

Abstract
SET domain-containing 5 (SETD5) is an uncharacterized member of the protein lysine methyltransferase family and is best known for its transcription machinery by methylating histone H3 on lysine 36 (H3K36). These well-characterized functions of SETD5 are transcription regulation, euchromatin formation, and RNA elongation and splicing. SETD5 is frequently mutated and hyperactive in both human neurodevelopmental disorders and cancer, and could be down-regulated by degradation through the ubiquitin-proteasome pathway, but the biochemical mechanisms underlying such dysregulation are rarely understood. Herein, we provide an update on the particularities of SETD5 enzymatic activity and substrate specificity concerning its biological importance, as well as its molecular and cellular impact on normal physiology and disease, with potential therapeutic options.
Keywords: SETD5, lysine methyltransferase, posttranslational modifications, methylation, neurodevelopmental disorder (NDD), cancer
1. Introduction
Methyltransferases are a superfamily of enzymes very present in nature, acting in the methylation of proteins, nucleic acids, and small molecules (1, 2). These enzymes work by catalyzing a methyl group for a receptor molecule, generating S-adenosylmethionine (SAM) and a modified methylated molecule (3). This methyl group conjugation not only affects the bioconversion pathways of many drugs but also affects the properties of endogenous neurotransmitters and hormones (4). Moreover, methylation is fundamental to regulating gene expression. Unlike DNA methylation which has been linked to gene silencing, RNA and protein methylation show differential patterns of activating and repressing gene transcription. Proteins can be methylated at different amino acids, primarily lysine and arginine residues (5, 6). Gene expression can be governed by lysine methylation on two levels: methylation of histones and non-histone proteins such as transcription factors and chromatin modifiers (7).
2. Structural features of SETD5
The human SETD5 gene (OMIM 615743), also known as MRD23, SETD5A, 2900045N06Rik or mKIAA1757, is located on the chromosome 3p25.3 and encodes the SETD5 protein composed of 1442 amino acids (8). The SETD5 gene consists of 31 exons and is ubiquitously expressed in human tissues such as the brain, thyroid, skin, ovary, lung and endometrium (9, 10). SETD5 contains a SET (Su(var)3-9, enhancer-of-zeste, trithorax) domain and is thus annotated as a candidate protein of lysine methyltransferase, which methylates H3K36 up to the tri-methyl form (H3K36me3) (9, 11, 12) ( Figure 1 ). It belongs to SET-domain lysine methyltransferase superfamily which functions to methylate certain histone lysine residues, resulting in regulating the expression of genes. However, there is evidence that SETD5 lacks the methyltransferase activity but scaffolds a co-repressor complex, including HDAC3, NCoR, G9a, and PAF1, which couples selective deacetylation of H3K9ac with methylation of this residue (13–15). The yeast SET3 and SET4, Drosophila UpSET, and human MLL5 are homologous to SETD5 over their SET domains and, except for SETD5, contain a PHD finger ( Figure 1 ). The PHD finger of MLL5 binds the H3K4me3 mark (16, 17), and Drosophila UpSET also recognizes H3K4me3 (16).
Figure 1.

SETD5 domain composition and homologue architecture. (A) Crystal structure of human SETD5 protein. (B) Schematic indicating the protein domain organization of human (h) SETD5 and MLL5, yeast (y) Set3 and Set4, and Drosophila (d) UpSET. SET domains are shown in red and PHD fingers are shown in green. The total number of amino acids is indicated for each protein. (C) SETD5 contains the SET domain and is annotated as a candidate protein of lysine methyltransferase, which methylates H3K36 residue. However, there is evidence that SETD5 lacks the methyltransferase activity but scaffolds the G9a/HDAC3 co-repressor complex, which couples methylation of H3K9 with deacetylation of this residue. Members of the Set3-Set4 SET domain subfamily are shown with known interacting partners and methyl-lysine binding activity of their PHD fingers. Known binding partners are shown in blue. Set4 is predicted to interact with other factors (shown in gray) that remain to be identified. MLL5 has known interactors, a subset of which are shown in blue, and other yet-to-be-determined factors are indicated in gray.
3. Biochemical features of SETD5
The main role of SETD5 is gene activation by trimethylating H3K36 residue ( Table 1 ). In this reaction, SETD5 utilizes the cofactor SAM as a methyl group donor, which binds to the substrate-binding site of the SET domain (9). In contrast, SETD5 can induce the methylation of H3K9 independently of its SET domain. This is achieved by binding to G9a histone methyltransferase and HDAC3 histone deacetylase complex, thus forming a SETD5-G9a-HDAC3 co-repressor complex (13). SETD5 also deacetylates H3K9ac; when partnered with HDAC3/NCoR1, SETD5 is converted from a relatively promiscuous enzyme into a selective one (13). This implies a model in which the SETD5-G9a-HDAC3-NCoR1 co-repressor complex couples selective methylation of H3K9 with deacetylation of this residue at target genes. Furthermore, SETD5 recruits the HDAC3 complex to the rDNA promoter, resulting in the removal of H4K16ac and its reader protein TIP5, a repressor of rDNA expression (18) ( Table 1 ). Another finding by Villain et al. was the connection of SETD5 with BRD2, a bromodomain protein that recruits transcription regulators onto the chromatin (19). In more detail, both SETD5 and BRD2 bind to upstream promoter regions of the Sema3A locus and BRD2 is necessary for regulating Sema3A expression by SETD5.
Table 1.
Summary of the identified SETD5 substrates.
| Complex | Substrate | Methylation sites | Acetylation sites | Effect of the modification | Reference |
|---|---|---|---|---|---|
| Unknown | Histone H3 | K36 | / | Preservation of global transcriptional fidelity during brain development and neuronal wiring | (9) |
| G9a, HDAC3, NCoR1 | Histone H3 | K9 | / | Promoting H3K9 methylation via interacting with G9a/HDAC3/NcoR1 complex and enhancing PDAC resistance to MEKi | (13) |
| HDAC3, NCoR, PAF1 | Histone H3 | / | K27 | Promoting H3K27 deacetylation via recruiting HDAC3/NCoR co-repressor and suppressing adipogenesis | (14) |
| HDAC3 | Histone H4 | / | K16 | Elevating rDNA expression via an HDAC3-mediated H4K16 deacetylation and promoting neural cell proliferation | (18) |
K, lysine; MEKi, MEK1/2 inhibition; PDAC, pancreatic ductal adenocarcinoma.
Several mechanisms have been proposed to regulate SETD5 expression and activity. The nuclear localization signal (NLS) motif in SETD5 protein can control its nuclear levels. Another interaction that can handle the nuclear levels of SETD5 is its degradation by the proteasome via the APC/C E3 ubiquitin ligase (14) ( Figure 2A ). Furthermore, SETD5 expression is inhibited by miR-139-5p, which may be sponged by circRNA PTPRM (circPTPRM) (20). SETD5 is also downregulated by miR-126-5p, which represses the expression of neuron-related genes in neurons (19, 21); however, the importance of this mechanism remains to be explored ( Figure 2B ).
Figure 2.
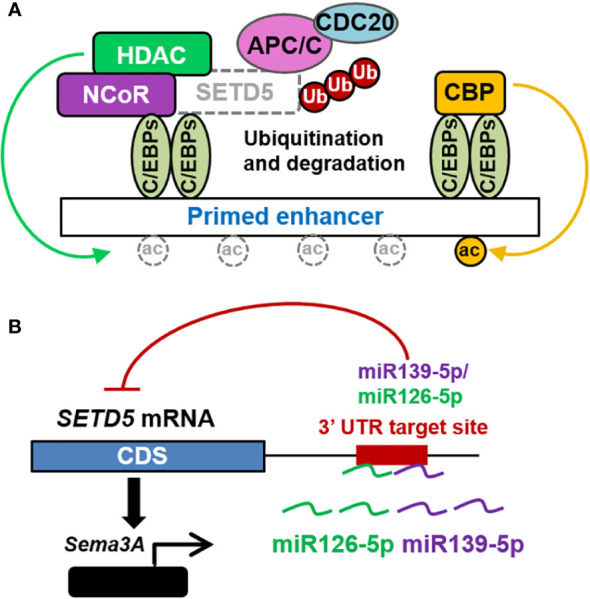
Regulatory mechanisms of SETD5 expression and activity. (A) SETD5 in NCoR-HDAC complex on primed enhancers is ubiquitinated and degraded by APC/C. The degradation of SETD5 from NCoR-HDAC3 co-repressor complex allows H3K27 acetylation and transits enhancers from primed to active state. (B) miR-139-5p or miR-126-5p binds to the SETD5 gene 3’ untranslated region (UTR) to repress the expression of SETD5 leading to low expression levels of Sema3A.
4. Physiologic functions of SETD5
4.1. SETD5, directly and indirectly, affects cellular functions
The cellular functions of SETD5 are primarily related to the trimethylation of H3K36, an active mark. Thus, SETD5 generates an “open”, more loose and accessible chromatin to transcription factors from a “closed” and inaccessible chromatin (9). SETD5 can also indirectly cause the deposition of repressive marks on histone tails by cross-talk with repressive methyltransferase. One indirect pathway of gene silencing is the interaction of SETD5 with the histone methyltransferase G9a, which dimethylated H3K9, establishing a repressive mark (13). Furthermore, the interaction of SETD5 with histone deacetylase HDAC3 causes the deposition of other repressive marks on histone tails (14). Therefore, SETD5 has the potential to interact indirectly with more pathways and repress a wider variety of genes. The change in chromatin architecture caused by SETD5, especially in gene enhancers or promoters, leads to the silencing of a vast array of genes. In these ways, SETD5 participates in several cellular functions, including regulation of the cell cycle and cell proliferation (22, 23), regulation of RNA elongation and splicing (9), control of brain and nervous system development (9, 19, 24–27), maintenance of tissue homeostasis (28–31), and embryonic development (22, 32, 33) ( Figure 3 ). Recently, SETD5 has been extensively associated with tumorigenesis (10, 13, 23, 34–39).
Figure 3.

Roles of SETD5 in regulation of nervous system development, embryonic development and tumorigenesis. ASD, autism spectrum disorder; BC, breast cancer; ESCC, esophageal squamous cell carcinoma; HCC, hepatocellular carcinoma; HSC, hematopoietic stem cell; ID, intellectual disability; NSCLC, non-small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma.
4.2. SETD5 coordinates the nervous system development
SETD5 has been demonstrated to participate in the early development of the nervous system. At different developmental stages, SETD5 exhibits a high expression level in the cerebral cortex (40, 41). The de novo mutation of the SETD5 gene has been identified as a genetic cause of neurodevelopmental disorders, such as intellectual disability (ID), autism spectrum disorder (ASD), and KBG syndrome (27, 42–46) ( Figure 3 ). Loss-of-function mutations in SETD5 lead to intellectual impairments often associated with speech, language, and developmental motor delays (8, 40, 47–49). Psychiatric manifestations of ASD-like behavior and obsessive-compulsive disorder (OCD) with hand flapping and ritualized movements have also been reported in SETD5 patients (40, 47, 50–52). Furthermore, the dysregulation of the axis SETD5-H3K36me3 is responsible for the alteration of neural progenitor proliferation and the synapse impairment that leads to neurological symptoms (9). It has been recently proposed that ASD may develop from altered mechanisms affecting neural progenitors (40), suggesting that SETD5 may act as a key regulator in ASD development.
4.3. SETD5 regulates the embryonic development
Another major cellular effect of SETD5 is the regulation of embryonic development. During early embryogenesis, SETD5 is required for maintaining the expression of germ cell-related genes and SETD5-associated protein complexes containing Tbl1xr1 and Ctr9, which in turn are involved in regulating the germ cell-related genes in murine ESCs (33) ( Figure 3 ). Deletion of SETD5 results in embryonic lethality at embryonic days 10.5 and 11.5 (22). In more detail, SETD5-deficient mouse embryos exhibit severe defects in neural tube formation, somitogenesis and cardiac development and have aberrant vasculogenesis in embryos, yolk sacs and placentas. Furthermore, the haploinsufficiency of SETD5 leads to disrupted developmental gene expression and cognition (41, 53). These data suggest a potential role of SETD5 in early embryonic development.
4.4. Connection of SETD5 with tumorigenesis
Knowledge about the function of SETD5 in tumors is sparse, and most of the information available is about its role in neurodevelopmental diseases ( Figure 3 ). SETD5 is located on chromosome 3p25.3 in a region linked to various diseases and amplified in primary tumors (8, 54, 55). Genomic alterations of SETD5 occur in multiple cancer types, implicating its cancer-promoting role (56, 57). In most cases, the upregulation of SETD5 is detected in pancreatic cancer, breast cancer, esophageal squamous cell carcinoma (ESCC), and non-small cell lung cancer (NSCLC) (13, 23, 56, 57). The high levels of the SETD5 gene are related to poor prognosis in patients with lung, bladder, and prostate cancer (35, 38, 56, 57). By contrast, the suppression of SETD5 expression leads to reduced cell growth and migration in pancreatic cancer, prostate cancer, and hepatocellular carcinoma (HCC) (13, 34, 58), as well as enhanced resistance to chemotherapeutic drugs (13). In terms of the mechanism, SETD5 is proposed to act as a tumor driver by inhibiting tumor suppressor gene transcription through H3K9 methylation via interacting with G9a/HDAC3 complex (13). Another mechanism of SETD5 involvement in cancer is the regulation of cell cycle-related genes through activating the PI3K/AKT signaling pathway (10, 23, 56) ( Figure 3 ).
In addition, mutations or amplification in the SET-domain proteins has been previously reported in various cancers. According to data in the PECAN database (https://pecan.stjude.cloud/home), high-grade gliomas and acute lymphoblastic leukemias present SETD5 mutations. SETD5 gene mutations are also associated with prostate cancer, colorectal cancer, and neuroblastoma (36, 59–61). Furthermore, SETD5 is identified with a rate of high-level amplification at around 10% in bladder cancer (38). Either mutation or amplification is demonstrated to promote the proliferation of cancer cells (38, 60).
Recent reports shed more light on how altered SETD5 activity promotes tumorigenesis and progression. These studies investigated the role of SETD5 in breast cancer, ESCC, and NSCLC (10, 23, 37). In more detail, SETD5 acts as a factor to reprogram stemness-related gene expression patterns. The deletion of SETD5 induces the inactivation of the PI3K/AKT pathway (10, 23). This leads to the repression of stemness-related genes like SOX2, CD44, and OCT4, which reduce stem cell-like properties and malignant transformation.
5. Outlook
Despite the recent achievements in the structural and biochemical analyses of SETD5 protein, not much information is available on its cellular functions. Nevertheless, the evidence that the methylation of H3K36 plays an important role in regulating enhancer activity and SETD5 is amplified in many cancers suggests that SETD5 must play a pivotal role in many different cellular processes. Epigenetic-based therapies are emerging as effective and valuable approaches in cancer, and targeting SETD5 may present a practical approach. Further research on the discovery and use of SETD5 inhibitors to combat cancer subtypes could help maximize the effects of current therapeutic regimens. First, a deeper understanding of the enzyme’s intracellular effects and affected genes is needed since there is evidence that SETD5 may also act as a tumor driver in some stages of cancer development. The cross-talk of SETD5 with other epigenetic enzymes also needs further exploration to minimize off-target side effects from its therapeutic targeting.
Author contributions
ML: writing original draft and editing. YH: writing-original draft. ZZ: investigation. BZ: writing original draft. TH: writing original draft. AS: review and editing. GS: writing-review and editing and supervision and funding acquisition. QL: editing and supervision and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding Statement
This work was supported by grants from The Key Research and Development Program of Jiangsu Province (grant no. BE2020678) and The National Natural Science Foundation of China (grant no. 81871888).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Banco MT, Mishra V, Greeley SC, Ronning DR. Direct cetection of products from s-adenosylmethionine-dependent enzymes using a competitive fluorescence polarization assay. Anal Chem (2018) 90:1740–47. doi: 10.1021/acs.analchem.7b03556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G, et al. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature (2019) 573:445–9. doi: 10.1038/s41586-019-1528-1 [DOI] [PubMed] [Google Scholar]
- 3. Boriack-Sjodin PA, Swinger KK. Protein methyltransferases: A distinct, diverse, and dynamic family of enzymes. Biochemistry (2016) 55:1557–69. doi: 10.1021/acs.biochem.5b01129 [DOI] [PubMed] [Google Scholar]
- 4. Xu J, Richard S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol Cell (2021) 81:4357–68. doi: 10.1016/j.molcel.2021.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol (2019) 26:880–9. doi: 10.1038/s41594-019-0298-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blanc RS, Richard S. Arginine methylation: The coming of age. Mol Cell (2017) 65:8–24. doi: 10.1016/j.molcel.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 7. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kellogg G, Sum J, Wallerstein R. Deletion of 3p25.3 in a patient with intellectual disability and dysmorphic features with further definition of a critical region. Am J Med Genet A (2013) 161:1405–8. doi: 10.1002/ajmg.a.35876 [DOI] [PubMed] [Google Scholar]
- 9. Sessa A, Fagnocchi L, Mastrototaro G, Massimino L, Zaghi M, Indrigo M, et al. SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron (2019) 104:271–289.e13. doi: 10.1016/j.neuron.2019.07.013 [DOI] [PubMed] [Google Scholar]
- 10. Chen Q, Sun Z, Li J, Zhang D, Guo B, Zhang T. SET domain-containing protein 5 enhances the cell stemness of non-small cell lung cancer via the PI3K/Akt/mTOR pathway. J Environ Pathol Toxicol Oncol (2021) 40:55–63. doi: 10.1615/JEnvironPatholToxicolOncol.2021036991 [DOI] [PubMed] [Google Scholar]
- 11. Tran K, Green EM. SET domains and stress: uncovering new functions for yeast Set4. Curr Genet (2019) 65:643–8. doi: 10.1007/s00294-018-0917-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol (2012) 13:115–26. doi: 10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Hausmann S, Lyu R, Li TM, Lofgren SM, Flores NM, et al. SETD5-coordinated chromatin reprogramming regulates adaptive resistance to targeted pancreatic cancer therapy. Cancer Cell (2020) 37:834–849.e13. doi: 10.1016/j.ccell.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsumura Y, Ito R, Yajima A, Yamaguchi R, Tanaka T, Kawamura T, et al. Spatiotemporal dynamics of SETD5-containing NCoR-HDAC3 complex determines enhancer activation for adipogenesis. Nat Commun (2021) 12:7045. doi: 10.1038/s41467-021-27321-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rincon-Arano H, Halow J, Delrow JJ, Parkhurst SM, Groudine M. UpSET recruits HDAC complexes and restricts chromatin accessibility and acetylation at promoter regions. Cell (2012) 151:1214–28. doi: 10.1016/j.cell.2012.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ali M, Rincon-Arano H, Zhao W, Rothbart SB, Tong Q, Parkhurst SM, et al. Molecular basis for chromatin binding and regulation of MLL5. Proc Natl Acad Sci U.S.A. (2013) 110:11296–301. doi: 10.1073/pnas.1310156110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X, Novera W, Zhang Y, Deng LW. MLL5 (KMT2E): structure, function, and clinical relevance. Cell Mol Life Sci (2017) 74:2333–44. doi: 10.1007/s00018-017-2470-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakagawa T, Hattori S, Nobuta R, Kimura R, Nakagawa M, Matsumoto M, et al. The autism-related protein SETD5 controls neural cell proliferation through epigenetic regulation of rDNA expression. iScience (2020) 23:101030. doi: 10.1016/j.isci.2020.101030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Villain G, Poissonnier L, Noueihed B, Bonfils G, Rivera JC, Chemtob S, et al. miR-126-5p promotes retinal endothelial cell survival through SetD5 regulation in neurons. Development (2018) 145:dev156232. doi: 10.1242/dev.156232 [DOI] [PubMed] [Google Scholar]
- 20. Jiang Z, Zhao J, Zou H, Cai K. CircRNA PTPRM promotes non-small cell lung cancer progression by modulating the miR-139-5p/SETD5 axis. Technol Cancer Res Treat (2022) 21:15330338221090090. doi: 10.1177/15330338221090090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poissonnier L, Villain G, Soncin F, Mattot V. miR126-5p repression of ALCAM and SetD5 in endothelial cells regulates leucocyte adhesion and transmigration. Cardiovasc Res (2014) 102:436–47. doi: 10.1093/cvr/cvu040 [DOI] [PubMed] [Google Scholar]
- 22. Osipovich AB, Gangula R, Vianna PG, Magnuson MA. Setd5 is essential for mammalian development and the co-transcriptional regulation of histone acetylation. Development (2016) 143:4595–607. doi: 10.1242/dev.141465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piao L, Li H, Feng Y, Yang Z, Kim S, Xuan Y. SET domain-containing 5 is a potential prognostic biomarker that promotes esophageal squamous cell carcinoma stemness. Exp Cell Res (2020) 389:111861. doi: 10.1016/j.yexcr.2020.111861 [DOI] [PubMed] [Google Scholar]
- 24. Pizzo L, Lasser M, Yusuff T, Jensen M, Ingraham P, Huber E, et al. Functional assessment of the "two-hit" model for neurodevelopmental defects in drosophila and x. laevis. PLos Genet (2021) 17:e1009112. doi: 10.1371/journal.pgen.1009112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miao SY, Miao SM, Cui RT, Yu AL, Miao ZJ. SETD5-AS1 stimulates neuron death in stroke via promoting PTEN expression. Eur Rev Med Pharmacol Sci (2018) 22:6035–41. doi: 10.26355/eurrev_201809_15940 [DOI] [PubMed] [Google Scholar]
- 26. Yagasaki H, Toda T, Koizumi K, Sugiyama T, Ohyama T, Hoshiai M, et al. A de novo 10.1-Mb 3p25 terminal deletion including SETD5 in a patient with ptosis and psychomotor retardation. Pediatr Neonatol (2018) 59:319–21. doi: 10.1016/j.pedneo.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 27. Rawlins LE, Stals KL, Eason JD, Turnpenny PD. De novo SETD5 nonsense mutation associated with diaphragmatic hernia and severe cerebral cortical dysplasia. Clin Dysmorphol (2017) 26:95–7. doi: 10.1097/MCD.0000000000000144 [DOI] [PubMed] [Google Scholar]
- 28. Li M, Qiu C, Bian Y, Shi D, Wang B, Ma Q, et al. SETD5 modulates homeostasis of hematopoietic stem cells by mediating RNA polymerase II pausing in cooperation with HCF-1. Leukemia (2022) 36:1111–22. doi: 10.1038/s41375-021-01481-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anderson E, Lam Z, Arundel P, Parker M, Balasubramanian M. Expanding the phenotype of SETD5-related disorder and presenting a novel association with bone fragility. Clin Genet (2021) 100:352–4. doi: 10.1111/cge.14014 [DOI] [PubMed] [Google Scholar]
- 30. Cheung MY, Roberts C, Scambler P, Stathopoulou A. Setd5 is required in cardiopharyngeal mesoderm for heart development and its haploinsufficiency is associated with outflow tract defects in mouse. Genesis (2021) 59:e23421. doi: 10.1002/dvg.23421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aoi H, Mizuguchi T, Ceroni JR, Kim VEH, Furquim I, Honjo RS, et al. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J Hum Genet (2019) 64:967–78. doi: 10.1038/s10038-019-0643-z [DOI] [PubMed] [Google Scholar]
- 32. Fellous A, Earley RL, Silvestre F. The Kdm/Kmt gene families in the self-fertilizing mangrove rivulus fish, kryptolebias marmoratus, suggest involvement of histone methylation machinery in development and reproduction. Gene (2019) 687:173–87. doi: 10.1016/j.gene.2018.11.046 [DOI] [PubMed] [Google Scholar]
- 33. Yu SE, Kim MS, Park SH, Yoo BC, Kim KH, Jang YK. SET domain-containing protein 5 is required for expression of primordial germ cell specification-associated genes in murine embryonic stem cells. Cell Biochem Funct (2017) 35:247–53. doi: 10.1002/cbf.3269 [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Yan L, Yao W, Chen K, Xu H, Ye Z. Integrated analysis of genetic abnormalities of the histone lysine methyltransferases in prostate cancer. Med Sci Monit (2019) 25:193–239. doi: 10.12659/MSM.912294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dmitriev AA, Rosenberg EE, Krasnov GS, Gerashchenko GV, Gordiyuk VV, Pavlova TV, et al. Identification of novel epigenetic markers of prostate cancer by NotI-microarray analysis. Dis Markers (2015) 2015:241301. doi: 10.1155/2015/241301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sowalsky AG, Xia Z, Wang L, Zhao H, Chen S, Bubley GJ, et al. Whole transcriptome sequencing reveals extensive unspliced mRNA in metastatic castration-resistant prostate cancer. Mol Cancer Res (2015) 13:98–106. doi: 10.1158/1541-7786.MCR-14-0273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z, Zhang C, Liu X, Che N, Feng Y, Xuan Y. SETD5 regulates glycolysis in breast cancer stem-like cells and fuels tumor growth. Am J Pathol (2022) 192:712–21. doi: 10.1016/j.ajpath.2021.12.006 [DOI] [PubMed] [Google Scholar]
- 38. Ding B, Yan L, Zhang Y, Wang Z, Zhang Y, Xia D, et al. Analysis of the role of mutations in the KMT2D histone lysine methyltransferase in bladder cancer. FEBS Open Bio (2019) 9:693–706. doi: 10.1002/2211-5463.12600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pinard A, Guey S, Guo D, Cecchi AC, Kharas N, Wallace S, et al. The pleiotropy associated with de novo variants in CHD4, CNOT3, and SETD5 extends to moyamoya angiopathy. Genet Med (2020) 22:427–31. doi: 10.1038/s41436-019-0639-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuechler A, Zink AM, Wieland T, Ludecke HJ, Cremer K, Salviati L, et al. Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur J Hum Genet (2015) 23:753–60. doi: 10.1038/ejhg.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deliu E, Arecco N, Morandell J, Dotter CP, Contreras X, Girardot C, et al. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat Neurosci (2018) 21:1717–27. doi: 10.1038/s41593-018-0266-2 [DOI] [PubMed] [Google Scholar]
- 42. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet (2012) 380:1674–82. doi: 10.1016/S0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- 43. Parenti I, Teresa-Rodrigo ME, Pozojevic J, Ruiz Gil S, Bader I, Braunholz D, et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum Genet (2017) 136:307–20. doi: 10.1007/s00439-017-1758-y [DOI] [PubMed] [Google Scholar]
- 44. Szczaluba K, Brzezinska M, Kot J, Rydzanicz M, Walczak A, Stawinski P, et al. SETD5 loss-of-function mutation as a likely cause of a familial syndromic intellectual disability with variable phenotypic expression. Am J Med Genet A (2016) 170:2322–27. doi: 10.1002/ajmg.a.37832 [DOI] [PubMed] [Google Scholar]
- 45. Pascolini G, Gnazzo M, Novelli A, Grammatico P. Clinical refinement of the SETD5-associated phenotype in a child displaying novel features and KBG syndrome-like appearance. Am J Med Genet A (2022) 188:1623–5. doi: 10.1002/ajmg.a.62679 [DOI] [PubMed] [Google Scholar]
- 46. Crippa M, Bestetti I, Maitz S, Weiss K, Spano A, Masciadri M, et al. SETD5 gene haploinsufficiency in three patients with suspected KBG syndrome. Front Neurol (2020) 11:631. doi: 10.3389/fneur.2020.00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grozeva D, Carss K, Spasic-Boskovic O, Parker MJ, Archer H, Firth HV, et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am J Hum Genet (2014) 94:618–24. doi: 10.1016/j.ajhg.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fernandes IR, Cruz ACP, Ferrasa A, Phan D, Herai RH, Muotri AR. Genetic variations on SETD5 underlying autistic conditions. Dev Neurobiol (2018) 78:500–18. doi: 10.1002/dneu.22584 [DOI] [PubMed] [Google Scholar]
- 49. Moore SM, Seidman JS, Ellegood J, Gao R, Savchenko A, Troutman TD, et al. Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl Psychiatry (2019) 9:24. doi: 10.1038/s41398-018-0344-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin GN, Song W, Wang W, Wang P, Yu H, Cai W, et al. De novo mutations identified by whole-genome sequencing implicate chromatin modifications in obsessive-compulsive disorder. Sci Adv (2022) 8:eabi6180. doi: 10.1126/sciadv.abi6180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet (2014) 94:677–94. doi: 10.1016/j.ajhg.2014.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen L, Li P, Zheng T, Luo M, Zhang S, Huang Y, et al. Comparative analysis of the autism-related variants between different autistic children in a family pedigree. Mol Med Rep (2021) 24:697. doi: 10.3892/mmr.2021.12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci (2019) 22:243–55. doi: 10.1038/s41593-018-0295-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Timmerman DM, Eleveld TF, Sriram S, Dorssers LCJ, Gillis AJM, Schmidtova S, et al. Chromosome 3p25.3 gain is associated with cisplatin resistance and is an independent predictor of poor outcome in Male malignant germ cell tumors. J Clin Oncol (2022) 40:3077–87. doi: 10.1200/JCO.21.02809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arcand SL, Provencher D, Mes-Masson AM, Tonin PN. OGG1 Cys326 variant, allelic imbalance of chromosome band 3p25.3 and TP53 mutations in ovarian cancer. Int J Oncol (2005) 27:1315–20. doi: 10.3892/ijo.27.5.1315 [DOI] [PubMed] [Google Scholar]
- 56. Yang Z, Zhang C, Che N, Feng Y, Li C, Xuan Y. Su(var)3-9, enhancer of zeste, and trithorax domain-containing 5 facilitates tumor growth and pulmonary metastasis through up-regulation of AKT1 signaling in breast cancer. Am J Pathol (2021) 191:180–93. doi: 10.1016/j.ajpath.2020.10.005 [DOI] [PubMed] [Google Scholar]
- 57. Yu H, Sun J, Zhao C, Wang H, Liu Y, Xiong J, et al. SET domain containing protein 5 (SETD5) enhances tumor cell invasion and is associated with a poor prognosis in non-small cell lung cancer patients. BMC Cancer (2019) 19:736. doi: 10.1186/s12885-019-5944-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park M, Moon B, Kim JH, Park SJ, Kim SK, Park K, et al. Downregulation of SETD5 suppresses the tumorigenicity of hepatocellular carcinoma cells. Mol Cells (2022) 45:550–63. doi: 10.14348/molcells.2022.0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim JC, Kim JH, Ha YJ, Kim CW, Tak KH, Yoon YS, et al. Analysis of genomic pathogenesis according to the revised Bethesda guidelines and additional criteria. J Cancer Res Clin Oncol (2021) 147:117–28. doi: 10.1007/s00432-020-03391-8 [DOI] [PubMed] [Google Scholar]
- 60. Pires SF, Tolezano GC, da Costa SS, Kawahira RSH, Kim CA, Rosenberg C, et al. Expanding the role of SETD5 haploinsufficiency in neurodevelopment and neuroblastoma. Pediatr Blood Cancer (2020) 67:e28376. doi: 10.1002/pbc.28376 [DOI] [PubMed] [Google Scholar]
- 61. Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med (2019) 21:816–25. doi: 10.1038/s41436-018-0266-3 [DOI] [PMC free article] [PubMed] [Google Scholar]