

Abstract

Hydrogen peroxide is an environmentally friendly oxidizing agent but current synthetic methods are wasteful. This is a result of the high flammability of H2/O2 mixtures and/or the requirement for cocatalysts. In this paper, we report the synthesis of H2O2 by means of a homogeneous catalyst, which allows a safe, one-pot synthesis in water, using only H2 and O2. This catalyst is capable of removing electrons from H2, storing them for the reduction of O2, and then permitting the protonation of the reduced oxygen to H2O2. The turnover number (TON) is 910 under an H2/O2 (95/5) atmosphere (1.9 MPa) for 12 h at 23 °C, which is the highest of any homogeneous catalyst. Furthermore, we propose a reaction mechanism based on two crystal structures.
Hydrogen peroxide (H2O2) is an important oxidizing agent but current synthetic methods are wasteful.1−5 This is because there is only a narrow range of H2/O2 mixtures that are not dangerously flammable, and the two gases must remain well-separated on-site.6−9 Furthermore, current methods often require diluents, cocatalysts, and/or organic solvents that must then be separated from the product stream and disposed of.10−15
If chemists were granted three wishes, they might desire (1) a safe reaction mixture, outside the flammability limit; (2) reaction in one vessel, without the need for transfer and separation; and (3) direct synthesis using only H2 and O2 (Table 1).
Table 1. H2O2 Synthesis under Safe Conditions.

See Table S2 in the Supporting Information for a detailed comparison of the anthraquinone process and this work.
See ref (10).
An example of just such a synthesis is thought to be conducted by extremophile microorganisms in nature.16−18 These organisms use hydrogenase enzymes that are usually degraded by O2, but it has been proposed that O2-tolerant hydrogenases might transfer electrons from H2 to O2, thereby reducing it to H2O2.
Inspired by this idea, we previously synthesized a complex with an iMP ligand {iMP = 2,6-bis(2-imidazolyl-1-methyl)pyridine} that stores electrons from H2 and, using a mixed gas of H2 and O2 outside the explosion range, investigated the direct synthesis of H2O2 in one flask.19 However, the reactivity of this electron storage complex with O2 was very low, and H2O2 was hardly produced catalytically. We synthesized a RhIII2 peroxide complex stepwise by irradiating a RhII2 complex and then showed the liberation of small amounts of H2O2.
The ligand of the desired catalyst requires electron-withdrawing properties to oxidize H2, but electron-donating properties to reduce O2. We have now improved the catalyst with a strongly electron-donating N-heterocyclic carbene ligand in order to catalytically reduce O2 without irradiation and, in this paper, describe its behavior and properties. We report how the catalyst allows the homogeneous synthesis of H2O2 starting with extracting electrons from H2, then using those electrons to reduce O2, and finally, acquiring protons from water to release the final product and return to the starting state.
This is a homogeneous H2O2 synthesis that meets the above three requirements: outside the flammability limit of the mixed gas (H2/O2 = 95/5),9 one flask, and homogeneous direct synthesis. The reaction mechanism of the homogeneous synthesis is discussed, starting from the structure of the RhII2 complex (1) that reacts with H2, then the structure of the RhI complex (2) that reacts with O2.
The initial rhodium dimer complex [RhII2(L)2(OH2)4](NO3)4 {[1](NO3)4, L = 2,6-bis(1-methylimidazol-2-ylidene)pyridine} was prepared from the reaction of a RhIII synthetic precursor [RhIII(L)(OH2)3](NO3)3 {[3](NO3)3} with H2 followed by oxygenation by O2 at 23 °C in water. Characterization of 1 was performed by 1H NMR and 13C NMR spectroscopies (Figures S4 and S5), C–H correlation spectroscopy (COSY) (Figure S6), ultraviolet–visible–near-infrared (UV–vis–NIR) absorption spectroscopy (Figure S7), X-ray photoelectron spectroscopy (XPS, Figure S8c), and elemental analysis. Single crystals of CH3CN-coordinated 1 [RhII2(L)2(OH2)2(CH3CN)2](CF3SO3)4 {[4](CF3SO3)4} were obtained from the slow evaporation of a CH3CN/H2O solution of 1 after replacing the NO3– ions with CF3SO3– ions. An ORTEP drawing shows that the two Rh metal centers adopt a distorted-octahedral geometry and are linked by a metal–metal bond to form the dimer structure (Figure 1). The Rh–Rh distance {2.7378(4) Å} of 4 was longer than those of other unsupported RhII dimer complexes {2.624(1)–2.7052(5) Å} (Table S3).19−21 A 1H NMR spectrum shows peaks in the diamagnetic region, which suggests that each RhII metal center is linked by a metal–metal bond (Figure S4). A UV–vis–NIR absorption spectrum of 1 shows a shoulder at 380 nm, which is similar to the spectrum of our previous RhII dimer with the iMP ligand (Figure S7).19 The XPS spectrum of 1 exhibits Rh 3d3/2 and 3d5/2 peaks at 313.3 and 308.7 eV (Figure S8c), which are similar to those of our previously reported RhII dimer complex and are lower than those of the related RhIII complex 3.19 These results, taken together, indicate that complex 1 adopts a dinuclear structure with divalent Rh centers.
Figure 1.
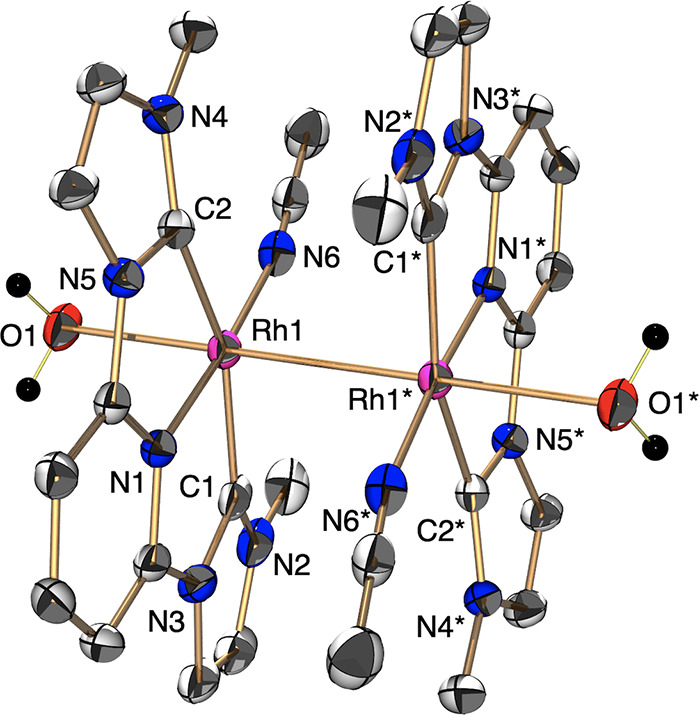
An ORTEP drawing of CH3CN-coordinated 1 [RhII2(L)2(OH2)2(CH3CN)2](CF3SO3)4 {[4](CF3SO3)4} with the ellipsoids at 50% probability. Counteranions (CF3SO3–) and hydrogen atoms (except for the aqueous ligands) are omitted for clarity.
Complex 1 reacts with H2 to form a low-valent RhI complex [RhI(L)(OH2)](NO3) {[2](NO3)} in water (eq 1). We characterized this complex by UV–vis–NIR absorption spectroscopy (Figure S9), XPS (Figure S8a), 1H NMR spectroscopy (Figure S10), and elemental analysis. A UV–vis–NIR absorption spectrum of 2 shows a broad absorption band at 650–1000 nm with/without CH3COONa (Figure S9), which is likely to arise from a charge transfer band, derived from metal–metal interactions, as seen in RhI polypyridyl complexes.25−30 An XPS spectrum of 2 shows Rh 3d3/2 and 3d5/2 peaks at 312.2 and 307.5 eV, respectively (Figure S8a). These binding energies were lower than those of RhII complex 1 (313.3 and 308.7 eV) (Figure S8c), but similar to those of the reported RhI complexes.19,28−32 Since the O2-sensitive 2 could not be crystallized, its structure was confirmed by X-ray analysis as a CO-adduct of 2, [RhI(L)(CO)](NO3) {[5](NO3)} (Figure S11). An ORTEP drawing of 5 shows that the Rh metal center has a square planar structure composed of L and the CO ligand. This geometry has been observed for other RhI complexes.19,22−24
These results indicate that complex 1 extracts two electrons from H2 to produce RhI complex 2 and that this complex is stabilized by the electron-withdrawing properties of L.
 |
1 |
Our next step was to investigate the recovery of 1 by the reaction of 2 with O2 under dark conditions (eq 2), with the reaction being monitored by UV–vis–NIR absorption spectroscopy (Figure S14). Injection of O2 gas into an aqueous solution of 2 led to the disappearance of the absorption bands at long wavelengths and the formation of a shoulder at around 380 nm with/without CH3COONa (Figure S14). The absorption spectrum of this reaction solution is similar to that of 1 (Figure S7).
 |
2 |
Quantitative analysis of the formed H2O2 was conducted by iodometric spectrophotometry (Figure S15), using NaI as the redox indicator.33 In order to separate the catalyst and product, after removing the Rh complexes by changing the counterion from NO3– to BPh4– and filtering, an excess of NaI was added to the remaining reaction solution in the presence of O2. The solution showed an absorption band at 353 nm, assigned to the absorption band of I3– generated from the two-electron oxidation of 3I– by H2O2 (Figure S15). The yield of H2O2 was determined as 53% based on the amount of I3– calculated using the absorbance at 353 nm (ε = 2.4 × 104 M–1 cm–1 for I3– in H2O).33
Quantitative analysis of the formed H2O2 was also conducted by titration with oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) complex (Ti-TPyP) (Figure S16).33,34 The UV–vis absorption spectrum of the following mixture was obtained: an aqueous HCl solution of Ti-TPyP, a reaction solution of 2 in the presence of O2 and an aqueous HClO4 solution. An absorption band at 433 nm was decreased compared with that of the control experiment, indicating the formation of a Ti peroxide species by the reaction of the Ti-TPyP reagent with H2O2. The yield of H2O2 was determined as 52% without CH3COONa (50% with CH3COONa) based on a calibration curve obtained from a standard H2O2 solution (Figure S16). The produced H2O2 decomposed less than 10% into H2O under stoichiometric and catalytic conditions, which was confirmed by the isotope labeling experiment using H218O2 (Figure S17).
The full catalytic cycle was performed with an aqueous CH3COONa solution of 1 (100 μM) under an H2/O2 (95/5) atmosphere (0.5–1.9 MPa) at 23 °C for 12 h under dark conditions–this is a nonexplosive gas mixture (Figure 2).9 The produced H2O2 was quantified by the Ti-TPyP reaction method. The turnover numbers (TONs) were increased depending on the total pressure of H2/O2 gas. The maximum TON was determined to be 910 under an H2/O2 (95/5) atmosphere (1.9 MPa) for 12 h at 23 °C, which is the highest TON in the direct synthesis of H2O2 from H2 and O2 using a homogeneous catalyst (Table 2). The initial turnover frequency of the catalytic reaction under an H2/O2 (95/5) atmosphere (1.9 MPa) for the first 1 h at 23 °C is 164 h–1 (163 mol kgcat–1 h–1), which is comparable to those of heterogeneous systems (60.8–180 mol kgcat–1 h–1).8 No H2O2 was formed without 1, H2, or O2.
Figure 2.

Pressure-dependent H2O2 production catalyzed by 1 under an H2/O2 (95/5) atmosphere for 12 h at 23 °C. The maximum TON is 910 under an H2/O2 (95/5) atmosphere (1.9 MPa).
Table 2. Requirements of Homogeneous H2O2 Synthesis.
| Requirement | Previous worka | This work | |
|---|---|---|---|
| Safe | H2 (%) | 95 | 95 |
| O2 (%) | 5 | 5 | |
| Temperature (°C) | 40 | 23 | |
| Solvent | Water | Water | |
| One-pot | Reaction vessel | High-pressure glass cylinder | High-pressure glass cylinder |
| Direct synthesis | H2 (MPa) | 0.76 | 0.475–1.8 |
| O2 (MPa) | 0.04 | 0.025–0.1 | |
| TON | 3.8 | 910 | |
| Catalyst (mmol %) | 0.11 | 0.22 | |
| Reactivity of RhI species to O2 | RhI species hardly reacts with O2 | RhI species reacts with O2 |
See ref (19).
The initial rate of H2O2 formation under catalytic conditions was investigated by varying the concentration of 1 at an H2/O2 (95/5) pressure of 1.9 MPa at 23 °C (Figure S18). It exhibits first-order kinetics, implying that the rate-determining step is the reaction of 1 with H2. A catalytic reaction using the in situ generated 2 under an H2/O2 (95/5) atmosphere (1.9 MPa) at 23 °C for 12 h under dark conditions gives TON as 908, suggesting complex 2 is part of the catalytic cycle. Under stoichiometric conditions, the pH was 4.9 with CH3COONa and 3.9 without CH3COONa. Under catalytic conditions, the pH was 8.3 with CH3COONa (0.5 M) and 3.9 without CH3COONa. The effect of CH3COONa on the formation of 1 and 2 was investigated by UV–vis absorption spectroscopy and 1H NMR spectroscopy, resulting in acetate ions interacting with 1 or 2 in a 1:1 or 1:2 ratio, respectively (Figures S4b and S19).
Qualitative analysis of H2O2 formed from the catalytic reaction was conducted with gas chromatography–mass spectrometry (GC–MS, Figure S20). The GC mass spectrum of the catalytic reaction solution, with Rh species removed, showed a signal at m/z = 34 assigned to H2O2 (Figure S20a). In an isotope labeling experiment using 18O2, this signal shifted to m/z = 38 (Figure S20c). During the catalytic reaction by 1, no metal nanoparticles were formed, as confirmed by a dynamic light scattering measurement. These results indicate that complex 1 catalyzes the synthesis of H2O2 as a single catalyst, even under a nonexplosive H2/O2 gas mixture.
The electrochemical properties of [3](NO3)3 and an analogue with the iMP ligand, [RhIII(iMP)(OH2)3](NO3)3 {[6](NO3)3}, were investigated by differential pulse voltammetry in an aqueous solution of CH3COONa (0.1 M) (Figure S21). The cathodic peak at −0.324 V versus Ag/AgCl for 3 is more negative than that for 6 (−0.164 V versus Ag/AgCl). This also shows that the electron-donating ability of L is much stronger than that of iMP.
Based on the above results, we propose the following catalytic reaction mechanism (Figure 3): First, RhII dimer complex 1 extracts electrons from H2 to form the electron storage catalyst 2. This step is aided by CH3COO– behaving as a Lewis base to abstract the protons and leave the electrons behind. Complex 2 then transfers two electrons to O2 to produce H2O2.
Figure 3.

Proposed catalytic reaction mechanism of a synthesis of H2O2 from H2 and O2 using 1 as a catalyst in water. Acetate ions could replace with aqua ligands, but this has no apparent effect on the catalytic mechanism.
In conclusion, we have synthesized a complex that stores electrons from hydrogen and transfers them to oxygen, thereby catalyzing the oxidation of the former and the reduction of the latter, in a safe, one-pot, aqueous process. The TON is the highest of any homogeneous catalyst. We are confident that our system points the way to all round savings in cost and materials, once the simplicity of the process is taken into account.
Acknowledgments
This work was supported by JST CREST Grant Number JPMJCR18R2, Japan, JSPS KAKENHI Grant Numbers JP26000008 (Specially Promoted Research), JP22K05130, and JP19K05503.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c13149.
Experimental details, characterization data of Rh complexes 1, 2, 3, 4, and 5 including NMR spectra, UV–vis–NIR absorption spectra, XPS spectra, an ORTEP drawing, and electrospray ionization (ESI) mass spectrum, and other experimental results including GC mass spectra, and differential pulse voltammograms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Eul W.; Moeller A.; Steiner N.. Hydrogen peroxide. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley, 2001. 10.1002/0471238961.0825041808051919.a01.pub2 [DOI] [Google Scholar]
- Jones C. W.; Clark J. H. In Applications of Hydrogen Peroxide and Derivatives; Jones C. W., Ed.; Royal Society of Chemistry: Cambridge, U.K., 1999; pp 1–35. [Google Scholar]
- Campos-Martin J. M.; Blanco-Brieva G.; Fierro J. L. G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem., Int. Ed. 2006, 45, 6962–6984. 10.1002/anie.200503779. [DOI] [PubMed] [Google Scholar]
- Chen Q. Development of an Anthraquinone Process for the Production of Hydrogen Peroxide in a Trickle Bed Reactor-From Bench Scale to Industrial Scale. Chem. Eng. Process. 2008, 47, 787–792. 10.1016/j.cep.2006.12.012. [DOI] [Google Scholar]
- Lewis R. J.; Hutchings G. J. Recent Advances in the Direct Synthesis of H2O2. ChemCatChem 2019, 11, 298–308. 10.1002/cctc.201801435. [DOI] [Google Scholar]
- Choudhary V. R.; Gaikwad A. G.; Sansare S. D. Nonhazardous Direct Oxidation of Hydrogen to Hydrogen Peroxide Using a Novel Membrane Catalyst. Angew. Chem., Int. Ed. 2001, 40, 1776–1779. 10.1002/1521-3773(20010504)40:9<1776::AID-ANIE17760>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Yamanaka I.; Onizawa T.; Takenaka S.; Otsuka K. Direct and Continuous Production of Hydrogen Peroxide with 93% Selectivity Using a Fuel-Cell System. Angew. Chem., Int. Ed. 2003, 42, 3653–3655. 10.1002/anie.200351343. [DOI] [PubMed] [Google Scholar]
- Xia C.; Xia Y.; Zhu P.; Fan L.; Wang H. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 2019, 366, 226–231. 10.1126/science.aay1844. [DOI] [PubMed] [Google Scholar]
- Schröder V.; Emonts B.; Janßen H.; Schulze H.-P. Explosion Limits of Hydrogen/Oxygen Mixtures at Initial Pressures up to 200 bar. Chem. Eng. Technol. 2004, 27, 847–851. 10.1002/ceat.200403174. [DOI] [Google Scholar]
- Edwards J. K.; Solsona B.; Ntainjua E. N.; Carley A. F.; Herzing A. A.; Kiely C. J.; Hutchings G. J. Switching Off Hydrogen Peroxide Hydrogenation in the Direct Synthesis Process. Science 2009, 323, 1037–1041. 10.1126/science.1168980. [DOI] [PubMed] [Google Scholar]
- Freakley S. J.; He Q.; Harrhy J. H.; Lu L.; Crole D. A.; Morgan D. J.; Ntainjua E. N.; Edwards J. K.; Carley A. F.; Borisevich A. Y.; Kiely C. J.; Hutchings G. J. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 965–968. 10.1126/science.aad5705. [DOI] [PubMed] [Google Scholar]
- Edwards J. K.; Freakley S. J.; Carley A. F.; Kiely C. J.; Hutchings G. J. Strategies for Designing Supported Gold–Palladium Bimetallic Catalysts for the Direct Synthesis of Hydrogen Peroxide. Acc. Chem. Res. 2014, 47, 845–854. 10.1021/ar400177c. [DOI] [PubMed] [Google Scholar]
- Wilson N. M.; Flaherty D. W. Mechanism for the direct synthesis of H2O2 on Pd clusters: heterolytic reaction pathways at the liquid–solid interface. J. Am. Chem. Soc. 2016, 138, 574–586. 10.1021/jacs.5b10669. [DOI] [PubMed] [Google Scholar]
- Shibata S.; Suenobu T.; Fukuzumi S. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen by Using a Water-Soluble Iridium Complex and Flavin Mononucleotide. Angew. Chem., Int. Ed. 2013, 52, 12327–12331. 10.1002/anie.201307273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage S. N.; James B. R. Catalytic Formation of an Amide Hydroperoxide and Hydrogen Peroxide using Rhodium Complexes and Dioxygen/Dihydrogen Mixtures. J. Chem. Soc., Chem. Commun. 1989, 1624–1626. 10.1039/c39890001624. [DOI] [Google Scholar]
- Wulff P.; Day C. C.; Sargent F.; Armstrong F. A. How oxygen reacts with oxygen-toletrant respiratory [NiFe]-hydrogenases. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 6606–6611. 10.1073/pnas.1322393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach L.; Lenz O. Catalytic Production of Hydrogen Peroxide and Water by Oxygen-Tolerant [NiFe]-Hydrogenase during H2 Cycling in the Presence of O2. J. Am. Chem. Soc. 2013, 135, 17897–17905. 10.1021/ja408420d. [DOI] [PubMed] [Google Scholar]
- Abou Hamdan A.; Burlat B.; Gutiérrez-Sanz O.; Liebgott P.-P.; Baffert C.; De Lacey A. L.; Rousset M.; Guigliarelli B.; Léger C.; Dementin S. O2-independent formation of the inactive states of NiFe hydrogenase. Nat. Chem. Biol. 2013, 9, 15–17. 10.1038/nchembio.1110. [DOI] [PubMed] [Google Scholar]
- Ogo S.; Minh L. T. T.; Kikunaga T.; Ando T.; Matsumoto T.; Yatabe T.; Kato K. Direct Synthesis of Hydrogen Peroxide in Water by Means of a Rh-Based Catalyst. Organometallics 2020, 39, 3731–3741. 10.1021/acs.organomet.0c00565. [DOI] [Google Scholar]
- Dunbar K. R. Spectroscopic and Structural Investigation of the Unbridged Dirhodium Cation [Rh2(CH3CN)10]4+. J. Am. Chem. Soc. 1988, 110, 8247–8249. 10.1021/ja00232a054. [DOI] [Google Scholar]
- Berry J. F.Metal-Metal Bonded Compounds of the Group IX Elements. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: 2021; pp 4–42. 10.1016/B978-0-08-102688-5.00103-3 [DOI] [Google Scholar]
- Mathieu R.; Esquius G.; Lugan N.; Pons J.; Ros J. Bis[(3,5-dimethyl-1-pyrazolyl)methyl]ethylamine – A Versatile Ligand for Complexation in RhI Cationic Complexes. Eur. J. Inorg. Chem. 2001, 2001, 2683–2688. 10.1002/1099-0682(200109)2001:10<2683::AID-EJIC2683>3.0.CO;2-T. [DOI] [Google Scholar]
- de Pater B. C.; Frühauf H.-W.; Vrieze K.; De Gelder R.; Baerends E. J.; McCormack D.; Lutz M.; Spek A. L.; Hartl F. Strongly Nucleophilic RhI Centre in Square-Planar Complexes with Terdentate (κ3) 2,2’:6’,2”-Terpyridine Ligands: Crystallographic, Electrochemical and Density Functional Theoretical Studies. Eur. J. Inorg. Chem. 2004, 2004, 1675–1686. 10.1002/ejic.200300699. [DOI] [Google Scholar]
- Gair J. J.; Qiu Y.; Chan N. H.; Filatov A. S.; Lewis J. C. Rhodium Complexes of 2,6-Bis(dialkylphosphinomethyl)pyridines: Improved C–H Activation, Expanded Reaction Scope, and Catalytic Direct Arylation. Organometallics 2017, 36, 4699–4706. 10.1021/acs.organomet.7b00532. [DOI] [Google Scholar]
- Chan A. K.-W.; Wu D.; Wong K. M.-C.; Yam V. W.-W. Rhodium(I) Complexes of Tridentate N-Donor Ligands and Their Supramolecular Assembly Studies. Inorg. Chem. 2016, 55, 3685–3691. 10.1021/acs.inorgchem.6b00289. [DOI] [PubMed] [Google Scholar]
- Chan A. K.-W.; Ng M.; Low K.-H.; Yam V. W.-W. Versatile Control of Directed Supramolecular Assembly via Subtle Changes of the Rhodium(I) Pincer Building Blocks. J. Am. Chem. Soc. 2018, 140, 8321–8329. 10.1021/jacs.8b04687. [DOI] [PubMed] [Google Scholar]
- Wan Q.; To W.-P.; Yang C.; Che C.-M. The Metal–Metal-to-Ligand Charge Transfer Excited State and Supramolecular Polymerization of Luminescent Pincer PdII–Isocyanide Complexes. Angew. Chem., Int. Ed. 2018, 57, 3089–3093. 10.1002/anie.201712249. [DOI] [PubMed] [Google Scholar]
- Yatabe T.; Kamitakahara K.; Higashijima K.; Ando T.; Matsumoto T.; Yoon K.-S.; Enomoto T.; Ogo S. Synthesis of acetic acid from CO2, CH3I and H2 using a water-soluble electron storage catalyst. Chem. Commun. 2021, 57, 4772–4774. 10.1039/D1CC01611C. [DOI] [PubMed] [Google Scholar]
- Yatabe T.; Futakuchi S.; Miyazawa K.; Shimauchi D.; Takahashi Y.; Yoon K.-S.; Nakai H.; Ogo S. Reductive C(sp3)–C(sp3) homo-coupling of benzyl or allyl halides with H2 using a water-soluble electron storage catalyst. RSC Adv. 2021, 11, 39450–39454. 10.1039/D1RA08596D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatabe T.; Tome T.; Takahashi Y.; Matsumoto T.; Yoon K.-S.; Nakai H.; Ogo S. C–H Arylation of Benzene with Aryl Halides using H2 and a Water-Soluble Rh-Based Electron Storage Catalyst. Chem.–Eur. J. 2021, 27, 17326–17330. 10.1002/chem.202102735. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Ishida N.; Imanaka T.; Teranishi S. Active States of Rhodium in Rhodium Exchanged Y Zeolite Catalysts for Hydrogenation of Ethylene and Acetylene and Dimerization of Ethylene Studied with X-Ray Photoelectron Spectroscopy. J. Catal. 1979, 58, 82–94. 10.1016/0021-9517(79)90247-1. [DOI] [Google Scholar]
- Stanger K. J.; Tang Y.; Anderegg J.; Angelici R. J. Arene hydrogenation using supported rhodium metal catalysts prepared from [Rh(COD)H]4, [Rh(COD)2]+BF4–, and [Rh(COD)Cl]2 adsorbed on SiO2 and Pd-SiO2. J. Mol. Catal. A: Chem. 2003, 202, 147–161. 10.1016/S1381-1169(03)00198-5. [DOI] [Google Scholar]
- Gobert S. R. L.; Kuhn S.; Braeken L.; Thomassen L. C. J. Characterization of Milli- and Microflow Reactors: Mixing Efficiency and Residence Time Distribution. Org. Process Res. Dev. 2017, 21, 531–542. 10.1021/acs.oprd.6b00359. [DOI] [Google Scholar]
- Matsubara C.; Kawamoto N.; Takamura K. Oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV): An Ultra-High Sensitivity Spectrophotometric Reagent for Hydrogen Peroxide. Analyst 1992, 117, 1781–1784. 10.1039/an9921701781. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.