

Abstract
Chronic disease results from the failure of tissues to maintain homeostasis. In the lung, coordinated repair of the epithelium is essential for preserving homeostasis. In animal models and human lung disease, airway epithelial cells mobilize in response to lung injury, resulting in the formation of airway-like cysts with persistent loss of functional cell types and parenchymal architecture. Using live-cell imaging of human lung epithelial cultures and mouse precision-cut lung slices, we demonstrated that distal airway epithelia are aberrantly fluidized both after injury and in fibrotic lung disease. Through transcriptomic profiling and pharmacologic stimulation of epithelial cultures, we identified interleukin-6 (IL-6) signaling as a driver of tissue fluidization. This signaling cascade occurred independently of canonical Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling but instead was dependent on a downstream SRC family kinase (SFK)–yes-associated protein (YAP) axis. Airway epithelial-fibroblast cocultures revealed that the fibrotic mesenchyme acts as a source of IL-6 family cytokines, which drive airway fluidization. Inhibition of the IL-6–SFK–YAP cascade was sufficient to prevent fluidization in both in vitro and ex vivo models. Last, we demonstrated a reduction in fibrotic lung remodeling in mice through genetic or pharmacologic targeting of IL-6–related signaling. Together, our findings illustrate the critical role of airway epithelial fluidization in coordinating the balance between homeostatic lung repair and fibrotic airspace remodeling.
INTRODUCTION
Lung injury and subsequent repair of the distal airspace require activation and coordination of diverse populations of stem cells to preserve respiratory function (1–4). Airway stem cells mobilize to the most distal airspaces after severe injury forming cystic structures (5–13). Similarly in fibrotic lung diseases, such as idiopathic pulmonary fibrosis (IPF), airway cells ectopically appear in the distal lung forming honeycomb cysts (14–17). Recent work has illustrated that the appearance of airway-like cells in the distal airspace may also occur through metaplastic differentiation of alveolar stem cells driven by dynamic epithelial-mesenchymal cross-talk (18, 19). These airway-centric cysts persist within the remodeled airspace, disrupting normal pulmonary structure and function by replacing gas exchange units. However, the active mechanisms regulating the development of this dysfunctional tissue-level reorganization and the nature of their conservation across the spectrum of lung injury and disease remain incompletely understood. In this work, we sought to identify the dynamic signatures and signaling modalities regulating distal lung remodeling using models of lung injury and fibrosis.
Mobilization of confluent tissues has been implicated in physiologic and pathologic tissue remodeling, including body axis elongation, cancer metastasis, and pulmonary disease (20–30). To achieve this reorganization, tissues undergo fluidization or a transition from a solid-like to a fluid-like phase. As tissues fluidize, they obtain the ability to collectively migrate and acquire distinct changes in cellular shape, cell-cell adhesion, and cell-matrix interaction, which can be actively quantified to describe this phase transition (20–30). The molecular mechanisms underlying this migratory program remain poorly understood. The epithelial-to-mesenchymal transition (EMT) is a commonly evoked program to explain cell migration and fibrosis, but it has a controversial role in pulmonary fibrosis pathogenesis (31–36). Work has demonstrated that EMT within the airway epithelium does not promote the large-scale tissue reorganization and the retention of epithelial cell properties in the same manner as tissue-wide epithelial fluidization programs (30). Yet, the mechanisms guiding organ-level remodeling observed in fibrotic lung disease remain largely unknown. The goal of this work was to determine whether airway epithelial fluidization acts as a regulator of lung remodeling after injury and during disease pathogenesis.
RESULTS
Distal airways fluidize after injury in mice
Consistent with previous reports (5–13, 37), we demonstrated that keratin-5 (Krt5)–positive cells were induced from diverse airway stem cell populations within the mouse lung after injury with either influenza (Fig. 1, A and B) or bleomycin (fig. S1, A and B). Using a Krt5-CreERT2/TdTomato mouse line, time-lapse imaging of influenza- or bleomycin-injured mouse precision cut lung slices (PCLS) revealed a structurally intact lung with airways lined by Krt5+ cells (Fig. 1B) that were collectively migrating (Fig. 1C, fig. S1C, and movies S1 to S3). This migratory program was accompanied by cellular elongation in the airway epithelium suggestive of transition from a solid-like to a fluid-like phase (Fig. 1, D to F). Further examination of the PCLS demonstrated that cell migration was confined to the airway, with no detectable extravasation of Krt5+ cells through the basement membrane as had been previously suggested (7). Instead, we found that this population of Krt5+ migratory cells left the airways as a collective at the airway-alveolar junction.
Fig. 1. Mouse and human airways share epithelial fluidization during repair.

(A) Timeline of Krt5-TdTomato mouse injury model and precision cut lung slicing. (B) Representative image of H1N1-injured mouse lung. Immunofluorescence staining of 4′,6-diamidino-2-phenylindole (DAPI; white), epithelial cells marked by EpCAM (blue), and Krt5+ cells (red) in the distal airways. Scale bars, 100 μm. (C) Mean root mean squared velocity (VRMS) of uninjured and H1N1-injured mouse airways. (D) Histogram of cellular aspect ratio (AR) of mouse airways from uninjured and injured mice. Mann-Whitney test used for statistical comparison. (E and F) Representative images of E-cadherin–stained mouse airways with corresponding AR segmentation. Scale bars, 100 μm. (G) Schematic of intra-airway epithelial cell isolation from patients with IPF. (H) Speed of proximal, distal, and honeycomb cultures across 28 days of air-liquid interface (ALI) with overlap parameter (Q) and mean squared displacement (MSD) measurements from day 14 of ALI. Error bars represent 95% confidence interval. (I) Representative images of F actin–stained proximal, distal, and honeycomb regions at early (day 4) and late (day 14) days of ALI. Scale bars, 100 μm. (J) Quantification of the epithelial shape index and AR from proximal, distal, and honeycomb regions across 28 days of ALI culture. Dashed line represents shape index of 3.81, the theoretical threshold between fluid and solid phases. Error bars represent 95% confidence interval. (A to J) For all statistical analysis, ****P < 0.0001. Mouse experiments were independently performed N ≥ 2 times with n ≥ 4 animals. Epithelial culture experiments were independently performed N ≥ 3 times with n ≥ 4 donors.
Diseased distal airway epithelia are aberrantly fluidized
Given the shared characteristics of mouse injury models and fibrotic lungs, including the ectopic presence of KRT5+ cells in microhoneycomb cysts (7, 8, 10–12, 15–19), we asked whether in vitro cultures of IPF epithelia could be used to model this phenotype. Using explanted IPF lung tissue from transplantation patients, we performed computed tomography (CT)–guided dissections of the airway tree, isolating airway epithelial cells from the proximal (cartilaginous airways), distal (respiratory airways), and honeycomb cyst epithelia (regions with microscopic honeycombing) within a single patient (Fig. 1G and table S1). When cultured in vitro, these epithelia maintained characteristics of their distinct anatomical regions within the IPF lung (fig. S2), including changes in barrier function (fig. S3A). These cultures displayed no differences in proliferation between regions (fig. S3B). The distal and honeycombed regions were marked by an expansion of mucin-5b (MUC5B+) secretory cells (fig. S3C), KRT5+ basal cells, and kertain-17 (KRT17+) basaloid cells with delayed multiciliation (fig. S3D) (20, 38–40). Solid-to-fluid phase changes have been well characterized in epithelia in development and disease and are accompanied by quantifiable changes in tissue-level dynamics and structure (20–30). To quantify these behaviors in the air-liquid interface (ALI) culture system, we used time lapse microscopy of the apical surface of the epithelium throughout epithelial maturation. Subsequent particle image velocimetry was used to assess cellular dynamics, including cellular speed, displacement, and overlap, of the epithelium. Structural changes in the epithelium were assessed using imaging of F-actin–labeled apical cell borders. These images were then segmented, and cell shapes were quantified on both an individual cell– and epithelial-wide basis. Paralleling our findings in the injured mouse airway epithelium, we found that distal and honeycomb IPF cultures displayed persistent fluidized phase dynamics (Fig. 1H) with accompanying elongation of cell shape (Fig. 1, I and J). In contrast, proximal airway cultures transitioned to a solid-like phase with accompanying diminished migratory dynamics (Fig. 1H) and characteristic acquisition of a cobblestone-like epithelium (Fig. 1, I and J) similar to uninjured airways.
IL-6 family cytokines are enriched during airway epithelial fluidization
We performed bulk RNA sequencing of cultured airway epithelia to identify the molecular regulators of this observed biophysical dysfunction along the intra-airway proximal-distal axis. When comparing genes down-regulated across this fluid-to-solid transition in proximal epithelia to genes persistently up-regulated in fluidized distal and honeycomb cultures, we identified 660 conserved transcripts (Fig. 2A and table S2).
Fig. 2. Activation of IL-6 family signaling drives epithelial fluidization.

(A) Venn diagram of genes down-regulated in proximal cultures between day 0 and 14 of air-liquid interface (ALI) compared with those up-regulated in distal and honeycomb cultures at day 14 of ALI. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) analysis of the shared 660 differentially regulated genes from (A). (C) Mean root-mean-squared velocity (VRMS) of IL-6 family cytokine–stimulated cultures over the course of 96 hours after their initial treatment. Treatments shown are IL-6 and soluble IL-6 receptor (IL-6 and sIL-6RA), IL-11, and oncostatin-M (OSM). (D) Histogram of cellular aspect ratio (AR) and shape index (q) with mean values from IL-6 family cytokine–stimulated cultures at 24 hours after initial stimulation with statistical comparisons to untreated proximal cultures. Dashed line represents shape index of 3.81, the theoretical threshold between fluid and solid phases. One-way ANOVA was used for statistical comparison. (E) Representative images of segmented cultures 24 hours after stimulation with IL-6 family cytokines color-coded on the basis of cell AR. Scale bar, 100 μm. (F) Representative images of YAP- and KRT5-stained cultures after IL-6 family cytokine stimulation. Scale bars, 100 μm. (A to F) For all statistical analysis, ****P < 0.0001. Epithelial culture experiments were independently performed N ≥ 3 times with n ≥ 4 donors.
Although these genes were enriched for previously identified pathways in IPF airway epithelia (such as HIPPO and ERBB) (20, 38–40), we identified a strong signature for genes involved in cytokine-mediated signaling and cellular responses to cytokine stimuli (Fig. 2B). When comparing this cytokine-enriched category from our dataset with genes enriched in airway cells after interleukin-6 (IL-6) treatment (GSE 113185), we identified robust overlap (table S3). The IL-6 family of cytokines includes IL-6, IL-11, oncostatin-M (OSM), and several other related ligands, which share a common coreceptor. Consequently, we asked whether IL-6 family cytokine stimulation could induce fluidization in proximal epithelial cultures.
We stimulated proximal, nonmigratory cultures with IL-6 family cytokines and found that cytokine stimulation induced epithelial fluidization (Fig. 2, C to E, and movie S4) without compromising epithelial barrier function (fig. S4). Treatment of distal or honeycomb cultures with IL-6 did not change epithelial dynamics because these epithelia were aberrantly fluidized at baseline (fig. S5). The preferential activation of IL-6 trans-signaling through costimulation of IL-6 and the IL-6 soluble receptor (sIL-6R) was able to produce a more robust and sustained phase change (Fig. 2, C to E) in the epithelia compared with treatment with IL-6 or sIL-6R alone (fig. S6).
IL-6–dependent airway epithelial fluidization requires SFK and YAP
IL-6 family cytokines signal through heterodimerization with a shared receptor, glycoprotein 130 (gp130), and a ligand-specific receptor. IL-6 canonically signals through a Janus kinase (JAK) and signal transducer and activator of transcription (STAT)–dependent mechanism, whereby receptor activation leads to JAK/STAT phosphorylation and activation (41, 42). However, a noncanonical signaling axis has been demonstrated to activate SRC family kinases (SFKs) upstream of yes-associated protein (YAP) activation (43, 44). We have previously demonstrated that IPF airway fluidization is YAP dependent, but it has been shown that JAK/STAT signaling is critical in a variety of collective migratory phenomena (20, 45). Hence, we asked whether JAK1/2 or STAT3 activation was required for IL-6 family cytokine–related fluidization or whether this induced phase was dependent on SFK/YAP signaling. Pretreatment of nonmigratory proximal cultures with ruxolitinib (JAK1/2 inhibitor), stattic (STAT3 inhibitor), saracatinib (SFK inhibitor), or verteporfin (YAP inhibitor) and subsequent stimulation of these cultures with IL-6 family cytokines revealed that JAK/STAT signaling was unnecessary for IL-6 family cytokine–induced fluidization (fig. S7A). Instead, SFK and YAP were essential for the acquisition of this fluid-like phenotype as measured by cell dynamics (fig. S7A). In addition, we identified increases in both KRT5+ and YAP nuclear–positive cells after IL-6 family cytokine stimulation (Fig. 2F), suggesting that IL-6 family cytokine stimulation induces the reemergence of progenitor cell markers. In addition, we found a concordant decrease in YAP phosphorylation at serine-127 (a marker of YAP cytoplasmic localization and targeting for degradation) and increased phosphorylation at serine-357 (a marker of SFK-dependent YAP activation) (fig. S7B). In aggregate, these data indicate that IL-6 family cytokine signaling is dependent on SFK/YAP activation to achieve epithelial fluidization.
Regardless of which IL-6 family cytokine was used to stimulate the epithelia, there was a robust increase in cytokeratin-, IL-, and extracellular matrix/integrin–related genes without a change in classic epithelial-to-mesenchymal markers (fig. S8). Further, we identified an increase in classic YAP-target genes (AREG, CTGF, and CYR61); however, we also identified an increase in ERBB-related genes (EGFR, ERBB2, and EGF) (fig. S8). This raised the possibility that IL-6 family cytokine stimulation was inducing fluidization through transcriptional regulation of the ERBB-related signaling pathways. To pursue this, we tested whether epidermal growth factor receptor (EGFR) or its ligand amphiregulin (AREG) was required for IL-6 family cytokine–induced fluidization (fig. S9A). When proximal cultures were pretreated with AG1478 (EGFR inhibitor) or an AREG neutralizing antibody and then stimulated these cultures with IL-6 family cytokines, fluidization still occurred (fig. S9B) without compromising barrier function (fig. S9C). Together, these data demonstrated that IL-6 family cytokine signaling depends upon SFK and YAP to promote biophysical dysfunction independent of other fluidization-associated axes.
Mesenchymal-derived IL-6 is sufficient to induce epithelial dysfunction
Mesenchymal-epithelial cross-talk is critical to lung homeostasis and repair (46, 47). Further, there have been multiple studies demonstrating that IL-6 family cytokines are increased in the pulmonary mesenchyme in the context of lung injury and disease (18, 48, 49). We sought to understand whether IL-6 family cytokines originating from the mesenchyme were sufficient to drive the biophysical dysfunction observed after lung injury and in IPF epithelia. Culture of primary human lung fibroblasts (HLF) from controls and patients with IPF revealed a disease-specific increase in IL-6 and IL-11 protein (Fig. 3A) and gene expression (Fig. 3B). Next, we used a coculture system in which primary airway epithelia from controls and HLF from controls and patients with IPF were cultured together for the duration of ALI (Fig. 3C). Epithelia cocultured with HLF began acquiring IPF-like characteristics as indicated by diminished barrier function (fig. S10A), change in cell types and proportions (fig. S11A), and persistent fluidization (Fig. 3, D to F). This phenotype was potentiated in cocultures where the HLFs originated from an IPF lung (Fig. 3E and fig. S10A). To determine the drivers of mesenchymal-induced dysfunction in the epithelia, we performed bulk RNA sequencing of the airway epithelia after 14 days of HLF coculture. We identified 411 shared genes up-regulated in HLF cocultures that were enriched for pathways involving cytokine-mediate signaling and regulation of cell migration (Fig. 3, G and H). We compared our epithelia coculture to a dataset from IL-6 and sIL-6R stimulated airway epithelia (GSE 113185) (50) and identified 100 genes shared in common (Fig. 3I). Many of these common genes were associated with canonical markers of epithelia injury/repair, growth factor signaling, and cytokine stimulation and were concordantly up-regulated in IPF monocultures (Fig. 3H). Furthermore, we found an increase in IL-6 protein expression in HLF cocultures compared with control airway epithelial monoculture (fig. S10B).
Fig. 3. Mesenchymal-derived cytokines modify epithelial phase.

(A) IL-6 and IL-11 protein secreted by control or IPF primary human lung fibroblast (HLF). Error bars represent SEM. Mann-Whitney test used for statistical comparison. (B) IL-6 and IL-11 gene expression by control or IPF HLF. Error bars represent SEM. Mann-Whitney test used for statistical comparison. (C) Schematic of HLF-airway epithelial coculture. (D) Epithelial speed across days of air-liquid interface (ALI) in monoculture and epithelial-HLF coculture conditions. Error bars represent 95% confidence interval. (E) Histogram of cellular aspect ratio (AR) with mean values from mono- and coculture epithelia at ALI day 14 with comparisons to control or IPF monocultures. One-way ANOVA was used for statistical comparison. (F) Representative images of segmented cultures at day 14 of ALI in mono- or co-culture conditions color coded on the basis of cell AR. Scale bar, 100 μm. (G) KEGG enrichment for genes up-regulated in epithelial cells under co-culture conditions when compared with control epithelial monocultures. (H) Top differentially regulated genes in mono- and coculture conditions selected for genes shared between cocultured and IPF monocultured epithelia. (I) Shared genes from HLF cocultured epithelia and IL-6/sIL-6R stimulated airway epithelial monocultures (GSE 113185). (J) Speed of cocultures at day 14 of ALI treated with IL-6–related inhibitors (anti–IL-6 antibody, anti–IL-6R antibody, and gp130-Fc chimera). Error bars represent SEM. One-way ANOVA used for statistical comparison. (A to J) For all statistical analysis n.s. (not significant), **P < 0.01, ***P < 0.001, ****P < 0.0001. Cell culture experiments were independently performed N ≥ 3 times with n = 4 donors.
To define the effect of IL-6/gp130–related signaling in HLF co-culture, we continuously treated our cocultures starting 4 days after establishing ALI with an anti–IL-6 antibody, anti–IL-6R antibody, or gp130-Fc chimera. Inhibition of the IL-6 cytokine axis prevented the extension of the epithelial fluid phase (Fig. 3J), indicating that fluidization was driven by HLF-derived IL-6 cytokines. This treatment was associated with improved barrier function specifically in IPF HLF coculture (fig. S10C), while also reducing aberrant cell type expansion (fig. S11B). Together, these findings indicated that epithelial dysfunction arising from mesenchymal-derived signaling can be abrogated through inhibition of IL-6/gp130 signaling.
Distal airway remodeling is IL-6/SFK/YAP dependent
We found that IL-6 family cytokines were persistently up-regulated in distal and honeycomb epithelial cultures (fig. S12) and questioned whether their presence was driving the prolonged epithelial fluidization. We found that distal and honeycomb epithelia treated with an anti–IL-6 antibody, anti–IL-6R antibody, or gp130-Fc chimera rapidly transitioned from a fluid-like to a solid-like phase characterized by a shift in epithelial dynamics (Fig. 4A, fig. S13A, and movies S5 and S6) and cell shape (Fig. 4, B and C, and fig. S13B). Further, IL-6/gp130 inhibition in distal or honeycomb cultures was able to reduce YAP target (CTGF, CYR61, and AREG) and IL-6 family cytokine (IL6, IL6R, and IL11) gene expression (fig. S14, A and B) without impairing barrier function (fig. S15). These data indicated that the persistence of this biophysical migratory phase was dependent on uninterrupted IL-6/gp130 signaling.
Fig. 4. Injured airways require IL-6/SFK for fluidization.

(A) Mean root-mean-squared velocity (VRMS) for distal and honeycomb cultures treated with anti–IL-6R antibody, ruxolitinib (JAK1/2 inhibitor), or saracatinib (SFK inhibitor). (B) Histogram of cellular aspect ratio (AR) and shape index (q) with mean values 48 hours after distal and honeycomb culture after inhibitor treatment. Dashed line represents shape index of 3.81, the theoretical threshold between fluid and solid phases. One-way ANOVA was used for statistical comparison. (C) Representative images of segmented distal and honeycomb cultures 48 hours after inhibitor treatment color coded on the basis of cell AR. Scale bars, 100 μm. (D) Western blot of inhibited distal and honeycomb cultures probed for YAP phospho-states after treatment with anti–IL-6R antibody, ruxolitinib (JAK1/2 inhibitor), or saracatinib (SFK inhibitor). Phospho-states shown signify YAP-cytoplasmic sequestration (S127) or SRC-dependent activation (S357). Error bars represent SEM. One-way ANOVA was used for statistical comparison. (E) Top: Timeline of Krt5-TdTomato mouse injury model and precision cut lung slicing (PCLS) with added inhibitor treatment. Bottom: Mean VRMS of PCLS treated with anti–IL-6/IL-6R antibodies, ruxolitinib, saracatinib, or blebbistatin. (A to E) For all statistical analysis n.s. (not significant), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Mouse experiments were independently performed N = 3 times with n ≥ 3 donors. Epithelial culture experiments were independently performed N ≥ 3 times with n ≥ 3 donors.
Concordantly, inhibition of the IL-6/gp130 effector protein JAK1/2 was unable to modify epithelial fluidization in either distal or honeycomb cultures; however, SFK activation was necessary for the persistence of the fluidized phase characterized by migratory dynamics (Fig. 4A) and cell shape (Fig. 4, B and C). In addition, blocking IL-6–related and SFK signaling led to a reduction in YAP activation, which was not observed with JAK1/2 inhibition (Fig. 4D and fig. S14, C and D). Together, these data indicated that IL-6–driven fluidization depends on a downstream SFK-YAP modality to modify epithelial phase dynamics.
To test whether noncanonical IL-6 family signaling axis was a conserved pathway driving distal airway fluidization in the mouse lung, we used our PCLS model (Fig. 4E). When PCLS were incubated continuously with anti–IL-6 and anti–IL-6R antibodies, we were able to prevent airway fluidization to the same degree as blebbistatin, a widely used inhibitor of cellular migration (Fig. 4E and movie S7) (51). We then asked whether downstream JAK1/2 or SFK played an important role in airway fluidization. Similar to our in vitro system, we demonstrated that JAK1/2 was uninvolved in airway fluidization, whereas SFK signaling was critical to the persistence of the collective migratory phenotype (Fig. 4E and movies S8 to S10). Together these data demonstrated that IL-6 signaling through an SFK-dependent axis is responsible for airway fluidization in multiple models of epithelial injury, in vitro and ex vivo.
IL-6 deficiency protects against bleomycin-induced fibrotic lung injury in mice
To address whether IL-6–dependent airway fluidization was ultimately responsible for the remodeling observed after lung injury, we injured IL-6–deficient mice (IL-6KO) with both single-dose and repeated-dose bleomycin. Single-dose bleomycin is the most commonly used model of pulmonary fibrosis in mice; however, repeated-dose bleomycin injury models have been shown to more robustly recapitulate features of human fibrotic lung disease, including markedly increased metaplastic honeycombing and sustained fibrosis (11, 37, 52). Although the influenza injury model produces the most dramatic induction of Krt5+ honeycombing, IL-6KO mice are unable to survive even sublethal doses of H1N1 because of the inability to recruit neutrophils (53). In both the single-dose (Fig. 5A) and repeated-dose (Fig. 5B) bleomycin models, we found that IL-6KO mice had a significant reduction in Krt5+ cells in the parenchyma after injury (single dose and repeated dose; P < 0.0001) with no change in the Krt5+ population in the airways (fig. S16), suggesting that blocking IL-6 signaling attenuated migration of Krt5+ cells through prevention of fluidization, resulting in a loss in cyst formation (Fig. 5, C to E). In addition, we found that these mice had significantly less lung fibrosis as measured by lung hydroxyproline content (single dose, P = 0.0051; repeated dose, P = 0.0003) (Fig. 5F).
Fig. 5. IL-6 deficiency prevents fibrotic lung remodeling.
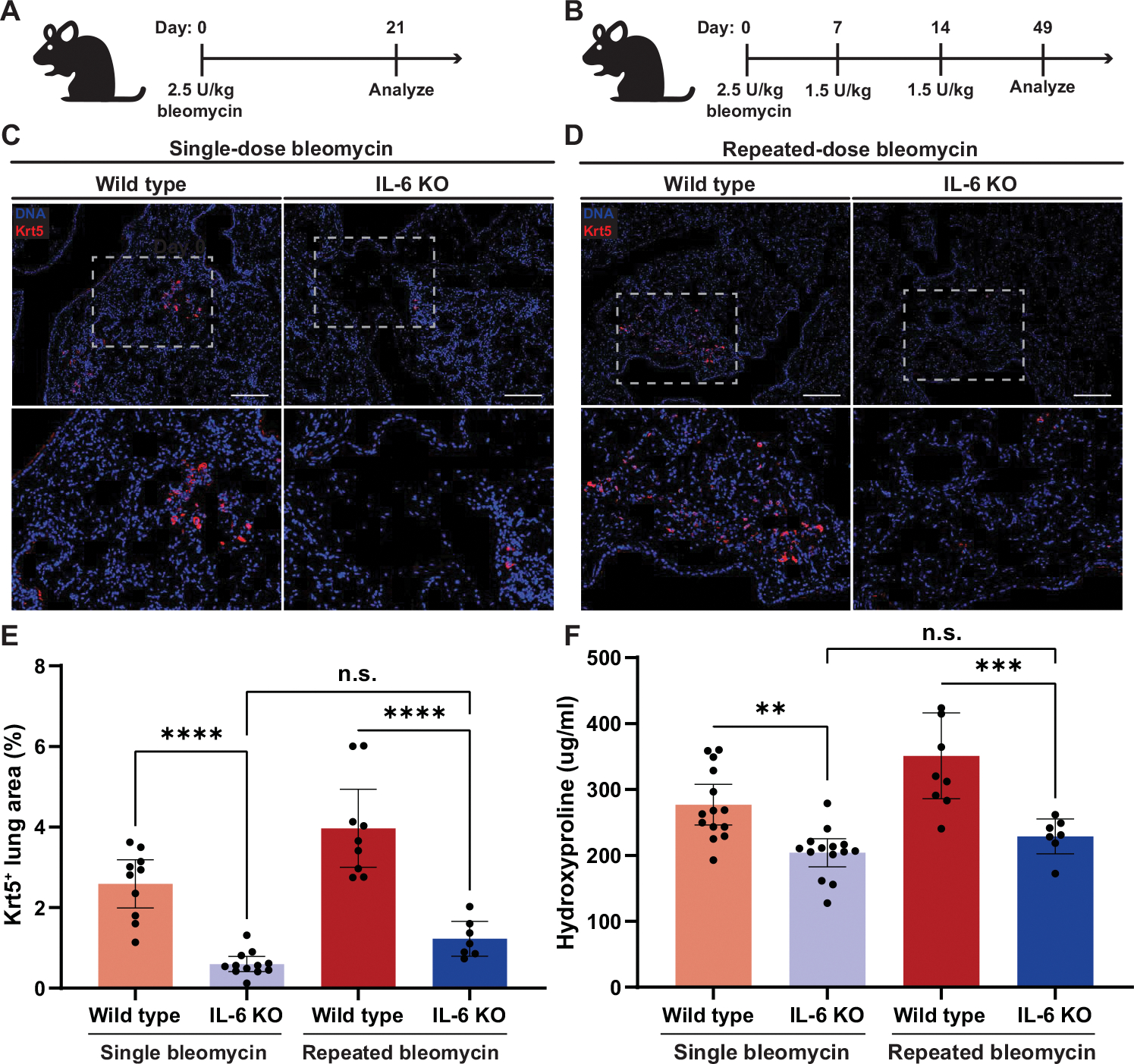
(A) Dosing schematic for single-dose bleomycin exposure for wild-type and IL-6–deficient (IL-6KO) mice. (B) Dosing schematic for repeated-dose bleomycin exposure for wild-type and IL-6KO mice. (C) Representative images for Krt5+-stained wild-type and IL-6KO mouse lungs after single-dose bleomycin injury. Immunofluorescence staining of DAPI (blue) and Krt5 (red) in the distal airways and parenchyma. Scale bars, 100 μm. (D) Representative images for Krt5+-stained wild-type and IL-6KO mouse lungs after repeated-dose bleomycin injury. Scale bars, 100 μm. (E) Quantification of Krt5+ area in the mouse lung after single- or repeated-dose bleomycin for wild-type (single dose, n = 10; repeat dose, n = 9) and IL-6KO (single dose, n = 12; repeat dose, n = 7) mice. Error bars represent 95% confidence interval. One-way ANOVA was used for statistical comparisons. (F) Quantification of hydroxyproline in the mouse lung after single- or repeated-dose bleomycin for wild-type (single dose, n = 14; repeat dose, n = 9) and IL-6KO mice (single dose, n = 14; repeat dose, n = 7). Error bars represent 95% confidence interval. One-way ANOVA was used for statistical comparisons. (A to E) For all statistical analysis n.s. (not significant), **P < 0.01, ***P < 0.001, ****P < 0.0001.
Furthermore, to determine whether these IL-6–dependent phenotypes could be prevented pharmacologically, we treated injured wild-type mice with a combined treatment of both anti–IL-6 and anti–IL-6R neutralizing antibodies for the duration of injury (fig. S17A). We reduced lung honeycombing and fibrosis to the same degree as in IL-6KO mice with treatment with neutralizing antibodies (fig. S17, B to D). Both genetic and pharmacologic targeting of IL-6 resulted in a reduction in neutrophils, macrophages, and total leukocytes collected from bronchoalveolar lavage fluid, consistent with previous findings (figs. S17E and S18) (53, 54). Together these findings demonstrated that IL-6–dependent epithelial fluidization serves as a critical mechanism of distal lung remodeling after injury.
DISCUSSION
Here, we illustrated that airway fluidization occurs after lung injury, is dependent on active IL-6 family signaling, and results in progenitor cell migration into the distal airspace. Whereas much of IL-6 signaling has been traditionally studied through the lens of immune cell responses (55), these findings provide valuable context for the role of cytokine signaling in epithelial biology and epithelial-mesenchymal cross-talk in the lung. The convergence of these signaling modalities on a common axis suggests that SFK/YAP signaling acts as a control switch in regulating airway epithelial fluidity in acute repair and chronic disease. Dysregulation and persistence of this prorepair fluid phase may underlie the shift from homeostatic repair to pathology in the setting of injury.
We have previously demonstrated that the EGFR-YAP axis can be modified to regulate airway epithelial fluidization (20). However, the possibility of targeting EGFR or YAP directly after injury or in chronic lung disease presents a considerable barrier given their near ubiquitous role in epithelial regeneration. IL-6 family cytokines seemingly converge on a conserved downstream pathway with EGFR in regulating similar pathophysiologic outcomes, but these cytokines outwardly appear to regulate a purely pathogenic arm of the repair cascade. The inhibition of IL-6–related signaling in fibrotic lung disease represents a previously unstudied approach to targeting lung remodeling without diminishing necessary growth factor signaling. Furthermore, our data suggest that trans–IL-6 signaling, in which the receptor and ligand are both solubilized, potentiates this injury response more markedly than classic signaling, indicating that there are subtleties to this signaling axis that remain important and unexplored.
Continued investigation of IL-6–targeted therapies for lung disease is required to further understand the nuances of preventing disease without disrupting homeostatic immune-driven repair. Work has demonstrated that inhibition of IL-6 after influenza infection in mouse models results in complete lethality because of their inability to clear the virus (53). Collectively, these findings highlight the context-specific role of IL-6 in modifying injury response and disease progression; however, it remains unclear how other members of this cytokine family differentially confer a protective or injurious role in the in vivo context.
Here, we have demonstrated both an autocrine and paracrine signaling axis underlying IL-6–dependent pathogenesis. Although it is clear that IL-6 family cytokines can emanate from immune, epithelial, and stromal populations, the relative role of each of these tissue types in establishing this dysregulated signaling network in the diseased lung remains unknown. We have reinforced here that there is a disease-specific increase in IL-6–related cytokine expression emanating from mesenchymal populations; however, in our coculture system, both control and IPF fibroblast populations were capable of inducing epithelial fluidization. Because we have shown that healthy distal airways are not fluidized at baseline, this may be in part due to the nonphysiologic nature of our in vitro systems; however, further investigation of the contributions of diverse cell types and subtypes in driving fluidization is paramount. Recent work has eloquently illustrated the capacity of stromal populations in the lung to induce nonautonomous differentiation of alveolar stem cells into airway lineages, which suggests an alternative mode of distal lung remodeling (18). This group and others have also identified subpopulations of mesenchymal cells that highly express multiple IL-6 cytokines (18, 38, 39). It remains unclear whether this IL-6–producing stromal population can contribute to metaplastic basal cell differentiation as has been shown with transforming growth factor–β–producing fibroblasts (18). Investigation of these questions on the necessity and sufficiency of tissue- and cell-specific IL-6 signaling would provide invaluable insight into possible therapeutic avenues to prevent disease progression.
Although our previous work has demonstrated a direct link between epithelial fluidization and the establishment of fibrosis (20), it is likely that IL-6 plays a myriad of roles outside of directing migration that result in the generation of a profibrotic environment within the lung. Further investigation into IL-6–driven fluidization and its ability to directly drive fibroproliferation and activation is necessary to clarify the relationship between honeycombing and fibrosis.
It is important to highlight several limitations of this study. First, although many of the models used in this work are commonplace in studying pulmonary fibrosis, they all fail to adequately recapitulate the complexity of IPF. We have attempted to overcome this shortcoming in this work by using multiple in vitro, ex vivo, and in vivo approaches to test our central hypothesis. Further, our current culture systems and use of primary tissue hinder our ability to sufficiently probe cell type–specific biology on a tissue-wide scale.
Advances in our understanding of the mechanisms regulating distal airspace changes in lung disease have been limited by the ability to actively monitor tissue-level repair and remodeling programs after injury. Investigation of the real-time dynamics of these processes provides a new lens for interpreting the spectrum of tissue remodeling in response to lung injury and progression of disease. Understanding this continuum between homeostasis and pathologic remodeling of the distal airspace is likely to lead to key therapeutic advances in fibrotic lung disease.
MATERIALS AND METHODS
Study design
The overall goal of this study was to investigate the mechanisms underlying lung remodeling through the use of both primary human airway cell cultures and murine models of lung injury and fibrosis. For in vivo experiments, mice were randomly assigned to treatment groups with littermates used as controls. Investigators were blinded to both mouse strain and group before treatment. Data collection and analysis were also conducted in a blinded manner. For in vitro experiments, samples were randomly selected for treatment. Investigators were blinded to treatment group during data collection and analysis. No statistical methods were used to predetermine sample size. No exclusion criteria were used, and no data were excluded from analysis. Primary human lung tissue was obtained from lung transplants at the University of Colorado Hospital (UCH) or from lung tissue not suitable for transplantation at National Jewish Health (NJH). Studies using human tissue were conducted in accordance with approved institutional review board protocols: 11-1664 (UCH), 18-057 (UCH), and HS-3209 (NJH). Studies involving animal experimentation adhered to all relevant ethical regulations and were conducted in accordance with an approved Institutional Animal Care and Use Committee protocol at the University of Colorado.
Animals
Krt5-CreERT2 mice were purchased from the Jackson Laboratory and crossed to B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J. IL-6–deficient (IL-6 KO) mice were generated as previously described (56). All mouse experiments used at least five mice per group unless otherwise noted. Studies involving animal experimentation adhered to all relevant ethical regulations. In accordance with the Institutional Animal Care and Use Committee at the University of Colorado, mice were housed in pathogen-free areas, which were monitored by institutional animal care staff. Mice were maintained on a 12-hour light/dark cycle. At weaning, mice were assigned nondescriptive numbers via an ear tag. Hunching, fur ruffing, lethargy, and/or a persistent loss in 15% of total body weight were used to identify moribund animals. Mice were euthanized by intraperitoneal injection of sodium pentobarbital followed by exsanguination.
Bleomycin and influenza exposure
For bleomycin injury experiments, mice were administered inhaled isoflurane (MWI Veterinary Supply Company) for anesthesia, and subsequently, tracheas were directly visualized using a rodent laryngoscope (Penn Century). Bleomycin (Meitheal Pharmaceuticals) or saline was instilled directly into the trachea using a gavage of 50 μl. For single-dose bleomycin experiments, bleomycin (2.5 U/kg) was administered on day 0. For repeated-dose bleomycin experiments, animals received bleomycin (2.5 U/kg) day 0, followed by 1.5 U/kg on days 7 and 14. Intraperitoneal tamoxifen (dissolved in sunflower oil at 250 mg/kg body weight) was administered as a single dose 16 days after single-dose bleomycin instillation when required.
For bleomycin injury experiments where mice also received an intraperitoneal injection of IL-6 (BioXCell, BE0046) and IL-6R (BioXCell, BE0047) monoclonal antibodies, the above single-dose bleomycin protocol was undertaken with the addition of intraperitoneal dosing of the monoclonal antibodies. Injections of IL-6 (400 μg per dose) and IL-6R (400 μg per dose) or an equivalent volume of vehicle (BioXCell, IP0070) were administered 48 hours before bleomycin exposure and then every 72 hours for 19 days after bleomycin exposure.
For H1N1 injury experiments, equal proportions of male and female mice were used. Following established protocols (37), mice were administered inhaled isoflurane and subsequently administered 4 × 104 plaque-forming units of culture-adapted influenza A/H1N1/Puerto Rico 8/34 [American Type Culture Collection (ATCC), VR-1469] dissolved in 20 μl of phosphate-buffered saline (PBS; Corning, 21-040-CV) via intranasal instillation. Intraperitoneal tamoxifen (dissolved in sunflower oil at 250 mg/kg body weight) was administered as a single dose 8 days after H1N1 instillation when required.
Precision cut lung slicing
Immediately after euthanasia, mouse tracheas were cannulated with a 20-gauge × ½-inch Luer stub (Instech, 50-811-22). An incision was made to the renal artery, and PBS was injected into the right ventricle using a 20-gauge needle on a 10-ml syringe to slowly perfuse the lungs. The lungs were then lavaged three times with 0.5 ml of PBS containing 0.6 mM EDTA. Cells were counted using a hemocytometer and spun on to slides using a Cytospin 4 (Thermo Fisher Scientific).
After perfusion, lungs were inflated with 1 to 2 ml of 3% (w/v) low–melting point agarose (Invitrogen, 16520-050) dissolved in Hanks’ balanced salt solution (HBSS) (Lonza. 10-553F) supplemented with penicillin-streptomycin (Gibco, 15140122). Lungs were carefully filled to avoid excessive inflation. The cannula was removed, and the trachea was subsequently occluded with a suture to prevent agarose leakage. Lungs were excised from the chest cavity and submerged in ice-cold HBSS for >30 min before slicing.
After cooling, the left and right lung lobes were isolated and sliced coronally at a width of 300 μm using a vibratome (Leica, VT1200S). The lungs were kept immersed in ice-cold HBSS for the duration of slicing to prevent agarose melting and tissue softening. Slices containing cyanoacrylate glue were discarded to ensure optimal PCLS quality. Slices were placed in 6-cm dishes containing ice-cold HBSS until slicing was completed. PCLS were incubated at 37°C for 1 hour and subsequently transferred to a new 6-cm dish containing warmed culture media. The PCLS culture media consisted of a 1:1 mixture of Dulbecco’s modified Eagle medium (DMEM) (Corning, 10-013-CV) and bronchial epithelial basal medium (BEBM) (Lonza, CC-3171) with accompanying BulletKit aliquots (Lonza, CC-4175) supplemented with the following: 50 nM retinoic acid (Sigma-Aldrich, R-2625), nystatin (20 U/ml; Sigma-Aldrich, N1638), bovine serum albumin (1.5 μg/ml; Thermo Fisher Scientific, BP9703100), recombinant mouse epidermal growth factor (50 μg/ml; Corning, 354001), and 1% fetal bovine serum (ATCC, 30-2020). For experiments using Krt5-CreERT2 mice, 500 nM 4-hydroxytamoxifen (Sigma-Aldrich, H7904) dissolved in molecular-grade ethanol (Thermo Fisher Scientific, BP2808100) was added to the culture media to induce further Cre recombination. Media was changed daily for the extent of the experiment, and PCLS were not cultured past 96 hours after initial slicing to optimize slice viability.
Live-cell staining and imaging of ex vivo mouse tissue
Following established protocols (57), PCLS were incubated with a fluorescein isothiocyanate–conjugated anti-CD326/EpCAM antibody (eBioscience, 11-5791-82) diluted at 1:200 in prewarmed PCLS media for 1 hour at 37°C. PCLS were then washed twice with fresh PCLS media containing no antibodies. Slices were then immediately used for live imaging or fixed with 4% formalin overnight at 4°C for immunofluorescence staining.
For live imaging of PCLS, EpCAM-labeled slices were placed into individual wells on a 12-well plate containing 1 ml of fresh PCLS media. Slice were centered within the wells and weighed down using a 12-mm Transwell (Corning, 3460) to prevent movement of the slice during imaging. Multichannel fluorescent images were acquired using a Keyence BZ-X810. PCLS were placed in a humidified stage incubator at 37°C, 5% CO2 for at least 2 hours before imaging to reduce focal drift. Images were acquired every 8 or 10 min for 18 hours. Fields of view were chosen by identifying EpCAM-positive airways containing TdTomato+ cells indicative of injury and Cre recombination. Postacquisition image processing was completed in ImageJ including optical drift correction using the StackReg plugin (58). Special care was taken to avoid TdTomato+ cells in incidentally included tracheal tissue as these have been robustly demonstrated to be a separate population from the distal Krt5+ cells that arise after injury (7).
Human epithelial cell culture
Primary human airway epithelial cells were obtained from lung transplants at the University of Colorado Hospital (institutional review board protocol: 11-1664 or 18-0572, approved by the Colorado Multiple Institutional Review Board). All patients provided informed consent before tissue collection. Airway cells from proximal (>10 mm internal diameter), distal (respiratory airways), or honeycomb regions (areas of microscopic honeycombing on histopathology) were obtained in parallel from the same patient sample using CT-guided dissection. Primary cell isolations were then expanded to passage 2 using established methods (20). Proximal (N = 7), distal (N = 7), and honeycomb (N = 6) samples were controlled for age, smoking history, and MUC5B promoter variant status (table S1). Passage 2 cells were seeded onto 24-Transwell plates (Corning, 3470) coated with type I collagen (Corning, 354236) and maintained under submerged conditions for about 5 to 7 days, or until confluence was achieved, before removing apical media to establish ALI. Culture media was standardized across all intra-airway epithelial cultures and was composed of a 1:1 mixture of DMEM and BEBM with accompanying BulletKit aliquots supplemented with retinoic acid, nystatin, and bovine serum albumin. We found that consistent culture media conditions were able to robustly recapitulate the in vivo characteristics of the regions from which the epithelial cells were isolated (figs. S2 and S3). Cells were switched to a starvation media (lacking epidermal growth factor, hydrocortisone, and bovine pituitary extract) 24 hours before experimentation.
In vitro ligand/inhibitor treatment
The following ligands were used in the assessment of epithelial function in vitro: recombinant human IL-6 at 0.5 ng/ml (R&D Systems, 206-IL), recombinant human IL-6R alpha at 10 ng/ml (R&D Systems, 227-SR), combined treatment of IL-6 and IL-6R alpha was done at 0.5 and 10 ng/ml, recombinant human IL-11 at 1 ng/ml (R&D Systems, 218-IL), and recombinant human OSM at 0.1 ng/ml (R&D Systems, 295-OM). Treatment concentrations were selected using ligand-specific effective dose (ED50) data as provided by the manufacturer. Ligand stimulation occurred before biophysical assessment. Briefly, cells were switched into ligand-containing media for 4 to 6 hours at 37°C, after which cells were moved to a starvation media for time-lapse imaging. Starvation media consisted of standard primary airway epithelial culture media and additives except for human recombinant EGF, bovine pituitary extract, and hydrocortisone. For conditions where cells were imaged for >48 hours, they were supplemented with fresh starvation media. The following were the inhibitors used in the assessment of epithelial function in vitro: AG-1478 at 100 nM (Millipore Sigma, T4182), AREG neutralizing antibody at 30 μg/ml (R&D Systems, MAB262), ruxolitinib at 5 μM (Selleckchem, S1378), stattic at 5 μM (Selleckchem, S7024), saracatinib at 1 μM (Selleckchem, S1006), verteporfin at 1 μM (Tocris, 3505), IL-6 monoclonal antibody at 10 ng/ml, IL-6R at 1 μg/ml, and recombinant human gp130 Fc chimera at 1 μg/ml. Inhibitors were added for 4 to 6 hours before biophysical assessment and/or subsequent ligand stimulation. Briefly, cells were switched into inhibitor-containing starvation media at 37°C, after which cells were moved to fresh starvation media for time-lapse imaging; the exception to this was when treating with neutralizing or monoclonal antibodies, and in that case, cells were continuously treated with these inhibitors.
Live-cell imaging and dynamic measurements
Epithelial dynamics of in vitro cultures were assessed via time-lapse microscopy on a Keyence BZ-X810 with stage incubator. Phase-contrast images were acquired via two distinct imaging schemes: (i) images throughout ALI differentiation captured every 5 min for 200 min or (ii) images after ligand and/or inhibitor treatment were captured every 20 min for 48 or 96 hours. Assessments of the epithelial fluidization were completed using established workflows. Briefly, instantaneous velocities were mapped using particle image velocimetry (PIV) in PIVlab, an open-source PIV algorithm in MATLAB (59). Velocity fields were obtained via a two-pass window (64 × 64 and 32 × 32 pixel size with 50% overlap) with a pixel size of 0.75488 μm using a fast Fourier transformation. PIV trajectories were seeded onto a grid acquired from forward integration of subsequent fields. These fields were then used to determine mean-squared displacement (MSD), self-overlap order parameter (Q), and instantaneous root mean-squared velocity (VRMS) as previously described (20, 24, 27).
Dynamics of the ex vivo airways followed a similar protocol as above with the modification that ≥3 regions of interest (ROI) were selected along the mouse airway using fluorescent images of EpCAM-labeled TdTomato+ cells. Each ROI was a nonoverlapping region of the airway and was subsequently averaged to represent an individual mouse airway with ≥3 airways imaged per PCLS. Three or more PCLS were imaged per treatment condition.
Statistical analysis
Data were analyzed in GraphPad Prism or MATLAB. Statistical significance was determined via either an ordinary one-way analysis of variance (ANOVA) or Mann-Whitney test. For the ANOVA, a Dunnett’s multiple-comparisons test with a single pooled variance was used (P < 0.05 was considered statistically significant). For Mann-Whitney tests, statistical significance of P < 0.05 was considered significant. Histograms were generated by graphing the frequency distribution of the data and subsequent Gaussian curve fitting. In vitro cultures were performed with at least N ≥ 3 donors, whereby all cells were grown under the same conditions. Table S1 describes donor information. Unless otherwise stated, assessment of epithelial dynamics was averaged over n ≥ 3 donors with N ≥ 2 independent wells. Experiments were repeated ≥3 independent times, and results are represented as the average across all repeats.
Supplementary Material
Acknowledgments:
We thank J. Cardwell for assistance in aligning RNA-sequencing data and the lab of A. Klug for providing access to the lab’s vibratome.
Funding:
These studies were supported by P01-HL0928701 (NIH/NHLBI to D.A.S.), UH2/3HL123442 (NIH/NHLBI to D.A.S.), R01-HL149836 (NIH/NHLBI to D.A.S. and I.V.Y.), W81XWH-17-1-0597 (Department of Defense to D.A.S. and I.V.Y.), and T32-AG000279 (NIH/NIA to I.T.S.).
Footnotes
Competing interests: D.A.S. is the founder and chief scientific officer of Eleven P15 Inc., a company focused on early diagnosis and treatment of pulmonary fibrosis. I.V.Y. is a consultant to Eleven P15 Inc. D.A.S. and I.V.Y. are inventors on patent “Biomarkers for the diagnosis and treatment of fibrotic lung disease” (U.S. patent nos. 62/525,087, 62/525,088, 16/624,500, and 63/123,246, WIPO - PCT/US2018/039573, Canada 3068546, Europe 18743190.3, and New Zealand 760756). The other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scitranslmed.abo5254
MDAR Reproducibility Checklist
View/request a protocol for this paper from Bio-protocol.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Source data are provided with this manuscript. All resources generated in this study are available upon request from the corresponding author upon providing a completed material transfer agreement. RNA-seq data have been deposited to the NCBI Gene Expression Omnibus (accession numbers GSE205523 and GSE205525).
REFERENCES AND NOTES
- 1.Rock JR, Randell SH, Hogan BL, Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 3, 545–556 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME, Airway epithelial cells: Current concepts and challenges. Proc. Am. Thorac. Soc. 5, 772–777 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck HW, Vunjak-Novakovic G, Whitsett JA, Morrisey EE, The cellular and physiological basis for lung repair and regeneration: Past, present, and future. Cell Stem Cell 26, 482–502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de M MF, A. I Costa, A. E Weiner. Vaughan, Basal-like progenitor cells: A review of dysplastic alveolar regeneration and remodeling in lung repair. Stem Cell Rep. 15, 1015–1025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang DY, Lim B, Chow VT, Crum CP, Xian W, McKeon F, Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F, p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature 517, 616–620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA, Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517, 621–625 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, Jackson JR, Xu J, Lee DK, Gotts JE, Matthay MA, Shannon JM, Chapman HA, Vaughan AE, Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 19, 904–914 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, Stripp BR, Rare SOX2+ airway progenitor cells generate KRT5+ cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Rep. 7, 817–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan T, Volckaert T, Redente EF, Hopkins S, Klinkhammer K, Wasnick R, Chao CM, Yuan J, Zhang JS, Yao C, Majka S, Stripp BR, Günther A, Riches DWH, Bellusci S, Thannickal VJ, De Langhe SP, FGF10-FGFR2B signaling generates basal cells and drives alveolar epithelial regeneration by bronchial epithelial stem cells after lung injury. Stem Cell Rep. 12, 1041–1055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassandras M, Wang C, Kathiriya J, Tsukui T, Matatia P, Matthay M, Wolters P, Molofsky A, Sheppard D, Chapman H, Peng T, Gli1+ mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung. Nat. Cell Biol. 22, 1295–1306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Königshoff M, Chapman HA, Theis FJ, Schiller HB, Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA, Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell 26, 346–358.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor MS, Chivukula RR, Myers LC, Jeck WR, Waghray A, Tata PR, Selig MK, O’Donnell WJ, Farver CF, Thompson BT, Rajagopal J, Kradin RL, A conserved distal lung regenerative pathway in acute lung injury. Am. J. Pathol. 188, 1149–1160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, Whitsett JA, Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 66, 651–657 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, Reynolds SD, The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLOS ONE 8, e58658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smirnova NF, Schamberger AC, Nayakanti S, Hatz R, Behr J, Eickelberg O, Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir. Res. 17, 83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kathiriya JJ, Wang C, Zhou M, Brumwell A, Cassandras M, Le Saux CJ, Cohen M, Alysandratos KD, Wang B, Wolters P, Matthay M, Kotton DN, Chapman HA, Peng T, Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell Biol. 24, 10–23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murthy PKL, Sontake V, Tata A, Kobayashi Y, Macadlo L, Okuda K, Conchola AS, Nakano S, Gregory S, Miller LA, Spence JR, Engelhardt JF, Boucher RC, Rock JR, Randell SH, Tata PR, Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 604, 111–119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stancil IT, Michalski JE, Davis-Hall D, Chu HW, Park JA, Magin CM, Yang IV, Smith BJ, Dobrinskikh E, Schwartz DA, Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat. Commun. 12, 4566 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spurlin JW, Siedlik MJ, Nerger BA, Pang MF, Jayaraman S, Zhang R, Nelson CM, Mesenchymal proteases and tissue fluidity remodel the extracellular matrix during airway epithelial branching in the embryonic avian lung. Development 146, dev175257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JA, Atia L, Mitchel JA, Fredberg JJ, Butler JP, Collective migration and cell jamming in asthma, cancer and development. J. Cell Sci. 129, 3375–3383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mongera A, Rowghanian P, Gustafson HJ, Shelton E, Kealhofer DA, Carn EK, Serwane F, Lucio AA, Giammona J, Campàs O, A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature 561, 401–405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malinverno C, Corallino S, Giavazzi F, Bergert M, Li Q, Leoni M, Disanza A, Frittoli E, Oldani A, Martini E, Lendenmann T, Deflorian G, Beznoussenko GV, Poulikakos D, Haur OK, Uroz M, Trepat X, Parazzoli D, Maiuri P, Yu W, Ferrari A, Cerbino R, Scita G, Endocytic reawakening of motility in jammed epithelia. Nat. Mater. 16, 587–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jain A, Ulman V, Mukherjee A, Prakash M, Cuenca MB, Pimpale LG, Münster S, Haase R, Panfilio KA, Jug F, Grill SW, Tomancak P, Pavlopoulos A, Regionalized tissue fluidization is required for epithelial gap closure during insect gastrulation. Nat. Commun. 11, 5604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JA, Kim JH, Bi D, Mitchel JA, Qazvini NT, Tantisira K, Park CY, McGill M, Kim SH, Gweon B, Notbohm J, Steward R Jr., Burger S, Randell SH, Kho AT, Tambe DT, Hardin C, Shore SA, Israel E, Weitz DA, Tschumperlin DJ, Henske EP, Weiss ST, Manning ML, Butler JP, Drazen JM, Fredberg JJ, Unjamming and cell shape in the asthmatic airway epithelium. Nat. Mater. 14, 1040–1048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bi D, Lopez JH, Schwarz JM, Manning ML, A density-independent rigidity transition in biological tissues. Nat. Phys. 11, 1074–1079 (2015). [Google Scholar]
- 28.Atia L, Bi D, Sharma Y, Mitchel JA, Gweon B, Koehler S, DeCamp SJ, Lan B, Kim JH, Hirsch R, Pegoraro AF, Lee KH, Starr JR, Weitz DA, Martin AC, Park JA, Butler JP, Fredberg JJ, Geometric constraints during epithelial jamming. Nat. Phys. 14, 613–620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stancil IT, Michalski JE, Schwartz DA, Integrin axis regulates airway biophysical dysfunction in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 66, 235–237 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchel JA, Das A, O’Sullivan MJ, Stancil IT, DeCamp SJ, Koehler S, Ocaña OH, Butler JP, Fredberg JJ, Nieto MA, Bi D, Park JA, In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat. Commun. 11, 5053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto MA, Huang RY, Jackson RA, Thiery JP, EMT: 2016. Cell 166, 21–45 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Campbell K, Casanova J, A common framework for EMT and collective cell migration. Development 143, 4291–4300 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Chapman HA, Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 73, 413–435 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Willis BC, Borok Z, TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L525–L534 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL, Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 108, E1475–E1483 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA, Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 103, 13180–13185 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurche JS, Dobrinskikh E, Hennessy CE, Huber J, Estrella A, Hancock LA, Schwarz MI, Okamoto T, Cool CD, Yang IV, Evans CM, Schwartz DA, Muc5b enhances murine honeycomb-like cyst formation. Am. J. Respir. Cell Mol. Biol. 61, 544–546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N, Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6, eaba1983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, Raju L, Roberson J, Ding G, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA, Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, Stripp BR, Whitsett JA, Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 1, e90558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson DE, O’Keefe RA, Grandis JR, Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rincon M, Irvin CG, Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int. J. Biol. Sci. 8, 1281–1290 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, Sandborn WJ, Hardiman G, Raz E, Maehara Y, Yoshimura A, Zucman-Rossi J, Guan KL, Karin M, A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taniguchi K, Moroishi T, de Jong PR, Krawczyk M, Grebbin BM, Luo H, Xu RH, Golob-Schwarzl N, Schweiger C, Wang K, Di Caro G, Feng Y, Fearon ER, Raz E, Kenner L, Farin HF, Guan KL, Haybaeck J, Datz C, Zhang K, Karin M, YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 114, 1643–1648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silver DL, Geisbrecht ER, Montell DJ, Requirement forJAK/STAT signaling throughout border cell migration inDrosophila. Development 132, 3483–3492 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Lewis KJR, Hall JK, Kiyotake EA, Christensen T, Balasubramaniam V, Anseth KS, Epithelial-mesenchymal crosstalk influences cellular behavior in a 3D alveolus-fibroblast model system. Biomaterials 155, 124–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hines EA, Sun X, Tissue crosstalk in lung development. J. Cell. Biochem. 115, 1469–1477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng B, Dong J, D’Agostino G, Viswanathan S, Widjaja AA, Lim WW, Ko NSJ, Tan J, Chothani SP, Huang B, Xie C, Pua CJ, Chacko AM, Guimarães-Camboa N, Evans SM, Byrne AJ, Maher TM, Liang J, Jiang D, Noble PW, Schafer S, Cook SA, Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 11, eaaw1237 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Tadokoro T, Wang Y, Barak LS, Bai Y, Randell SH, Hogan BL, IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E3641–E3649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jevnikar Z, Östling J, Ax E, Calvén J, Thörn K, Israelsson E, Öberg L, Singhania A, Lau LCK, Wilson SJ, Ward JA, Chauhan A, Sousa AR, De Meulder B, Loza MJ, Baribaud F, Sterk PJ, Chung KF, Sun K, Guo Y, Adcock IM, Payne D, Dahlen B, Chanez P, Shaw DE, Krug N, Hohlfeld JM, Sandström T, Djukanovic R, James A, Hinks TSC, Howarth PH, Vaarala O, van Geest M, Olsson H, Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J. Allergy Clin. Immunol. 143, 577–590 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Vedula SR, Leong MC, Lai TL, Hersen P, Kabla AJ, Lim CT, Ladoux B, Emerging modes of collective cell migration induced by geometrical constraints. Proc. Natl. Acad. Sci. U.S.A. 109, 12974–12979 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE, Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L442–L452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M, Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 5, 258–266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saito F, Tasaka S, Inoue K, Miyamoto K, Nakano Y, Ogawa Y, Yamada W, Shiraishi Y, Hasegawa N, Fujishima S, Takano H, Ishizaka A, Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am. J. Respir. Cell Mol. Biol. 38, 566–571 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Hunter CA, Jones SA, IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan GA, Costantini F, Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 13, 1189–1196 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akram KM, Yates LL, Mongey R, Rothery S, Gaboriau DCA, Sanderson J, Hind M, Griffiths M, Dean CH, Live imaging of alveologenesis in precision-cut lung slices reveals dynamic epithelial cell behaviour. Nat. Commun. 10, 1178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thévenaz P, Ruttimann UE, Unser M, A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Thielicke W, Stamhuis EJ, PIVlab—Towards user-friendly, affordable and accurate digital particle image velocimetry in MATLAB. J. Open Res. Softw. 2, e30 (2014). [Google Scholar]
- 60.Mashburn DN, Lynch HE, Ma X, Hutson MS, Enabling user-guided segmentation and tracking of surface-labeled cells in time-lapse image sets of living tissues. Cytometry A 81A, 409–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Source data are provided with this manuscript. All resources generated in this study are available upon request from the corresponding author upon providing a completed material transfer agreement. RNA-seq data have been deposited to the NCBI Gene Expression Omnibus (accession numbers GSE205523 and GSE205525).