


Abstract
Three highly conserved aromatic residues in RNA recognition motifs (RRM) participate in stacking interactions with RNA bases upon binding RNA. We have investigated the contribution of one of these aromatic residues, Phe56, to the complex formed between the N-terminal RRM of the spliceosomal protein U1A and stem–loop 2 of U1 snRNA. Previous work showed that the aromatic group is important for high affinity binding. Here we probe how mutation of Phe56 affects the kinetics of complex dissociation, the strength of the hydrogen bonds formed between U1A and the base that stacks with Phe56 (A6) and specific target site recognition. Substitution of Phe56 with Trp or Tyr increased the rate of dissociation of the complex, consistent with previously reported results. However, substitution of Phe56 with His decreased the rate of complex association, implying a change in the initial formation of the complex. Simultaneous modification of residue 56 and A6 revealed energetic coupling between the aromatic group and the functional groups of A6 that hydrogen bond to U1A. Finally, mutation of Phe56 to Leu reduced the ability of U1A to recognize stem–loop 2 correctly. Taken together, these experiments suggest that Phe56 contributes to binding affinity by stacking with A6 and participating in networks of energetically coupled interactions that enable this conserved aromatic amino acid to play a complex role in target site recognition.
INTRODUCTION
The RNA recognition motif (RRM), also known as the ribonucleoprotein domain (RNP) or the RNA-binding domain (RBD), is one of the most common RNA-binding domains and is found in proteins that function in nearly every step of post-transcriptional gene expression (1). The RRM comprises 90–100 amino acids that form an antiparallel β-sheet flanked by two α-helices (2). The RRM binds single-stranded RNA through interactions with the surface of the β-sheet. Two sequences, called RNP-1 and RNP-2, in the central two strands of the β-sheet contain the most conserved sequences that contact RNA (3). Residues in the variable loops joining the strands of the β-sheet and in the C-terminal α-helix are important for specific target site recognition. Often two or more RRMs cooperate to recognize the RNA target site (4). As a result of the modular design of the RRM scaffold and the ability of RRMs to cooperate to bind RNA, a large diversity of RNA target site sequences and structures are recognized by different RRMs.
Three of the most highly conserved residues in RNP-1 and RNP-2 that contact RNA are aromatic (3). These aromatic residues are observed to stack with RNA bases in all structurally characterized RRM–RNA complexes (5–10). Stacking interactions are fundamental non-covalent interactions that are more common in the recognition of single-stranded than helical nucleic acids. For example, stacking interactions are important for single-stranded nucleic acid binding by the OB fold (11–14), single-stranded DNA-binding proteins (15–17) and other RNA-binding proteins (18–20). They are also used by DNA repair proteins to recognize DNA damaged sites (21–24) and by mRNA cap-binding proteins to recognize methylated guanosine (25). Stacking interactions probably contribute primarily to affinity in nucleic acid–protein complexes, rather than specificity, because all of the nucleic acid bases are capable of stacking and a variety of orientations of the aromatic rings can occur in stable stacking interactions (26,27). However, recent investigations of RNA–protein complexes have shown that intricate cooperative networks of different interactions can be essential to the high affinity and specificity of binding (28–32). Therefore, stacking interactions may contribute to specific target site recognition indirectly through their participation in these cooperative networks.
U1A is a spliceosomal protein that contains two RRMs, but only the N-terminal RRM binds RNA (33,34). U1A binds to stem–loop 2 of U1 snRNA in U1 snRNP, a subunit of the spliceosome. U1A also binds to two adjacent internal loops in the 3′-untranslated region (UTR) of its own pre-mRNA to inhibit polyadenylation (35). All three of these target sites contain nearly identical sequences in the loop, AUUGCAC closed by a CG base pair (Fig. 1). The N-terminal RRM of U1A has been structurally characterized by X-ray crystallography and NMR spectroscopy alone and bound to stem–loop 2 and the UTR target sites (Fig. 2) (5,6,36,37). These structural studies demonstrated that recognition of the stem–loop and internal loop target sites is nearly identical. The U1A–stem–loop 2 complex has also been extensively characterized biochemically and, as a result, is one of the best characterized RNA–protein complexes (28,29,38–47).
Figure 1.

(A) Stem–loop 2 RNA used in these experiments. The adenine that stacks with Phe56 is shown in red. (B) The U1A binding site in the 3′-UTR of U1A pre-mRNA. (C) The internal loop U1A binding site from the UTR used in these experiments.
Figure 2.

(A) Diagram of the complex formed between the N-terminal RRM of U1A and stem–loop 2 from the X-ray co-crystal structure (5). (B) Close-up of the complex showing the stacking interactions between Phe56, A6 and C7 and between Tyr13 and C5.
U1A contains two of the three conserved aromatic residues, Phe56 in RNP-1 and Tyr 13 in RNP-2. These are observed to stack with A6 and C5, respectively, in structural studies with both stem–loop 2 and the internal loop binding sites in the UTR (Fig. 2) (5,6,37). To estimate the contribution of Phe56 to the stability of the complex formed between the N-terminal RRM of U1A and stem–loop 2, we substituted Phe56 with Tyr, Trp, His, Leu and Ala (40). Substitution of Phe with any of the aromatic amino acids led to little destabilization of the complex, while substitution with Leu or Ala resulted in a loss of 4 and 5.5 kcal/mol, respectively, in binding energy. It was surprising that the aromatic mutants all bound with high affinity to stem–loop 2 since this position corresponds to Phe in 74% of RRMs, to Tyr in 10% of RRMs and only rarely to His or Trp (3). In U1A from different species, Trp occasionally replaces Phe at this position, but His and Tyr do not (48).
Our initial experiments, described above, demonstrated that an aromatic residue at position 56 is essential for U1A to bind RNA with high affinity. Here we describe experiments, using the same set of Phe56 mutants, that more precisely probe the contribution of Phe56 to target site recognition. We have found that the identity of the aromatic group at position 56 can change the kinetics of association of the complex, which may help to explain the high conservation of Phe at this position. We have probed the interdependence between the highly conserved stacking interaction and the network of hydrogen bonds around A6, demonstrating energetic coupling between conserved and variable interactions in this high affinity complex. Finally, experiments with mutated RNA target sites have shown that the stacking interaction can contribute, albeit indirectly, to specific recognition of RNA.
MATERIALS AND METHODS
RNA synthesis
1-Deaza-adenosine was synthesized by published methods (49), while purine riboside and tubercidin were purchased from Sigma. The phosphoramidites were synthesized and incorporated into the RNA stem–loop by chemical synthesis (49,50).
RNA sequences were synthesized on an Applied Biosystems ABI 394 automated DNA/RNA synthesizer using Pac-A, iPr-Pac-G, Ac-C and U phosphoramidites from Glen Research. Samples were cleaved and deprotected for 4 h at 55°C using 3:1 NH4OH/CH3CH2OH or for 10 min at 65°C using 40% methylamine in H2O (Aldrich), followed by washing with 3:1:1 EtOH/MeCN/H2O. The supernatant was removed and dried in vacuo. The silyl groups were removed with 250 µl of triethylamine:trihydrofluoride overnight at room temperature. An equal volume of H2O was added and the RNA was precipitated with 2 ml of cold butanol. After centrifugation at 16 000 g for 15 min, the butanol was removed and the pellet was dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0) and ethanol precipitated. RNA was purified using denaturing gel electrophoresis [20% acrylamide, 20:1 mono:bisacrylamide, 7 M urea in TBE (89 mM Tris–borate, 2 mM EDTA), 15 cm × 40 cm × 0.75 mm, 3 h at 50 W]. The major band, which was the slowest band, was visualized by UV shadowing and was excised. RNA was eluted from the gel with three 8 h extractions in 3 ml TE each. The pooled extracts were dialyzed against 0.1× TE buffer, concentrated to ∼150 µl in vacuo and ethanol precipitated. The RNA was dissolved in TE buffer and stored at –20°C. Concentrations were determined by UV at 260 nm. Correct composition was confirmed by MALDI mass spectrometry and enzymatic hydrolysis.
Peptide purification
An expression vector for the N-terminal RRM of U1A, U1A101, was obtained from Nagai (45). Phe56His, Phe56Leu, Phe56Tyr, Phe56Trp and Phe56Ala mutations were introduced using standard Kunkel mutagenesis procedures (51). The proteins were expressed in BL21DE3(pLysS) grown in LB medium and induced with 2 mM IPTG at mid-log phase. The cultures were grown for an additional 6 h after induction. The cells were pelleted and resuspended in 30 ml of lysis buffer (50 mM Tris–HCl, pH 8.0, 20 mM NaCl, 2 mM EDTA, 1 mM PMSF). The cells were lysed by a freeze–thaw cycle followed by ultrasonication. The lysate was precipitated with successive 30 and 55% (NH4)2SO4 precipitations. The final precipitate was resuspended in 5 ml of buffer 1 (100 mM NaCl, 25 mM Tris–HCl, pH 8.0) and dialyzed against this buffer. The sample was applied to a CM Sepharose column and eluted with a linear gradient from 100 to 500 mM NaCl. Peak fractions were pooled and dialyzed against buffer 2 (50 mM KCl, 10 mM potassium phosphate, pH 7.4). The sample was applied to a hydroxyapatite column and eluted with a linear gradient from 0 to 6% (NH4)2SO4. Peak fractions were pooled and dialyzed against buffer 2. Each protein was concentrated to 2 ml, glycerol was added to a final concentration of 20% and fractions were stored at –80°C. The concentration of each protein was determined by amino acid analysis and the molecular weight by MALDI mass spectrometry.
Gel mobility shift assays
Peptide–RNA equilibrium dissociation constants were measured by gel mobility shift assays (42). Binding reactions were performed at 25°C for at least 30 min in 10 mM Tris–HCl, pH 7.4, 250 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mg/ml tRNA in a total volume of 10 µl. RNA concentration was 25 pM. After addition of glycerol to a final concentration of 5%, the reactions were loaded onto an 8% polyacrylamide gel (80:1 mono:bisacrylamide, 15 cm × 40 cm × 1.5 mm) in 100 mM Tris–borate, pH 8.3, 1 mM EDTA and 0.1% Triton X-100 for 1 h at 350 V. The temperature of the gel was maintained at 25°C by a circulating water bath. Gels were analyzed on a Molecular Dynamics Storm phosphorimager. Fraction RNA bound versus protein concentration was plotted and curves were fitted to the equation: fraction bound = 1/(1 + Kd/[P]). Some of the equilibrium dissociation constants reported here are lower than in our previous work (40). We believe that the lower binding affinities are due to more rigorous control of temperature during the gel shift assay.
Dissociation kinetics
Dissociation rates were measured using a gel mobility shift competition assay (52). 32P-labeled stem–loop 2 RNA (∼0.02 nM) was equilibrated with 4 nM protein for 60 min at 25°C in a buffer containing 10 mM Tris–HCl, pH 7.4, 1 mM EDTA, 125 mM NaCl, 1 mg/ml tRNA, 5% glycerol and 0.5% Triton X-100 in a total volume of 100 µl. A 10 µl aliquot was loaded onto a running polyacrylamide gel identical to those used in the gel mobility shift assays. Depending on the protein mutant, 90 µl of 600–800 nM unlabeled stem–loop 2 RNA was added to the solution. Another aliquot, which served as the t = 0 sample, was loaded onto the gel. Aliquots of 10 µl were then loaded at various intervals over a 60–90 min period. The fastest time points obtainable using this method were 30 s. Gels were analyzed on a Molecular Dynamics Storm phosphorimager. Fraction bound versus time was plotted and curves were fitted to the equation: fraction bound = A0e–kt + C, where k = koff.
RNA melts
The melting curves (absorbance versus temperature) were measured at 260 nm on a Beckman DU 650. Samples were heated from 25 to 85°C at 0.5°C/min in a buffer comprised of 250 mM NaCl, 20 mM sodium cacodylate, pH 6.5, 0.5 mM EDTA, 1 mM MgCl2. Absorbance data were collected at 1 min intervals. The first derivative of the melting curve was used to calculate the melting temperature (Tm) assuming a two-state model (53,54).
RESULTS
Rate of dissociation
The rates of dissociation of complexes formed between stem–loop 2 and the wild-type, Phe56Trp, Phe56His and Phe56Tyr proteins were measured using gel mobility shift competition assays. Similar assays have been used to determine the dissociation rate constants of other nucleic acid–protein complexes, including the Tat–TAR complex, bZIP–DNA complexes and the 5S rRNA–TFIIIA complex (55–57). Representative plots are shown in Figure 3 and the koff values are reported in Table 1. The competition assays were performed at a lower salt concentration (125 mM NaCl) than the equilibrium binding assays reported in the remainder of this paper so that the rate of dissociation for all of the aromatic mutants could be accurately determined. Nevertheless, the rates of dissociation of Phe56Ala and Phe56Leu were too fast to be measured by this method. Equilibrium binding assays were also performed with 125 mM NaCl so that calculated association rate constants could be obtained from the measured dissociation rate constants and equilibrium dissociation constants. Whether these calculated association rate constants are identical to the actual association rate constants depends on the mechanisms of association and dissociation, which are not known at this time. However, the calculated rate constants are useful for comparing the wild-type, Phe56Trp, Phe56His and Phe56Tyr proteins. Tighter RNA binding was observed for all of the proteins at 125 than at 250 mM NaCl, consistent with previously published data (58).
Figure 3.
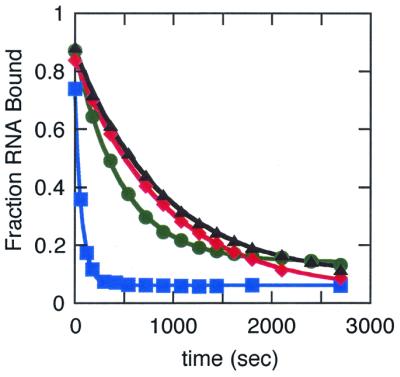
Representative plots of the fraction RNA bound by the wild type (triangle), Phe56His (diamond), Phe56Trp (circle) and Phe56Tyr (square) proteins as a function of time obtained from the competition assays.
Table 1. Kinetic and dissociation constants for U1A–stem–loop 2 RNA complexes in 150 mM NaCl.
| Protein | koff (s–1) | kon (M–1s–1)a | Kd (M) |
|---|---|---|---|
| Wild-type | 0.96 (± 0.4) × 10–3 | 3.1 × 107 | 3 (± 2) × 10–11 |
| F56H | 1.2 (± 0.2) × 10–3 | 0.60 × 107 | 20 (± 3) × 10–11 |
| F56Y | 14 (± 2) × 10–3 | 1.8 × 107 | 80 (± 14) × 10–11 |
| F56W | 2 (± 1) × 10–3 | 2.9 × 107 | 7 (± 2) × 10–11 |
akon was calculated from the measured koff and Kd.
The rates of dissociation of the wild-type, Phe56Trp and Phe56His proteins were within experimental error of the same value, 1 × 10–3s–1. Similar dissociation rates for the wild-type–stem–loop 2 complex have been previously measured by competition gel shift assays, competition filter binding assays and surface plasmon resonance (38,44,58). The rate of dissociation of the Phe56Tyr–stem–loop 2 complex was 14-fold faster than that of the wild-type complex, 1.4 × 10–2s–1. This increase in dissociation rate is responsible for the majority of the 26-fold lower affinity of Phe56Tyr for stem–loop 2. In contrast, the lower affinity of Phe56His for stem–loop 2 is not due to an increase in the rate of dissociation but must be due to a decrease in the rate of association.
Interdependence of the stacking interaction and the hydrogen bonding network formed with A6
The adenine (A6) that stacks with Phe56 also participates in three hydrogen bonds with U1A (Fig. 4). N1 forms a hydrogen bond with the side chain of Ser91, the 6-amino group forms a hydrogen bond with the carbonyl of Thr89 and N7 forms a water-mediated hydrogen bond with Thr89 (5). Previously we reported the synthesis of stem–loop 2 RNAs in which the modified bases N1-deaza-adenine (c1A), tubercidin and purine were substituted for A6 (Fig. 5) (40). With each substitution, one of the three hydrogen bonds was eliminated from the complex. Although stacking interactions may be altered by these base substitutions, previous work has shown that subtle modifications of the aromatic ring minimally effect stacking interactions (59–61). In the experiments reported in this paper we measured the affinity of Phe56His, Phe56Trp and Phe56Tyr for stem–loop 2 target sites containing the modified adenines in order to probe whether changes in the identity of the conserved aromatic group stacking with A6 alters the hydrogen bonding network formed between A6 and U1A.
Figure 4.
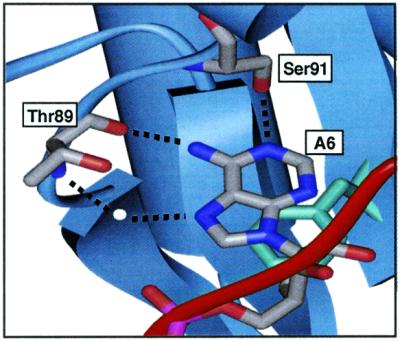
Diagram of the hydrogen bonding interactions between A6, Ser91 and Thr89 in the U1A–stem–loop 2 complex (5).
Figure 5.

Modified bases used to probe the hydrogen bonds formed between A6, Ser91 and Thr89.
The equilibrium dissociation constants for complexes formed between the wild-type, Phe56His, Phe56Trp and Phe56Tyr proteins and stem–loop 2 target sites containing the modified adenines are listed in Table 2. The affinities of Phe56His, Phe56Trp and Phe56Tyr for variants of the stem–loop containing c1A, purine or tubercidin instead of A6 were reduced (Table 2). The energetic penalties for eliminating each functional group were greater, in general, for the aromatic mutants than for the wild-type protein, indicating energetic coupling between the functional groups on A6 and aromatic substitutions at position 56 (Table 3). The most significant coupling was observed between N7 and position 56 for the Phe56Trp and Phe56Tyr mutations. The deletion of N7 resulted in a 1 kcal/mol greater destabilization of the Phe56Trp–stem–loop 2 complex and a 1.6 kcal/mol greater destabilization of the Phe56Tyr–stem–loop 2 complex than was observed for the wild-type complex. These results suggest that subtle changes in the aromatic group can change the interactions between A6 and amino acids in U1A.
Table 2. Binding affinities of wild-type and mutant proteins for wild-type and modified stem–loop 2 RNA target sites.
| RNA | Protein Wild-type | F56W | F56Y | F56H | F56L | |
|---|---|---|---|---|---|---|
| Wild-type | Kd (M) | 5 (± 3) × 10–10 | 5 (± 4) × 10–10 | 2 (± 1) × 10–9 | 9 (± 7) × 10–10 | 5 (± 2) × 10–7 |
| ΔG (kcal/mol)a | –12.7 ± 0.3 | –12.7 ± 0.3 | –11.9 ± 0.3 | –12.3 ± 0.5 | –8.6 ± 0.3 | |
| A6purine | Kd (M) | 1.0 (± 0.5) × 10–8 | 3 (± 2) × 10–8 | 1.2 (± 0.6) × 10–7 | 6 (± 4) × 10–8 | 8 (± 5) × 10–6 |
| ΔG (kcal/mol)a | –10.9 ± 0.3 | –10.3 ± 0.5 | –9.4 ± 0.3 | –9.8 ± 0.4 | –7.0 ± 0.3 | |
| A6tubercidin | Kd (M) | 2 (± 1) × 10–9 | 1.0 (± 0.6) × 10–8 | 1.0 (± 0.4) × 10–7 | 4 (± 2) × 10–9 | 3 (± 1) × 10–6 |
| ΔG (kcal/mol)a | –11.9 ± 0.4 | –10.9 ± 0.4 | –9.5 ± 0.3 | –11.4 ± 0.3 | –7.6 ± 0.3 | |
| c1A6 | Kd (M) | 1.2 (± 0.6) × 10–8 | 2 (± 1) × 10–8 | 1.3 (± 0.4) × 10–7 | 3 (± 2) × 10–8 | 6 (± 1) × 10–6 |
| ΔG (kcal/mol)a | –10.8 ± 0.3 | –10.5 ± 0.4 | –9.4 ± 0.2 | –10.3 ± 0.4 | –7.2 ± 0.1 | |
| UTR | Kd (M) | 7 (± 4) × 10–10 | 2 (± 1) × 10–9 | 4 (± 2) × 10–9 | 1.5 (± 0.7) × 10–9 | |
| ΔG (kcal/mol)a | –12.5 ± 0.3 | –11.9 ± 0.3 | –11.4 ± 0.5 | –12.0 ± 0.3 | ||
| A6U | Kd (M) | 2 (± 1) × 10 –5 | 1.1 (± 0.8) × 10–5 | 7 (± 4) × 10–5 | 2.8 (± 0.7) × 10–5 | 5 (± 3) × 10–5 |
| ΔG (kcal/mol)a | –6.4 ± 0.4 | –6.8 ± 0.4 | –5.7 ± 0.4 | –6.2 ± 0.2 | –5.9 ± 0.3 | |
| A6C | Kd (M) | 4 (± 2) × 10–5 | 5 (± 3) × 10–5 | 4 (± 2) × 10–4 | 4 (± 1) × 10–5 | 3 (± 1) × 10–4 |
| ΔG (kcal/mol)a | –6.0 ± 0.4 | –5.9 ± 0.3 | –4.6 ± 0.3 | –6.0 ± 0.2 | –4.8 ± 0.2 | |
| A6G | Kd (M) | 3 (± 2) × 10–5 | 3 (± 2) × 10–5 | 7 (± 5) × 10–5 | 4 (± 1) × 10–5 | 6 (± 2) × 10–5 |
| ΔG (kcal/mol)a | –6.2 ± 0.4 | –6.2 ± 0.4 | –5.7 ± 0.5 | –6.0 ± 0.3 | –5.8 ± 0.2 |
aΔG is the free energy of association of the complex.
Table 3. Comparison of the destabilization (ΔΔG) resulting from each base modification in complexes formed with the wild-type and mutant U1A proteins.
| Protein | ΔΔG (kcal/mol)a |
|
|
|---|---|---|---|
| A6purine | A6tubercidin | c1A6 | |
| Wild-type | 1.8 | 0.8 | 1.9 |
| F56W | 2.4 | 1.8 | 2.2 |
| F56Y | 2.5 | 2.4 | 2.5 |
| F56H | 2.5 | 0.9 | 2 |
| F56L | 1.6 | 1 | 1.4 |
aΔΔG is the difference in binding energy between a stem–loop 2 containing the indicated base modification and the wild-type stem–loop 2.
Equilibrium binding of the Phe56 mutants to a UTR target site
The stem–loop 2 and UTR RNA target sites have similar loop sequences, AUUGCAC closed by a CG base pair, presented in the context of different secondary structures. Although structural studies have been performed on both complexes, biochemical data is essentially limited to complex formation with stem–loop 2. The structure of U1A bound to the UTR target sites is similar to that of U1A bound to stem–loop 2 (5,6,37). However, structural studies cannot reveal the energetic contribution of individual interactions to the recognition of these two target sites that have significantly different secondary structures. Therefore, we have investigated whether mutation of Phe56 causes the same alterations in binding affinity for the U1A–UTR complex as for the U1A–stem–loop 2 complex. The UTR target site in these experiments was identical to that used in NMR structural studies of the U1A–UTR complex and is shown in Figure 1 (6).
The affinity of the wild-type protein for the UTR and stem–loop 2 target sites was similar (Table 2). Phe56His and Phe56Tyr also bound with comparable affinities to the two target sites. The Phe56Trp–UTR complex was destabilized by 0.6 kcal/mol compared with the wild-type complex, while no destabilization of the Phe56Trp–stem–loop 2 complex was observed. Overall, these data suggest that the roles of these aromatic residues in binding stem–loop 2 and the UTR target site are comparable, as expected from the similarity of the structures of the complexes (5,6,37). In contrast, Phe56Leu and Phe56Ala bound with much lower affinity to the UTR than to stem–loop 2. The binding was not tight enough to calculate an equilibrium dissociation constant. Even at protein concentrations of 100 µM, the fraction of UTR bound did not exceed 25–50%. Thus, the presence of an aromatic residue at position 56 is more important for the recognition of the UTR target site than it is for the recognition of stem–loop 2. Even though the structures of U1A bound to the UTR and stem–loop 2 target sites are similar, the energetic contribution of individual interactions to the stability of each complex is different.
Equilibrium binding of the Phe56 mutants to stem–loop 2 target sites containing A6G, A6C and A6U substitutions
U1A binds to stem–loop 2 and the UTR target sites with high specificity (46). Previous experiments showed that U1A is selective for A at position 6 and that mutation of A6 to G in stem–loop 2 destabilized the complex by 5–7 kcal/mol (41,62). Since Phe56 stacks with A6, any modification of the stacking interaction might alter the ability of U1A to recognize A6 specifically. Therefore, we probed whether the mutants in which the aromatic group is changed (Phe56Tyr, Phe56His and Phe56Trp) or replaced with an aliphatic group (Phe56Leu and Phe56Ala) would be as able as the wild-type protein to correctly distinguish A from G, C or U at this position.
Three stem–loop 2 target sites were synthesized in which A6 was replaced by U, C or G. Absorbance versus temperature profiles were obtained and used to calculate the Tm of each stem–loop. The wild-type, A6U and A6C stem–loop melting profiles were similar. The melting profile for the A6G stem–loop differed from the other three stem–loops, showing two sharp transitions. Using the program mfold, a dimer structure was found to be more stable for the A6G stem–loop than for the wild-type, A6C or A6U stem–loops (63,64). However, at the very low concentrations of RNA used in the binding experiments described below, the stem–loop structure was observed exclusively by native polyacrylamide gel electrophoresis.
U1A is specific for A at position 6 in the loop. Binding of the wild-type U1A protein was reduced 100 000-fold or by 6.3–6.5 kcal/mol when A6 was substituted with C, G or U (Table 2). The dramatic destabilization of the complex presumably results from changes in the hydrogen bond donor and acceptor pattern of the base, the size of the base for C and U and the strength of the stacking interactions with Phe56 and C7. The aromatic mutants, Phe56Tyr, Phe56Trp and Phe56His, were almost as specific for A at position 6 in the loop as the wild-type protein. In general, the destabilization of the complex observed as a result of each base substitution was slightly smaller for the aromatic mutants than that observed with the wild-type protein (Table 4).
Table 4. Comparison of the destabilization (ΔΔG) resulting from each base substitution in complexes formed with the wild-type and mutant U1A proteins.
| Protein | ΔΔG (kcal/mol)a |
||
|---|---|---|---|
| A6U | A6C | A6G | |
| Wild-type | 6.3 | 6.7 | 6.5 |
| F56W | 5.9 | 6.8 | 6.5 |
| F56Y | 6.2 | 7.3 | 6.2 |
| F56H | 6.1 | 6.3 | 6.3 |
| F56L | 2.7 | 3.8 | 2.8 |
aΔΔG is the difference in binding energy between a stem–loop 2 containing the indicated base substitution and the wild-type stem–loop 2.
The Phe56Leu–stem–loop 2 complex was destabilized by 2.7–3.8 kcal/mol upon replacing A6 with U, C or G (Table 4). This destabilization is 2.6–3.7 kcal/mol less than that observed for the wild-type–stem–loop 2 complex. Therefore, Phe56Leu is 100- to 500-fold less effective than the wild-type protein or any of the conservative aromatic mutants at correctly recognizing adenine at position 6 in the loop. If Phe56Leu were as specific as the wild-type protein, the affinity of Phe56Leu for the A6U, A6C and A6G stem–loop 2 sequences would have been below the detection limit of the gel mobility shift assay. We were unable to measure binding of Phe56Ala to any of the stem–loop 2 target sites substituted at position A6. However, the affinity of Phe56Ala for stem–loop 2 is so low that a decrease in binding affinity of even 3 kcal/mol would not be detectable by gel mobility shift assay. The data with Phe56Leu suggest that replacing the aromatic residue with an aliphatic residue alters the complex or the free protein so that the ability of the protein to distinguish between A and C, G or U at position 6 in the loop is diminished.
DISCUSSION
Phenylalanine is found at the position equivalent to Phe56 in 74% of RRMs (3). Although Phe is sometimes replaced with Tyr, it is rarely replaced with His or Trp. In some species this position in U1A is occupied by Trp, but not by His or Tyr (48). Therefore, it was surprising that the substitution of Phe56 with Tyr and His changed the stability of the U1A–stem–loop 2 complex so little. However, equilibrium binding measurements do not reveal whether these mutations have changed the rates of complex dissociation or association, disrupted cooperative networks of interactions involving Phe56 or altered specific target site recognition. Changes in any of these elements of binding upon mutation of Phe56 could be responsible for the high conservation of Phe at this position.
The rate of dissociation of the complex increased when Phe56 was replaced with Trp or Tyr. This increase in rate was comparable to the observed loss in equilibrium binding affinity. Although we were not able to measure the rate of dissociation of the Phe56Ala– or Phe56Leu–stem–loop 2 complexes, Katsamba et al. measured the rates of association and dissociation of the Phe56Ala protein with stem–loop 2 (38). They found that the rate of dissociation was substantially faster, while the rate of association was only slightly slower than the wild-type–stem–loop 2 complex. These data suggest that the rate determining step of association does not involve residue 56. It is known from NMR studies that helix C must move off the face of the β-sheet, where it contacts residue 56, in order for RNA to bind (36). Since the rate of association does not depend on the identity of residue 56, initial association probably does not require this conformational change in the protein.
In contrast to the results obtained with Phe56Trp, Phe56Tyr and Phe56Ala, substitution of His for Phe56 did not affect the rate of dissociation of the complex. Therefore, the rate of association of the protein must decrease to account for the decrease in binding affinity. Katsamba et al. measured a decrease in the rate of association of the U1A–stem–loop 2 complex upon substitution of three lysines that participate in electrostatic interactions with alanine (38). This work suggested that electrostatic interactions are important for complex formation. The substitution of a polar, and possibly charged, His for the hydrophobic Phe56 may disrupt this normal mechanism of association. If this change of mechanism were general, it could contribute to the absence of His from this position in RRMs.
A6 of stem–loop 2 is recognized by U1A by stacking with Phe56 and by participating in three hydrogen bonds with side chain or backbone functional groups (5). We deleted single functional groups from A6 in order to eliminate each hydrogen bond and probe whether the conserved stacking interaction cooperates with the hydrogen bonding network. We found that the destabilization of the complex that resulted from each functional group deletion depended on the identity of the amino acid in position 56. Therefore, the functional groups on A6 that form hydrogen bonds with U1A are coupled energetically to this aromatic amino acid. The effect on binding affinity of mutating Phe56 to either Tyr, Trp or His, while simultaneously eliminating a hydrogen bonding functional group from A6, was greater than the sum of the effects on binding of the Phe56 mutation and the base modification implemented separately. The largest energetic coupling was observed between N7 and position 56 in the stem–loop 2 complexes of Phe56Tyr and Phe56Trp. In contrast to the aromatic substitutions, substitution of Leu for Phe56 reduced or did not alter the effects of A6 functional group deletions on complex stability (40). Any destabilization originating from the substitution of Phe56 with Tyr, Trp or His may be compensated for, at least in part, by the increased contribution of the functional groups on A6 to complex stability. This increased energetic contribution may result from strengthening the hydrogen bonds formed between A6 and U1A or from the modulation of other interactions coupled to the functional groups on A6.
Stacking interactions are not usually thought to contribute to specific target site recognition. The bases do not participate equally well in stacking interactions; the extended aromatic systems of the purines form stronger stacking interactions than those of the pyimidines in the context of an RNA helix (65). However, the bases that stack well in the complex are also likely to stack well in the free RNA and the strength of the binding interaction is the difference in stability between the free RNA and protein and the RNA–protein complex. Nevertheless, the results obtained with the UTR target site and the A6U, A6C and A6G stem–loop 2 sequences suggest that Phe56 is important for specific target site recognition in the U1A–RNA complex.
The structure of the UTR–U1A complex is similar to that of the U1A–stem–loop 2 complex (5,6,37). In particular, the stacking interactions appear to be nearly identical in the two structures. Therefore, it is surprising that Phe56Leu and Phe56Ala were unable to bind to the UTR target site. These data suggest that the stacking interaction is more important to binding affinity in the complex with the UTR target site than in the complex with stem–loop 2, implying that the stacking interactions differ depending on the RNA target site. Phe56Ala and Phe56Leu can distinguish between the stem–loop and UTR target sites, while the wild-type protein cannot. Since the UTR target site contains A6, other interactions besides those that form with A6 must cooperate with the stacking interaction to destabilize the complexes of Phe56Ala and Phe56Leu.
The wild-type protein bound poorly to stem–loop 2 sites containing base substitutions at A6. Phe56Tyr, Phe56His and Phe56Trp were nearly as specific as the wild-type protein for the correct sequence. Therefore, the identity of the aromatic group is not important for specific recognition of A6, even though experiments discussed earlier showed that the aromatic group does affect the energetic contributions of A6 functional groups to complex stability. However, many stabilizing interactions are lost with the mutation of A to U, G or C, and the structure of the complex may be substantially altered. In contrast, Phe56Leu is significantly less specific for the correct base at A6 than the wild-type protein, losing only 2.6–3.7 kcal/mol binding energy with each base substitution.
One obvious explanation for the loss of specificity of Phe56Leu is that the hydrogen bonds formed between A6 and U1A are broken in the Phe56Leu–RNA complex and, therefore, the bonds responsible for sequence specificity are missing. However, the binding experiments with the RNA target sites containing purine, tubercidin and c1A suggested that these hydrogen bonds are still present in the Phe56Leu–stem–loop 2 complex (40). In fact, the destabilization resulting from the elimination of each hydrogen bonding functional group from A6 was nearly identical in the Phe56Leu– and wild-type–stem–loop 2 complexes. Therefore, other groups that cooperate with the aromatic side chain, either in the free protein or in the complex, must contribute to the correct recognition of A6. Molecular dynamics simulations performed on Phe56Ala suggested that the mutation altered the interactions of helix C with the surface of the β-sheet in the free protein and also affected the dynamics of loop 3 in both the free protein and the complex (66). Both of these regions of U1A are known to be important for specific target site recognition and to cooperate with residues on the surface of the β-sheet (28,29,44,67). Similar structural changes in Phe56Leu could contribute to its diminished specificity.
The experiments reported here have shown that the conserved aromatic residue Phe56 does not simply contribute to affinity in the U1A–RNA complex. Phe56 is involved in networks of cooperative interactions, probably in both the free protein and the complex. These cooperative interactions allow Phe56 to contribute to specific target site recognition, even though the formation of the stacking interaction itself is unlikely to be specific for a particular base. The experiments with the single atom mutants, tubercidin, purine and c1A, demonstrated that the highly conserved stacking interaction is coupled to the hydrogen bonding network surrounding A6. Such coupling between conserved and variable interactions may be a common feature of specific, high affinity RNA–protein complexes. Finally, our experiments suggest that modification of energetically coupled interactions and changes in the rates of complex association and dissociation upon mutation of Phe56 to other aromatic groups may contribute to the high conservation of Phe at this position in RRMs.
The RRM is an adaptable RNA-binding scaffold in which highly conserved residues maintain the fold of the RRM and provide a low level of affinity for many RNA sequences, while variable regions allow different RNA sites to be bound by different RRMs. Induced conformational changes in both the RNA and protein can occur upon binding and assist specific complex formation (6,7,10,36,68). The three aromatic residues that stack with RNA bases are among the most conserved residues that contact RNA, suggesting that the stacking interactions contribute non-specifically to complex stability (3). Our experiments show that the conserved aromatic residues can also play an important role in target site recognition by participating in cooperative networks of interactions. Since these cooperative networks will differ among RRM–RNA complexes, the aromatic residues probably do not contribute equivalently to RNA recognition in different RRM–RNA complexes. Thus, the stacking interaction, although highly conserved, can contribute to the ability of RRMs to target diverse RNA sequences and structures.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Prof. K. Nagai for the expression vector for U1A101. Funding was provided by the NIH (grant GM-56857). A.M.B. is an Alfred P. Sloan Research Fellow. J.C.S. and J.B.T. were supported by a NIH Training Grant in Molecular Biophysics (GM-08271).
REFERENCES
- 1.Varani G. and Nagai,K. (1998) RNA recognition by RNP proteins during RNA processing. Annu. Rev. Biophys. Biomol. Struct., 27, 407–445. [DOI] [PubMed] [Google Scholar]
- 2.Burd C.G. and Dreyfuss,G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 3.Birney E., Kumar,S. and Krainer,A.R. (1993) Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res., 21, 5803–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pérez-Cañadillas J.-M. and Varani,G. (2001) Recent advances in RNA-protein recognition. Curr. Opin. Struct. Biol., 11, 53–58. [DOI] [PubMed] [Google Scholar]
- 5.Oubridge C., Ito,N., Evans,P.R., Teo,C.H. and Nagai,K. (1994) Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature, 372, 432–438. [DOI] [PubMed] [Google Scholar]
- 6.Allain F.H., Howe,P.W.A., Neuhaus,D. and Varani,G. (1997) Structural basis of the RNA-binding specificity of human U1A protein. EMBO J., 16, 5764–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deo R.C., Bonanno,J.B., Sonenberg,N. and Burley,S.K. (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell, 98, 835–845. [DOI] [PubMed] [Google Scholar]
- 8.Price S.R., Evans,P.R. and Nagai,K. (1998) Crystal structure of the spliceosomal U2B″-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature, 394, 645–650. [DOI] [PubMed] [Google Scholar]
- 9.Ding J., Hayashi,M.K., Zhang,Y., Manche,L., Krainer,A.R. and Xu,R.J. (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev., 13, 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handa N., Nureki,O., Kuimoto,K., Kim,I., Sakamoto,H., Shimura,Y., Muto,Y. and Yokoyama,S. (1999) Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature, 398, 579–585. [DOI] [PubMed] [Google Scholar]
- 11.Murzin A.G. (1993) OB (oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J., 12, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavarelli J., Rees,B., Ruff,M., Thierry,J.-C. and Moras,D. (1993) Yeast tRNAAsp recognition by its cognate class II aminoacyl-tRNA synthetase. Nature, 362, 181–184. [DOI] [PubMed] [Google Scholar]
- 13.Bogden C.E., Fass,D., Bergman,N., Nichols,M.D. and Berger,J.M. (1999) The structural basis for terminator recognition by the Rho transcription termination factor. Mol. Cell, 3, 487–493. [DOI] [PubMed] [Google Scholar]
- 14.Hillier B.J., Rodriguez,J.M. and Gregoret,L.M. (1998) Coupling protein stability and protein function in Escherichia coli CspA. Folding Des., 3, 87–93. [DOI] [PubMed] [Google Scholar]
- 15.Bochkarev A., Pfeutzner,R.A., Edwards,A.M. and Frappier,L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- 16.Folmer R.H.A., Nilges,M., Papavoine,C.H.M., Harmsen,B.J.M., Konings,R.N.H. and Hilbers,C.W. (1997) Refined structure, DNA binding studies and dynamics of the bacteriophage Pf3 encoded single-stranded DNA binding protein. Biochemistry, 36, 9120–9135. [DOI] [PubMed] [Google Scholar]
- 17.Raghunathan S., Kozlov,A.G., Lohman,T.M. and Waksman,G. (2000) Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nature Struct. Biol., 7, 648–652. [DOI] [PubMed] [Google Scholar]
- 18.Antson A.A., Dodson,E.J., Dodson,G., Greaves,R.B., Chen,X. and Gollnick,P. (1999) Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature, 401, 235–242. [DOI] [PubMed] [Google Scholar]
- 19.Morellet N., Déméné,H., Teilleux,V., Huynh-Dinh,T., Rocquigny,H., Fournié-Zaluski,M. and Roques,B.P. (1998) Structure of the complex between the HIV-1 nucleocapsid protein NCp7 and single-stranded pentanucleotide d(ACGCC). J. Mol. Biol., 283, 419–434. [DOI] [PubMed] [Google Scholar]
- 20.Valegard K., Murray,J.B., Stockley,P.G., Stonehouse,N.J. and Liljas,L. (1994) Crystal structure of an RNA bacteriophage coat protein-operator complex. Nature, 371, 623–626. [DOI] [PubMed] [Google Scholar]
- 21.Obmolova G., Ban,C., Hsieh,P. and Yang,W. (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature, 407, 703–710. [DOI] [PubMed] [Google Scholar]
- 22.Slupphaug G., Mol,C.D., Kavli,B., Arvai,A.S., Krokan,H.E. and Tainer,J.A. (1996) A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature, 384, 87–92. [DOI] [PubMed] [Google Scholar]
- 23.Lau A.Y., Scharer,O.D., Samson,L., Verdine,G.L. and Ellenberger,T. (1998) Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell, 95, 249–258. [DOI] [PubMed] [Google Scholar]
- 24.Bruner S.D., Norman,D.P.G. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- 25.Qiocho F.A., Hu,G. and Gershon,P.D. (2000) Structural basis of mRNA cap recognition by proteins. Curr. Opin. Struct. Biol., 10, 78–86. [DOI] [PubMed] [Google Scholar]
- 26.Hunter C.A. and Sanders,J.K.M. (1990) The nature of π–π interactions. J. Am. Chem. Soc., 112, 5525–5534. [Google Scholar]
- 27.Norberg J. and Nilsson,L. (1995) Stacking free energy profiles for all 16 natural ribodinucleoside monophosphates in aqueous solution. J. Am. Chem. Soc., 117, 10832–10840. [Google Scholar]
- 28.Kranz J.K. and Hall,K.B. (1998) RNA binding mediates the local cooperativity between the β-sheet and the C-terminal tail of the human U1A RBD1 protein. J. Mol. Biol., 275, 465–481. [DOI] [PubMed] [Google Scholar]
- 29.Kranz J.K. and Hall,K.B. (1999) RNA recognition by the human U1A protein is mediated by a network of local cooperative interactions that create the optimal binding surface. J. Mol. Biol., 285, 215–231. [DOI] [PubMed] [Google Scholar]
- 30.Johansson H.E., Dertinger,D., LeCuyer,K.A., Behlen,L.S., Greef,C.H. and Uhlenbeck,O.C. (1998) A thermodynamic analysis of the sequence-specific binding of RNA by bacteriophage MS2 coat protein. Proc. Natl Acad. Sci. USA, 95, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steer B.A. and Schimmel,P. (1999) Different adaptations of the same peptide motif for tRNA functional contacts by closely homologous tRNA synthetases. Biochemistry, 38, 4965–4971. [DOI] [PubMed] [Google Scholar]
- 32.Hawko S.A. and Francklyn,C.S. (2001) Covariation of a specificity-determining structural motif in an aminoacyl-tRNA synthetase and a tRNA identity element. Biochemistry, 40, 1930–1936. [DOI] [PubMed] [Google Scholar]
- 33.Scherly D., Boelens,W., van Venrooij,W.J., Dathan,N.A., Hamm,J. and Mattaj,I.W. (1989) Identification of the RNA binding segment of human U1 A protein and definition of its binding site on U1 snRNA. EMBO J., 8, 4163–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu J. and Hall,K.B. (1995) An RBD that does not bind RNA: NMR secondary structure determination and biochemical properties of the C-terminal RNA binding domain from the human U1A protein. J. Mol. Biol., 247, 739–752. [DOI] [PubMed] [Google Scholar]
- 35.Boelens W.C., Jansen,E.J.R., van Venrooij,W.J., Stripecke,R., Mattaj,I.W. and Gunderson,S.I. (1993) The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell, 72, 881–892. [DOI] [PubMed] [Google Scholar]
- 36.Avis J.M., Allain,F.H.-T., Howe,P.W.A., Varani,G., Nagai,K. and Neuhaus,D. (1996) Solution structure of the N-terminal RNP domain of U1A protein: the role of C-terminal residues in structure stability and RNA binding. J. Mol. Biol., 257, 398–411. [DOI] [PubMed] [Google Scholar]
- 37.Varani L., Gunderson,S.I., Mattaj,I.W., Kay,L.E., Neuhaus,D. and Varani,G. (2000) The NMR structure of the 38 kDa U1A protein-PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nature Struct. Biol., 7, 329–335. [DOI] [PubMed] [Google Scholar]
- 38.Katsamba P.S., Myszka,D.G. and Laird-Offringa,I.A. (2001) Two functionally distinct steps mediate high affinity binding of U1A protein to U1 hairpin II RNA. J. Biol. Chem., 276, 21476–21481. [DOI] [PubMed] [Google Scholar]
- 39.Luchansky S.J., Nolan,S.J. and Baranger,A.M. (2000) Contribution of RNA conformation to the stability of a high-affinity RNA-protein complex. J. Am. Chem. Soc., 122, 7130–7131. [Google Scholar]
- 40.Nolan S.J., Shiels,J.C., Tuite,J.B., Cecere,K.L. and Baranger,A.M. (1999) Recognition of an essential adenine at a protein-RNA interface: comparison of the contributions of hydrogen bonds and a stacking interaction. J. Am. Chem. Soc., 121, 8951–8952. [Google Scholar]
- 41.Hall K.B. (1994) Interaction of RNA hairpins with the human U1A N-terminal RNA binding domain. Biochemistry, 33, 10076–10088. [DOI] [PubMed] [Google Scholar]
- 42.Rimmele M. and Belasco,J. (1998) Target discrimination by RNA-binding proteins: role of the ancillary protein U2A′ and a critical leucine residue in differentiating the RNA-binding specificity of spliceosomal proteins U1A and U2B″. RNA, 4, 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grainger R.J., Murchi,A.I.H., Norman,D.G. and Lilley,D.M.J. (1997) Severe axial bending of RNA induced by the U1A binding element present in the 3′ untranslated region of the U1A mRNA. J. Mol. Biol., 273, 84–92. [DOI] [PubMed] [Google Scholar]
- 44.Laird-Offringa I.A. and Belasco,J.G. (1995) Analysis of RNA-binding proteins by in vitro genetic selection: identification of an amino acid residue important for locking U1A onto its RNA target. Proc. Natl Acad. Sci. USA, 92, 11859–11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jessen T.-H., Oubridge,C., Teo,C.H., Pritchard,C. and Nagai,K. (1991) Identification of molecular contacts between the U1 A small nuclear ribonuleoprotein and U1 RNA. EMBO J., 10, 3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai D.E., Harper,D.S. and Keene,J.D. (1991) U1-snRNP-A selects a ten nucleotide consensus sequence from a degenerate RNA pool presented in various structural contexts. Nucleic Acids Res., 19, 4931–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bentley R.C. and Keene,J.D. (1991) Recognition of U1 and U2 small nuclear RNAs can be altered by a 5-amino-acid segment in the U2 small nuclear ribonucleoprotein particle (snRNP) B″ protein and through interactions with U2 snRNP-A′ protein. Mol. Cell. Biol., 11, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simpson G.G., Clark,G.P., Rothnie,H.M., Boelens,W., van Venrooij,W. and Brown,J.W.S. (1995) Molecular characterization of the spliceosomal proteins U1A and U2B″ from higher plants. EMBO J., 14, 4540–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seela F., Debelak,H., Usman,N., Burgin,A. and Beigelman,L. (1998) 1-Deazaadenosine: synthesis and activity of base-modified hammerhead ribozymes. Nucleic Acids Res., 26, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scaringe S.A., Francklyn,C. and Usman,N. (1990) Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidites. Nucleic Acids Res., 18, 5433–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kunkel T.A., Bebenek,K. and McClary,J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol., 204, 125–139. [DOI] [PubMed] [Google Scholar]
- 52.Setzer D.R. (1999) Measuring equilibirum and kinetic constants using gel retardation assays. In Haynes,S.R. (ed.), RNA–Protein Interaction Protocols. Humana Press, Totowa, NJ, pp. 115–128. [DOI] [PubMed]
- 53.Laing L.G. and Draper,D.E. (1994) Thermodynamics of RNA folding in a conserved ribosomal RNA domain. J. Mol. Biol., 238, 560–576. [DOI] [PubMed] [Google Scholar]
- 54.Theimer C.A., Wang,Y., Hoffman,D.W., Krisch,H.M. and Giedroc,D.P. (1998) Non-nearest neighbor effects on the thermodynamics of unfolding of a model mRNA pseudoknot. J. Mol. Biol., 279, 545–564. [DOI] [PubMed] [Google Scholar]
- 55.Setzer D.R., Menezes,S.R., Del Rio,S., Hung,V.S. and Subramanyan,G. (1996) Functional interactions between the zinc fingers of Xenopus transcription factor IIIA during rRNA binding. RNA, 2, 1254–1269. [PMC free article] [PubMed] [Google Scholar]
- 56.Metallo S.J. and Schepartz,A. (1997) Certain bZIP peptides bind DNA sequentially as monomers and dimerize on the DNA. Nature Struct. Biol., 4, 115–117. [DOI] [PubMed] [Google Scholar]
- 57.Weeks K.M. and Crothers,D.M. (1991) RNA recognition by Tat-derived peptides: interaction in the major groove? Cell, 66, 577–588. [DOI] [PubMed] [Google Scholar]
- 58.Hall K.B. and Stump,W.T. (1992) Interaction of N-terminal domain of U1A protein with an RNA stem/loop. Nucleic Acids Res., 20, 4283–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.SantaLucia J., Kierzek,R. and Turner,D.H. (1991) Functional group substitutions as probes of hydrogen bonding between GA mismatches in RNA internal loops. J. Am. Chem. Soc., 113, 4312–4322. [Google Scholar]
- 60.McKay S.L., Haptonstall,B. and Gellman,S.H. (2000) Beyond the hydrophobic effect: attractions involving heteroaromatic rings in aqueous solution. J. Am. Chem. Soc., 123, 1244–1245. [DOI] [PubMed] [Google Scholar]
- 61.Guckian K.M., Schweitzer,B.A., Ren,R.X.-F., Sheils,C.J., Tahmassebi,D.C. and Kool,E.T. (2001) Factors contributing to aromatic stacking in water: evaluation in the context of DNA. J. Am. Chem. Soc., 122, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeng Q. and Hall,K.B. (1997) Contribution of the C-terminal tail of U1A RBD1 to RNA recognition and protein stability. RNA, 3, 303–314. [PMC free article] [PubMed] [Google Scholar]
- 63.Zuker M., Mathews,D.H. and Turner,D.H. (1999) Algorithms and thermodynamics for RNA secondary structure prediciton: a practical guide. In Barciszewski,J. and Clark,B.F.C. (eds), RNA Biochemistry and Biotechnology. Kluwer Academic, Dordrecht, the Netherlands, pp. 11–43.
- 64.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 65.Turner D.H., Sugimoto,N. and Freier,S.M. (1988) RNA structure determination. Annu. Rev. Biophys. Biophys. Chem., 17, 167–192. [DOI] [PubMed] [Google Scholar]
- 66.Blakaj D.M., McConnell,K.J., Beveridge,D.L. and Baranger,A.M. (2001) Molecular dynamics and thermodynamics of protein-RNA interactions: mutation of a conserved aromatic residue modifies stacking interactions and structural adaptation in the U1A-stem loop 2 RNA complex. J. Am. Chem. Soc., 123, 2548–2551. [DOI] [PubMed] [Google Scholar]
- 67.Scherly D., Kambach,C., Boelens,W., van Venrooij,W.J. and Mattaj,I.W. (1991) Conserved amino acid residues within and outside of the N-terminal ribonucleoprotein motif of U1A small nuclear ribonucleoprotein involved in U1 RNA binding. J. Mol. Biol., 219, 577–584. [DOI] [PubMed] [Google Scholar]
- 68.Gubser C.C. and Varani,G. (1996) Structure of the polyadenylation regulatory element of the human U1A pre-mRNA 3′-untranslated region and interaction with the U1A protein. Biochemistry, 35, 2253–2267. [DOI] [PubMed] [Google Scholar]