Compernolle G, Gils A, et al. Prediction and Reduction of the Aggregation of Monoclonal Antibodies. J Mol Biol. 2017;429:1244–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Crawford Y, Zhou M, Hu Z, Joly J, Snedecor B, Shen A, Gao A. Fast identification of reliable hosts for targeted cell line development from a limited-genome screening using combined φC31 integrase and CRE-Lox technologies. Biotechnol Prog. 2013;29(5):1307–15. [DOI] [PubMed] [Google Scholar]
- 230.Scarcelli JJ, Shang TQ, Iskra T, Allen MJ, Zhang L. Strategic deployment of CHO expression platforms to deliver Pfizer’s Monoclonal Antibody Portfolio. Biotechnol Prog. 2017;33(6):1463–67. [DOI] [PubMed] [Google Scholar]
- 231.Shen Y, Burakov D, Chen G, Fandl JP. CHO integration sites and uses thereof. US Patent2017. https://patentimages.storage.googleapis.com/da/c7/31/8fda4baae3bd0d/US9816110.pdf [Google Scholar]
- 232.Chen K, Zhu Y, Zhang Y, Hamza T, Yu H, Saint Fleur A, Galen J, Yang Z, Feng H. A probiotic yeast-based immunotherapy against Clostridioides difficile infection. Sci Transl Med. 2020;12(567):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zhao H, Tasch M, Dodds M, Gewe M, Martinez A, Hutton M, Keeney K, Pollock A, Jester B, Khuong N, et al. Using antibody synergy to engineer a high potency biologic cocktail against C. difficile. bioRxiv. 2022; doi: 10.1101/2021.12.21.473715. [DOI] [Google Scholar]
- 234.Sarker SA, Jäkel M, Sultana S, Alam NH, Bardhan PK, Chisti MJ, Salam MA, Theis W, Hammarström L, Frenken LGJ. Anti-rotavirus protein reduces stool output in infants with diarrhea: a randomized placebo-controlled trial. Gastroenterology. 2013;145(4):740–8.e8. [DOI] [PubMed] [Google Scholar]
- 235.Fiil BK, Thrane SW, Pichler M, Kittilä T, Ledsgaard L, Ahmadi S, Hermansen GMM, Jelsbak L, Lauridsen C, Brix S, et al. Orally active bivalent VHH construct prevents proliferation of F4+ enterotoxigenic Escherichia coli in weaned piglets. iScience. 2022;25(4):104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Butler JE, Wertz N, Deschacht N, Porcine Igg: KI. structure, genetics, and evolution. Immunogenetics. 2009;61(3):209–30. [DOI] [PubMed] [Google Scholar]
- 237.Virdi V, Palaci J, Laukens B, Ryckaert S, Cox E, Vanderbeke E, Depicker A, Callewaert N. Yeast-secreted, dried and food-admixed monomeric IgA prevents gastrointestinal infection in a piglet model. Nat Biotechnol. 2019;37(5):527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pellis M, Pardon E, Zolghadr K, Rothbauer U, Vincke C, Kinne J, Dierynck I, Hertogs K, Leonhardt H, Messens J, et al. A bacterial-two-hybrid selection system for one-step isolation of intracellularly functional Nanobodies. Arch Biochem Biophys. 2012;526(2):114–23. [DOI] [PubMed] [Google Scholar]
- 239.Kunz P, Ortale A, Mücke N, Zinner K, Hoheisel JD. Nanobody stability engineering by employing the ΔTm shift; a comparison with apparent rate constants of heat-induced aggregation. Protein Eng Des Sel. 2019;32(5):241–49. [DOI] [PubMed] [Google Scholar]
- 240.Hagihara Y, Mine S, Uegaki K. Stabilization of an immunoglobulin fold domain by an engineered disulfide bond at the buried hydrophobic region. J Biol Chem. 2007;282(50):36489–95. [DOI] [PubMed] [Google Scholar]
- 241.Saerens D, Conrath K, Govaert J, Muyldermans S. Disulfide bond introduction for general stabilization of immunoglobulin heavy-chain variable domains. J Mol Biol. 2008;377(2):478–88. [DOI] [PubMed] [Google Scholar]
- 242.Hussack G, Hirama T, Ding W, Mackenzie R, Tanha J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS One. 2011;6(11):e28218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jester BW, Zhao H, Gewe M, Adame T, Perruzza L, Bolick DT, Agosti J, Khuong N, Kuestner R, Gamble C, et al. Development of spirulina for the manufacture and oral delivery of protein therapeutics. Nat Biotechnol. 2022;40(6):956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Saberianfar R, Chin-Fatt A, Scott A, Henry KA, Topp E, Plant-Produced Chimeric MR. VHH-sIgA Against Enterohemorrhagic E. coli Intimin Shows Cross-Serotype Inhibition of Bacterial Adhesion to Epithelial Cells . Front Plant Sci. 2019;10:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Shanmugaraj B, I Bulaon CJ, Phoolcharoen W. Plant Molecular Farming: A Viable Platform for Recombinant Biopharmaceutical Production. Plants. 2020;9(7):842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.De Meyer T, Laukens B, Nolf J, Van Lerberge E, De Rycke R, De Beuckelaer A, De Buck S, Callewaert N, Depicker A. Comparison of VHH-Fc antibody production in Arabidopsis thaliana, Nicotiana benthamiana and Pichia pastoris. Plant Biotechnol J. 2015;13(7):938–47. [DOI] [PubMed] [Google Scholar]