

Abstract
Actin and its dynamic structural remodelings are involved in multiple cellular functions, including maintaining cell shape and integrity, cytokinesis, motility, navigation, and muscle contraction. Many actin-binding proteins regulate the cytoskeleton to facilitate these functions. Recently, actin’s post-translational modifications (PTMs) and their importance to actin functions have gained increasing recognition. The MICAL family of proteins has emerged as important actin regulatory oxidation-reduction (Redox) enzymes, influencing actin’s properties both in vitro and in vivo. MICALs specifically bind to actin filaments and selectively oxidize actin’s methionine residues 44 and 47, which perturbs filaments’ structure and leads to their disassembly. This review provides an overview of the MICALs and the impact of MICAL-mediated oxidation on actin’s properties, including its assembly and disassembly, effects on other actin-binding proteins, and on cells and tissue systems.
Keywords: MICAL1, MICAL2, MICAL3, MsrB, SelR, semaphorin, plexin, rab
1 Introduction
Actin is one of the main components of the cytoskeleton, playing an essential role in muscle contraction, cell division, motility, navigation, mechanosensing, and in maintaining cellular structure (Blanchoin et al., 2014; Pollard, 2016; Rottner et al., 2017; Lappalainen et al., 2022). Actins share high sequence homology (∼90%) across species from invertebrates to vertebrates. Six distinct isoforms of actin are present in invertebrates such as Drosophila and vertebrates (called α-cardiac, α-skeletal, α-smooth, γ-smooth, β-cytoplasmic, and γ-cytoplasmic actins in mammals), which vary primarily at their N-terminus (Kashina, 2020). The actin cytoskeleton is dynamic and constantly remodels to perform various cellular functions (Blanchoin et al., 2014; Pollard, 2016; Lappalainen et al., 2022). Many actin-binding proteins (ABPs) dynamically fine-tune actin structures in response to cell needs (Gupta et al., 2020; Kadzik et al., 2020; Lappalainen et al., 2022). These proteins help in actin assembly from its monomers (G-actin) to filaments (F-actin) and then to higher-order structures, such as actin bundles and networks (Pollard, 2016; Merino et al., 2020; Gautreau et al., 2022). Among others, ABPs are categorized as proteins assisting in actin nucleation and elongation (e.g., formins and Ena/VASP family), network formation (e.g., Arp2/3 complex), severing and depolymerization (e.g., ADF/cofilin, gelsolin superfamily, and twinfilin), bundling (e.g., fascin, espin, α-actinin), G-actin binding (e.g., profilin), and barbed-end/pointed-end capping (e.g., CapZ and tropomodulin) (Siripala and Welch, 2007a; Siripala and Welch, 2007b; Pollard, 2016; Lappalainen et al., 2022).
It is now well-established that besides classical ABPs that regulate actin’s properties and dynamics via physical, non-covalent mechanisms, actin is also regulated by alterations of its amino acids (Terman and Kashina, 2013; Varland et al., 2019). These co-translational and post-translational modifications (PTMs) of actin, such as phosphorylation, oxidation, acetylation, arginylation, SUMOlytion, ubiquitination, and others, also dynamically control actin’s properties, including F-actin’s stability, functions, and interaction with other ABPs (Terman and Kashina, 2013; Varland et al., 2019). Since actin is one of the most ubiquitous proteins, some of these PTMs are likely to occur non-specifically, including as by-products of enzymatic reactions or via non-enzymatic (including environmental) mechanisms (Terman and Kashina, 2013; Varland et al., 2019). Yet, specific enzymes have also now been identified that selectively target actin as a substrate. Defining these enzymes and their modification of actin is therefore a critical biomedical goal.
MICAL (Molecule Interacting with CasL) proteins have recently emerged as a family of enzymes that selectively target actin as a substrate. MICALs specifically bind and oxidize F-actin to posttranslationally alter specific amino acids within filament subunits, and by this, MICALs robustly alter the properties of actin (Hung et al., 2010a; Hung et al., 2011). Herein, we review the MICALs, first describing their protein organization, enzymatic region, and structural properties. Then, we highlight MICAL-mediated post-translational oxidation of actin and its impact on actin structures and dynamics. Lastly, we focus on the modulation of MICAL-mediated actin regulation by other proteins and the consequence of the MICAL’s oxidation of actin on cellular and tissue functions and dysfunctions.
2 The MICAL family of proteins
MICAL proteins were found independently as a binding partner for the SH3-domain containing adaptor protein CasL (Suzuki et al., 2002) and as a functional driver of cellular morphology/neuronal guidance downstream of Plexin cell-surface receptors (Terman et al., 2002). It was the work in (Terman et al., 2002) that first identified MICALs as having an enzymatic domain and indicated they were oxidation-reduction (Redox) enzymes. That work (Terman et al., 2002) also identified multiple different MICAL genes and that they comprise a family of phylogenetically-conserved proteins with one member in Drosophila (called Mical) and three in vertebrates, including humans [named MICAL-1, MICAL-2, and MICAL-3 (or also known as MICAL1, MICAL2, and MICAL3)] (Figure 1A). MICALs are large proteins (>1,000 amino acids) consisting of a highly conserved N-terminal flavoprotein monooxygenase (also called hydroxylase, MO, FM, or Redox) domain, followed by several other notable regions, including a calponin homology (CH) domain, a Lin-11, Isl-1, Mec-3 (LIM) domain, a proline-rich region with PxxP ligands for SH3-domain containing proteins, and a region that shares homology to the alpha (α) region of Ezrin, Radixin, and Moesin (ERM) proteins (this region is now often referred to as the Plexin-interacting region (PIR), Rab binding domain (RBD), bMERB, or CC) [Figure 1A (Terman et al., 2002; Alto and Terman, 2018; Rouyère et al., 2022)]. Each MICAL family member, including those in Drosophila and humans, has multiple different splice forms/isoforms (Terman et al., 2002), which impact their enzymatic and cellular functions [Figure 1A; e.g., (Terman et al., 2002; Weide et al., 2003; Pasterkamp et al., 2006; Hung and Terman, 2011; Wilson et al., 2016; Rouyère et al., 2022)]. Among the different family members, the MICALs are expressed in most, if not all, tissues (see below).
FIGURE 1.
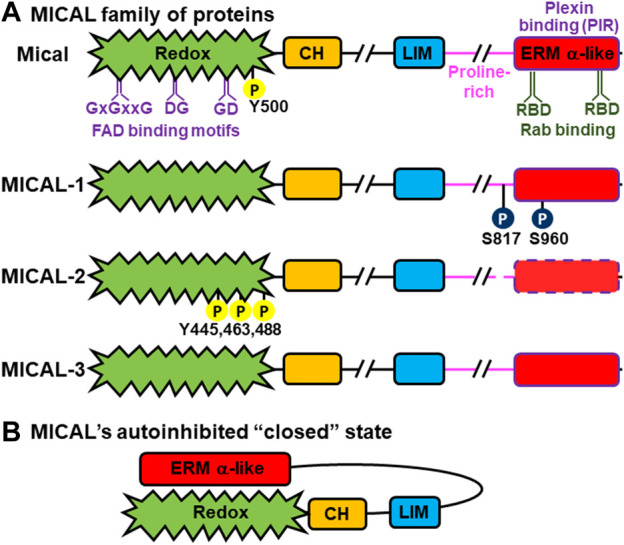
MICAL family proteins: domain organization and allosteric interaction. (A) Domain organization of Drosophila Mical and human/mammalian MICAL-1, MICAL-2, and MICAL-3. All MICALs contain the N-terminal flavoprotein monooxygenase (Redox) domain (green), followed by a calponin homology (CH) domain (orange), a LIM domain (blue), a proline-rich region (pink), and an ERM alpha (α)-like domain (red). These domains are linked by regions of variable length (//). The FAD binding (GxxGxxG, DG, and GD) motifs in the Redox domain are denoted in purple. The two Rab binding (RBD) regions (dark green) and plexin binding (PIR) region (purple) within the ERM α-like domain are also shown. Sites of phosphorylation by Abl kinase are shown as yellow circles [Y (tyrosine)], while those of PAK1 kinase are in dark blue circles [S (Serine)]. It is notable that many annotated cDNAs for MICAL-2 do not contain the C-terminal ERM α-like region, but the MICAL-2 genomic locus includes an ERM α-like region that is similar to MICAL-1 and MICAL-3. This region has independently been called MICAL-CL and Ebitein and is denoted here with dashed lines. (B) Full-length MICAL family members have been found to exist in autoinhibited (inactive) forms. In the autoinhibited form, the C-terminus ERM α-like domain of the MICAL’s folds in antiparallel fashion towards the N-terminus, and interacts with the Redox and LIM domains to inhibit the Redox enzymatic activity of the MICALs.
The Redox region of the MICALs consists of three motifs that bind specifically to FAD (flavin adenine dinucleotide): a distinct dinucleotide binding Rossman fold GxGxxG motif (where G is glycine and x is any amino acid), and both GD (glycine-aspartic acid) and DG (aspartic acid-glycine) motifs that interact with the ribose and pyrophosphate moieties of flavin, respectively [Figure 1A (Terman et al., 2002)]. The presence of these three motifs defined MICALs as flavoprotein monooxygenases, versus other types of Redox enzymes such as oxidases (Terman et al., 2002). Structural studies have also confirmed that the Redox region of MICALs is most similar to flavoprotein monooxygenases, including to the classical flavoprotein monooxygenase p-hydroxybenzoate hydroxylase (pHBH) (Nadella et al., 2005; Siebold et al., 2005; Alqassim et al., 2016; Kim et al., 2020). Biochemical assays have also confirmed MICALs’ catalytic activity. In particular, MICALs non-covalently bind the cofactor FAD and use NADPH as a coenzyme and oxygen (O2) in Redox reactions (Terman et al., 2002; Nadella et al., 2005; Hung et al., 2010a; Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018). Further, unlike oxidases, flavoprotein monooxygenases such as pHBH have substrates that they physically interact with, are activated by, and specifically modify [i.e., direct substrates (Huijbers et al., 2014)]. So too, MICALs’ Redox region has substrates that they physically interact with and whose binding activates MICALs to specifically modify them. In particular, MICALs modify specific methionine residues within these direct substrates: F-actin being the best defined [(Hung et al., 2011); see below]. Of note, in this way, MICAL family proteins are the first flavoprotein monooxygenases found to have a direct protein substrate. Another protein, the Ca2+/calmodulin-dependent protein kinase II (CaMKII), has also recently been linked to being a direct substrate for MICAL-1 (Konstantinidis et al., 2020). In the absence of a substrate, flavoprotein monooxygenases such as pHBH produce hydrogen peroxide (H2O2) (Entsch and van Berkel, 1995; Huijbers et al., 2014), and can therefore modify molecules indirectly (i.e., as indirect substrates). The MICALs’ Redox domain also produces H2O2 in the absence of a direct substrate (Terman et al., 2002; Nadella et al., 2005; Schmidt et al., 2008; Hung et al., 2010a; Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018). This H2O2 affects the activity of the collapsin response mediator protein (CRMP) (Schmidt et al., 2008; Morinaka et al., 2011; Tominaga et al., 2019) and the tau protein (Prifti et al., 2022), with which MICALs are known to interact, by oxidizing specific cysteine residues in them (Schmidt et al., 2008; Morinaka et al., 2011; Tominaga et al., 2019; Prifti et al., 2022). A member of the actin nucleator Arp2/3 complex, Arp3B, has also been linked to being modified by the MICALs on a specific methionine residue (Galloni et al., 2021), but it has not yet been determined whether this is via direct or indirect mechanisms. It is also notable that the amount of H2O2 produced differs between different MICALs, such that MICAL-1 produces higher amounts of H2O2, as compared to other known monooxygenases and other MICALs (Nadella et al., 2005; Zucchini et al., 2011; Wu et al., 2018). In this regard, MICAL-1 has a substitution of a critical aspartate (Asp) to alanine (Ala) in the flavin-binding DG motif, which is responsible for this increased catalytic activity in the absence of a substrate (Wu et al., 2018).
Unlike classical flavoprotein monooxygenases such as pHBH, MICALs contain other domains besides their Redox domains. Therefore, MICALs are referred to as multidomain flavoprotein monooxygenases (Terman et al., 2002) and these other domains, including the proteins they interact with, are essential for regulating the activity of the MICAL’s Redox domain (Schmidt et al., 2008; Hung et al., 2010a; Alto and Terman, 2018; Rouyère et al., 2022). In particular, the MICAL’s other domains have been found to regulate its catalytic activity and its effects in cells/in vivo – including to induce it to exist in an autoinhibited state, such that the C-terminal portion of the MICALs autoinhibits their N-terminal Redox activity [Figure 1B; (Schmidt et al., 2008; Hung et al., 2010a; Giridharan et al., 2012; Vitali et al., 2016; Frémont et al., 2017)]. The MICAL’s Redox activity therefore is under the spatiotemporal instruction of other specific proteins (Terman et al., 2002; Schmidt et al., 2008; Hung et al., 2010a; Frémont et al., 2017) (see below).
The CH domain of each of the MICALs (Figure 1A) is structurally similar to the CH type-2 domain present in several other actin-binding proteins (such as smoothelin) (Terman et al., 2002). While CH type-1 domains are known to directly bind to F-actin, CH type-2 domains, including those present in MICALs, do not appear to directly bind to F-actin (Sun et al., 2006; Ishida et al., 2008; Yin et al., 2020). Indeed, work with purified proteins has revealed that the Redox domain of the MICALs alone is sufficient for its binding to F-actin (Hung et al., 2010a; Yoon et al., 2021). So too, the catalytic activity (Kcat) of the MICAL’s Redox domain and its effects on F-actin are similar with or without the CH domain (Hung et al., 2010a; Vitali et al., 2016; Yoon et al., 2021). Yet, the MICAL’s CH domain may help facilitate the MICAL’s F-actin binding, including the possibility that the MICAL’s CH domain might interact with the CH domain of other proteins to further promote the MICAL’s F-actin binding (Alqassim et al., 2016; Kim et al., 2020). The MICAL’s CH domain may also provide the means to interact with non-CH domain-containing proteins (Suzuki et al., 2002). Based on analogy to other proteins, the MICAL’s CH domain (or residues nearby) may also be involved in its self-association (e.g., (Liu et al., 2016; Rai et al., 2020)). Notably, in vivo work reveals that the CH domain helps localize the MICALs subcellularly (through unknown means) to exert its F-actin effects (Hung et al., 2010a).
The LIM domain of each of the MICALs (Figure 1A) is highly similar to other LIM domains, indicating it contains Zn2+ finger motifs (Terman et al., 2002). As with other LIM domains, the MICAL’s LIM domain may serve to bring it into contact with other proteins (Matthews et al., 2009) — including that it may contribute to the MICAL’s interaction with CRMP, although no direct interactions between MICALs’ LIM domain and CRMP have been observed (Schmidt et al., 2008). The MICAL’s LIM domain may also be involved in its self-association (Schmidt et al., 2008; Liu et al., 2016; Miyake et al., 2019). The MICAL’s proline-rich PxxP region (Figure 1A) is known to mediate its interactions with other proteins, including with SH3 domain-containing proteins such as the adaptor protein CasL (Suzuki et al., 2002) and the non-receptor tyrosine kinase Abl (Yoon et al., 2017). The MICAL’s C-terminal ERM alpha (α)-like domain (also called PIR, CC, RBD, bMERB) [Figure 1A (Terman et al., 2002; Alto and Terman, 2018; Rouyère et al., 2022)] serves an essential function in regulating the enzymatic activity of the MICALs. It is through this region that MICALs interact with the cytoplasmic portion of the Plexin transmembrane receptor (Terman et al., 2002; Schmidt et al., 2008) and also with the activated (GTP-bound form) of Rab GTPases (Weide et al., 2003; Fukuda et al., 2008; Deng et al., 2016; Rai et al., 2016; Frémont et al., 2017). It is also notable that many annotated cDNAs for MICAL-2 do not contain the C-terminal ERM α-like region, but the MICAL-2 genomic locus includes an ERM α-like domain that is similar to invertebrate Mical, MICAL-1, and MICAL-3 (Terman et al., 2002; Hung et al., 2011; Giridharan and Caplan, 2014; Alto and Terman, 2018; Rouyère et al., 2022). This region has independently been called MICAL-CL and Ebitein (Figure 1A, dashed box; see (Hung and Terman, 2011) for further discussion).
Another family of proteins known as the Mical-like (MICAL-L) has also been identified (Terman et al., 2002). They share similar domain composition with the MICALs but lack the Redox domain. Humans have two MICAL-like proteins [MICAL-L1 and MICAL-L2 (also called JRAB)], while Drosophila has one Mical-like protein (Terman et al., 2002). MICAL-Ls have key cellular functions, including in endocytosis and vesicle trafficking [e.g., (Farmer et al., 2021; Malinova et al., 2021; Sikora et al., 2021)], but since they do not have the actin-modifying Redox domain, they will not be further discussed herein.
3 The MICAL’s effects on F-actin properties
The Redox region of all MICALs directly interacts with F-actin (Hung et al., 2010a; Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018) (Figures 2A, B). Moreover, this binding to F-actin substantially enhances each MICAL family member’s ability to interact with and consume its coenzyme NADPH (Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018) (Figure 2C). In particular, the direct binding of each MICAL family member to F-actin (not to G-actin) substantially increases its Redox activity (e.g., including >100-fold, in an F-actin and MICALs concentration-dependent manner) (Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018). Each MICAL family member then oxidizes two specific methionine (Met) residues in actin (Met44 and Met47) in a stereoselective manner (in the Met R-isomer conformation) to generate actin Met-44, 47-R-sulfoxide [actin Met(R)O-44,47] [Figures 2B, C (Hung et al., 2011; Hung et al., 2013; Lee et al., 2013; Wu et al., 2018)]. Notably, it is the stereospecific oxidation of these two Met residues that underlies the MICALs’ F-actin effects. In particular, a high rate of actin disassembly is achieved at very low, substoichiometric levels of MICALs, pointing to the catalytic activity of MICALs in disassembling F-actin (Hung et al., 2011; Wu et al., 2018). So too, work with purified proteins and in vivo reveals that mutating the Met44 and Met47 residues of actin prevents MICALs from exerting their effects on F-actin (Hung et al., 2011; Lundquist et al., 2014; Grintsevich et al., 2017; Orr et al., 2017; Wu et al., 2018).
FIGURE 2.
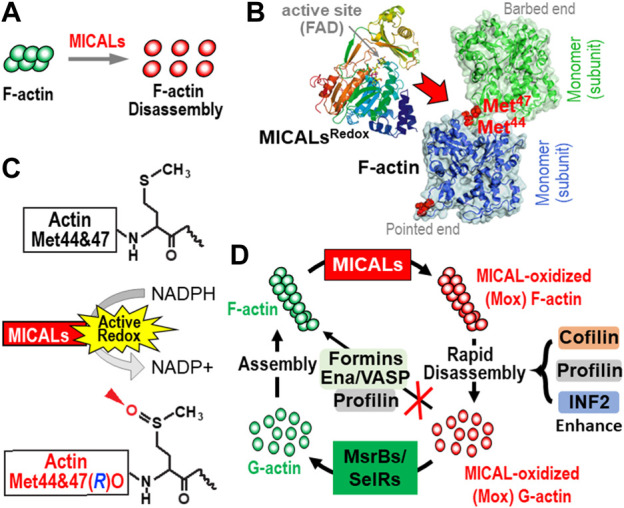
The MICAL’s activity and its effects on actin dynamics. (A–C) MICALs posttranslationally modify specific methionine (Met) residues in F-actin (red), which triggers F-actin disassembly (A). More specifically, MICALs bind to F-actin [(B), red arrow)] and in the presence of their coenzyme, NADPH (C), selectively and stereospecifically oxidize (O) actin’s Met44 and Met47 in the R-conformation [(C), arrowhead]. This oxidation of Met44 and Met47 [see (B), red] occurs in the D-loop, at the pointed end of individual actin filament subunits, which disrupts the interprotomers interactions in F-actin and leads to their rapid disassembly. (D) MICAL-mediated F-actin disassembly is regulated by other proteins. From top and clockwise: following MICALs’ oxidation of F-actin to generate MICAL-oxidized (Mox) F-actin, MICAL-triggered F-actin disassembly is enhanced by other proteins, including cofilin, profilin, and INF2. The Mox-G-actin that is formed does not readily re-polymerize even in the presence of profilin, formins, and Ena/VASP. Yet, Mox-G-actin is reduced specifically by selective methionine sulfoxide reductases (MsrBs/SelRs), and this G-actin can then re-polymerize normally. In this way, MICALs and MsrBs/SelRs create a reversible system for Redox regulation of actin dynamics in cells.
The identification of the MICALs and their mechanism for affecting actin also provides new insight into the regulation of actin by oxidative means. In particular, oxidation has long been known to affect actin filament dynamics [Reviewed in (Terman and Kashina, 2013)]. What was not clear was whether these effects were specific, selective, and/or locally controlled versus simply being a random by-product of enzymatic reactions or non-enzymatic mechanisms (Shacter, 2000; Stadtman et al., 2003; Kim and Gladyshev, 2007). The identification, as described above, that the MICAL’s enzymatic modification of actin is substrate specific (i.e., F-actin, but not G-actin), residues specific (i.e., Met44, Met47), and stereo specific (i.e., in the R-isomer versus the S-isomer conformation), thus reveals that actin dynamics are controlled by distinct oxidative mechanisms. In the same way, the MICALs do not simply release a diffusible oxidant, such as H2O2, to cause widespread effects on F-actin – since such oxidants, including H2O2, do not mimic the effects of the MICALs [e.g., cell-lethal levels of H2O2 have no effect on F-actin disassembly in the assays used to define the MICALs (Hung et al., 2010a; Hung et al., 2011; Wu et al., 2018)]. Furthermore, the H2O2 scavenger catalase (and other types of reductants such as DTT and thioredoxin/thioredoxin reductase) do not alter the MICAL’s F-actin effects (Hung et al., 2011; Hung et al., 2013). Additionally, the MICAL’s substrate residues (Met44 and Met47) are buried within F-actin, in a hydrophobic pocket that is poorly accessible to diffusible oxidants (Dalle-Donne et al., 2002; Guan et al., 2003; Guan et al., 2005; Takamoto et al., 2007; Hung et al., 2011; Chou and Pollard, 2019). Moreover, MICALs need to be in close proximity to F-actin to exert their effects (Hung et al., 2011) — and such binding to F-actin is needed to activate MICALs’ enzymatic activity (Hung et al., 2010a; Hung et al., 2011; Zucchini et al., 2011; McDonald et al., 2013; Wu et al., 2018) to then oxidize actin filament subunits (Hung et al., 2011; Wu et al., 2018). Thus, the MICAL family forms a class of monooxygenases that modify actin by direct interactions. Moreover, as the MICALs directly/physically interact with F-actin, further work will seek to capture/determine whether the MICALs directly interact with the Met44 and Met47 residues to add oxygen to them [which would be similar to how flavoprotein monooxygenases such as pHBH work on their substrate residues (Entsch and van Berkel, 1995; Huijbers et al., 2014)]. MICALs’ ability to stereospecifically modify Met44 and Met47 in the R, but not the S, conformation indicates this type of selective modification within the MICALs’ – F-actin substrate interaction pocket. Alternatively, the substrate binding pocket between the MICALs and F-actin (i.e., the in situ enzyme–substrate interaction region) may provide an environment where MICALs release an oxidant that selectively modifies Met44 and Met47.
The Met44 and Met47 residues are present in the DNaseI-binding loop (D-loop) of actin at the pointed-end of actin filament subunits (Figure 2B). Actin’s D-loop is essential for forming longitudinal contacts between actin subunits in F-actin and regulates filaments’ stability (Oztug Durer et al., 2010; Dominguez and Holmes, 2011; Durer et al., 2012; Chou and Pollard, 2019; Das et al., 2020). More specifically, the side chain of Met44 is thought to be an important residue for inter-subunit interactions along the long-pitch helix of filaments (Oztug Durer et al., 2010; Dominguez and Holmes, 2011; Durer et al., 2012; Chou and Pollard, 2019; Chou and Pollard, 2020; Das et al., 2020). Oxidation of these two methionine residues increases the local charge of F-actin (Hung et al., 2011) and alters the filaments’ D-loops positions (Grintsevich et al., 2017). This loosening of inter-subunit interactions upon MICAL-mediated oxidation destabilizes the F-actin structures, making them prone to fragmentation even under a mild mechanical force such as pipetting, and ultimately disassembles them (Hung et al., 2011; Grintsevich et al., 2016; Grintsevich et al., 2017). Of note, actin’s Met44 and Met47 are phylogenetically conserved in all actin isoforms, suggesting that MICALs are likely to exert similar effects on all actins (Hung et al., 2011). Furthermore, the importance of Met44 in actin structures can be judged by the fact that a mutation of Met44 is lethal in yeast (Oztug Durer et al., 2010). Likewise, dominant de novo (heterozygous missense) mutations in actin’s Met44 and Met47 result in human diseases, including those defined by an accumulation of F-actin (Laing et al., 2009; Hung et al., 2010b; Hoffjan et al., 2011; Zou et al., 2013; Regalado et al., 2014; Wangler et al., 2014; Yates et al., 2017; Zhang et al., 2019).
The MICAL-mediated modification of F-actin subunits induces filaments to disassemble [Figure 2A (Hung et al., 2010a; Hung et al., 2011; Wu et al., 2018)]. Real-time analysis of immobilized actin filaments by TIRF microscopy reveals that MICALs induce depolymerization and severing (fragmentation) of filaments [e.g., (Hung et al., 2011; Grintsevich et al., 2016; Grintsevich et al., 2017)]. MICALs depolymerize barbed ends of filaments with disassembly rates that reach as high as >84 subunits/sec, as opposed to disassembly rates that on rare occasions can reach as high as ∼5.4 subunits/sec for unmodified F-actin (Grintsevich et al., 2017). At the same time, the pointed-end depolymerization rate is ∼1.44 subunits/sec in the presence of MICAL, in contrast to ∼0.17 subunits/sec in control (no MICAL) conditions (Frémont et al., 2017; Grintsevich et al., 2017). Thus, the prime cause of MICAL-oxidized F-actin’s rapid depolymerization are their unstable barbed ends. Intriguingly, the nucleotide state of F-actin also plays a considerable role in its susceptibility to MICAL-mediated depolymerization. ADP-Pi·F-actin is less prone to MICAL-mediated depolymerization than ADP·F-actin (Grintsevich et al., 2017). Incubation of MICAL-oxidized F-actin with either BeFx (which mimics the ADP-Pi cap at the barbed ends) or a heterodimeric capping protein (CP) abolishes the rapid depolymerization event (Grintsevich et al., 2017). Overall, this reveals that MICALs affect aged actin filaments much more than new ones. This is significant at the cellular level because rapid actin remodeling is required to bring morphological changes to cells – and these changes are achieved by disassembling existing actin structures (filaments and bundles). Indeed, MICALs dramatically decrease and remodel F-actin structures in cultured cells and in vivo, both in the cytosol [(Hung et al., 2010a; Hung et al., 2011; Hung et al., 2013; Lee et al., 2013; Wu et al., 2018); see below] and in the nucleus (Lundquist et al., 2014) — and exert disassembling effects on different networks of actin including bundled actin (Hung et al., 2010a), branched actin (Galloni et al., 2021), and actin bound to decorating proteins — including tropomyosin (Wioland et al., 2021). Loss of MICALs in vivo results in the accumulation of F-actin [e.g., (Beuchle et al., 2007; Hung et al., 2010a; Hung et al., 2011; Hung et al., 2013; Lundquist et al., 2014); see below].
Actin disassembly proteins, such as cofilin, gelsolin, twinfilin, etc., sever or depolymerize filaments by inducing conformational changes in F-actin upon their binding (Pollard, 2016; Lappalainen et al., 2022). MICALs, on the other hand, chemically modify (oxidize) actin filaments to cause their severing and depolymerization (Hung et al., 2010a; Hung et al., 2011; Grintsevich et al., 2016; Frémont et al., 2017; Grintsevich et al., 2017; Wu et al., 2018). Additionally, in cofilin-mediated severing/depolymerization, the end products are newly formed barbed ends or G-actin, which promote actin polymerization (Chin et al., 2016). In contrast to that, the end product of the MICAL’s disassembly is MICAL-oxidized actin – and it is ineffectively reused for filaments’ elongation due to its poor polymerization properties (Hung et al., 2011; Hung et al., 2013; Lee et al., 2013; Wu et al., 2018; Grintsevich et al., 2021). Thus, in contrast to other well-known actin-binding proteins that disassemble F-actin but also promote actin polymerization, MICALs both disassemble F-actin and inhibit actin polymerization.
4 Structural and dynamic properties of MICAL-oxidized actin (Mox-actin)
MICAL-oxidized actin (Mox-actin) is the end product of the site-specific F-actin oxidation by the MICALs. Mox-actin has different polymerization properties than unoxidized actin. ATP-bound Mox-actin monomers (Mox-G-actin) form filaments with altered polymerization kinetics and length compared to unoxidized actin. They have ∼10-fold higher critical concentration for polymerization than unoxidized actin (∼1 μM for Mox-actin versus 0.1 μM for unoxidized actin), with a prolonged nucleation phase that reaches saturation at a lower level than unoxidized actin (Grintsevich et al., 2016). This results in ∼3-fold slower rate of Mox-G-actin elongation than that of unoxidized actin. Intriguingly, ADP-bound Mox-actin monomers cannot polymerize even at high concentrations (>30 µM) (Grintsevich et al., 2017). Notably, a decrease in the critical concentration of Mox-actin was observed when BeFx was present (∼0.24 µM), suggesting that the protection of barbed ends of filaments reduces their disassembly (Grintsevich et al., 2017). In addition to that, the filaments formed by Mox-actin are also generally very short and highly fragile, breaking even under small force as mentioned above (Grintsevich et al., 2016; Grintsevich et al., 2021). Altogether, Mox-G-actin is unlikely to participate in active actin polymerization in the cell, and even if it does, those filaments would not be stable enough to withstand any applied force.
Mox-F-actin also readily disassembles — by steps of slow (∼2.6 subunits/sec compared to ∼0.2 subunits/sec for unmodified actin) and catastrophic depolymerization (>84 subunits/sec compared to rare events of ∼5.4 subunits/sec for unmodified actin) (Grintsevich et al., 2017). Cryo-EM structural analysis showed two Mox-F-actin conformations (Class 1, PDB ID: 6AV9, and Class 2 PDB ID: 6AVB), supporting the two different disassembly modes (Grintsevich et al., 2017). Most of the structural changes were limited to the D-loop. In Class 1, Met44-O (MICAL-oxidized Met44) moves out from its canonical position (hydrophobic cleft of actin), which ablates the interprotomer longitudinal contacts in Mox-F-actin. In contrast to that, Met47-O in Class 1 forms a new hydrogen bond with threonine (Thr) 351, thereby twisting the D-loop and further destabilizing F-actin (Grintsevich et al., 2017). Thus, the Class 1 conformation supports the catastrophic (>84 subunits/sec) disassembly of F-actin (Grintsevich et al., 2017). In Class 2, Met44-O remains in its position and forms close interactions with the cleft. Met47-O in this class forms a hydrogen bond with tyrosine (Tyr) 169 instead, and the Mox-F-actin structure remains similar to that of unmodified F-actin (Grintsevich et al., 2017). These structural observations complement the enzymology and site-directed mutagenesis results described above, i.e. that Met44 is the main site through which MICAL-mediated oxidation exerts its effects on F-actin but Met47 oxidation also impacts F-actin (Hung et al., 2011; Lundquist et al., 2014; Grintsevich et al., 2017; Orr et al., 2017; Wu et al., 2018) and support the notion that MICALs rapidly disassemble F-actin by modifying the Met44 and Met47 residues present in the D-loop of F-actin subunits.
5 The MICAL’s and Mox-actin’s effects on the function of actin-binding proteins, and vice versa
The MICAL’s Redox actin regulatory system is therefore an unusual system for robustly regulating actin dynamics. Yet, recent results also reveal that it should not be thought of as working independently from other classical actin-binding proteins but that it interacts/works together with them. In particular, the binding of ABPs to F-actin is controlled by its nucleotide-bound state (Chou and Pollard, 2019), its tension and torque (Hayakawa et al., 2011; Risca et al., 2012), and structural impacts due to the presence of other actin-binding proteins (Reymann et al., 2012; Ngo et al., 2016; Mizuno et al., 2018). Furthermore, since MICALs oxidize F-actin specifically at its two Met residues in the D-loop (Hung et al., 2011), this alters F-actin’s structure (Grintsevich et al., 2017) and, thereby F-actin’s interactions with other ABPs (see below).
Cofilin is a well-studied actin depolymerization and severing protein. It binds cooperatively to actin filaments to form clusters of cofilin-decorated regions (Wioland et al., 2017). The boundaries between these clusters are the filaments’ severing sites (Andrianantoandro and Pollard, 2006; Suarez et al., 2011). Severing occurs because the cofilin-decorated regions are over-twisted compared to the bare F-actin, leading to its structural instability (Bobkov et al., 2004; Bobkov et al., 2006; Huehn et al., 2020). MICAL-mediated oxidation of actin subunits weakens inter-longitudinal interactions in F-actin and makes them more fragile and susceptible to severing/depolymerization-inducing conditions [Figure 2D (Hung et al., 2011; Grintsevich et al., 2016; Grintsevich et al., 2017)]. Even low concentrations of cofilin, which are harmless to unmodified F-actin, disrupt Mox-F-actin by quickly disassembling it [Figure 2D (Grintsevich et al., 2016; Wioland et al., 2021)]. Thus, despite cofilin having a low affinity for ADP. Pi-actin, and/or the presence of inorganic phosphate inhibiting F-actin depolymerization, the MICAL’s oxidation of F-actin counteracts these effects (Muhlrad et al., 2006; Grintsevich et al., 2017; Wioland et al., 2021). MICALs do this by increasing cofilin’s binding and favoring the growth of cofilin domains on actin filaments (Grintsevich et al., 2016; Wioland et al., 2021). The MICAL’s oxidation of actin allows cofilin to exert its effects also in the presence of at least some of its well-known inhibitory modifications, such as a phosphomimetic substitution at Ser3 [that is thought to mimic the effects of phosphorylation by LIM Kinase, a well-known inhibitor of cofilin (Wioland et al., 2021)].
Profilin, a well-known G-actin binding protein, also increases F-actin depolymerization (Kinosian et al., 2002; Jégou et al., 2011; Courtemanche and Pollard, 2013). Interestingly, its depolymerization effect is more profound on Mox-F-actin, for which even low profilin concentrations are sufficient to induce its disassembly [Figure 2D (Grintsevich et al., 2021)]. The exact mechanism for how profilin causes this increased disassembly is not known, but several mechanisms are in line with the data (Grintsevich et al., 2021). In particular, modeling studies suggest that profilin’s binding to the barbed-end of F-actin requires reduced flattening of terminal actin subunits to prevent steric clashes (Courtemanche and Pollard, 2013). This effect would increase the rate of subunits dissociation from the barbed ends of actin by inducing more of a G-actin-like conformation – and thus further enhance the depolymerization of Mox-actin, which is known to be substantially faster than “normal” actin even in the absence of profilin (Grintsevich et al., 2017; Grintsevich et al., 2021). It is also possible, given the unusual structure of Mox-F-actin (Grintsevich et al., 2017), that free profilin binds to Mox-actin’s barbed-ends with higher affinity compared to “normal” actin, therefore, blocking new subunit addition (Kinosian et al., 2002; Jégou et al., 2011; Courtemanche and Pollard, 2013). Furthermore, a loss of an ATP cap at the barbed-end of Mox-F-actin may also contribute to profilin-induced destabilization of Mox-F-actin by exposing ADP-bound Mox-F-actin segments that are intrinsically unstable and undergo catastrophic collapse (Grintsevich et al., 2017). This intrinsic/profilin-induced instability of Mox-F-actin at its barbed ends would also be enhanced if the association of profilin–Mox-ATP-G-actin complexes with barbed ends is greatly inhibited/abolished.
The inverted formin (INF2) is an atypical formin that can both polymerize and depolymerize actin in a concentration-dependent manner (Chhabra and Higgs, 2006; Gurel et al., 2014; Hegsted et al., 2017). Mox-F-actin is also more vulnerable to INF2 [the non-autoinhibited form of INF2 (INF2-FFC)]-mediated depolymerization than unoxidized actin [Figure 2D (Das et al., 2022)]. The exact mechanism of Mox-F-actin disassembly by INF2-FFC is unknown, but it could be due to an INF2-induced enhanced destabilization of its D-loop upon MICAL-mediated oxidation, and a further weakening of interprotomer actin contacts in Mox-F-actin (Grintsevich et al., 2017). Overall, the oxidation of filaments by the MICALs sensitizes them for rapid disassembly by other more classical severing/depolymerizing actin-binding proteins (Figure 2D).
Mox-G-actin is the end product of MICAL-mediated rapid F-actin disassembly — with or without the assistance of other actin severing/depolymerizing proteins. In cells, G-actin is the fuel for actin polymerization and is required for maintaining cellular structures. Formins and Ena/VASP are actin nucleation/polymerization-promoting proteins that bind directly, or through profilin, to G-actin to accelerate the polymerization process (Higgs, 2005; Goode and Eck, 2007; Breitsprecher et al., 2011; Courtemanche and Pollard, 2013; Courtemanche, 2018). Profilin regulates actin remodeling in cells by delivering G-actin to formin-bound actin filaments, while also inhibiting spontaneous actin polymerization by shielding the barbed ends of G-actin (Kovar et al., 2006). Interestingly, despite being modified, Mox-G-actin binds normally to profilin (Grintsevich et al., 2021). However, profilin-Mox-G-actin complexes do not fuel formin-mediated polymerization [Figure 2D (Grintsevich et al., 2021)]. Moreover, profilin alone inhibits the polymerization of Mox-actin, even when unoxidized actin seeds are present (Figure 2D (Grintsevich et al., 2021)). Thus, when combined with Mox-G-actin, profilin does not facilitate actin polymerization but inhibits it and further promotes F-actin disassembly (Grintsevich et al., 2021).
The Arp2/3 complex is a well-known nucleator and regulator of branched actin networks (Pollard, 2007; Pizarro-Cerda et al., 2017). The Arp2/3 complex consists of seven proteins, with Arp2 and Arp3/Arp3B being unconventional actin-related proteins that mimic a filamentous actin dimer (Pizarro-Cerda et al., 2017; Narvaez-Ortiz and Nolen, 2022). To attain an active actin dimer state, which acts as a nucleation site for polymerization of a new filament on the side of the mother filament, Arp2 and Arp3/Arp3B need to undergo conformation changes. These changes are mediated by cortactin and Wiskott Aldrich Syndrome protein (WASP) (Pollard, 2007; Pizarro-Cerda et al., 2017; Narvaez-Ortiz and Nolen, 2022). Notably, Arp3B is a target for MICAL-mediated oxidation, such that MICAL-2 oxidizes the Met293 of Arp3B (Galloni et al., 2021). This oxidation renders Arp3B inactive and promotes disassembly of branched actin networks (Galloni et al., 2021). The exact mechanism of debranching is unknown, but it could be due to an altered structure of the Arp2 and Arp3B complex, which no longer acts as a nucleation site for branch formation.
Overall, MICALs collaborate with other actin-binding proteins, changing actin polymerization/depolymerization dynamics and shifting the cell to impaired actin assembly conditions. Moreover, because of its reduced polymerization capacity, the MICAL-oxidized actin monomer is not reused in the actin assembly cycle. Thus, MICALs are potent actin disassemblers that combine with other proteins to dynamically tune their functional effectiveness.
6 Reversing the MICAL’s effects on F-actin: MsrB/SelR family reductases
In light of the identification of this new direct oxidation-dependent means to regulate actin cytoskeletal dynamics it was of great interest to determine whether these MICAL-mediated actin alterations were reversible. Notably, it was found that the MICAL’s actin modification (stereospecific oxidation of Met44 and Met47) was selectively counteracted by a family of stereospecific methionine sulfoxide reductase enzymes called MsrB/SelR (Hung et al., 2013; Lee et al., 2013). There are three MsrB/SelR enzymes in mammals (MsrB1, 2, and 3) and one in invertebrates (called SelR). In particular, MsrBs/SelRs can specifically reverse each of the MICALs effects on actin (Figure 2D), while other reductases, including other methionine sulfoxide reductase enzymes (MsrA) and chemical reducing agents such as DTT, cannot do that (Hung et al., 2013; Lee et al., 2013; Grintsevich et al., 2016; Wu et al., 2018). In vitro experiments with purified proteins reveal that MsrBs/SelRs restore Mox-actin polymerization and formin-assisted polymerization kinetics (Hung et al., 2013; Lee et al., 2013; Grintsevich et al., 2016; Wu et al., 2018; Bai et al., 2020; Grintsevich et al., 2021). MsrBs/SelRs also counteract MICALs in vivo effects, including on F-actin remodeling and actin-dependent cellular functions such as cell morphology, axon guidance, synaptogenesis, muscle organization, trafficking, and cytokinesis (Hung et al., 2013; Wu et al., 2018; Bai et al., 2020; Hamdan et al., 2020). Results of experiments support the point that while F-actin is a substrate for the MICALs, the MsrB/SelR’s reversing effect occurs on G-actin (Figure 2D (Hung et al., 2013; Lee et al., 2013)). Other points to consider are the relative rates of the MICAL’s oxidation reaction on actin versus the MsrB/SelR’s reduction reaction, and whether the catalytic activity of the MICAL’s versus the MsrB/SelR’s favors one reaction over the other. In short, experimental observations with purified proteins do not reveal major differences in the relative rates of the MICAL’s oxidation of actin versus the MsrB/SelR’s reduction of MICAL-oxidized actin. In other words, although the exact rates of the MICAL’s oxidation of actin have not been published, experimental studies (using antibodies to MICAL-oxidized actin and subtilisin digestion of F-actin after MICAL treatment) reveal that MICAL rapidly (essentially ∼ instantaneously) oxidizes filament subunits in a concentration-dependent manner (Hung et al., 2011; Hung et al., 2013; Grintsevich et al., 2016). MsrBs/SelRs quickly reverse this MICAL’s-oxidation of actin, and then actin polymerizes normally (Hung et al., 2013; Lee et al., 2013; Wu et al., 2018). In vivo observations are also consistent with these results with purified proteins. In particular, increasing MICAL levels induces dramatic effects on F-actin in vivo (Hung et al., 2010a; Wu et al., 2018), but the organization of F-actin in cells – with simultaneous increases in the MsrB/SelRs – indicates that the MsrB/SelRs serve to negate the MICAL’s effect (Hung et al., 2013; Wu et al., 2018). Lastly, although proteins (such as Plexins and Rabs) have been identified that prominently control/regulate the MICAL’s actions in cells (i.e., control the MICAL’s side of the reaction; see below for more details), little is known of what controls the MsrB/SelR side of the reaction in cells. Related to this, MICALs appear to exhibit a specific subcellular localization pattern (Hung et al., 2010a), while MsrB/SelRs are more broadly localized (Hung et al., 2013). This difference in localization may play a role in regulating MsrB/SelRs activity and effects on MICALs (including favoring one side of the reaction versus the other). The MICALs and MsrB/SelRs therefore comprise a specific reversible Redox system for robustly regulating actin dynamics.
7 Regulation of the MICAL’s activity
7.1 Activation of the MICALs: Relieving the MICAL’s autoinhibited state
MICAL Redox enzymes do not induce their effects on actin indirectly (e.g., via general effects on the cellular redox state or global H2O2 production), but locally target F-actin for disassembly (Hung et al., 2010a; Hung et al., 2013; Wu et al., 2018). So, an important question in this regard is how is the activity of the MICALs regulated in a localized manner. Work with purified proteins and in vivo reveals that MICALs are self-regulated through intramolecular interactions between their C-terminal ERM α-like domain and the N-terminal Redox and LIM domains [Figure 1B (Schmidt et al., 2008; Hung et al., 2010a; Giridharan et al., 2012; Vitali et al., 2016; Frémont et al., 2017)]. These intramolecular interactions mask the active site in the F-actin regulatory Redox domain of MICALs to render them catalytically inactive (Figure 1B).
So how do MICALs become active? Physical interactions between MICALs and proteins in specific signaling pathways spatiotemporally relieve MICALs autoinhibition. In particular, the ERM α-like/PIR region of the MICALs directly interacts with the cytoplasmic region of Plexin transmembrane cell-surface receptors [Figure 3A (Terman et al., 2002)]. Plexins are receptors for one of the largest protein families of extracellular guidance cues, semaphorins (Semas), with over twenty family members conserved from invertebrates to humans (Alto and Terman, 2017). In the absence of Semas, Plexins’ cytoplasmic region is also autoinhibited, and results support a model that Sema binding to the extracellular portion of Plexin induces an allosteric change, which relieves this autoinhibition and activates Plexin (Pascoe et al., 2015). Activated Plexin then binds to the MICALs to relieve the MICAL’s autoinhibition and allow for the activation of its Redox domain to spatiotemporally regulate actin dynamics [Figures 3A,B (Terman et al., 2002; Schmidt et al., 2008)]. Further specificity, including precise regulatory mechanisms, is gained by different semaphorins and plexins utilizing different MICALs to exert their cellular effects, including on F-actin disassembly (Terman et al., 2002; Ayoob et al., 2006; Schmidt et al., 2008; Hung et al., 2010a; Morinaka et al., 2011; Aggarwal et al., 2015; Hou et al., 2015; Loria et al., 2015; Orr et al., 2017; Tominaga et al., 2019).
FIGURE 3.
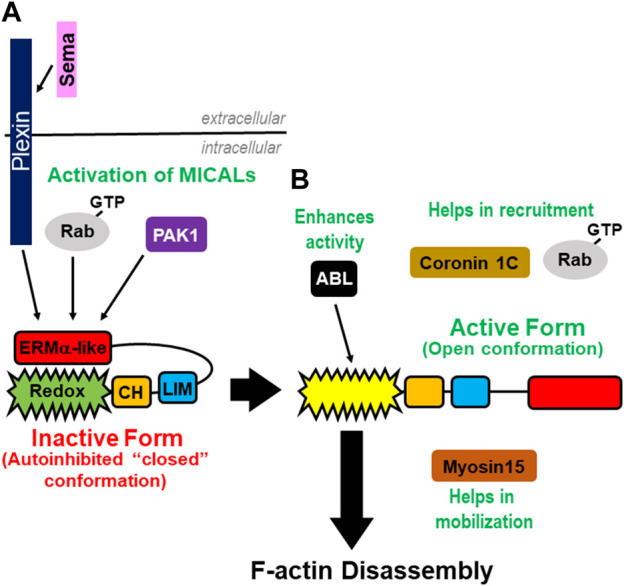
Regulation of the MICAL’s activity. (A) Plexins and GTP-bound Rab proteins bind the MICAL’s ERM α-like domain to relieve the MICAL’s autoinhibition. PAK1 kinases also play a role in relieving the MICAL’s autoinhibition by phosphorylating residues within the MICAL’s C-terminus. (B) Rabs and Coronin 1C are involved in the MICAL’s translocation and positioning. Myosin15 is involved in moving the MICAL’s into a new region to propagate its F-actin disassembly effects.
Specific Rab family small GTPases also bind and relieve the MICAL’s intramolecular autoinhibition (Figure 3A). In particular, multiple different Rab GTPases (including Rab1A/B, Rab7A, Rab8A/B, Rab10, Rab13, Rab15, Rab33B, Rab35, and Rab36) directly interact with different MICALs via MICALs’ ERM α-like/RBD region, and they do this primarily in their active GTP-bound state [Figure 3A (Weide et al., 2003; Fischer et al., 2005; Fukuda et al., 2008; Rai et al., 2016; Frémont et al., 2017; Liu et al., 2017; Tian et al., 2018; Gillingham et al., 2019; Hamdan et al., 2020; McGarry et al., 2022)]. Rab GTPases are well-known regulators of cellular functions, including as key vesicle trafficking proteins in endocytosis and exocytosis, and their direct interaction with MICALs releases the MICAL’s autoinhibition (Frémont et al., 2017; Esposito et al., 2019). Further specificity is gained by different MICALs interacting and utilizing different Rabs to recruit and activate them at specific locations to exert their cellular effects, including on F-actin disassembly [Figures 3A,B (Grigoriev et al., 2011; Deng et al., 2016; Frémont et al., 2017; Hamdan et al., 2020)]. Different Rab and MICAL-interacting proteins, including ELKS active-zone proteins (Grigoriev et al., 2011; Liu et al., 2017), NINL (Bachmann-Gagescu et al., 2015), MKLP1/Kif23 centralspindlin components/kinesin family proteins (Maliga et al., 2013; Liu et al., 2016; Liu et al., 2017), GRAF membrane tubulating proteins (Lucken-Ardjomande Hasler et al., 2020), TBC1D1 Rab GTPase activating proteins (GAPs) (Hook et al., 2020), alpha2-spectrin (Liu et al., 2016; Hamdan et al., 2020), and the MuSK and PAK1 serine/threonine kinases (Budayeva et al., 2022; McGarry et al., 2022) are also linked to Rab and MICAL interactions/cellular effects.
Thus, different modes of regulation help in the precise spatiotemporal activation of the MICALs, which is a prerequisite for normal cellular forms and functions [e.g., (Terman et al., 2002; Schmidt et al., 2008; Hung et al., 2010a; Frémont et al., 2017)]. Further, the dysregulation of the MICALs has a detrimental effect on cellular forms and functions. For example, constitutively-active forms of MICAL (e.g., those lacking the ERM α-like domain that serves an autoinhibitory function) induce widespread and excessive F-actin disassembly and marked abnormalities in cellular morphology/structure (Hung et al., 2010a; Giridharan et al., 2012; Wu et al., 2018) and disease (Dazzo et al., 2018). Related to this, without a C-terminal ERM α-like domain, MICAL-2 would not be autoinhibited in cells (i.e., it would be constitutively active in cells). Thus, further study is needed to determine if the MICAL-CL/Ebitein region/ERM α-like domain that is a part of the MICAL-2 locus, is involved in MICAL-2’s autoinhibition (Terman et al., 2002; Hung and Terman, 2011).
7.2 Additional positive effectors of the MICALs
The MICAL’s effects have also been found to be enhanced through the action of specific signaling proteins. For example, vascular endothial-derived growth factor (VEGF), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF) and their receptors have been linked to increasing the MICAL’s F-actin disassembly effects (Hou et al., 2015; Deng et al., 2016; Evans et al., 2017; Yoon et al., 2017; Barravecchia et al., 2019; McGarry et al., 2022). Further results reveal that they enhance the MICAL’s F-actin disassembly effects by working with Abl/Arg non-receptor tyrosine kinases, which phosphorylate specific tyrosine (Y) residues within the Redox domain of the MICALs (Y500 in Mical and Y445, Y463 in MICAL-2) [Figure 1A (Yoon et al., 2017; Yoon and Terman, 2018a; Shi et al., 2020; Zhang et al., 2022)]. Furthermore, at least in the case of Mical, this Abl phosphorylation increases its NADPH consumption activity in the presence of F-actin and potentiates its F-actin disassembly/repulsive activity [Figure 3B (Yoon et al., 2017)]. PAK1 kinase also works downstream of growth factors, and its activated form binds to the Redox and CH regions of MICAL-1 and phosphorylates MICAL-1 at specific serine (S) residues (S817, S960) within the PxxP and ERM α-like domain, respectively [Figure 1A (McGarry et al., 2022)]. This phosphorylation relieves MICAL-1’s autoinhibition and also increases Rab7A and Rab10’s interaction with MICAL-1 [Figure 3A (McGarry et al., 2022)]. These results (McGarry et al., 2022) also add to the work of others (Wang et al., 2018; Zhao et al., 2019; Wang et al., 2021a; Wang et al., 2021b; Qi et al., 2021) and link Rho family GTPases to the regulation of the MICAL’s effects. MICALs have also been associated with nerve growth factor (NGF) signaling, such that it induces MICAL-2’s F-actin disassembly activity in the nucleus to promote gene transcription through the serum response factor (SRF)/MRTF-A (Lundquist et al., 2014). MICALs have also been linked to regulating the effects of other extracellular ligands/receptors, including those of TGFβ/TGFR and Ephrins/Eph, on cell migration and F-actin disassembly (Liu et al., 2019; Shi et al., 2020; Jiang et al., 2021; Pu et al., 2021).
MICALs’ localization and action in specific subcellular regions is important for their F-actin/cellular effects. Towards this end, MICALs were recently found to interact with specific myosins, which are well-known regulators of cellular behaviors. Myosins regulate cell functions in two ways: they move/hold F-actin (i.e., myosins act as force generators/mechanical tethers) and/or they move processively along F-actin (i.e., myosins act as intracellular transports) (Coluccio and Biol, 2020). A specific myosin, Myosin 15 (Myo15), physically associates, transports, and broadens MICAL’s distribution to expand and directionally orient the MICAL’s F-actin effects [Figure 3B (Rich et al., 2021)]. MICALs also interact with members of another class of myosins, MyoVa (and MyoVb) (Niu et al., 2020). Yet, interestingly, in contrast to Myo15, which expands the MICAL’s distribution/F-actin disassembly, MICAL-1 was found not to be transported by MyoVa, but to be tethered to a specific spot and derail MyoVa and its cargo as MyoVa passed through that specific site (Niu et al., 2020). Thus, myosins are involved in at least two functions as it relates to the MICALs: 1) myosins expand MICALs-mediated F-actin disassembly and cellular remodeling and 2) myosins restrict MICALs-mediated F-actin disassembly and cargo unloading. Specific myosins may also interact with specific MICALs [e.g., MyoV interacts with MICAL-1 but not with MICAL-2 and 3 (Niu et al., 2020)] and control other aspects of MICALs’ functions [e.g., another myosin, Myo9, binds and promotes the nuclear export of MICAL-2 (Zhou et al., 2020)]. Other intracellular proteins have also been found to bind and recruit MICALs to specific subcellular regions. Coronin 1C, an actin regulatory protein, facilitates the recruitment of MICAL-2 to vaccinia-induced actin tails [Figure 3B (Galloni et al., 2021)]. This type of recruitment may be specific to different MICALs since coronin 1C recruits MICAL-2 but not MICAL-1 (Galloni et al., 2021).
MICALs have been found to interact also with other proteins, including most notably CasL/Nedd9 (Suzuki et al., 2002; Evans et al., 2017; Grauzam et al., 2018; Zhao et al., 2019) and the intermediate filament protein vimentin (Suzuki et al., 2002; Grauzam et al., 2018; Semelakova et al., 2019), but the specific roles of these interactions are unclear. Cortactin’s presence on actin branches is required for the enhanced MICAL-2’s effects on the dissociation of Arp3B-containing Arp2/3 complexes and the increased rate of actin network disassembly via yet unknown mechanisms (Galloni et al., 2021). MICALs have also been functionally linked to other proteins and signaling pathways, including ERK, PP2A, and RanBPM, among others, although the roles of these proteins/signaling pathways in regulating MICALs is unclear [e.g., (Togashi et al., 2006; Deng et al., 2018; Tao et al., 2019; Wolterhoff et al., 2020)]. Moreover, in vitro studies have suggested that MICALs’ activity depends on ionic strength and pH (Zucchini et al., 2011; Grintsevich et al., 2016), including that MICAL-1 shows higher catalytic turnover with increasing pH (Zucchini et al., 2011). Such changes may regulate MICALs’ effects in vivo – particularly given that intracellular alkaline pH promotes tumor progression and growth (White et al., 2017), and H+ transporters NBCn1 and NHE1 (and thereby intracellular alkaline pH) are upregulated in specific cancers [e.g., (Flinck et al., 2018)], and increased MICAL activity is associated with specific cancers (Yoon and Terman, 2018b) (see below).
7.3 Dampening the MICAL’s effects
Factors that dampen MICALs’ functions are also likely to play prominent roles in regulating the MICAL’s effects. Yet, besides MsrB/SelR, as described above, so far these negative regulators have been poorly defined. For example, an antagonistic relationship between MICAL-1 and NDR Kinase apoptotic signaling has been identified, but NDR kinases do not negatively regulate MICAL-1’s effects (Zhou et al., 2011). Notably, micro RNAs have been found that specifically target MICALs, including to promote actin polymerization (Tao et al., 2019; Torrini et al., 2019; Han et al., 2022). Future work is likely to reveal more effectors that work in opposition to MICALs.
8 The MICAL’s physiological and pathological functions
The MICAL’s role in robustly altering actin dynamics is critical for regulating specific cellular and tissue behaviors in the cytosol (often in close proximity to the plasma membrane) and in the nucleus. Furthermore, as described below, it is important to note that while the MICAL’s role in vivo was first identified in the nervous system (Terman et al., 2002), MICALs are broadly expressed and regulate the form, function, and dysfunction of multiple different types of cells and tissues. Yet, it is also clear that much remains to be learned about how broadly the MICALs are utilized in vivo, including a full understanding of their use by nature in different tissue systems, cell types, and specific cellular events. Below we provide a general overview of what is known of the MICAL’s in vivo functions. For additional coverage including detailed specifics of the MICAL’s in vivo functions, we refer readers to (Hung and Terman, 2011; Wilson et al., 2016; Manta and Gladyshev, 2017; Vanoni, 2017; Alto and Terman, 2018; Ortegón Salas et al., 2020; Haikazian and Olson, 2022; Rouyère et al., 2022).
8.1 Physiology: Cell biology, neurobiology, musculoskeletal biology, cardiovascular biology, and more
Cells undergo rapid reorganization during division, polarity, motility, navigation, and other behaviors by robustly inducing actin remodeling (Pollard and Borisy, 2003; Blanchoin et al., 2014). MICALs and their effects on F-actin structures have now emerged as robust modulators of these behaviors – being required and sufficient to alter the form and function of multiple different cells and tissues (Figure 4). In particular, MICALs were first identified for their functions in the nervous system – being required for the guidance of neuronal axons in vivo (Terman et al., 2002). Subsequent work has added to these guidance roles, as well as defined roles for the MICALs in neuronal growth cone morphology and axon extension (Beuchle et al., 2007; Schmidt et al., 2008; Hung et al., 2010a; Morinaka et al., 2011; Hashimoto et al., 2012; Hung et al., 2013; Lundquist et al., 2014; Van Battum et al., 2014). MICALs also regulate the morphology/complexity of neuronal dendrites (Kirilly et al., 2009; Loncle and Williams, 2012; Rumpf et al., 2014; Rode et al., 2018) and are important for establishing and regulating the connection between neurons (i.e., synaptic formation, organization, and activity) (Beuchle et al., 2007; Hung et al., 2013; Van Battum et al., 2014; Orr et al., 2017; Schaukowitch et al., 2017). MICALs regulate the migration of neurons (Bron et al., 2007), the trafficking of proteins into axons (Hamdan et al., 2020), the docking and fusing of vesicles at the plasma membrane (Grigoriev et al., 2011; Van Battum et al., 2014; Bachmann-Gagescu et al., 2015), and the morphology of neuro-mechanosensory system cells (e.g., Drosophila bristle cells, which are akin to the inner ear hair cells required for hearing in mammals) (Hung et al., 2010a; Hung et al., 2013).
FIGURE 4.
Cellular behavior controlled by the MICAL’s activity contributes to multiple functions and dysfunctions in numerous tissues. Modified from (Alto and Terman, 2018).
MICALs also play important non-neuronal functions. They have been associated with regulating smooth, skeletal, and cardiac muscle organization, including being required to control F-actin organization in skeletal muscle (Beuchle et al., 2007; Schnorrer et al., 2010; Hung et al., 2013; Tao et al., 2019; Giarratana et al., 2020; Konstantinidis et al., 2020). They regulate the cardiovasculature, including in angiogenesis, vessel integrity, heart development/function, and lymphatic remodeling (Lundquist et al., 2014; Hou et al., 2015; Williams et al., 2017; Barravecchia et al., 2019; Konstantinidis et al., 2020; Erdmann et al., 2021; Francis et al., 2022). MICALs have been linked to kidney function (Aggarwal et al., 2015) and immune response (Lee et al., 2013). MICALs play important roles in other cell biological events in different cells, including specifying cell morphology [e.g., (Hung et al., 2010a; Hung et al., 2011; Giridharan et al., 2012; Hung et al., 2013; Hou et al., 2015; Wu et al., 2018)], migration [e.g., (Liu et al., 2019; Jiang et al., 2021; Pu et al., 2021)], proliferation [e.g., (Tao et al., 2019; Pu et al., 2021)], wound healing (Williams et al., 2017), membrane tubulation (Lucken-Ardjomande Hasler et al., 2020; Wang et al., 2021b), endocytosis/exocytosis/vesicle trafficking (Grigoriev et al., 2011; Bachmann-Gagescu et al., 2015; Liu et al., 2016), and cytokinesis, where they are required to control the F-actin cytoskeleton at abscission sites (Liu et al., 2016; Frémont et al., 2017; Bai et al., 2020). MICALs have also been defined for effects on cell-cell repulsion mediated by one of the largest families of cellular guidance cues, Semaphorins and their Plexin receptors (Terman et al., 2002; Schmidt et al., 2008; Hung et al., 2010a; Morinaka et al., 2011; Aggarwal et al., 2015; Hou et al., 2015; Yoo et al., 2016; Tominaga et al., 2019).
8.2 Pathology: Cancer, brain disorders, cardiovascular defects, and more
MICALs and their ability to robustly control the cytoskeleton have become increasingly linked to different pathologies, including that altered expression levels and SNPs/missense mutations for MICALs have been associated with numerous cellular dysfunctions (Figure 4) and disease. Below, we highlight a few of these studies in which functional analysis has been carried out. In particular, MICALs have been functionally linked to different types of cancers/cancer cells, including bladder (Ho et al., 2012), blood/leukemia (Corces-Zimmerman et al., 2014), brain/glioma (Pu et al., 2021), breast (Deng et al., 2016; Mariotti et al., 2016; Wang et al., 2017; Yoon et al., 2017; Deng et al., 2018; Tominaga et al., 2019; McGarry et al., 2021), colorectal (Gu et al., 2022), gastric (Mariotti et al., 2016; Zhao et al., 2019; Wang et al., 2021a; Qi et al., 2021), lung-related (Mariotti et al., 2016; Liu et al., 2019; Zhou et al., 2020; Wang et al., 2021b), pancreatic (Cai et al., 2022), prostate (Ashida et al., 2006), renal (Mariotti et al., 2016), and skin/mucous membrane (Loria et al., 2015; Grauzam et al., 2018; Zhang et al., 2022). Notably, the MICAL’s involvement in at least some of these cancers/cancer cells has been linked to its actions on F-actin. The MICALs have also been prominently linked to neurological and mental health disorders, including neurodegeneration (Prifti et al., 2022), spinal cord injury (Pasterkamp et al., 2006; Xu et al., 2021), and epilepsy (Dazzo et al., 2018). Most notably, heterozygous missense mutations in the Redox domain (Gly150Ser) and C-terminal ERM α-like domain (Ala1065fs, deletion of last three amino acids, and addition of 59 extra residues) generate changes in MICAL-1 that have been linked to Autosomal-Dominant Lateral Temporal Epilepsy (Dazzo et al., 2018). In particular, it is thought that these dominant mutants generate constitutively active MICAL-1 and its effects on cells are consistent with increased F-actin cytoskeletal disassembly (Dazzo et al., 2018). Since MICAL-1 is also expressed in other tissues besides the brain, it is interesting to consider if these heterozygous missense mutations lead to comorbid pathologies in other tissues/behaviors, or whether the brain may be particularly susceptible to having these heterozygous missense MICAL-1 alleles (i.e., only one normal copy of MICAL-1 present). MICALs have been functionally linked to cardiovascular abnormalities, including heart arrhythmias, pathologic responses to cardiac stress, myocardial hypertrophy, endothelial and blood-brain barrier (BBB) permeability, and contributing to effects seen with mutations in cardiac actin (Hou et al., 2015; Konstantinidis et al., 2020; Erdmann et al., 2021; Zhao et al., 2021). Notably, the MICAL’s involvement in at least some of these cardiovascular defects has been linked to its actions on F-actin. MICALs have also been functionally linked to renal disease/diabetic nephropathy through effects on the F-actin cytoskeleton (Aggarwal et al., 2015). Less well-defined functional links between MICALs and muscular dystrophy (Marotta et al., 2009), fibrosis (Zhao et al., 2020; Jiang et al., 2021), autoimmune disorders (Johar et al., 2015), skin aging (Han et al., 2022), and viral infection (Shapira et al., 2009; Mitchell et al., 2013) have been observed. Lastly, as described above, the sites that the MICALs modify on actin (Met44 and/or Met47) have been linked to different diseases, including nemaline myopathy, CAP myopathy, aortic aneurisms, hypertrophic cardiomyopathy, intestinal hypoperistalsis, Baraitser–Winter cerebrofrontofacial syndrome, and ductus arteriosus (Laing et al., 2009; Hung et al., 2010b; Hoffjan et al., 2011; Zou et al., 2013; Regalado et al., 2014; Wangler et al., 2014; Yates et al., 2017; Zhang et al., 2019). It is also worth considering that some of the defects associated with mutations in the family of enzymes that reverse the MICAL’s effects, MsrBs/SelR’s, may result from increased effects of the MICALs on F-actin [see (Tarrago et al., 2022) for coverage of those defects].
9 Summary and conclusion
The MICAL family of proteins was discovered a little over 20 years ago, and with the uncovering of their enzymatic domain and activity, specific targeting of F-actin as a substrate, and their role in numerous cellular and tissues systems, they have now matured from proteins of unknown function to ones of significant interest in biomedical research that are being pursued by multiple laboratories. Yet, much remains to be learned about the MICALs and the reversible Redox-driven mechanism they use to regulate F-actin dynamics. We need to further define MICALs’ interactions with F-actin and 1) their effects on different F-actin networks, 2) their roles in different cells, tissues, and biological events, 3) their ability to interact with and regulate the effects of classical actin regulatory and signaling proteins, and 4) their interactions with proteins that may dampen their effects. Numerous studies have revealed altered expression levels and SNPs/missense mutations for MICALs in diverse diseases, but our understanding of these changes (including if, how, and the molecular/cellular basis for the MICAL’s possible involvement in these pathologies) is still poor and in need of further exploration. Thus, future studies should also focus on these areas of critical biomedical importance. MICALs have now emerged as a phylogenetically-conserved family of proteins with surprising functions, unexpected mechanisms of action, and crucial in vivo importance. The continued study of these biomedically significant proteins is likely to yield further surprising and unexpected discoveries.
Acknowledgments
We thank ER and JT lab members for discussions.
Funding Statement
Supported by grants from the NIH (NS073968) to JRT and (GM077190) to ER.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Aggarwal P. K., Veron D., Thomas D. B., Siegel D., Moeckel G., Kashgarian M. (2015). Semaphorin3a promotes advanced diabetic nephropathy. Diabetes 64 (5), 1743–1759. 10.2337/db14-0719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alqassim S. S., Urquiza M., Borgnia E., Nagib M., Amzel L. M., Bianchet M. A. (2016). Modulation of MICAL monooxygenase activity by its calponin homology domain: Structural and mechanistic insights. Sci. Rep. 6, 22176. 10.1038/srep22176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto L. T., Terman J. R. (2018). MICALs. Curr. Biol. 28 (9), R538–R541. 10.1016/j.cub.2018.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto L. T., Terman J. R. (2017). Semaphorins and their signaling mechanisms. Methods Mol. Biol. 1493, 1–25. 10.1007/978-1-4939-6448-2_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianantoandro E., Pollard T. D. (2006). Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24 (1), 13–23. 10.1016/j.molcel.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Ashida S., Furihata M., Katagiri T., Tamura K., Anazawa Y., Yoshioka H. (2006). Expression of novel molecules, MICAL2-PV (MICAL2 prostate cancer variants), increases with high Gleason score and prostate cancer progression. Clin. Cancer Res. 12 (9), 2767–2773. 10.1158/1078-0432.CCR-05-1995 [DOI] [PubMed] [Google Scholar]
- Ayoob J. C., Terman J. R., Kolodkin A. L. (2006). Drosophila Plexin B is a Sema-2a receptor required for axon guidance. Development 133 (11), 2125–2135. 10.1242/dev.02380 [DOI] [PubMed] [Google Scholar]
- Bachmann-Gagescu R., Dona M., Hetterschijt L., Tonnaer E., Peters T., de Vrieze E., et al. (2015). The ciliopathy protein CC2D2A associates with NINL and functions in RAB8-MICAL3-regulated vesicle trafficking. PLoS Genet. 11 (10), e1005575. 10.1371/journal.pgen.1005575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J., Wioland H., Advedissian T., Cuvelier F., Romet-Lemonne G., Echard A. (2020). Actin reduction by MsrB2 is a key component of the cytokinetic abscission checkpoint and prevents tetraploidy. Proc. Natl. Acad. Sci. U. S. A. 117 (8), 4169–4179. 10.1073/pnas.1911629117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barravecchia I., Mariotti S., Pucci A., Scebba F., De Cesari C., Bicciato S., et al. (2019). MICAL2 is expressed in cancer associated neo-angiogenic capillary endothelia and it is required for endothelial cell viability, motility and VEGF response. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (9), 2111–2124. 10.1016/j.bbadis.2019.04.008 [DOI] [PubMed] [Google Scholar]
- Beuchle D., Schwarz H., Langegger M., Koch I., Aberle H. (2007). Drosophila MICAL regulates myofilament organization and synaptic structure. Mech. Dev. 124 (5), 390–406. 10.1016/j.mod.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Blanchoin L., Boujemaa-Paterski R., Sykes C., Plastino J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 94 (1), 235–263. 10.1152/physrev.00018.2013 [DOI] [PubMed] [Google Scholar]
- Bobkov A. A., Muhlrad A., Pavlov D. A., Kokabi K., Yilmaz A., Reisler E., et al. (2006). Cooperative effects of cofilin (ADF) on actin structure suggest allosteric mechanism of cofilin function. J. Mol. Biol. 356 (2), 325–334. 10.1016/j.jmb.2005.11.072 [DOI] [PubMed] [Google Scholar]
- Bobkov A. A., Muhlrad A., Shvetsov A., Benchaar S., Scoville D., Almo S. C. (2004). Cofilin (ADF) affects lateral contacts in F-actin. J. Mol. Biol. 337 (1), 93–104. 10.1016/j.jmb.2004.01.014 [DOI] [PubMed] [Google Scholar]
- Breitsprecher D., Kiesewetter A. K., Linkner J., Vinzenz M., Stradal T. E., Small J. V., et al. (2011). Molecular mechanism of Ena/VASP-mediated actin-filament elongation. Embo J. 30 (3), 456–467. 10.1038/emboj.2010.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R., Vermeren M., Kokot N., Andrews W., Little G. E., Mitchell K. J., et al. (2007). Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev. 2, 21. 10.1186/1749-8104-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budayeva H. G., Sengupta-Ghosh A., Phu L., Moffat J. G., Ayalon G., Kirkpatrick D. S. (2022). Phosphoproteome profiling of the receptor tyrosine kinase MuSK identifies tyrosine phosphorylation of rab GTPases. Mol. Cell Proteomics 21 (4), 100221. 10.1016/j.mcpro.2022.100221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K., Deng L., Zheng D., Li L., He Z., Yu C., et al. (2022). MICAL1 facilitates pancreatic cancer proliferation, migration, and invasion by activating WNT/β-catenin pathway. J. Transl. Med. 20 (1), 528. 10.1186/s12967-022-03749-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra E. S., Higgs H. N. (2006). INF2 Is a WASP homology 2 motif-containing formin that severs actin filaments and accelerates both polymerization and depolymerization. J. Biol. Chem. 281 (36), 26754–26767. 10.1074/jbc.M604666200 [DOI] [PubMed] [Google Scholar]
- Chin S. M., Jansen S., Goode B. L. (2016). TIRF microscopy analysis of human Cof1, Cof2, and ADF effects on actin filament severing and turnover. J. Mol. Biol. 428 (8), 1604–1616. 10.1016/j.jmb.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. Z., Pollard T. D. (2020). Cryo-electron microscopy structures of pyrene-labeled ADP-Pi- and ADP-actin filaments. Nat. Commun. 11 (1), 5897. 10.1038/s41467-020-19762-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. Z., Pollard T. D. (2019). Mechanism of actin polymerization revealed by cryo-EM structures of actin filaments with three different bound nucleotides. Proc. Natl. Acad. Sci. U. S. A. 116 (10), 4265–4274. 10.1073/pnas.1807028115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio L. M. (2020). in Myosins: A superfamily of molecular motors Editor Biol A. E. M. 2nd ed (Cham, Switzerland: Springer Nature; ). [Google Scholar]
- Corces-Zimmerman M. R., Hong W. J., Weissman I. L., Medeiros B. C., Majeti R. (2014). Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. U. S. A. 111 (7), 2548–2553. 10.1073/pnas.1324297111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche N. (2018). Mechanisms of formin-mediated actin assembly and dynamics. Biophys. Rev. 10 (6), 1553–1569. 10.1007/s12551-018-0468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche N., Pollard T. D. (2013). Interaction of profilin with the barbed end of actin filaments. Biochemistry 52 (37), 6456–6466. 10.1021/bi400682n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I., Rossi R., Giustarini D., Gagliano N., Di Simplicio P., Colombo R., et al. (2002). Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic. Biol. Med. 32 (9), 927–937. 10.1016/s0891-5849(02)00799-2 [DOI] [PubMed] [Google Scholar]
- Das S., Ge P., Oztug Durer Z. A., Grintsevich E. E., Zhou Z. H., Reisler E. (2020). D-Loop dynamics and near-Atomic-Resolution cryo-EM structure of phalloidin-bound F-actin. Structure 28 (5), 586–593. 10.1016/j.str.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Zhang Z., Kalvakota S., Soto R., Jr., Phillips M. L., Terman J. R., et al. (2022). Parallel actin monomers in the 8S complex of actin-INF2. J. Biomol. Struct. Dyn. 28, 1–10. 10.1080/07391102.2022.2050947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazzo E., Rehberg K., Michelucci R., Passarelli D., Boniver C., Vianello Dri V., et al. (2018). Mutations in MICAL1 cause autosomal-dominant lateral temporal epilepsy. Ann. Neurol. 83 (3), 483–493. 10.1002/ana.25167 [DOI] [PubMed] [Google Scholar]
- Deng W., Wang Y., Gu L., Duan B., Cui J., Zhang Y., et al. (2016). MICAL1 controls cell invasive phenotype via regulating oxidative stress in breast cancer cells. BMC cancer 16, 489. 10.1186/s12885-016-2553-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Wang Y., Zhao S., Zhang Y., Chen Y., Zhao X., et al. (2018). MICAL1 facilitates breast cancer cell proliferation via ROS-sensitive ERK/cyclin D pathway. J. Cell Mol. Med. 22 (6), 3108–3118. 10.1111/jcmm.13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R., Holmes K. C. (2011). Actin structure and function. Annu. Rev. Biophys. 40, 169–186. 10.1146/annurev-biophys-042910-155359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durer Z. A., Kudryashov D. S., Sawaya M. R., Altenbach C., Hubbell W., Reisler E. (2012). Structural states and dynamics of the D-loop in actin. Biophys. J. 103 (5), 930–939. 10.1016/j.bpj.2012.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entsch B., van Berkel W. J. (1995). Structure and mechanism of para-hydroxybenzoate hydroxylase. Faseb J. 9 (7), 476–483. 10.1096/fasebj.9.7.7737455 [DOI] [PubMed] [Google Scholar]
- Erdmann C., Hassoun R., Schmitt S., Kikuti C., Houdusse A., Mazur A. J., et al. (2021). Integration of cardiac actin mutants causing hypertrophic (p.A295S) and dilated cardiomyopathy (p.R312H and p.E361G) into cellular structures. Antioxidants (Basel). 10 (7), 1082. 10.3390/antiox10071082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito A., Ventura V., Petoukhov M. V., Rai A., Svergun D. I., Vanoni M. A. (2019). Human MICAL1: Activation by the small GTPase Rab8 and small-angle X-ray scattering studies on the oligomerization state of MICAL1 and its complex with Rab8. Protein Sci. 28 (1), 150–166. 10.1002/pro.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans I. M., Kennedy S. A., Paliashvili K., Santra T., Yamaji M., Lovering R. C., et al. (2017). Vascular endothelial growth factor (VEGF) promotes assembly of the p130Cas interactome to drive endothelial chemotactic signaling and angiogenesis. Mol. Cell Proteomics 16 (2), 168–180. 10.1074/mcp.M116.064428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer T., Xie S., Naslavsky N., Stockli J., James D. E., Caplan S. (2021). Defining the protein and lipid constituents of tubular recycling endosomes. J. Biol. Chem. 296, 100190. 10.1074/jbc.RA120.015992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J., Weide T., Barnekow A. (2005). The MICAL proteins and rab1: A possible link to the cytoskeleton? Biochem. Biophys. Res. Commun. 328 (2), 415–423. 10.1016/j.bbrc.2004.12.182 [DOI] [PubMed] [Google Scholar]
- Flinck M., Kramer S. H., Schnipper J., Andersen A. P., Pedersen S. F. (2018). The acid-base transport proteins NHE1 and NBCn1 regulate cell cycle progression in human breast cancer cells. Cell Cycle 17 (9), 1056–1067. 10.1080/15384101.2018.1464850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis C. R., Kincross H., Kushner E. J. (2022). Rab35 governs apicobasal polarity through regulation of actin dynamics during sprouting angiogenesis. Nat. Commun. 13 (1), 5276. 10.1038/s41467-022-32853-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frémont S., Hammich H., Bai J., Wioland H., Klinkert K., Rocancourt M., et al. (2017). Oxidation of F-actin controls the terminal steps of cytokinesis. Nat. Commun. 8, 14528. 10.1038/ncomms14528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Kanno E., Ishibashi K., Itoh T. (2008). Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol. Cell Proteomics 7 (6), 1031–1042. 10.1074/mcp.M700569-MCP200 [DOI] [PubMed] [Google Scholar]
- Galloni C., Carra D., Abella J. V. G., Kjaer S., Singaravelu P., Barry D. J., et al. (2021). MICAL2 enhances branched actin network disassembly by oxidizing Arp3B-containing Arp2/3 complexes. J. Cell Biol. 220 (8), e202102043. 10.1083/jcb.202102043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau A. M., Fregoso F. E., Simanov G., Dominguez R. (2022). Nucleation, stabilization, and disassembly of branched actin networks. Trends Cell Biol. 32 (5), 421–432. 10.1016/j.tcb.2021.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giarratana N., Conti F., La Rovere R., Gijsbers R., Carai P., Duelen R. (2020). MICAL2 is essential for myogenic lineage commitment. Cell Death Dis. 11 (8), 654. 10.1038/s41419-020-02886-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham A. K., Bertram J., Begum F., Munro S. (2019). In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation. Elife 8, e45916. 10.7554/eLife.45916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridharan S. S., Caplan S. (2014). MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid. Redox Signal 20 (13), 2059–2073. 10.1089/ars.2013.5487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridharan S. S., Rohn J. L., Naslavsky N., Caplan S. (2012). Differential regulation of actin microfilaments by human MICAL proteins. J. Cell Sci. 125 (3), 614–624. 10.1242/jcs.089367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode B. L., Eck M. J. (2007). Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 76, 593–627. 10.1146/annurev.biochem.75.103004.142647 [DOI] [PubMed] [Google Scholar]
- Grauzam S., Brock A. M., Holmes C. O., Tiedeken J. A., Boniface S. G., Pierson B. N. (2018). NEDD9 stimulated MMP9 secretion is required for invadopodia formation in oral squamous cell carcinoma. Oncotarget 9 (39), 25503–25516. 10.18632/oncotarget.25347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I., Yu K. L., Martinez-Sanchez E., Serra-Marques A., Smal I., Meijering E., et al. (2011). Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr. Biol. 21 (11), 967–974. 10.1016/j.cub.2011.04.030 [DOI] [PubMed] [Google Scholar]
- Grintsevich E. E., Ahmed G., Ginosyan A. A., Wu H., Rich S. K., Reisler E., et al. (2021). Profilin and Mical combine to impair F-actin assembly and promote disassembly and remodeling. Nat. Commun. 12 (1), 5542. 10.1038/s41467-021-25781-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grintsevich E. E., Ge P., Sawaya M. R., Yesilyurt H. G., Terman J. R., Zhou Z. H., et al. (2017). Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 8 (1), 2183. 10.1038/s41467-017-02357-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grintsevich E. E., Yesilyurt H. G., Rich S. K., Hung R. J., Terman J. R., Reisler E. (2016). F-actin dismantling through a redox-driven synergy between Mical and cofilin. Nat. Cell Biol. 18 (8), 876–885. 10.1038/ncb3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Li Y., Cui X., Cao H., Hou Z., Ti Y., et al. (2022). MICAL1 inhibits colorectal cancer cell migration and proliferation by regulating the EGR1/β-catenin signaling pathway. Biochem. Pharmacol. 195, 114870. 10.1016/j.bcp.2021.114870 [DOI] [PubMed] [Google Scholar]
- Guan J. Q., Almo S. C., Reisler E., Chance M. R. (2003). Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-Actin solution structure is divalent cation dependent. Biochemistry 42, 11992–12000. 10.1021/bi034914k [DOI] [PubMed] [Google Scholar]
- Guan J. Q., Takamoto K., Almo S. C., Reisler E., Chance M. R. (2005). Structure and dynamics of the actin filament. Biochemistry 44 (9), 3166–3175. 10.1021/bi048021j [DOI] [PubMed] [Google Scholar]
- Gupta C. M., Ambaru B., Bajaj R. (2020). Emerging functions of actins and actin binding proteins in trypanosomatids. Front. Cell Dev. Biol. 8, 587685. 10.3389/fcell.2020.587685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel P. S., Ge P., Grintsevich E. E., Shu R., Blanchoin L., Zhou Z. H., et al. (2014). INF2-mediated severing through actin filament encirclement and disruption. Curr. Biol. 24 (2), 156–164. 10.1016/j.cub.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haikazian S., Olson M. F. (2022). MICAL1 monooxygenase in autosomal dominant lateral temporal epilepsy: Role in cytoskeletal regulation and relation to cancer. Genes (Basel) 13 (5), 715. 10.3390/genes13050715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan H., Lim B. C., Torii T., Joshi A., Konning M., Smith C., et al. (2020). Mapping axon initial segment structure and function by multiplexed proximity biotinylation. Nat. Commun. 11 (1), 100. 10.1038/s41467-019-13658-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Wu T., Jin J., Li Z., Cheng W., Dai X. (2022). Exosome-like nanovesicles derived from Phellinus linteus inhibit Mical2 expression through cross-kingdom regulation and inhibit ultraviolet-induced skin aging. J. Nanobiotechnology 20 (1), 455. 10.1186/s12951-022-01657-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Muramatsu K., Kunii M., Yoshimura S., Yamada M., Sato T., et al. (2012). Uncovering genes required for neuronal morphology by morphology-based gene trap screening with a revertible retrovirus vector. FASEB J. 26 (11), 4662–4674. 10.1096/fj.12-207530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Tatsumi H., Sokabe M. (2011). Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J. Cell Biol. 195 (5), 721–727. 10.1083/jcb.201102039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegsted A., Yingling C. V., Pruyne D. (2017). Inverted formins: A subfamily of atypical formins. Cytoskelet. Hob. 74 (11), 405–419. 10.1002/cm.21409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs H. N. (2005). Formin proteins: A domain-based approach. Trends Biochem. Sci. 30 (6), 342–353. 10.1016/j.tibs.2005.04.014 [DOI] [PubMed] [Google Scholar]
- Ho J. R., Chapeaublanc E., Kirkwood L., Nicolle R., Benhamou S., Lebret T., et al. (2012). Deregulation of Rab and Rab effector genes in bladder cancer. PLoS One 7 (6), e39469. 10.1371/journal.pone.0039469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffjan S., Waldmuller S., Blankenfeldt W., Kotting J., Gehle P., Binner P., et al. (2011). Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 19, 520–524. 10.1038/ejhg.2010.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook S. C., Chadt A., Heesom K. J., Kishida S., Al-Hasani H., Tavare J. M., et al. (2020). TBC1D1 interacting proteins, VPS13A and VPS13C, regulate GLUT4 homeostasis in C2C12 myotubes. Sci. Rep. 10, 17953. 10.1038/s41598-020-74661-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S. T., Nilchi L., Li X., Gangaraju S., Jiang S. X., Aylsworth A., et al. (2015). Semaphorin3A elevates vascular permeability and contributes to cerebral ischemia-induced brain damage. Sci. Rep. 5, 7890. 10.1038/srep07890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huehn A. R., Bibeau J. P., Schramm A. C., Cao W., De La Cruz E. M., Sindelar C. V. (2020). Structures of cofilin-induced structural changes reveal local and asymmetric perturbations of actin filaments. Proc. Natl. Acad. Sci. U. S. A. 117 (3), 1478–1484. 10.1073/pnas.1915987117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers M. M., Montersino S., Westphal A. H., Tischler D., van Berkel W. J. (2014). Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17. 10.1016/j.abb.2013.12.005 [DOI] [PubMed] [Google Scholar]
- Hung R. J., Pak C. W., Terman J. R. (2011). Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334 (6063), 1710–1713. 10.1126/science.1211956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R. J., Spaeth C. S., Yesilyurt H. G., Terman J. R. (2013). SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15 (12), 1445–1454. 10.1038/ncb2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R. J., Terman J. R. (2011). Extracellular inhibitors, repellents, and semaphorin/plexin/MICAL-mediated actin filament disassembly. Cytoskelet. Hob. 68 (8), 415–433. 10.1002/cm.20527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R. J., Yazdani U., Yoon J., Wu H., Yang T., Gupta N., et al. (2010). Mical links semaphorins to F-actin disassembly. Nature 463 (7282), 823–827. 10.1038/nature08724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R. M., Yoon G., Hawkins C. E., Halliday W., Biggar D., Vajsar J. (2010). Cap myopathy caused by a mutation of the skeletal alpha-actin gene ACTA1. Neuromuscul. Disord. 20 (4), 238–240. 10.1016/j.nmd.2010.01.011 [DOI] [PubMed] [Google Scholar]
- Ishida H., Borman M. A., Ostrander J., Vogel H. J., MacDonald J. A. (2008). Solution structure of the calponin homology (CH) domain from the smoothelin-like 1 protein: A unique apocalmodulin-binding mode and the possible role of the C-terminal type-2 CH-domain in smooth muscle relaxation. J. Biol. Chem. 283, 20569–20578. 10.1074/jbc.M800627200 [DOI] [PubMed] [Google Scholar]
- Jégou A., Niedermayer T., Orbán J., Didry D., Lipowsky R., Carlier M. F., et al. (2011). Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biol. 9 (9), e1001161. 10.1371/journal.pbio.1001161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F., Cao J., Kong R., Fang L., Wang B., Zhang S., et al. (2021). MICAL2 regulates myofibroblasts differentiation in epidural fibrosis via SRF/MRTF-A signaling pathway. Life Sci. 269, 119045. 10.1016/j.lfs.2021.119045 [DOI] [PubMed] [Google Scholar]
- Johar A. S., Mastronardi C., Rojas-Villarraga A., Patel H. R., Chuah A., Peng K., et al. (2015). Novel and rare functional genomic variants in multiple autoimmune syndrome and Sjogren's syndrome. J. Transl. Med. 13, 173. 10.1186/s12967-015-0525-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadzik R. S., Homa K. E., Kovar D. R. (2020). F-actin cytoskeleton network self-organization through competition and cooperation. Annu. Rev. Cell Dev. Biol. 36, 35–60. 10.1146/annurev-cellbio-032320-094706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashina A. S. (2020). Regulation of actin isoforms in cellular and developmental processes. Semin. Cell Dev. Biol. 102, 113–121. 10.1016/j.semcdb.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y., Gladyshev V. N. (2007). Methionine sulfoxide reductases: Selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem. J. 407 (3), 321–329. 10.1042/BJ20070929 [DOI] [PubMed] [Google Scholar]
- Kim J., Lee H., Roh Y. J., Kim H. U., Shin D., Kim S., et al. (2020). Structural and kinetic insights into flavin-containing monooxygenase and calponin-homology domains in human MICAL3. IUCrJ 7 (1), 90–99. 10.1107/S2052252519015409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinosian H. J., Selden L. A., Gershman L. C., Estes J. E. (2002). Actin filament barbed end elongation with nonmuscle MgATP-actin and MgADP-actin in the presence of profilin. Biochemistry 41 (21), 6734–6743. 10.1021/bi016083t [DOI] [PubMed] [Google Scholar]
- Kirilly D., Gu Y., Huang Y., Wu Z., Bashirullah A., Low B. C., et al. (2009). A genetic pathway composed of Sox14 and Mical governs severing of dendrites during pruning. Nat. Neurosci. 12 (12), 1497–1505. 10.1038/nn.2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis K., Bezzerides V. J., Lai L., Isbell H. M., Wei A. C., Wu Y., et al. (2020). MICAL1 constrains cardiac stress responses and protects against disease by oxidizing CaMKII. J. Clin. Invest. 130 (9), 4663–4678. 10.1172/JCI133181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar D. R., Harris E. S., Mahaffy R., Higgs H. N., Pollard T. D. (2006). Control of the assembly of ATP- and ADPactin by formins and profilin. Cell 124 (2), 423–435. 10.1016/j.cell.2005.11.038 [DOI] [PubMed] [Google Scholar]
- Laing N. G., Dye D. E., Wallgren-Pettersson C., Richard G., Monnier N., Lillis S., et al. (2009). Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum. Mutat. 30 (9), 1267–1277. 10.1002/humu.21059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen P., Kotila T., Jegou A., Romet-Lemonne G. (2022). Biochemical and mechanical regulation of actin dynamics. Nat. Rev. Mol. Cell Biol. 23 (12), 836–852. 10.1038/s41580-022-00508-4 [DOI] [PubMed] [Google Scholar]
- Lee B. C., Péterfi Z., Hoffmann F. W., Moore R. E., Kaya A., Avanesov A., et al. (2013). MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell 51, 397–404. 10.1016/j.molcel.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Borcherding N., Shao P., Cao H., Zhang W. (2019). Qi HH. Identification of novel TGF-beta regulated genes with pro-migratory roles. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (12), 165537. 10.1016/j.bbadis.2019.165537 [DOI] [PubMed] [Google Scholar]
- Liu Q., Liu F., Yu K. L., Tas R., Grigoriev I., Remmelzwaal S., et al. (2016). MICAL3 flavoprotein monooxygenase forms a complex with centralspindlin and regulates cytokinesis. J. Biol. Chem. 291 (39), 20617–20629. 10.1074/jbc.M116.748186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Remmelzwaal S., Heck A. J. R., Akhmanova A., Liu F. (2017). Facilitating identification of minimal protein binding domains by cross-linking mass spectrometry. Sci. Rep. 7 (1), 13453. 10.1038/s41598-017-13663-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncle N., Williams D. W. (2012). An interaction screen identifies headcase as a regulator of large-scale pruning. J. Neurosci. 32 (48), 17086–17096. 10.1523/JNEUROSCI.1391-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria R., Bon G., Perotti V., Gallo E., Bersani I., Baldassari P., et al. (2015). Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget 6 (5), 2779–2793. 10.18632/oncotarget.2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucken-Ardjomande Hasler S., Vallis Y., Pasche M., McMahon H. T. (2020). GRAF2, WDR44, and MICAL1 mediate Rab8/10/11-dependent export of E-cadherin, MMP14, and CFTR DeltaF508. J. Cell Biol. 219, e201811014. 10.1083/jcb.201811014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist M. R., Storaska A. J., Liu T. C., Larsen S. D., Evans T., Neubig R. R., et al. (2014). Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell 156 (3), 563–576. 10.1016/j.cell.2013.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliga Z., Junqueira M., Toyoda Y., Ettinger A., Mora-Bermudez F., Klemm R. W., et al. (2013). A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat. Cell Biol. 15 (3), 325–334. 10.1038/ncb2689 [DOI] [PubMed] [Google Scholar]
- Malinova T. S., Angulo-Urarte A., Nuchel J., Tauber M., van der Stoel M. M., Janssen V., et al. (2021). A junctional PACSIN2/EHD4/MICAL-L1 complex coordinates VE-cadherin trafficking for endothelial migration and angiogenesis. Nat. Commun. 12 (1), 2610. 10.1038/s41467-021-22873-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manta B., Gladyshev V. N. (2017). Regulated methionine oxidation by monooxygenases. Free Radic. Biol. Med. 109, 141–155. 10.1016/j.freeradbiomed.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti S., Barravecchia I., Vindigni C., Pucci A., Balsamo M., Libro R., et al. (2016). MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget 7 (2), 1808–1825. 10.18632/oncotarget.6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta M., Ruiz-Roig C., Sarria Y., Peiro J. L., Nunez F., Ceron J., et al. (2009). Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol. Genomics 37 (2), 119–132. 10.1152/physiolgenomics.90370.2008 [DOI] [PubMed] [Google Scholar]
- Matthews J. M., Bhati M., Lehtomaki E., Mansfield R. E., Cubeddu L., Mackay J. P. (2009). It takes two to tango: The structure and function of LIM, RING, PHD and MYND domains. Curr. Pharm. Des. 15 (31), 3681–3696. 10.2174/138161209789271861 [DOI] [PubMed] [Google Scholar]
- McDonald C. A., Liu Y. Y., Palfey B. A. (2013). Actin stimulates reduction of the MICAL-2 monooxygenase domain. Biochemistry 52 (35), 6076–6084. 10.1021/bi4008462 [DOI] [PubMed] [Google Scholar]
- McGarry D. J., Armstrong G., Castino G., Mason S., Clark W., Shaw R. (2021). MICAL1 regulates actin cytoskeleton organization, directional cell migration and the growth of human breast cancer cells as orthotopic xenograft tumours. Cancer Lett. 519, 226–236. 10.1016/j.canlet.2021.07.039 [DOI] [PubMed] [Google Scholar]
- McGarry D. J., Castino G., Lilla S., Carnet A., Kelly L., Micovic K., et al. (2022). MICAL1 activation by PAK1 mediates actin filament disassembly. Cell Rep. 41 (1), 111442. 10.1016/j.celrep.2022.111442 [DOI] [PubMed] [Google Scholar]
- Merino F., Pospich S., Raunser S. (2020). Towards a structural understanding of the remodeling of the actin cytoskeleton. Semin. Cell Dev. Biol. 102, 51–64. 10.1016/j.semcdb.2019.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell H. D., Eisfeld A. J., Sims A. C., McDermott J. E., Matzke M. M., Webb-Robertson B. J. (2013). A network integration approach to predict conserved regulators related to pathogenicity of influenza and SARS-CoV respiratory viruses. PLoS One 8 (7), e69374. 10.1371/journal.pone.0069374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K., Sakane A., Tsuchiya Y., Sagawa I., Tomida Y., Kasahara J. (2019). Actin cytoskeletal reorganization function of JRAB/MICAL-L2 is fine-tuned by intramolecular interaction between first LIM zinc finger and C-terminal coiled-coil domains. Sci. Rep. 9 (1), 12794. 10.1038/s41598-019-49232-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H., Tanaka K., Yamashiro S., Narita A., Watanabe N. (2018). Helical rotation of the diaphanous-related formin mDia1 generates actin filaments resistant to cofilin. Proc. Natl. Acad. Sci. U. S. A. 115, E5000–E5007. 10.1073/pnas.1803415115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka A., Yamada M., Itofusa R., Funato Y., Yoshimura Y., Nakamura F., et al. (2011). Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal 4 (170), ra26. 10.1126/scisignal.2001127 [DOI] [PubMed] [Google Scholar]
- Muhlrad A., Pavlov D., Peyser Y. M., Reisler E. (2006). Inorganic phosphate regulates the binding of cofilin to actin filaments. FEBS J. 273 (7), 1488–1496. 10.1111/j.1742-4658.2006.05169.x [DOI] [PubMed] [Google Scholar]
- Nadella M., Bianchet M. A., Gabelli S. B., Barrila J., Amzel L. M. (2005). Structure and activity of the axon guidance protein MICAL. Proc. Natl. Acad. Sci. U. S. A. 102 (46), 16830–16835. 10.1073/pnas.0504838102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez-Ortiz H. Y., Nolen B. J. (2022). Unconcerted conformational changes in Arp2/3 complex integrate multiple activating signals to assemble functional actin networks. Curr. Biol. 32 (5), 975–987. 10.1016/j.cub.2022.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo K. X., Umeki N., Kijima S. T., Kodera N., Ueno H., Furutani-Umezu N., et al. (2016). Allosteric regulation by cooperative conformational changes of actin filaments drives mutually exclusive binding with cofilin and myosin. Sci. Rep. 6, 35449. 10.1038/srep35449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu F., Sun K., Wei W., Yu C., Wei Z. (2020). F-actin disassembly factor MICAL1 binding to Myosin Va mediates cargo unloading during cytokinesis. Sci. Adv. 6 (45), eabb1307. 10.1126/sciadv.abb1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr B. O., Fetter R. D., Davis G. W. (2017). Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550 (7674), 109–113. 10.1038/nature24017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortegón Salas C., Schneider K., Lillig C. H., Gellert M. (2020). Signal-regulated oxidation of proteins via MICAL. Biochem. Soc. Trans. 48 (2), 613–620. 10.1042/BST20190866 [DOI] [PubMed] [Google Scholar]
- Oztug Durer Z. A., Diraviyam K., Sept D., Kudryashov D. S., Reisler E. (2010). F-Actin structure destabilization and DNase I binding loop: Fluctuations mutational cross-linking and electron microscopy analysis of loop states and effects on F-actin. J. Mol. Biol. 395 (3), 544–557. 10.1016/j.jmb.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoe H. G., Wang Y., Zhang X. (2015). Structural mechanisms of plexin signaling. Prog. Biophys. Mol. Biol. 118, 161–168. 10.1016/j.pbiomolbio.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp R. J., Dai H. N., Terman J. R., Wahlin K. J., Kim B., Bregman B. S., et al. (2006). MICAL flavoprotein monooxygenases: Expression during neural development and following spinal cord injuries in the rat. Mol. Cell Neurosci. 31 (1), 52–69. 10.1016/j.mcn.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J., Chorev D. S., Geiger B., Cossart P. (2017). The diverse family of arp2/3 complexes. Trends Cell Biol. 27 (2), 93–100. 10.1016/j.tcb.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T. D. (2016). Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 8 (8), a018226. 10.1101/cshperspect.a018226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T. D., Borisy G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. 10.1016/s0092-8674(03)00120-x [DOI] [PubMed] [Google Scholar]
- Pollard T. D. (2007). Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 36, 451–477. 10.1146/annurev.biophys.35.040405.101936 [DOI] [PubMed] [Google Scholar]
- Prifti E., Tsakiri E. N., Vourkou E., Stamatakis G., Samiotaki M., Skoulakis E. M. C., et al. (2022). Mical modulates Tau toxicity via cysteine oxidation in vivo . Acta Neuropathol. Commun. 10 (1), 44. 10.1186/s40478-022-01348-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu B., Zhang X., Yan T., Li Y., Liu B., Jian Z. (2021). MICAL2 promotes proliferation and migration of glioblastoma cells through TGF-beta/p-smad2/EMT-like signaling pathway. Front. Oncol. 11, 735180. 10.3389/fonc.2021.735180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C., Min P., Wang Q., Wang Y., Song Y., Zhang Y., et al. (2021). MICAL2 contributes to gastric cancer cell proliferation by promoting YAP dephosphorylation and nuclear translocation. Oxid. Med. Cell Longev. 2021, 9955717. 10.1155/2021/9955717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A., Bleimling N., Vetter I. R., Goody R. S. (2020). The mechanism of activation of the actin binding protein EHBP1 by Rab8 family members. Nat. Commun. 11 (1), 4187. 10.1038/s41467-020-17792-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A., Oprisko A., Campos J., Fu Y., Friese T., Itzen A., et al. (2016). bMERB domains are bivalent Rab8 family effectors evolved by gene duplication. Elife 5, e18675. 10.7554/eLife.18675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regalado E. S., Guo D. C., Estrera A. L., Buja L. M., Milewicz D. M. (2014). Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am. J. Med. Genet. A 164 (1), 106–112. 10.1002/ajmg.a.36208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymann A. C., Boujemaa-Paterski R., Martiel J. L., Guerin C., Cao W., Chin H. F., et al. (2012). Actin network architecture can determine myosin motor activity. Science 336 (6086), 1310–1314. 10.1126/science.1221708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich S. K., Baskar R., Terman J. R. (2021). Propagation of F-actin disassembly via Myosin15-Mical interactions. Sci. Adv. 7 (20), eabg0147. 10.1126/sciadv.abg0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risca V. I., Wang E. B., Chaudhuri O., Chia J. J., Geissler P. L., Fletcher D. A. (2012). Actin filament curvature biases branching direction. Proc. Natl. Acad. Sci. U. S. A. 109 (8), 2913–2918. 10.1073/pnas.1114292109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode S., Ohm H., Anhauser L., Wagner M., Rosing M., Deng X., et al. (2018). Differential requirement for translation initiation factor pathways during ecdysone-dependent neuronal remodeling in Drosophila. Cell Rep. 24 (9), 2287–2299. 10.1016/j.celrep.2018.07.074 [DOI] [PubMed] [Google Scholar]
- Rottner K., Faix J., Bogdan S., Linder S., Kerkhoff E. (2017). Actin assembly mechanisms at a glance. J. Cell Sci. 130 (20), 3427–3435. 10.1242/jcs.206433 [DOI] [PubMed] [Google Scholar]
- Rouyère C., Serrano T., Frémont S., Echard A. (2022). Oxidation and reduction of actin: Origin, impact in vitro and functional consequences in vivo . Eur. J. Cell Biol. 101 (3), 151249. 10.1016/j.ejcb.2022.151249 [DOI] [PubMed] [Google Scholar]
- Rumpf S., Bagley J. A., Thompson-Peer K. L., Zhu S., Gorczyca D., Beckstead R. B., et al. (2014). Drosophila Valosin-Containing Protein is required for dendrite pruning through a regulatory role in mRNA metabolism. Proc. Natl. Acad. Sci. U. S. A. 111 (20), 7331–7336. 10.1073/pnas.1406898111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K., Reese A. L., Kim S. K., Kilaru G., Joo J. Y., Kavalali E. T., et al. (2017). An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Rep. 18 (6), 1512–1526. 10.1016/j.celrep.2017.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E. F., Shim S. O., Strittmatter S. M. (2008). Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J. Neurosci. 28 (9), 2287–2297. 10.1523/JNEUROSCI.5646-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorrer F., Schonbauer C., Langer C. C., Dietzl G., Novatchkova M., Schernhuber K., et al. (2010). Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464 (7286), 287–291. 10.1038/nature08799 [DOI] [PubMed] [Google Scholar]
- Semelakova M., Grauzam S., Betadthunga P., Tiedeken J., Coaxum S., Neskey D. M., et al. (2019). Vimentin and non-muscle myosin IIA are members of the neural precursor cell expressed developmentally DownRegulated 9 (NEDD9) interactome in head and neck squamous cell carcinoma cells. Transl. Oncol. 12, 49–61. 10.1016/j.tranon.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacter E. (2000). Quantification and significance of protein oxidation in biological samples. Drug metab. Rev. 32 (3-4), 307–326. 10.1081/dmr-100102336 [DOI] [PubMed] [Google Scholar]
- Shapira S. D., Gat-Viks I., Shum B. O., Dricot A., de Grace M. M., Wu L., et al. (2009). A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139 (7), 1255–1267. 10.1016/j.cell.2009.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Ye B., Rame M., Wang Y., Cioca D., Reibel S. (2020). The receptor tyrosine kinase EPHB6 regulates catecholamine exocytosis in adrenal gland chromaffin cells. J. Biol. Chem. 295 (22), 7653–7668. 10.1074/jbc.RA120.013251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebold C., Berrow N., Walter T. S., Harlos K., Owens R. J., Stuart D. I., et al. (2005). High-resolution structure of the catalytic region of MICAL (molecule interacting with CasL), a multidomain flavoenzyme-signaling molecule. Proc. Natl. Acad. Sci. U. S. A. 102 (46), 16836–16841. 10.1073/pnas.0504997102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora R., Bun P., Danglot L., Alqabandi M., Bassereau P., Niedergang F., et al. (2021). MICAL-L1 is required for cargo protein delivery to the cell surface. Biol. Open 10 (6), bio058008. 10.1242/bio.058008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siripala A. D., Welch M. D. (2007a). SnapShot: Actin regulators I. Cell 128 (3), 626. 10.1016/j.cell.2007.02.001 [DOI] [PubMed] [Google Scholar]
- Siripala A. D., Welch M. D. (2007b). SnapShot: Actin regulators II. Cell 128 (5), 1014. 10.1016/j.cell.2007.02.021 [DOI] [PubMed] [Google Scholar]
- Stadtman E. R., Moskovitz J., Levine R. L. (2003). Oxidation of methionine residues of proteins: Biological consequences. Antioxid. Redox Signal 5 (5), 577–582. 10.1089/152308603770310239 [DOI] [PubMed] [Google Scholar]
- Suarez C., Roland J., Boujemaa-Paterski R., Kang H., McCullough B. R., Reymann A. C., et al. (2011). Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr. Biol. 21 (10), 862–868. 10.1016/j.cub.2011.03.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Dai H., Zhang J., Jin X., Xiong S., Xu J., et al. (2006). Solution structure of calponin homology domain of Human MICAL-1. J. Biomol. NMR 36 (4), 295–300. 10.1007/s10858-006-9062-5 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Nakamoto T., Ogawa S., Seo S., Matsumura T., Tachibana K., et al. (2002). MICAL, a novel CasL interacting molecule, associates with vimentin. J. Biol. Chem. 277 (17), 14933–14941. 10.1074/jbc.M111842200 [DOI] [PubMed] [Google Scholar]
- Takamoto K., Kamal J. K., Chance M. R. (2007). Biochemical implications of a three-dimensional model of monomeric actin bound to magnesium-chelated ATP. Structure 15 (1), 39–51. 10.1016/j.str.2006.11.005 [DOI] [PubMed] [Google Scholar]
- Tao W., Sun W., Zhu H., Zhang J. (2019). miR-205-5p suppresses pulmonary vascular smooth muscle cell proliferation by targeting MICAL2-mediated Erk1/2 signaling. Microvasc. Res. 124, 43–50. 10.1016/j.mvr.2019.03.001 [DOI] [PubMed] [Google Scholar]
- Tarrago L., Kaya A., Kim H. Y., Manta B., Lee B. C., Gladyshev V. N. (2022). The selenoprotein methionine sulfoxide reductase B1 (MSRB1). Free Radic. Biol. Med. 191, 228–240. 10.1016/j.freeradbiomed.2022.08.043 [DOI] [PubMed] [Google Scholar]
- Terman J. R., Kashina A. (2013). Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25 (1), 30–38. 10.1016/j.ceb.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman J. R., Mao T., Pasterkamp R. J., Yu H. H., Kolodkin A. L. (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109 (7), 887–900. 10.1016/s0092-8674(02)00794-8 [DOI] [PubMed] [Google Scholar]
- Tian A. L., Lu M., Zhang F. K., Calderon-Mantilla G., Petsalaki E., Tian X., et al. (2018). The pervasive effects of recombinant Fasciola gigantica Ras-related protein Rab10 on the functions of goat peripheral blood mononuclear cells. Parasit. Vectors 11 (1), 579. 10.1186/s13071-018-3148-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H., Schmidt E. F., Strittmatter S. M. (2006). RanBPM contributes to Semaphorin3A signaling through plexin-A receptors. J. Neurosci. 26 (18), 4961–4969. 10.1523/JNEUROSCI.0704-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K., Minato H., Murayama T., Sasahara A., Nishimura T., Kiyokawa E., et al. (2019). Semaphorin signaling via MICAL3 induces symmetric cell division to expand breast cancer stem-like cells. Proc. Natl. Acad. Sci. U. S. A. 116 (2), 625–630. 10.1073/pnas.1806851116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrini C., Cubero R. J., Dirkx E., Braga L., Ali H., Prosdocimo G., et al. (2019). Common regulatory pathways mediate activity of MicroRNAs inducing cardiomyocyte proliferation. Cell Rep. 27 (9), 2759–2771. 10.1016/j.celrep.2019.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Battum E. Y., Gunput R. A., Lemstra S., Groen E. J., Yu K. L., Adolfs Y., et al. (2014). The intracellular redox protein MICAL-1 regulates the development of hippocampal mossy fibre connections. Nat. Commun. 5, 4317. 10.1038/ncomms5317 [DOI] [PubMed] [Google Scholar]
- Vanoni M. A. (2017). Structure-function studies of MICAL, the unusual multidomain flavoenzyme involved in actin cytoskeleton dynamics. Arch. Biochem. Biophys. 632, 118–141. 10.1016/j.abb.2017.06.004 [DOI] [PubMed] [Google Scholar]
- Varland S., Vandekerckhove J., Drazic A. (2019). Actin post-translational modifications: The cinderella of cytoskeletal control. Trends Biochem. Sci. 44 (6), 502–516. 10.1016/j.tibs.2018.11.010 [DOI] [PubMed] [Google Scholar]
- Vitali T., Maffioli E., Tedeschi G., Vanoni M. A. (2016). Properties and catalytic activities of MICAL1, the flavoenzyme involved in cytoskeleton dynamics, and modulation by its CH, LIM and C-terminal domains. Arch. Biochem. Biophys. 593, 24–37. 10.1016/j.abb.2016.01.016 [DOI] [PubMed] [Google Scholar]
- Wang F., Chen X., Cheng H., Song L., Liu J., Caplan S., et al. (2021). MICAL2PV suppresses the formation of tunneling nanotubes and modulates mitochondrial trafficking. EMBO Rep. 22 (7), e52006. 10.15252/embr.202052006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Deng W., Zhang Y., Sun S., Zhao S., Chen Y., et al. (2017). Adaptations to endurance training depend on exercise-induced oxidative stress: Exploiting redox interindividual variability. Acta Physiol. (Oxf) 222, 12898. 10.1111/apha.12898 [DOI] [PubMed] [Google Scholar]
- Wang Y., Deng W., Zhang Y., Sun S., Zhao S., Chen Y. (2018). MICAL2 promotes breast cancer cell migration by maintaining epidermal growth factor receptor (EGFR) stability and EGFR/P38 signalling activation. Acta Physiol. (Oxf) 222 (2), 12920. 10.1111/apha.12920 [DOI] [PubMed] [Google Scholar]
- Wang Y., Min P., Qi C., Zhao S., Yu M., Zhang Y., et al. (2021). MICAL2 facilitates gastric cancer cell migration via MRTF-A-mediated CDC42 activation. Front. Mol. Biosci. 8, 568868. 10.3389/fmolb.2021.568868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangler M. F., Gonzaga-Jauregui C., Gambin T., Penney S., Moss T., Chopra A., et al. (2014). Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolonintestinal hypoperistalsis syndrome. PLoS Genet. 10 (3), e1004258. 10.1371/journal.pgen.1004258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide T., Teuber J., Bayer M., Barnekow A. (2003). MICAL-1 isoforms, novel rab1 interacting proteins. Biochem. Biophys. Res. Commun. 306 (1), 79–86. 10.1016/s0006-291x(03)00918-5 [DOI] [PubMed] [Google Scholar]
- White K. A., Grillo-Hill B. K., Barber D. L. (2017). Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 130 (4), 663–669. 10.1242/jcs.195297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. P., Odell A. F., Karnezis T., Farnsworth R. H., Gould C. M., Li J., et al. (2017). Genome-wide functional analysis reveals central signaling regulators of lymphatic endothelial cell migration and remodeling. Sci. Signal 10 (499), eaal2987. 10.1126/scisignal.aal2987 [DOI] [PubMed] [Google Scholar]
- Wilson C., Terman J. R., González-Billault C., Ahmed G. (2016). Actin filaments-A target for redox regulation. Cytoskelet. Hob. 73 (10), 577–595. 10.1002/cm.21315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wioland H., Frémont S., Guichard B., Echard A., Jégou A., Romet-Lemonne G. (2021). Actin filament oxidation by MICAL1 suppresses protections from cofilin-induced disassembly. EMBO Rep. 22 (2), e50965. 10.15252/embr.202050965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wioland H., Guichard B., Senju Y., Myram S., Lappalainen P., Jégou A., et al. (2017). ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Curr. Biol. 27, 1956–1967. 10.1016/j.cub.2017.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolterhoff N., Gigengack U., Rumpf S. (2020). PP2A phosphatase is required for dendrite pruning via actin regulation in Drosophila. EMBO Rep. 21 (5), e48870. 10.15252/embr.201948870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Yesilyurt H. G., Yoon J., Terman J. R. (2018). The MICALs are a family of F-actin dismantling oxidoreductases conserved from Drosophila to humans. Sci. Rep. 8 (1), 937. 10.1038/s41598-017-17943-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Mao L., Tian H., Lin S., Zhao X., Lin J., et al. (2021). MICAL1 (molecule interacting with CasL 1) protects oligodendrocyte cells from oxidative injury through regulating apoptosis, autophagy in spinal cord injury. Neurosci. Lett. 750, 135712. 10.1016/j.neulet.2021.135712 [DOI] [PubMed] [Google Scholar]
- Yates T. M., Turner C. L., Firth H. V., Berg J., Pilz D. T. (2017). Baraitser-Winter cerebrofrontofacial syndrome. Clin. Genet. 92 (1), 3–9. 10.1111/cge.12864 [DOI] [PubMed] [Google Scholar]
- Yin L. M., Schnoor M., Jun C. D. (2020). Structural characteristics, binding partners and related diseases of the calponin homology (CH) domain. Front. Cell Dev. Biol. 8, 342. 10.3389/fcell.2020.00342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S. K., Pascoe H. G., Pereira T., Kondo S., Jacinto A., Zhang X., et al. (2016). Plexins function in epithelial repair in both Drosophila and zebrafish. Nat. Commun. 7, 12282. 10.1038/ncomms12282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J., Kim S. B., Ahmed G., Shay J. W., Terman J. R. (2017). Amplification of F-actin disassembly and cellular repulsion by growth factor signaling. Dev. Cell 42 (2), 117–129. 10.1016/j.devcel.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J., Terman J. R. (2018). Common effects of attractive and repulsive signaling: Further analysis of Mical-mediated F-actin disassembly and regulation by Abl. Commun. Integr. Biol. 11 (1), e1405197. 10.1080/19420889.2017.1405197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J., Terman J. R. (2018). MICAL redox enzymes and actin remodeling: New links to classical tumorigenic and cancer pathways. Mol. Cell Oncol. 5 (1), e1384881. 10.1080/23723556.2017.1384881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J., Wu H., Hung R. J., Terman J. R. (2021). Enhanced production of the Mical redox domain for enzymology and F-actin disassembly assays. Int. J. Mol. Sci. 22 (4), 1991. 10.3390/ijms22041991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A., Jo A., Grajewski K., Kim J. (2019). Characteristic cerebrovascular findings associated with ACTA2 gene mutations. Can. J. Neurol. Sci. 46 (3), 342–343. 10.1017/cjn.2019.20 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Liu R., Wang Y., Wang Y., Shuai Y., Ke C., et al. (2022). Phosphorylation of MICAL2 by ARG promotes head and neck cancer tumorigenesis by regulating skeletal rearrangement. Oncogene 41 (3), 334–346. 10.1038/s41388-021-02101-z [DOI] [PubMed] [Google Scholar]
- Zhao S., Min P., Liu L., Zhang L., Zhang Y., Wang Y., et al. (2019). NEDD9 facilitates hypoxia-induced gastric cancer cell migration via MICAL1 related Rac1 activation. Front. Pharmacol. 10, 291. 10.3389/fphar.2019.00291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Wang Z., Zhou J., Feng D., Li Y., Hu Y., et al. (2020). LncRNA Mical2/miR-203a-3p sponge participates in epithelial-mesenchymal transition by targeting p66Shc in liver fibrosis. Toxicol. Appl. Pharmacol. 403, 115125. 10.1016/j.taap.2020.115125 [DOI] [PubMed] [Google Scholar]
- Zhao Z., Zheng W., Bai H., Feng T. (2021). MICAL3 affects myocardial hypertrophy by regulating CK2A1/HDAC2 pathway. Rev. Argent. Clínica Psicológica 21 (2), 846–852. [Google Scholar]
- Zhou W., Liu Y., Gao Y., Cheng Y., Chang R., Li X., et al. (2020). MICAL2 is a novel nucleocytoplasmic shuttling protein promoting cancer invasion and growth of lung adenocarcinoma. Cancer Lett. 483, 75–86. 10.1016/j.canlet.2020.04.019 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Adolfs Y., Pijnappel W. W., Fuller S. J., Van der Schors R. C., Li K. W., et al. (2011). MICAL-1 is a negative regulator of MST-NDR kinase signaling and apoptosis. Mol. Cell Biol. 31 (17), 3603–3615. 10.1128/MCB.01389-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Wang J., Liu X., Wang Y., Chen Y., Sun K., et al. (2013). Multiple gene mutations, not the type of mutation, are the modifier of left ventricle hypertrophy in patients with hypertrophic cardiomyopathy. Mol. Biol. Rep. 40 (6), 3969–3976. 10.1007/s11033-012-2474-2 [DOI] [PubMed] [Google Scholar]
- Zucchini D., Caprini G., Pasterkamp R. J., Tedeschi G., Vanoni M. A. (2011). Kinetic and spectroscopic characterization of the putative monooxygenase domain of human MICAL-1. Arch. Biochem. Biophys. 515 (1-2), 1–13. 10.1016/j.abb.2011.08.004 [DOI] [PubMed] [Google Scholar]