

Abstract
Purpose:
We aimed to identify the underlying genetic cause for a novel form of distal arthrogryposis.
Methods:
Rare variant family-based genomics, exome and disease-specific panel sequencing were used to detect ADAMTS15 variants in affected individuals. Adamts15 expression was analyzed at the single-cell level during murine embryogenesis. Expression patterns were characterized by in situ hybridization and RNAscope.
Results:
We identified homozygous rare variant alleles of ADAMTS15 in five affected individuals from four unrelated consanguineous families, presenting with congenital flexion contractures of the interphalangeal joints and hypoplastic or absent palmar creases. Radiographic investigations showed physiological interphalangeal joint morphology. Additional features included knee, Achilles tendon, and toe contractures, spinal stiffness, scoliosis, and orthodontic abnormalities. Analysis of mouse whole-embryo single-cell sequencing data revealed a tightly regulated Adamts15 expression in the limb mesenchyme between embryonic stages E11.5 and E15.0. A perimuscular and peritendinous expression was evident by in situ hybridization in the developing mouse limb. In accordance, RNAscope analysis detected a significant co-expression with Osr1, but not with markers for skeletal muscle or joint formation.
Conclusion:
In aggregate, our findings provide evidence that rare biallelic recessive trait variants in ADAMTS15 cause a novel autosomal recessive connective tissue disorder resulting in a distal arthrogryposis syndrome.
Keywords: distal arthrogryposis, ADAMTS15, exome sequencing, connective tissue
Introduction
The distal arthrogryposes (DA) are characterized by contractures involving two or more body parts, primarily affecting the wrists, hands, ankles and feet.1 The contractures can vary in severity, but usually are not progressive and do not affect previously unaffected joints.2 To date, more than ten DA types and 15 associated genes have been identified.1,3 Most DA types are caused by heterozygous pathogenic variants in genes encoding sarcomeric components of skeletal muscle fibers.4 Besides this “myopathic” forms, arthrogryposis can be categorized etiologically into “neuropathic” or “connective tissue” associated types.5
The ADAMTS/L superfamily comprises 19 metalloproteases and seven structurally related glycoproteins (i.e., ADAMTS-like proteins) that play prominent roles in connective tissue homeostasis.6 The ADAMTS proteases share a similar structure containing a conserved N-terminal protease domain and a modular organized C-terminal ancillary domain, which mediates binding to extracellular matrix (ECM) components.7 While most of these enzymes participate in cleaving various substrates, such as procollagen, versican and aggrecan, some proteases appear to function more as ADAMTS-like proteins and are implicated in microfibril assembly and ECM regulating pathways such as TGF-ß- and BMP-signaling.6,7 To date, eight members of the superfamily have been linked to different autosomal recessive (AR) disease traits.8 Using exome sequencing (ES) and disease-specific panel analysis, we detected biallelic variants in ADAMTS15 (HGNC:16305, MIM 607509), in five affected individuals from four independent consanguineous families with congenital distal contractures. ADAMTS15 is located on chromosome 11q24.3. The canonical transcript (ENST00000299164.3; NM_139055.3) consists of 8 exons and encodes a 950 residue protein that presumably acts as an extracellularly activated protease that hydrolyses versican (versicanase).9 Detailed knowledge about the function and tissue-specific expression is lacking.
Our data implicate biallelic ADAMTS15 pathogenic variants as causative for a Mendelian disease trait involving distal contractures. Given our RNA data, we hypothesize that ADAMTS15 plays a critical role in perimuscular connective tissue and tendon development, which is disturbed in distal arthrogryposis caused by ADAMTS15 loss of function.
Methods and Materials
Parental consent was obtained for all clinical and molecular studies in this report and for the publication of clinical photographs. Next generation sequencing (NGS) was performed at Charité-Universitätsmedizin Berlin (Individual 1: Trio-ES and Individual 5: Trio-ES), Baylor College of Medicine (Individual 3: Single-ES) and University Medical Center Göttingen (Individual 4: Panel sequencing). Given the virtual absence of ADAMTS15 expression in all typical cell lines and accessible human tissues (for an overview consult https://www.proteinatlas.org/ENSG00000166106-ADAMTS15), we investigated the cell types expressing the gene during embryogenesis at the single-cell level using the Mouse Organogenesis Cell Atlas (MOCA).10 Furthermore, we performed droplet digital PCR (ddPCR), in situ hybridization, and RNAscope for further functional characterization of Adamts15 expression. Further details can be found in Supplemental Material.
Results
Clinical data
All five affected participants displayed similar distal congenital contractures of the fingers and toes (Fig. 1c). The fingers were bent in the proximal interphalangeal joints (PIPs), while the distal parts were tapered and had hypoplastic or absent flexion creases. The musculature of the hands was partially atrophic, and all had a mild appearance of fetal finger pads and clinodactyly of the fifth finger. Further clinical findings included contractures of knee, Achilles tendon and ankle (4/5), spine involvement (kyphoscoliosis and/- or spinal stiffness) (4/5), and orthodontic features (small mouth, dental crowding, missing teeth or arched palate) (4/5). No involvement of the central nervous system or other organs was noted, and radiographs of the hands and feet excluded a primary involvement of the bones or joints (Fig. 1c). Detailed clinical descriptions are available in the Supplemental Material, summarized as Table S1, and compared to overlapping entities in Table S2.
Figure 1. Pedigrees, structure of ADAMTS15 protein and location of the identified variants and clinical features of individuals with biallelic variants in ADAMTS15.
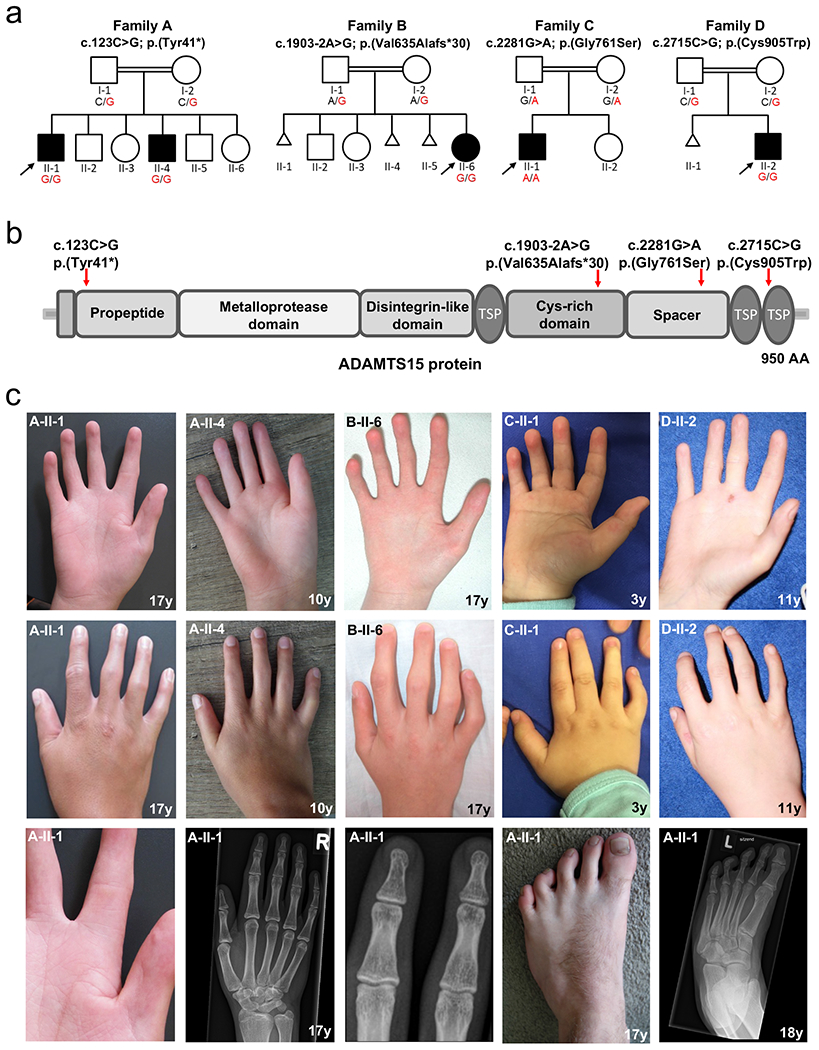
(a) Pedigrees of the four consanguineous families included in this study. Affected and unaffected individuals are indicated by filled and open squares (males) and circles (females). (b) Identified variants and schematic overview of their location within the ADAMTS15 protein. Red arrows point to the locations of the four variants identified within this study. TSP = thrombospondin type 1 domain. (c) Clinical pictures and radiographs of affected individuals 1-5, all showing congenital flexion contractures of the interphalangeal joints and hypoplastic or absent palmar creases. Additional pictures of individual 1 (family A: II-1) (bottom row) show a close-up to highlight the reduction of palmar creases and flexion contractures of the toes. Radiographs of the hands and feet indicate absence of any bony abnormalities that could explain the stiffening of the affected joints. Mild appearance of fetal finger pads and clinodactyly of the fifth finger were present in all affected individuals.
Molecular findings
Using ES, rare variant family-based genomics, and disease-specific panel analyses, we identified four rare homozygous variants in ADAMTS15 (NM_139055.3). The variant c.123C>G, p.(Tyr41*) is predicted to result in a premature termination codon (PTC) in the first exon. On the cDNA level (synthesized from RNA extracted from patient-derived fibroblasts), the variant c.1903-2A>G was shown by droplet digital PCR (ddPCR) and Sanger sequencing of an RT-PCR amplicon to lead to a complete skipping of exon 7 (r.1903_2078del), which is predicted to result in a frameshift and premature stop of translation p.(Val635Alafs*30) (Fig. S1). The missense variants c.2281G>A, p.(Gly761Ser) and c.2715C>G, p.(Cys905Trp) affect highly conserved amino acids within the ADAMTS spacer 1 and thrombospondin type 1 domain, respectively (Fig. S2). All variants except c.2281G>A are not found in gnomAD. The location of the identified variants and bioinformatic in silico predictions are summarized in Fig. 1b and Table S3. Segregation analysis is provided in Fig. S3.
Calculations for individuals 1, 2 and 5 revealed large absence of heterozygosity (AOH) blocks surrounding ADAMTS15, likely resulting from homozygosity on recently configured haplotypes due to parental consanguinity. Inbreeding coefficients calculated from ES data confirmed known family histories of consanguinity (Table S4).
Adamts15 expression during embryogenesis
Using the MOCA database, we observed that Adamts15 expression is mainly restricted to the mesenchyme (Fig. S4a). Therefore, we performed further characterization of the mesenchyme trajectory, which showed that most of the Adamts15-positive cells mapped to the connective tissue, skeletal muscle and chondrocyte trajectories (Fig. 2a).
Figure 2. Expression of Adamts15 RNA during mouse embryonic development.
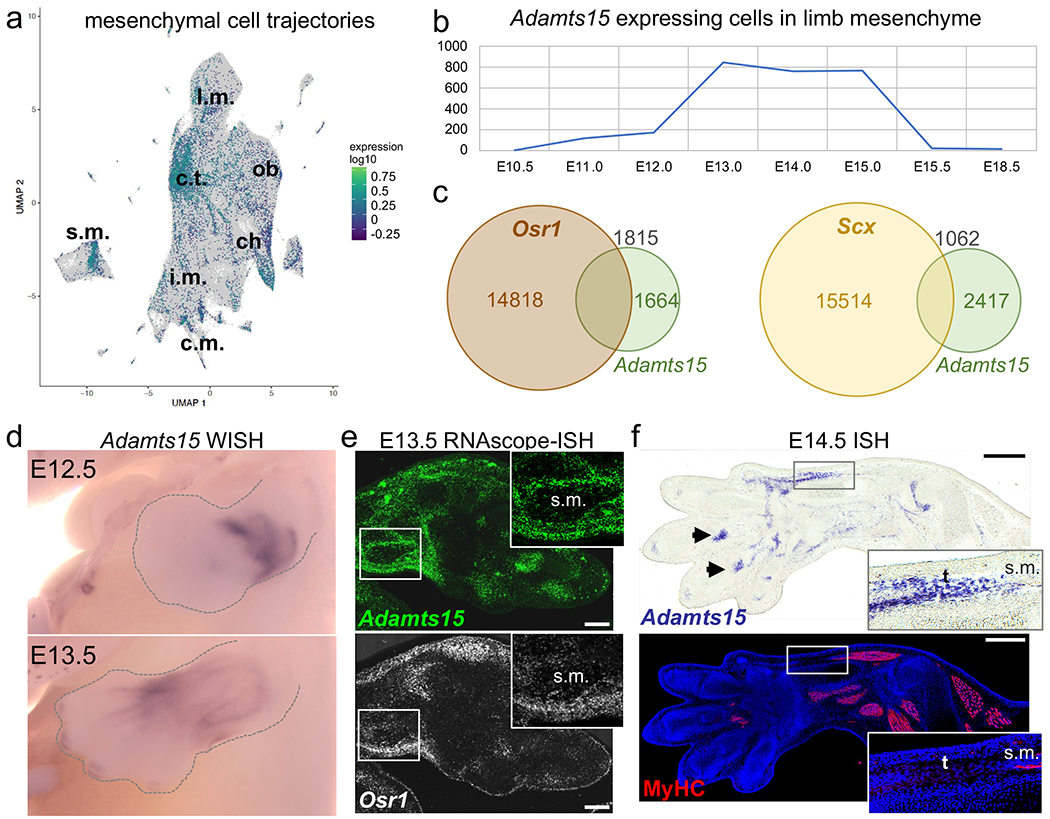
(a) Whole mouse embryo single-cell analysis of mesenchymal cells showing Adamts15 expression. The strongest accumulation of positive cells was found in the connective tissue (c.t.) trajectory. (b) Single-cell analysis of Adamts15-expressing cells among mesenchymal cells in the developing limbs. Highest expression was found between E13.0 and E15.0. (c) Co-expression of Adamts15 and marker genes Scx and Osr1 RNA in cells from developing limbs. Colored numbers correspond to cells displaying expression for the respective gene, black numbers indicate double positive cells. (d) Whole mount in situ hybridization (WISH) for Adamts15 in developing limbs at E12.5 and E13.5. Note perimuscular and -tendinous expression pattern at E13.5. (e) RNAscope co-labeling of Adamst15 (green) and Osr1 (white) in the distal limb at E13.5. Scale bar = 200 μm. (f) In situ hybridization (ISH) for Adamts15 in E14.5 forelimb in comparison to pan-MyHC immunostaining (red) for muscles and DAPI (blue) for nuclei on the same section. Inserts show muscle-tendon connection and arrowheads indicate tendon attachment sites at digits. Scale bar = 400 μm. ch = chondrocytes, c.m. = cardiac muscle, c.t. = connective tissue, i.m. = intermediate mesoderm, l.m. = limb mesenchyme, ob = osteoblasts, s.m. = skeletal muscle, t = tendon.
In the developing limb, which is pathogenetically most relevant, Adamts15 expression plateaued between E13.0 and E15.0 and disappeared almost completely by E15.5 (Fig. 2b). Also, in the developing limb most positive cells were mesenchymal, only few were of muscle or vascular origin (Fig. S4b). In order to gain insight into the cell types potentially affected by loss of Adamts15, we analyzed its co-expression with marker genes within the limb mesenchyme. While almost no co-expression was found for muscle, cartilage and joint interzone markers Myh7, Acan and Gdf5, versican (Vcan), one of the predicted substrates of Adamts15, was strongly co-expressed as well as Fbn2 associated with an overlapping disorder (Fig. S4c). Moreover, significant overlaps were detected with Osr1 and Osr2, transcription factors expressed in muscle connective tissue, as well as the tendon markers Scx and Tnmd (Fig. 2c, S4b).
A whole mount in situ hybridization confirmed that Adamts15 is expressed at perimuscular and peritendious areas in the developing limbs (Fig. 2d). Additional RNAscope analysis corroborated the partial co-localization of Adamts15 and Osr1 (Fig. 2e). At E14.5, strong Adamts15 signals were observed around tendons and at tendon attachment sites (Fig. 2f). These data indicate that the pathomechanism in this AR trait could involve muscle and tendon development.
Discussion
Our study identified four different homozygous variants in ADAMTS15 in five individuals from four unrelated families that share similar rare disease traits. The phenotype is characterized by congenital contractures, primarily affecting the small joints of the fingers and toes. Additional features included contractures of the knee, Achilles tendon, spinal stiffness, scoliosis, and orthodontic abnormalities. Radiographic investigations excluded bony abnormalities of the affected joints. The sequence changes included an early nonsense variant: c.123C>G; p.(Tyr41*), a splice variant: c.1903-2A>G, and two missense variants: c.2281G>A; p.(Gly761Ser) and c.2715C>G; p.(Cys905Trp).
Interestingly, other ADAMTS superfamily members and direct interaction partners have been described in association with congenital contractures. Clinical features of ADAMTS10- and ADAMTS17- associated Weill-Marchesani-Syndrome Types 1 (WMS1; MIM: 277600) and 4 (WMS4; MIM: 613195) include short stature, microspherophakia, brachydactyly, thickened skin and joint stiffness of small and large joints.11,12 Besides strabismus, our affected participants did not show any additional ophthalmological features. Biallelic pathogenic variants of ADAMTSL2 are associated with geleophysic dysplasia type 1 (GPHYSD1, MIM: 231050), a progressive musculoskeletal disorder characterized by severe short stature, brachydactyly, progressive joint contractures, cardiac valvular involvement and thickened skin.13 In contrast to WMS and GPHYSD1, the herein reported individuals do not consistently show skeletal anomalies, skin stiffening or pulmonary involvement. Compared to FBN2-associated congenital contractual arachnodactyly syndrome (CCA, MIM: 121050), also previously known as DA type 9, our participants did not display arachnodactyly, ear deformities or dolichostenomelia.14 A detailed list of the clinical features in our participants compared to the typical clinical findings of different types of WMS, FBN2-associated DA Type 9 (CCA) and GPHYSD1 is available in Table S2.
The variants identified here presumably result in an ADAMTS15 loss of function (LoF). Remarkably, no homozygous individuals for likely damaging predicted LoF variants are present in the gnomAD database (last access date 03/03/2022). The variant c.123C>G, p.(Tyr41*) in family A causes a PTC in the first exon and the intronic variant c.1903-2A>G leads to skipping of exon 7, resulting in a frameshift and PTC: p.(Val635Alafs*30). In both cases, the PTCs presumably result in nonsense-mediated decay (NMD) or a truncated protein, effectively deleting many functional domains. The missense variant p.(Gly761Ser) in family C affects a highly conserved amino acid residue within the ADAMTS spacer region, while the missense variant in Family D p.(Cys905Trp) localizes to the second thrombospondin type 1 repeat. Notably, a variant at a homologous position in ADAMTS13 p.(Cys1024Gly) is listed as pathogenic in ClinVar (ID: 5803). Analysis of ES-derived AOH/ROH data revealed, that the ultra-rare variants are located within long-sized runs of ROH, further supporting the Clan Genomics hypothesis.15 The AR inheritance and presence of two likely LoF alleles as the underlying disease mechanism are compatible with enzymatic disease and the thus far reported spectrum of pathogenic variants in the ADAMTS family members.8
In general, our understanding of the biological mechanisms of ECM regulation by ADAMTS proteoglycanases is only emerging and essentially depend on their substrates and other ECM-binding partners.16 ADAMTS15 is one of seven members (ADAMTS1, -4, -5, -8, -9, -15 and -20) that belong to an evolutionary distinct subset of proteoglycanases that are implicated in versican turnover.17,18 Since the other members of the ADAMTS/L superfamily that are already associated with joint contractures play a pivotal role in fibrillin microfibril assembly and ECM-associated signaling pathways (TGF-ß, BMP), a functional interaction between these members and ADAMTS15 seems plausible and parsimoniously explains the aggregate data.
Analysis of cell lines frequently used for functional in vitro investigations indicated a surprising restriction of ADAMTS15 expression. This was corroborated by single-cell sequencing data of whole mouse embryos showing an expression in the mouse limb only between E11.5 and E15.0. During this developmental phase, mesenchymal condensations of the skeletal elements are converted into cartilage and endochondral ossification begins.19 In parallel, the musculotendinous apparatus develops and tendon-bone connections are formed. While in the whole embryo Adamts15 expression is partially found also in chondrocytes and muscle cells, in the limb the strongest co-expression is with the tendon marker scleraxis (Scx), the perimuscular connective tissue marker Osr1 and with versican (Vcan). The latter co-expression is in line with the suggested function of Adamts15 as a versicanase, although co-expression is not conclusive evidence for enzyme function.9 A direct role of Adamts15 in the fusion of myoblasts was suggested based on expression analysis in C2C12 cells but has never been experimentally proven.18 The expression data presented here does not indicate significant expression in muscle cells. Co-expression with Osr1 and Scx suggests a role of Adamts15 in the formation of perimuscular connective tissue and tendons. Since depletion of Osr1 in the limb leads to abnormal skeletal muscle development through altered ECM production, the loss of ADAMTS15 might also have a similar, non-cell autonomous effect on muscle cells.20
In conclusion, these studies describe a new rare syndrome with a distal arthrogryposis phenotype that is associated with biallelic pathogenic variants in ADAMTS15. Due to the clinical features and the fact that ADAMTS15 belongs to the ADAMTS/L family, we would classify this disease trait as a novel connective-tissue related DA type. We hypothesize that impaired ADAMTS15 function causes tissue specific ECM dysregulation. Further experimental investigations are necessary to identify these tissue-specific molecular mechanisms.
Supplementary Material
Table S1. Clinical features of affected individuals with biallelic ADAMTS15 pathogenic variants.
Table S2. Comparison of the clinical findings of affected individuals carrying biallelic variants in ADAMTS15 with the main features of different types of Weill-Marchesani syndrome (WMS), congenital contractural arachnodactyly (CCA) and geleophysic dysplasia 1 (GPHYSD1).
Table S3. Molecular data and in-silico prediction of ADAMTS15 variants. All variants are described using the NM_139055.3 (GRCh37/hg19) transcript of ADAMTS15 in accordance to the HGVS recommendations.
Table S4. Absence of heterozygosity (AOH) at ADAMTS15 locus and inbreeding coefficients.
Figure S1. Splicing analysis. (a) Schematic overview of the ADAMTS15 gene. (b) Localization of the intronic variant and the probe used for ddPCR at the junction between exon 6 and exon 7. ddPCR revealed no expression in skin fibroblasts of the affected individual 3 compared to controls. (c) Sanger sequencing of a cDNA amplicon confirmed skipping of exon 7 leading to the indicated transcript.
Figure S2. Structural modeling of missense variants c.2281G>A, p.(Gly761Ser) and c.2715C>G, p.(Cys905Trp). (a) Overview over ADAMTS15 domain structure. (b) Residue Gly761 forms hydrogen bonds and (c) Cys905 a disulfide bridge important for stability of the 3D structure.
Figure S3. Segregation analysis.
Figure S4. (a) Analysis of Adamts15 and Osr1 expression on single-cell level from whole embryo single-cell sequencing data. (b) Single-cell analysis of Adamts15-expressing cells of ectodermal, muscular, and vascular origin in the developing limb. Note that cell counts are much lower than those for mesenchymal cells (Figure 2b). (c) Co-expression of Adamts15 and additional markers for muscles (Myh7), joints (Gdf5), fibrillin-1 (Fbn1), fibrillin-2 (Fbn2), and the proteoglycans aggrecan (Acan) and versican (Vcan). Osr2 is a marker for muscle connective tissue and early tendon progenitors with expression domains overlapping or adjacent with Osr1. Tnmd encodes for tenomodulin, a tendon marker.
Figure S5. Additional phenotypic aspects.
Acknowledgements
We are grateful to the families for their participation in this study. We thank Aris. N. Economides and Manuel Holtgrewe for their valuable suggestions and support. JRL laboratory is supported by the United States (US) National Institutes of Neurological Disorders and Stroke (NINDS; R35 NS 105078), and in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542), the National Institute of General Medical Sciences (NIGMS; R01 GM106373), the NHGRI Baylor College of Medicine Genomics Research Elucidates Genetics of Rare disease (BCM-GREGoR; U01 HG011758), the Muscular Dystrophy Association (MDA) (512848), and the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) of the National Institutes of Health under Award Number P50HD103555 for use of the Clinical Translation Core facilities. D.P. is supported by International Rett Syndrome Foundation (IRSF grant #3701-1). J.E.P. was supported by NHGRI K08 HG008986. U.K. obtained funding from the German Research Council (DFG)(KO 2891/9-1) and the BIH Center for Regenerative Therapies (BCRT)(cross-field project GenoPro).
Footnotes
Conflict of Interest
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic and genomic testing conducted at Baylor Genetics (BG); J.R.L. serves on the Scientific Advisory Board (SAB) of BG.
Ethics declaration
This study adheres to the principles in the Declaration of Helsinki. Permission for the study was obtained from the Ethics Committee of the University Medical Center Göttingen (proposal no. 10/4/21 Ü). Written informed consent was obtained from all participants including consent for publication of photographs. Consent forms are archived and available upon request.
Data Availability
Data supporting this paper are contained within the article and Supplementary information. Any additional data not compromised by ethical issues will be available upon request.
References:
- 1.Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009;91 Suppl 4:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dahan-Oliel N, Cachecho S, Barnes D, et al. International multidisciplinary collaboration toward an annotated definition of arthrogryposis multiplex congenita. Am J Med Genet C Semin Med Genet. 2019;181(3):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pehlivan D, Bayram Y, Gunes N, et al. The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am J Hum Genet. 2019;105(1):132–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whittle J, Johnson A, Dobbs MB, Gurnett CA. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes (Basel). 2021;12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall JG, Kimber E, Dieterich K. Classification of arthrogryposis. Am J Med Genet C Semin Med Genet. 2019;181(3):300–303. [DOI] [PubMed] [Google Scholar]
- 6.Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284(46):31493–31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubmacher D, Apte SS. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015;47:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rim JH, Choi YJ, Gee HY. Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation. Biomolecules. 2020;10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dancevic CM, Fraser FW, Smith AD, Stupka N, Ward AC, McCulloch DR. Biosynthesis and expression of a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats-15: a novel versican-cleaving proteoglycanase. J Biol Chem. 2013;288(52):37267–37276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566(7745):496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales J, Al-Sharif L, Khalil DS, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85(5):558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagoneau N, Benoist-Lasselin C, Huber C, et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75(5):801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Goff C, Morice-Picard F, Dagoneau N, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40(9):1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callewaert BL, Loeys BL, Ficcadenti A, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum Mutat. 2009;30(3):334–341. [DOI] [PubMed] [Google Scholar]
- 15.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apte SS. ADAMTS Proteins: Concepts, Challenges, and Prospects. Methods Mol Biol. 2020;2043:1–12. [DOI] [PubMed] [Google Scholar]
- 17.McCulloch DR, Nelson CM, Dixon LJ, et al. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell. 2009;17(5):687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stupka N, Kintakas C, White JD, et al. Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and −15 facilitates myoblast fusion. J Biol Chem. 2013;288(3):1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rafipay A, Berg ALR, Erskine L, Vargesson N. Expression analysis of limb element markers during mouse embryonic development. Developmental Dynamics. 2018;247(11):1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallecillo-Garcia P, Orgeur M, Vom Hofe-Schneider S, et al. Odd skipped-related 1 identifies a population of embryonic fibro-adipogenic progenitors regulating myogenesis during limb development. Nat Commun. 2017;8(1):1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical features of affected individuals with biallelic ADAMTS15 pathogenic variants.
Table S2. Comparison of the clinical findings of affected individuals carrying biallelic variants in ADAMTS15 with the main features of different types of Weill-Marchesani syndrome (WMS), congenital contractural arachnodactyly (CCA) and geleophysic dysplasia 1 (GPHYSD1).
Table S3. Molecular data and in-silico prediction of ADAMTS15 variants. All variants are described using the NM_139055.3 (GRCh37/hg19) transcript of ADAMTS15 in accordance to the HGVS recommendations.
Table S4. Absence of heterozygosity (AOH) at ADAMTS15 locus and inbreeding coefficients.
Figure S1. Splicing analysis. (a) Schematic overview of the ADAMTS15 gene. (b) Localization of the intronic variant and the probe used for ddPCR at the junction between exon 6 and exon 7. ddPCR revealed no expression in skin fibroblasts of the affected individual 3 compared to controls. (c) Sanger sequencing of a cDNA amplicon confirmed skipping of exon 7 leading to the indicated transcript.
Figure S2. Structural modeling of missense variants c.2281G>A, p.(Gly761Ser) and c.2715C>G, p.(Cys905Trp). (a) Overview over ADAMTS15 domain structure. (b) Residue Gly761 forms hydrogen bonds and (c) Cys905 a disulfide bridge important for stability of the 3D structure.
Figure S3. Segregation analysis.
Figure S4. (a) Analysis of Adamts15 and Osr1 expression on single-cell level from whole embryo single-cell sequencing data. (b) Single-cell analysis of Adamts15-expressing cells of ectodermal, muscular, and vascular origin in the developing limb. Note that cell counts are much lower than those for mesenchymal cells (Figure 2b). (c) Co-expression of Adamts15 and additional markers for muscles (Myh7), joints (Gdf5), fibrillin-1 (Fbn1), fibrillin-2 (Fbn2), and the proteoglycans aggrecan (Acan) and versican (Vcan). Osr2 is a marker for muscle connective tissue and early tendon progenitors with expression domains overlapping or adjacent with Osr1. Tnmd encodes for tenomodulin, a tendon marker.
Figure S5. Additional phenotypic aspects.
Data Availability Statement
Data supporting this paper are contained within the article and Supplementary information. Any additional data not compromised by ethical issues will be available upon request.