


Abstract
The first 11 nt at the 5′ end of influenza virus genomic RNA were shown to be both necessary and sufficient for specific binding by the influenza virus polymerase. A novel in vitro transcription assay, in which the polymerase was bound to paramagnetic beads via a biotinylated 5′-vRNA oligonucleotide, was used to study the activities of different forms of the polymerase. Complexes composed of co-expressed PB1/PB2/PA proteins and a sub-complex composed of PB1/PA bound to the 5′-vRNA oligonucleotide, whereas PB1 expressed alone did not. The enriched 5′-vRNA/PB1/PB2/PA complex was highly active for ApG and globin mRNA primed transcription on a model 3′-vRNA template. RNA synthesis in the absence of added primers produced products with 5′-terminal tri- or diphosphate groups, indicating that genuine unprimed initiation of transcription also occurred. No transcriptase activity was detected for the PB1/PA complex. These results demonstrate a role for PA in the enhancement of 5′ end binding activity of PB1, a role for PB2 in the assembly of a polymerase complex able to perform both cap-dependent and -independent synthesis and that NP is not required for the initiation of replicative transcription.
INTRODUCTION
The influenza virus RNA polymerase is a multifunctional complex composed of the three viral proteins PB1, PB2 and PA, which, together with the viral nucleoprotein NP, form the minimum complement required for viral mRNA synthesis and replication (1). PB1 contains the polymerase active site for nucleotide addition (2,3), the sites for sequence specific binding to the conserved 5′ and 3′ terminal sequences of the negative and positive sense viral RNAs (4,5) and independent binding sites for the PB2 and PA polymerase subunits (6). PB2 binds to host cell capped mRNAs (7,8) and has been proposed as the viral endonuclease responsible for cleavage of these RNAs to form the primers for viral mRNA synthesis (9,10). However, other data strongly indicate that PB1 is the endonuclease (11). PB1 and PB2 also bind NP (12,13) and, while the function of these P–NP interactions is unknown, they are apparent in an electron-microscopic reconstruction of a recombinant mini-RNP (14). Genetic evidence from temperature sensitive mutations in the PA segment suggests that this protein may be involved in the transition from mRNA transcription to cRNA and vRNA synthesis (reviewed in 15). However, a clear role for PA in RNA synthesis has yet to be defined.
The polymerase complex must synthesise three different viral RNA species: viral mRNA; the cRNA replication intermediate; and negative sense vRNA. The 5′ end of the viral mRNA is composed of a cap structure and 10–15 nt derived from the host cell mRNA, which are used as primers for initiation (16). mRNA transcription terminates 15–17 nt before the end of the vRNA template, at which point the RNA is polyadenylated (17). cRNA is the full length complement of vRNA. The mechanism of mRNA synthesis is relatively well understood (reviewed in 18) and quite distinct from that of v- and cRNA. Much of this understanding has come from the use of in vitro systems for studying the polymerase. One such system uses recombinant vaccinia viruses to express the polymerase. This has provided important insights into the composition and formation of the active transcription complex (1,19,20). It has been used primarily to study the early events of transcription such as template binding, cap-binding and endonuclease activities. This work forms the basis of the model for the ordered assembly of the viral transcription complex responsible for mRNA synthesis (21). Unfortunately the extracts used in this system contain a substantial amount of ribonuclease activity that precludes its use for studying transcriptional elongation, termination or polyadenylation.
The mechanisms responsible for c- and vRNA synthesis remain very poorly characterised. The 5′ terminal base of vRNA and cRNA is adenosine triphosphate (22–24), which implies that initiation does not involve the use of a capped primer but is instead initiated de novo. It is not known if different forms of the polymerase perform different transcriptional functions and the precise subunit requirements for replicative transcription remain a matter of controversy. Evidence for complexes of PB1 and PA or even PB1 alone being capable of performing some of the transcriptional activities associated with genome replication (25,26) is contradicted by other studies that have concluded that PB2 is an essential part of the polymerase complex for all forms of transcription (27). The form of polymerase packaged into virions is unable to synthesise cRNA (28,29) and after infection an initial round of viral mRNA synthesis and translation is required before cRNA synthesis can occur (30). Although the involvement of host-cell proteins has been proposed (31,32), it is likely that this requirement for protein synthesis reflects the obligatory synthesis of a viral protein (e.g. NP and/or polymerase) for replicative transcription. Synthesis of a full-length cRNA requires readthrough of the polyadenylation signal (anti-termination), which in vitro, using infected cell extracts, is dependent upon the availability of NP (33,34). Consistent with an essential role for NP, several NP mutants have been isolated that are selectively defective for c- and/or vRNA synthesis (15,35–38). Two such mutants have RNA binding defects indicating that an NP–RNA interaction may be required for the control of replicative synthesis (13). Nevertheless, the precise role of NP in replicative transcription remains unclear. In particular, it is not known what causes the polymerase to swap from cap-primed transcription initiation to unprimed synthesis and whether NP is required for this step.
Here we report the identification of the minimal 5′-vRNA oligonucleotide capable of being bound by recombinant polymerase and its use as an affinity ligand to circumvent the limitations of the vaccinia virus-expression system to allow the transcriptional activities of a highly active recombinant polymerase to be measured. We show for the first time that transcription initiation in the absence of added primers, in vitro, produces RNA products with the authentic terminal structure expected for a true cRNA replicative intermediate produced in vivo. Furthermore, whereas the PB2 protein is required for this form of transcription initiation, the NP protein is not.
MATERIALS AND METHODS
Plasmids and antisera
Plasmid pMLV3′ contains the T7 promoter upstream of the 3′ terminal sequence of segment 8 of the A/PR/8/34 strain of influenza virus. Plasmid pPH-V has been described previously (19). It encodes a model pan handle template RNA composed of the 5′ and 3′ ends of segment 8 separated by a short linker.
Rabbit antisera against C-terminal peptides of PB1, PB2, PA, monoclonal antibody (10.2) specific for PB1 and polyclonal serum raised against amino acids 50–370 of PB1 (anti-PB1-F1) have been described previously (6,19). Hyperimmune polyclonal anti-PB2 serum (anti-MBP-2N580) was raised in rabbits using a maltose-binding protein fusion polypeptide containing amino acids 580–759 of PB2. Polyclonal anti-PA sera were raised in rabbits (6) with bacterially expressed β-galactosidase fusion proteins containing amino acids 16–213 (anti-PA-F2) or 187–340 (anti-PA-F3) of A/PR/8/34 PA.
Virus and cell culture
Recombinant vaccinia viruses that individually express the three influenza virus RNA polymerase proteins (VacPB1, VacPB2 and VacPA) (39) or express the T7 RNA polymerase protein (VTF 7.3) (40) were propagated on HeLa cell monolayers and titrated on BSC-1 cells using standard procedures (41).
Preparation of virus infected cell extracts
Nuclear extracts from vaccinia-infected HeLa cells were prepared as previously described (42,43). Briefly, HeLa cells grown as monolayers on 800 cm2 roller bottles were infected with the recombinant vaccinia viruses that express PB1, PB2 or PA (m.o.i. = 5 for each virus, for triple, double or single infections) or T7 RNA polymerase (m.o.i. = 10). Cells were harvested 18 h post infection and lysed in hypotonic buffer containing 0.2% Nonidet P-40. The nuclei were recovered, lysed in buffered 0.3 M ammonium sulphate and soluble nuclear proteins precipitated by raising the concentration of ammonium sulphate to 1.5 M. Precipitated proteins were redissolved in 50 mM HEPES pH 7.6, 50 mM KCl, 0.1 mM EDTA, 1mM dithiothreitol (DTT) at ∼10 mg/ml and stored in small aliquots at –70°C. The presence (or absence) of the expected P proteins in the extracts was confirmed by western blot analysis (data not shown).
Synthesis of template RNAs and RNA probes
Plasmids pPH-V and pMLV3′ were linearised with MboII and transcribed using T7 RNA polymerase to generate RNAs designated: PHV, 5′-AGUAGAAACAAGGGUGUUUUUUCAGAUCUAUUAAACUUCACCCUGCUUUUGCU-3′; and 3′V, 5′-GGAGACGAAUUCGGAUCCACACCCUGCUUUUGCU-3′, respectively, where the underlined letters correspond to the conserved 5′ and/or 3′ terminal vRNA sequences. The transcripts were purified by electrophoresis through 18% polyacrylamide gels containing 7 M urea. The RNA bands were located by UV shadowing, excised and recovered by passive elution into 1 mM EDTA. The RNAs were then ethanol precipitated and quantitated using the Ribogreen™ assay (Molecular Probes Inc.). Radiolabelled RNAs were prepared by phosphorylation of the 5′ end of the RNA transcripts using polynucleotide kinase (Boehringer Mannheim, Indianapolis, IN) and [γ-32P]ATP (3000 Ci/mmol) and were repurified as described above. A synthetic RNA oligonucleotide (5′V16B) was synthesised (Cruachem Ltd) with the sequence: 5′-AGUAGAAACAAGGGUG(CH2)12-biotin.
Defining the minimal length requirements for 5′ end binding
5′ end labelled PHV RNA (~5 × 106 c.p.m.) was subjected to partial alkaline hydrolysis to produce a probe mixture comprised of nested 5′ co-terminal labelled RNAs. The hydrolysis reaction was terminated by adding sufficient 0.1 N hydrochloric acid to neutralise the sample. Binding reactions (20 µl) containing 5 × 105 c.p.m. of probe RNA and 20 µg of nuclear extract were prepared as described above. The samples were then centrifuged through 10–20% w/v sucrose gradients at 37 000 r.p.m. (164 000 g) for 18 h at 3°C using a Beckman SW50.1 rotor as described previously (19). Fractions (200 µl) were collected drop-wise from the bottom of the tube. Aliquots of 10 µl from each fraction were analysed in a scintillation counter to determine the migration of the probe RNA. 2000 c.p.m. from the bound and free peaks on the gradient were then analysed on a 20% 7 M urea sequencing gel and compared to the original hydrolysis ladder, and to an RNA sequencing ladder produced using an RNA sequencing kit (Pharmacia LKB).
Affinity purification of the polymerase and in vitro transcription reactions
Streptavidin coated paramagnetic beads (SA-beads, Dynal, M280) were prepared by washing twice with 0.1 M NaOH/50 mM NaCl, once with 100 mM NaCl and once with minimal binding buffer (MBB: 10 mM HEPES pH 7.5, 0.5 mM EGTA, 2 mM MgCl2, 120 mM KCl, 10% glycerol) + 0.1% BSA and then resuspended in MBB. SA-beads, 5 µl per reaction, were incubated with 1 pmol of the biotinylated RNA oligonucleotide (5′V16B) in 10 µl 50 mM Tris pH 7.5, 0.5 mM EDTA, 1 M NaCl (TEN buffer) at room temperature for 10 min to allow binding of the oligonucleotide to the SA-beads. The reaction was washed twice with TEN buffer and once with MBB. The SA-beads were finally resuspended in 10 µl complete binding buffer (10 mM HEPES pH 7.5, 0.5 mM EDTA, 1.8 mM MgCl2, 120 mM KCl, 10% glycerol, 10 U RNasin, 1 mM DTT) containing 10 µg of nuclear extract and incubated at room temperature for 15 min. The SA-beads were then washed twice with 100 µl MBB. Samples for western blotting and silver staining experiments were resuspended in 1× SDS–PAGE loading buffer. Samples intended for transcription reactions were resuspended in 10 µl transcription buffer containing the appropriate primer, nucleotides and template. Typical reaction conditions were 50 mM Tris pH 8.2, 100 mM KCl, 2 mM MgCl2, 100 ng tRNA, 10 mM DTT, 10 pmol 3′V template, 5 µCi [α-32P]GTP (400 Ci/mmol; Amersham Pharmacia Biotech Ltd), 0.8 mM ATP, 0.4 mM CTP, 0.2 mM UTP, 1 µM GTP and 0.5 mM ApG or 20 ng globin mRNA (Gibco-BRL). Reactions were incubated at 30°C for 30 min and terminated by adding 10 µl of RNA loading dye mix (80% formamide, 0.2× TBE, 0.05% bromophenol blue, xylene cyanol, Orange G). Transcription products were resolved by electrophoresis on an 18% (19:1 acrylamide:bisacrylamide) 7 M urea gel maintained accurately at 55°C using a heat exchanger and visualised by autoradiography at –70°C without prior drying of the gel. Exposure times ranged from 16 to 120 h as indicated in the figure legends.
Gel shift assays
Assays were performed as described previously (19). Binding reactions of 10 µl contained 1–2 × 104 c.p.m. of RNA probe and 10 µg of nuclear extract. The final buffer components were: 20 mM HEPES pH 7.6, 0.5 mM EGTA, 20 µM EDTA, 2 mM MgCl2, 7.5 mM NaCl, 110 mM KCl, 1.2 mM DTT, 27.5 µg/ml RNase free BSA (Boehringer Mannheim), 1 U/µl RNAGuard (Pharmacia) and 12% glycerol. Binding reactions were incubated at room temperature for 30 min and terminated by the addition of heparin (Sigma Chemical Co.) to a final concentration of 5 mg/ml. Complexes were resolved by native gel electrophoresis (4%; 79:1 acrylamide:bis acrylamide, 50 mM Tris–HCl pH 8.4, 50 mM glycine, 3% glycerol, 12 V/cm), and visualised by autoradiography.
SDS–PAGE and western blot analysis
Samples from total nuclear extracts or SA-bead purified polymerase were resuspended in SDS–PAGE loading buffer containing 10% 2-mercaptoethanol, boiled for 5 min and then analysed by discontinuous PAGE (4.5% stacking gel and 7.5% resolving gel). Resolved proteins were visualised by silver staining (Silver Stain Plus, Bio-Rad Laboratories). For western blots, proteins were transferred to PVDF or nitrocellulose membranes (Amersham Life Science or Schleicher and Schuell, respectively) by semi-dry electroblotting (Bio-Rad Laboratories). Membranes were probed with antisera as indicated in the figure legends. Bound antibodies were detected using biotin conjugated goat, anti-rabbit or anti-mouse IgG secondary antibody as appropriate (Sigma-Aldrich Co.) followed by either streptavidin conjugated alkaline phosphatase/BCIP/NBT development (Sigma-Aldrich Co.) for Figure 2 only; or streptavidin conjugated horseradish peroxidase (Pierce) chemiluminescent development according to the manufacturer’s protocol (BM Chemiluminescence Blotting substrate, Roche Diagnostics Ltd).
Figure 2.
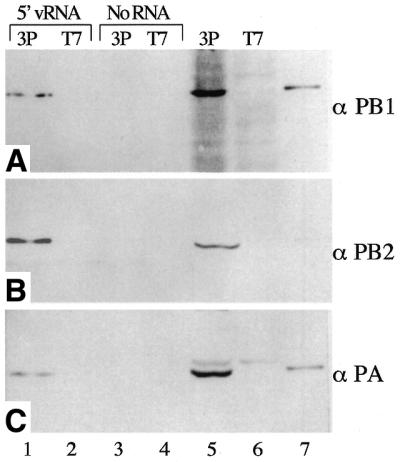
Composition of the P protein complex bound to 5′-vRNA. Nuclear extracts from Vac3P or VacT7 infected HeLa cells were incubated with a biotinylated RNA oligonucleotide containing the conserved 5′ terminal sequence of vRNA bound to streptavidin coated paramagnetic beads. Bound proteins were then analysed by western blot using C-terminal peptide antibodies specific for (A) PB1, (B) PB2 and (C) PA. Lane 1, 5′-vRNA coated SA-beads incubated with Vac3P extract. Lane 2, 5′-vRNA coated SA-beads incubated with VacT7 extract. Lanes 3 and 4, uncoated SA-beads incubated with Vac3P or VacT7 extracts, respectively. Lane 5, total Vac3P extract. Lane 6, total VacT7 extract. Lane 7, purified A/PR/8/34 virus marker.
Analysis of 5′ end groups
Standard transcription assays were scaled up proportionately to 200 µl for unprimed synthesis or 80 µl for ApG, GpppG and globin mRNA primed synthesis. The reactions were performed as described above except that UTP was omitted from the reaction mixture to prevent the polymerase from transcribing beyond nucleotide 13 of the 3′V template (first A occurs at position 14). The reactions were terminated by addition of an equal volume of phenol/chloroform, phenol extracted and ethanol precipitated twice from 2.5 M ammonium acetate to reduce NTP levels. Each sample was finally redissolved in 20 µl of water. Five microlitres of each sample was then treated with 3 U of calf intestinal phosphatase (CIP; Roche Diagnostics Ltd) or tobacco acid pyrophosphatase (TAP; Epicenter Technologies) (44) for 1 h at 50 or 37°C, respectively, in the reaction buffers supplied with the enzymes. For analysis using guanylyl transferase (GTase) it was necessary to disrupt the RNA/RNA duplex formed by the template 3′-vRNA and the 5′-cRNA product RNA. The samples were prepared as described above, except that after ethanol precipitation the samples were redissolved in GTase reaction buffer (50 mM Tris–HCl pH 7.9, 1.2 mM MgCl2, 6 mM KCl) incubated with a 30-fold excess of biotinylated 5′-cRNA oligonucleotide at 80°C for 2 min and then cooled to 37°C to allow the RNA to anneal to the 3′-vRNA template RNA. The samples were then incubated with SA-beads to remove the annealed RNAs. The samples were supplemented with 3 mM DTT, 2 U RNasin and GTase (45), and incubated for 2 h at 37°C. To avoid any possible electrophoresis artifacts due to differences in the reaction components, mock treated controls were incubated under identical conditions in the absence of the enzyme for each sample. All reactions were terminated by the addition of 80% formamide/1 mM EDTA RNA loading buffer and heated to 90°C for 2 min. The reaction products were analysed by 7 M urea, 18% PAGE as described above.
RESULTS
Definition of a minimal length 5′-vRNA sequence for polymerase binding
Mutational and modification interference analysis of the 5′-vRNA sequence has defined the specific bases required for binding by the polymerase. Nucleotides A1, G2, U3, A8, C9, A10 and A11 of the wild-type sequence are involved in binding, either directly or through short range interactions, to form a hooked RNA secondary structure (19,46–49). However, it remained to be demonstrated what constituted the minimal polymerase binding region on the RNA and whether any non-base-specific interactions outside this region were involved. To this end, a synthetic 5′ end-labelled vRNA was subjected to partial alkaline hydrolysis to generate a nested set of 5′ co-terminal labelled RNAs that run as a ladder of products on a denaturing gel (Fig. 1, lane 5). This population of RNAs was incubated with sufficient Vac3P nuclear extract to bind ∼30% of the input RNA and bound and free populations of RNA separated by sucrose gradient centrifugation. Under the conditions used, binding is specific, and no complex was detected for control extracts lacking the polymerase (19). RNA remaining at the top of the gradient was almost totally degraded by the nucleases present in the extract (Fig. 1, lane 6). However, RNA that migrated through the gradient to the position associated with the 3P polymerase (Fig. 1, lane 7) was not degraded, as evidenced by the predominant band corresponding to the full length RNA and the distribution of smaller RNAs corresponding to alkaline hydrolysis products that were still able to bind to the polymerase. RNAs below a certain size no longer bound and the cut-off point could be identified by comparison with the corresponding sequencing ladder (Fig. 1, lanes 1–3, migration of the bound RNA was altered slightly by the presence of sucrose from the gradient). This showed that RNAs including at least the region from the 5′ end to base A11 of vRNA bound to the polymerase but smaller fragments of the 5′-terminus did not. The presence of a band corresponding to G2 in the bound fraction (Fig. 1, lane 7) may indicate an affinity of the polymerase for ApG, but is most likely explained as a trace contaminant from the abundant G2 degradation product found in the free population (Fig. 1, lane 6). The smallest fragment of vRNA bound by the polymerase contains all the bases previously shown to be critical for polymerase binding (19,46) thus demonstrating that no other RNA contacts outside this region are necessary for this interaction.
Figure 1.
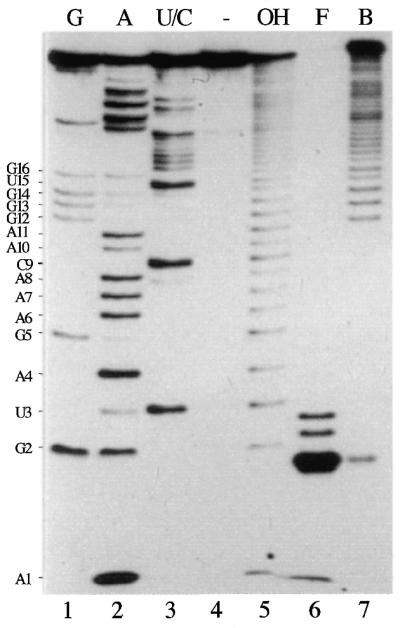
Defining the minimum length 5′ end binding site for the influenza polymerase. 5′ end labelled PHV RNA was subjected to limited alkaline hydrolysis to produce a ladder of labelled 5′ co-terminal RNA species (lane 5). 5 × 105 c.p.m. of hydrolysed RNA was mixed with Vac3P extract and bound and free populations separated by sucrose gradient centrifugation. Equal amounts of RNA from the bound and free peaks were then analysed on a 7 M urea, 20% acrylamide gel. Lanes 1–3, RNA sequencing ladder using base-specific ribonucleases T1 (G), U2 (A) and B.cereus (U/C). Lane 4, untreated RNA. Lane 6, free (non-bound) RNA. Lane 7, bound RNA.
Identification of polymerase subunits required for 5′ end binding
The polymerase packaged into influenza virions consists of one copy each of the three P proteins (50), but it is less clear if a trimeric polymerase is the minimal functional unit in infected cells (25,26). Previous studies on recombinant polymerase in vitro demonstrated that co-expression of all three P proteins was necessary to form an enzymatically active complex (9,19,20). Analysis of template binding activity by gel shift assays combined with P-specific antibody supershifts confirmed the presence of PB1 and PB2 in the complex, but failed to show the presence of PA (19). This was attributed to the PA antibodies being unable to bind PA under the native conditions used in the assay. To overcome this limitation, an alternative strategy was used where the RNA/protein complexes were recovered and the protein component analysed by western blot, for which there are many suitable antibodies. To facilitate recovery of P-protein/RNA complexes, a short RNA molecule containing the essential 5′-vRNA binding site was bound to streptavidin-coated paramagnetic beads (SA-beads) via a 3′ terminal biotin group. The SA-beads were then incubated with nuclear extracts containing the P proteins or T7 RNA polymerase as a negative control. After thorough washing, the bound polypeptides were analysed by western blotting using antibodies to PB1, PB2 or PA (Fig. 2). All three P proteins were detected in the total Vac3P extract, but not in the negative control VacT7 extract (Fig. 2, lanes 5 and 6). The recombinant P proteins were seen to co-migrate with the authentic P proteins in purified A/PR/8/34 virions (lanes 5 and 7), although in this experiment the antipeptide antiserum to the C-terminus of PB2 reacted poorly with the A/PR/8/34 sample (Fig 2, lane 7). Cross reactivity of the anti-PA serum with an unidentified polypeptide with slightly slower electrophoretic mobility than PA was also noted (Fig. 2C, lanes 5 and 6). No detectable binding of the P proteins to the SA-beads was observed in the absence of the biotinylated 5′-vRNA oligonucleotide (Fig. 2, lane 3). However, in the presence of the 5′-vRNA sequence, PB1, PB2 and PA all bound to the SA-beads (Fig. 2, lane 1). This confirms that PA is indeed a component of the binding complex(es) present in the nuclear extracts.
PB1 has been shown by cross-linking and northwestern blot analysis to be the protein most directly involved in 5′ end binding (4,51), but other experiments suggest that all three P proteins play a role (19,20,52). The minimum complement of P proteins required for binding to a model panhandle vRNA was determined using a gel-shift assay with extracts prepared from cells infected with pair-wise combinations of P proteins. Binding of a protein complex to the RNA causes the migration of the probe RNA to be retarded. A slower migrating complex was observed when the probe was incubated with extracts from Vac3P or VacPB1/PA infected cells (Fig. 3A, lanes 1 and 4). The extract containing only PB2 and PA did not cause retardation of the RNA probe significantly above that seen for the control extract (Fig. 3A, lanes 5 and 6). This is consistent with a major role for PB1 in template binding. However, extracts containing PB1 alone, or PB1 and PB2 also did not bind (Fig. 3A, lanes 2 and 3). This implies that both PB1 and PA are required for template binding. The 3P complex and the PB1/PA complex had very similar electrophoretic mobilities, although it might have been expected that the 3P complex containing PB2 would migrate more slowly. The presence or absence of PB1, PB2 and PA in the appropriate extracts and complexes was confirmed by western blot and antibody supershift experiments, respectively (data not shown). Because none of the available antibodies against PA caused super-shifting, further confirmation of the composition of the complexes was therefore obtained using the SA-bead binding assay. The results were in complete agreement with those from the gel shift assay. No binding of P proteins was observed with extracts containing PB1 alone, PB2 and PA, or PB1 and PB2 (Fig. 3B, lanes 3, 4 and 6). In contrast, when PB1 and PA were co-expressed, either with or without PB2, both proteins bound to the RNA-coated SA-beads (Fig. 3B, lanes 1 and 5). We therefore conclude that PA is required for efficient binding of PB1 to the 5′-terminus of vRNA.
Figure 3.

Binding of polymerase sub-complexes to 5′-vRNA sequences. (A) Gel shift analysis of polymerase sub-complexes. Nuclear extracts (10 µg total protein) from cells infected with vaccinia viruses expressing the indicated proteins were incubated with radiolabelled PHV RNA and analysed by non-denaturing gel electrophoresis. The migration of the bound and the free probe is indicated. (B) 5′-vRNA bound to SA-beads was incubated with nuclear extracts (5 µg total protein) from HeLa cells infected with: Vac3P (lane 1), VacT7 (lane 2), VacPB1 (lane 3), VacPB2 + VacPA (lane 4), VacPB1 + VacPA (lane 5) and VacPB1 + VacPB2 (lane 6). Bound proteins were resolved by SDS–PAGE and analysed by chemiluminescent western blotting using antibodies specific for the individual P proteins as indicated (PB1, monoclonal antibody 10.2; PB2, anti-MBP-2N580; PA, 1:1 mixture of anti-PA-F2 and anti-PA-F3). Lane 7, purified A/PR/8/34 virus marker.
Affinity purification of RNA polymerase using a synthetic RNA ligand
The ability of the polymerase to bind stably to short RNA molecules containing the 5′-vRNA terminal sequences bound to a solid matrix presented an avenue for selective enrichment of the polymerase. To assess the degree of purification, proteins that bound the immobilised 5′-vRNA were analysed by SDS–PAGE and silver staining. Comparison between the unfractionated Vac3P and VacT7 extracts (Fig. 4A, lanes 4 and 5) demonstrated that the proportion of P protein to total protein was low, with no identifiable P-specific polypeptides in the Vac3P extract. Analysis of material bound to the 5′-vRNA-coated SA-beads showed a selective enrichment of the polymerase such that polypeptides of the expected molecular weights for PB1 and PB2/PA (PB2 and PA co-migrate in this gel system) were now clearly visible in the Vac3P but not the VacT7 extract (Fig. 4A, lanes 1 and 3, respectively). Parallel western blot analysis with anti-PB1 and -PB2 sera showed that these polypeptides co-migrated with PB1 and PB2, respectively (Fig. 4B and C, lanes 1). Heparin is known to inhibit binding of the polymerase to the 5′ end of vRNA, although it does not efficiently displace polymerase that has already bound (19). Inclusion of heparin in the binding reaction strongly inhibited binding of the polymerase to the SA-beads but did not substantially alter the binding of the background polypeptides (Fig. 4A–C, lanes 2) indicating that they are not bound to the bead through any affinity for the polymerase itself. In addition, binding of the polymerase (PB1) to the immobilised 5′ end could be inhibited completely by a 20-fold excess of a panhandle RNA containing the 5′-vRNA sequence, thus confirming the specificity of the interaction (Fig. 4D, lane 7). To determine the approximate amount of polymerase that bound to the bead, a comparison was made between the amount of bound PB1 and a titration of virion associated PB1. Based on the estimate that each P protein comprises ∼1% of the total protein in virions (53), ∼5 ng of PB1 bound to the SA-beads (Fig. 4D, lane 8). This is a small fraction of the PB1 present in the total extract (∼75 ng; Fig. 4D, lane 1).
Figure 4.
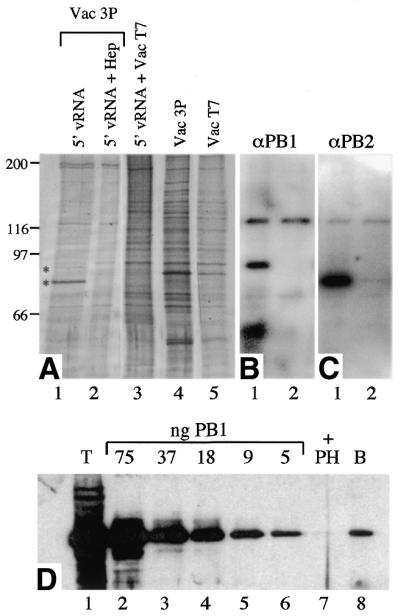
Selective enrichment of influenza virus RNA polymerase using immobilised 5′-vRNA oligonucleotide. (A) Nuclear extracts from Vac3P or VacT7 infected HeLa cells were analysed by SDS–PAGE and silver stained before or after binding to SA-beads coated with the 5′vRNA oligonucleotide in the presence or absence of 5 mg/ml heparin (Hep) as indicated. Identities of the protein bands were determined by comparison between silver stained gel and duplicate western blots probed with antibodies specific for PB1, anti-PB1-F1 (B) or PB2, anti-MBP-2N580 (C). Lane 1, Vac3P incubated with SA-beads coated in 5′-vRNA. Lane 2, Vac3P in the presence of 5 mg/ml heparin incubated with SA-beads coated in 5′-vRNA. Lane 3, VacT7 incubated with SA-beads coated in 5′vRNA. Lane 4, crude Vac3P extract. Lane 5, crude VacT7 extract. (D) Estimation of the quantity of PB1 bound to the immobilised vRNA oligonucleotide. Vac3P nuclear extract was analysed by SDS–PAGE and western blotting with anti-PB1 MAb 10.2 before (lane 1, T) or after binding to RNA-coated SA-beads in the presence (lane 7, +PH) or absence (lane 8, B) of a non-biotinylated competitor panhandle RNA in parallel with a dilution series of purified A/PR/8/34 virions (lanes 2–6). Also indicated are the approximate quantities of PB1 present in each virion sample.
Transcriptional activity of the immobilised polymerase
The sequence of the oligonucleotide used for the affinity purification was deliberately chosen to include four bases 3′ to the minimal binding site defined in Figure 1. This was done for two reasons. First, it was possible that the physical proximity of the SA-bead relative to the polymerase binding sequence could cause steric problems. Extending the sequence by a few bases would allow more flexibility for binding. Secondly, these additional bases are known to be crucial for efficient transcription of template RNAs, where they are probably involved in 3′ template recognition and binding (47,54). Therefore, by including these sequences, it was realistic to expect that the polymerase might be transcriptionally active while still bound to the SA-bead. In addition, enrichment of the polymerase should reduce the level of nucleases that prevented transcriptional activity of the vaccinia virus-expressed polymerase from being studied satisfactorily in the past. To test this, the bound polymerase was resuspended in transcription buffer containing ribonucleotides, [α-32P]GTP, a 34 nt in vitro transcribed RNA containing the 3′-terminal sequence from A/PR/8/34 segment 8 vRNA, and either ApG, globin mRNA (as a model cellular capped RNA) or no primer. After incubation at 30°C for 30 min the reaction products were analysed by denaturing PAGE. ApG primed transcription produced a major reaction product 34 nt long that corresponds to the predicted full-length transcription product for the template used in this assay (Fig. 5A, lane 1 and 5B, lane 2). In contrast, no full-length products were synthesised by the 3P nuclear extract incubated with the same reaction components if the polymerase enrichment step was omitted (Fig. 5B, lane 8), providing a clear illustration of the improvement this new assay represents. Reactions primed with globin mRNA gave rise to a longer product of ∼46 nt as expected for a full length product containing an additional 12 globin mRNA derived bases and cap-structure at the 5′ end (Fig. 5A, lane 2 and 5B, lane 3). Synthesis of these products was dependent on the inclusion of polymerase, biotinylated 5′ vRNA and 3′ template as omission of any single component ablated activity (Fig. 5A, lanes 3, 4 and 7).
Figure 5.

Immobilised affinity purified polymerase is transcriptionally active. Nuclear extracts from Vac3P or VacT7 infected HeLa cells were mixed with SA-beads coated with the 5′-v16B oligonucleotide. The SA-beads were then resuspended in transcription buffer containing nucleotides, the indicated primer and 3′-vRNA template. Transcription products incorporated [α- 32P]GTP and were resolved on an 18% 7 M urea denaturing polyacrylamide gel and visualised by autoradiography for (A) 16 or (B) 120 h. (A) Lane 1: ApG primed reaction. Lane 2, globin mRNA primed reaction. Lane 3, ApG primed reaction with the omission of the 5′-v16B oligonucleotide. Lane 4, ApG primed reaction with the omission of 3′V template RNA. Lane 5, unprimed reaction. Lane 6, globin mRNA primed reaction using VacT7 nuclear extract. Lane 7, ApG primed reaction using VacT7 extract. (B) Co-expression of PB2 is required for unprimed, ApG primed, globin mRNA primed transcription. Lanes 1–3, SA beads coated with Vac3P. Lanes 4–6, SA beads coated with VacPB1/PA. Lane 7, SA beads coated with VacT7. Lane 8, Vac3P extract plus 5′-v16B (omitting bead selection procedure). Lanes 1 and 4, unprimed transcription. Lanes 2, 5, 7 and 8, ApG primed transcription. Lanes 3 and 6, globin mRNA primed. Lane 4, VacPB1/PA, unprimed synthesis.
Significantly, low level synthesis of two specific products was also observed in the absence of added primer (Fig. 5B, lane 1 with lane 4 serving as a control). One transcription product migrated slightly faster than the 34 nt ApG primed product (Fig. 5B, compare lanes 1 and 2). This is consistent with the increased electrophoretic mobility expected for an RNA with a 5′-triphosphate characteristic of unprimed replicative synthesis initiated with ATP (ApG-primed synthesis gives rise to a 5′-hydroxyl). A second product of the predicted size for mRNA primed transcripts (~46 nt) was also observed in the absence of added primer (Fig. 5B, lane 1) suggesting that some capped mRNA-primed transcription might be occurring even though no exogenous primer had been added. One explanation for this is that there are low levels of capped RNAs present in the nuclear extract that associate with the polymerase after it has bound to the immobilised 5′ end and can function as primers. Several diffuse background bands were observed even in the absence of influenza virus polymerase, primer, 5′-vRNA or template (Fig. 5A, lanes 3–7) and are not considered to be significant. However, a prominent group of prematurely terminated influenza virus specific transcription products, most notably ~14 nt were obtained in ApG primed reactions, although interestingly, not in those primed with globin mRNA (Fig. 5A, lanes 1 and 2 and 5B, lanes 2, 3 and 8).
PB2 is essential for in vitro transcription activity
There is some evidence that polymerase complexes lacking PB2 have residual transcriptional activity (25,26). Having shown that complexes of PB1 and PA are able to bind efficiently to the 5′ end of vRNA (Fig. 3), the same extracts containing incomplete combinations of P proteins were analysed for their transcriptional activity. As expected, no activity was detected from combinations lacking PB1 and/or PA (data not shown). However, despite the substantial level of vRNA binding activity present in the PB1/PA extract shown in Figure 3, no significant transcriptional activity was detected for either ApG, mRNA or unprimed reactions (Fig. 5B, lanes 4–6). No bands of the predicted sizes were observed and any trace products were also present in the T7 control (Fig. 5B, lane 7). Therefore, we conclude that PB2 is an essential component of the polymerase complex for all forms of transcription in vitro.
Characterisation of the unprimed transcription products
To confirm the identity of the two species of products observed in unprimed transcription reactions, it was necessary to examine the nature of their 5′ ends. Unprimed synthesis is expected to produce a product with a 5′-terminal adenosine triphosphate, ApG primed synthesis would have a 5′ hydroxyl, and mRNA primed products would have a terminal m7GpppG cap structure. These can readily be distinguished by the differential effects of treatment with CIP, GTase and TAP. CIP enzyme removes the 5′ phosphate of polynucleotides, leaving a 5′ hydroxyl that causes the RNA to migrate slightly more slowly on PAGE. GTase catalyses the transfer of GMP from GTP onto RNA possessing a 5′ di- or triphosphate group (55), thus decreasing the mobility of the product. TAP removes the 5′ capping G residue, increasing the electrophoretic mobility of the RNA. It also hydrolyses 5′ nucleotide triphosphates to the monophosphate (44). To maximise the changes in relative mobility of the RNA products, transcription reactions were modified by omitting UTP. This causes transcription to terminate when the polymerase encounters the first adenine base at position 14 in the template, thus producing a 13 nt product that can be more readily resolved. This also rules out the possibility that the product being analysed is merely a small fraction of the input (34 nt) template RNA that has been labelled by some spurious mechanism. Under these conditions, the 13 nt product of unprimed transcription clearly migrated faster than the major product of ApG primed transcription (Fig. 6A, lanes 1 and 5). Treatment with CIP altered the migration of this RNA such that it now co-migrated with the ApG primed product that was itself left unaffected by CIP treatment (Fig. 6A, lanes 2 and 6). This confirmed that the major product synthesised in the absence of added primer has a 5′ terminal phosphate group. Migration of the 13 nt product of unprimed transcription was not affected by TAP treatment (Fig 6A, lane 4) because 5′ triphosphate and monophosphate RNA oligonucleotides are not resolved under these conditions (data not shown). Therefore, to examine the number of 5′ phosphate groups present on the 13 nt unprimed transcription product we tested the effect of GTase treatment. A substantial fraction of the product migrated more slowly after incubation with GTase (Fig 6B, lanes 3 and 4), consistent with the addition of a capping guanosine residue. In contrast, no shift in mobility was observed for the ApG primed product (Fig. 6B, lanes 1 and 2). Therefore unprimed synthesis by the polymerase gives rise to a product that contains a 5′ di- or triphosphate group. Unprimed transcription also gave rise to a larger product that was potentially the result of priming with host cell mRNAs present in the nuclear extracts. Consistent with this, under the UTP-deficient transcription reaction conditions these minor transcription products resolved into several distinct bands, as would be predicted for priming by a heterogeneous mixture of host cell mRNAs (indicated by asterisk, Fig. 6A, lanes 1–4). Furthermore, while treatment with CIP did not affect these RNAs (Fig. 6A, lane 2), TAP treatment increased their electrophoretic mobility as predicted for the removal of the m7GDP group (Fig. 6A, lane 4). TAP treatment of the globin mRNA primed product similarly confirmed that it possessed a 5′ terminal cap structure (Fig. 6A, lanes 15 and 16), as did two larger, minor products of the transcription reaction. CIP treatment did not alter the migration of these products, but did however affect a minor RNA species that co-migrated with the 13 nt major unprimed transcription product (Fig. 6A, lanes 13 and 14). This RNA is probably the result of unprimed transcription initiation occurring even in the presence of a donor mRNA. However, another possible explanation is that it corresponds to the 3′ endonucleolytic fragment of a globin mRNA primed product that has subsequently been rebound and cleaved by the polymerase (56), as this would be expected to have a 5′ monophosphate. The same argument could also be applied to the fraction of the 13 nt product obtained in the absence of exogenous primer that was not capped by GT (Fig. 6B, lane 4). This is unlikely, given the low level of capped products present in the unprimed reactions and the very different ratios of capped to uncapped products obtained between unprimed and globin mRNA primed reactions (Fig. 6A, lanes 1 and 13). In addition, control experiments indicated that GTase mediated capping reactions proceed inefficiently under the conditions used here because of the formation of RNA duplexes between product and template (data not shown). Nevertheless, to test the possibility that the polymerase was endonucleolytically cleaving its own mRNA products, we examined the effects of adding a large excess of free cap analogue to the transcription reactions to compete out polymerase binding to capped RNA oligonucleotides. The addition of 0.4 mM m7GpppG completely abolished synthesis of the ∼25 nt TAP sensitive capped RNA products (Fig. 6A, lanes 9–12). However, a 13 nt CIP sensitive species was still synthesised (Fig. 6A, lanes 9 and 10). This strongly suggests that this RNA is not derived from cleavage of previously synthesised viral mRNAs but instead is an authentic product resulting from unprimed transcription initiation. Curiously, reactions containing m7GpppG also gave rise to a product that co-migrated with the major ApG primed RNA (Fig. 6A, lane 9). However, this RNA species was insensitive to TAP and CIP treatment (Fig. 6A, lanes 7 and 8), indicating that it is not capped or 5′-phosphorylated and is therefore most plausibly the result of a contaminant in the m7GpppG preparation, possibly GpG.
Figure 6.

Analysis of the 5′ terminal groups of the product RNAs. Transcription was performed in the absence of UTP to stall the polymerase at position 14 of the template. The reaction products were recovered by phenol extraction and ethanol precipitation, resuspended in the appropriate buffer and incubated in the presence (+) or absence (–) of CIP, TAP or GTase as indicated. The reaction products were then analysed by denaturing electrophoresis using 7 M urea 18% PAGE. The bands corresponding to the major 13 nt unprimed product and mRNA primed products (*) are indicated. (A) Lanes 1–4, transcription in the absence of any added primer. Lanes 5–8, ApG primed transcription. Lanes 9–12, unprimed transcription in the presence of 0.4 mM m7GpppG. Lanes 13–16, globin mRNA primed transcription. (B) Product RNAs from ApG (lanes 1 and 2) or unprimed reactions (lanes 3 and 4) were prepared as above and depleted of template RNA as described in the Materials and Methods. The samples were then treated with GTase (lanes 2 and 4) or mock treated (lanes 1 and 3) and analysed by PAGE as above.
DISCUSSION
Expression of influenza virus RNA polymerase using vaccinia virus vectors has been key to elucidating the template-binding, cap-binding and endonuclease activities of the polymerase (1,19–21,39,57). However, because the extracts contain significant levels of nuclease activity they have not been suitable for studying the transcriptional activities of the polymerase. This limitation has now been overcome by the development of a novel assay system that uses the affinity of the enzyme for the conserved 5′-terminal sequences of the viral genome to purify it away from the deleterious nucleases. An important advantage this strategy has over conventional purification approaches is that the polymerase is purified by virtue of one of its functional properties, namely template binding. Previous studies using polymerase expressed from heterologous systems have shown that the polymerase has a tendency to form a wide spectrum of complexes ranging from monomers to higher order aggregates (6,52). The inclusion of an activity-based affinity purification step is an effective way to remove non-functional polymerase molecules. Repetitive depletion experiments using the SA-beads have shown that the majority of the transcriptionally active polymerase that is able to bind to the beads does so during the first round (data not shown). We estimate that <10% of the Vac3P polymerase is transcriptionally active. Although the bead enrichment is very significant from a qualitative aspect (i.e. in terms of transcriptional activity) the polymerase is clearly not pure (Fig. 4, lane 1). The purity achieved is comparable to that of recent attempts using His-tagged proteins expressed in Pichia pastoris (58) or baculovirus systems (59). However, neither of these systems displays appreciable levels of transcription initiation in the absence of an added primer, which precludes their use for the enzymatic analyses of 5′ end structure reported here. In addition, the affinity selection system permits the analysis of wild-type, non-tagged polymerase.
Efficient binding to the 5′ end of vRNA was shown to be dependent on the formation of a complex between PB1 and PA, with or without the inclusion of PB2. No binding was observed for PB1 alone using either a gel shift assay or SA-bead immobilised 5′-vRNA sequences. This is in contrast to what has been observed using co-immunoprecipitation assays and northwestern blotting where binding of PB1 alone has been clearly demonstrated (51). This discrepancy is not due to differences between the A/PR/8/34 and A/Victoria/72 virus strains used in the two studies, because we find that the A/PR/8/34 PB1 protein is capable of binding 5′-terminal sequences in northwestern blot experiments (E.Medcalf and P.Digard, unpublished experiments). However, it may arise from differences between the assay systems used. One possibility is that PA increases the affinity of PB1 for 5′-terminal sequences, and that in our system, the concentration of PB1 is too low to detect binding in the absence of PA. The estimated apparent Kd for the PB1–5′ end interaction is ∼2 × 10–8 M (51) and this is slightly lower than the concentration of PB1 we estimate to be present in our binding reactions (∼8 × 10–8 M). One would therefore expect to see some binding of PB1 alone to the immobilised 5′-vRNA oligonucleotide. However, the estimates of PB1 concentration in both studies are subject to a significant degree of error, and furthermore, it is possible that the proportion of functional PB1 in the extracts varies from one preparation to another. The finding that PA is necessary for a stable interaction with the 5′ end of vRNA under the conditions used here could be explained by a chaperone effect on PB1, or alternatively by a PA–RNA interaction that increases the avidity of the protein–RNA interaction. The latter possibility is consistent with earlier work that showed PA could be UV cross-linked to a radiolabelled 5′-vRNA molecule (46).
The purified 3P complex was transcriptionally active when bound to the immobilised 5′ vRNA sequence, and could perform ApG, capped mRNA-primed and unprimed synthesis of a 34 nt template RNA. The PB1/PA complex had no measurable transcriptase activity (at least under the current conditions of the assay). Thus, PB2 was essential for the formation of a transcriptionally active complex. However, NP was not necessary for any of these activities. In a cellular environment, NP is essential for viral RNA gene expression (1) and in vitro studies based on authentic viral RNPs have suggested at least two mechanisms to explain this dependency. Removal of NP from RNPs by treatment with high salt concentrations left polymerase–RNA complexes that were able to initiate ApG primed transcription but were unable to synthesise full- length products (60). Thus, it was suggested that NP acts as a processivity factor for the polymerase. In our recombinant system, NP is clearly not essential for transcription of 34 or 53 nt templates (Fig. 5 and data not shown). Experiments are in progress to determine if longer templates can also be used in the absence of NP. Another identified role for NP in transcription is during replicative transcription. RNP complexes from infected cells will synthesise c- and vRNA in vitro, but only in the presence of non-RNP associated NP. In the case of cRNA synthesis, free NP was shown to be necessary for readthrough of the polyadenylation signal, and for vRNA synthesis, it was required for the production of full-length transcripts (33,34). However, technical limitations prevented examination of the role of NP in the initiation of unprimed transcription and of the nature of the 5′-terminal groups of the in vitro synthesised c- and vRNA molecules (34). We show here, for the first time, unambiguous in vitro transcription initiation in the absence of NP and added primers to produce an RNA molecule with a 5′ di- or triphosphate group (Fig. 6). This suggests that the role of NP in the switch from mRNA to replicative synthesis is not a direct effect on the ability of the polymerase to initiate unprimed synthesis. It has been proposed that NP must co-transcriptionally encapsidate the nascent RNA for the polymerase to continue to replicate its template RNA (34). The 34 nt template used in this present study may be too short to reveal such a requirement. One other mechanism for NP control of replication is its possible role in the shift from pan handle to linear conformations of the template RNA. In the pan handle conformation, the polymerase is able to bind to 5′ and 3′ ends simultaneously, whereas in the linear conformation it must first bind to the 5′ end alone before subsequently interacting with the 3′ end of the same or different template RNA. This latter scenario is analogous to the assembly steps followed in the bead assay in its current form. It will be interesting to test in future whether the initial conformation of the template RNA affects the different transcriptional activities of the polymerase and whether NP exerts any influence upon this.
ApG primed synthesis gave rise to a predominant 14 nt premature termination product (with lesser amounts of smaller products) that was absent from globin mRNA primed synthesis. These products appear to be distinct from those reported previously (29) in that their synthesis is clearly template-dependent and they are smaller (ranging from 11 to 14 nt, compared to the 15–21 nt template-independent products reported previously). This previous study used micrococcal nuclease treated viral cores as the source of polymerase that undoubtedly contains residual degraded 5′ and 3′ template sequences that could give rise to such products. This is not the case with the vaccinia-expressed polymerase used in this present study. The cause or significance of these presumed premature termination products is not known. However, the region where termination occurs corresponds to the clamp region of the putative RNA fork that is important for the association between the 5′ and 3′ ends (54). Possibly the polymerase struggles to disrupt this base paired region, and this results in product and/or template release. Lack of premature termination for the cap-primed reactions may reflect the more stable association of the product RNA with the polymerase due to binding to the cap structure. Alternatively it may reflect a cap induced conformational change in the polymerase (akin to the allosteric modulation effect where ApG primed synthesis is increased as a result of occupation of the cap-binding site by a cap analogue) (61) whereby the polymerase no longer terminates prematurely.
The assay reported here is a significant advance for studying the activities of the viral polymerase. In particular it offers tremendous potential for studying the various stages of the transcription process. Because the influenza polymerase does not release from the bead during transcription it is possible to manipulate the reaction conditions sequentially by exchanging the composition of the transcription buffer. By using subsets of nucleotides the polymerase can be ‘walked’ to defined points along its template to study its behaviour as it encounters putative signals at different stages of the transcription process, e.g. initiation, cap release, termination and polyadenylation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Mark Krystal for generously providing plasmid pPHV and antisera to the P proteins. This work is supported by grants from the Wellcome Trust (059151 to L.T. and P.D.), MRC (G9901213 to P.D.) and Royal Society (to P.D.). P.D. is a Royal Society University Research Fellow.
REFERENCES
- 1.Huang T.S., Palese,P. and Krystal,M. (1990) Determination of influenza virus proteins required for genome replication. J. Virol., 64, 5669–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braam J., Ulmanen,I. and Krug,R.M. (1983) Molecular model of a eukaryotic transcription complex: Functions and movements of influenza P proteins during capped RNA-primed transcription. Cell, 34, 609–618. [DOI] [PubMed] [Google Scholar]
- 3.Biswas S.K. and Nayak,D.P. (1994) Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol., 68, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M.L., Ramirez,B.C. and Krug,R.M. (1998) RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J., 17, 5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez S. and Ortin,J. (1999) Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J., 18, 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Digard P., Blok,V.C. and Inglis,S.C. (1989) Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology, 171, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulmanen I., Broni,B.A. and Krug,R.M. (1981) Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl Acad. Sci. USA, 78, 7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaas D., Patzelt,E. and Kuechler,E. (1982) Identification of the cap binding protein of influenza virus. Nucleic Acids Res., 10, 4803–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi L., Summers,D.F., Peng,Q. and Galarza,J.M. (1995) Influenza virus RNA polymerase subunit PB2 is the endonuclease which cleaves host cell mRNA and functions only as the trimeric enzyme. Virology, 208, 38–47. [DOI] [PubMed] [Google Scholar]
- 10.Blok V., Cianci,C., Tibbles,K.W., Inglis,S.C., Krystal,M. and Digard,P. (1996) Inhibition of the influenza virus RNA-dependent RNA polymerase by antisera directed against the carboxy-terminal region of the PB2 subunit. J. Gen. Virol., 77, 1025–1033. [DOI] [PubMed] [Google Scholar]
- 11.Li M.L., Rao,P. and Krug,R.M. (2001) The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J., 20, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas S.K., Boutz,P.L. and Nayak,D.P. (1998) Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol., 72, 5493–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medcalf L., Poole,E., Elton,D. and Digard,P. (1999) Temperature-sensitive lesions in two influenza A viruses defective for replicative transcription disrupt RNA binding by the nucleoprotein. J. Virol., 73, 7349–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Benito J., Area,E., Ortega,J., Llorca,O., Valpuesta,J.M., Carrascosa,J.L. and Ortin,J. (2001) Three-dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO Rep., 2, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahy B.W.J. (1983) The viruses and their replication. In Kingsbury,D.W. (ed.), Genetics of Influenza Viruses. Springer-Verlag, Vienna, Austria, pp. 192–253.
- 16.Plotch S.J., Bouloy,M., Ulmanen,I. and Krug,R.M. (1981) A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- 17.Robertson J.S., Schubert,M. and Lazzarini,R.A. (1981) Polyadenylation sites for influenza virus mRNA. J. Virol., 38, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamb R.A. and Krug,R.M. (1996) Mutants of influenza virus. In Howley,P.M. (ed.), Fields Virology, 3rd Edn. Raven Press, Philadelphia, PA, Vol. 1, pp. 1353–1395.
- 19.Tiley L.S., Hagen,M., Matthews,J.T. and Krystal,M. (1994) Sequence-specific binding of the influenza virus RNA polymerase to sequences located at the 5′ ends of the viral RNAs. J. Virol., 68, 5108–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagen M., Chung,T.D., Butcher,J.A. and Krystal,M. (1994) Recombinant influenza virus polymerase: requirement of both 5′ and 3′ viral ends for endonuclease activity. J. Virol., 68, 1509–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cianci C., Tiley,L. and Krystal,M. (1995) Differential activation of the influenza virus polymerase via template RNA binding. J. Virol., 69, 3995–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young R.J. and Content,J. (1971) 5′-terminus of influenza virus RNA. Nature New Biol., 230, 140–142. [DOI] [PubMed] [Google Scholar]
- 23.Hay A.J., Skehel,J.J. and McCauley,J. (1982) Characterization of influenza virus RNA complete transcripts. Virology, 116, 517–522. [DOI] [PubMed] [Google Scholar]
- 24.Honda A., Mizumoto,K. and Ishihama,A. (1998) Identification of the 5′ terminal structure of influenza virus genome RNA by a newly developed enzymatic method. Virus Res., 55, 199–206. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa Y., Kimura,N., Toyoda,T., Mizumoto,K., Ishihama,A., Oda,K. and Nakada,S. (1995) The RNA polymerase PB2 subunit is not required for replication of the influenza virus genome but is involved in capped mRNA synthesis. J. Virol., 69, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagawa Y., Oda,K. and Nakada,S. (1996) The PB1 subunit alone can catalyze cRNA synthesis and the PA subunit in addition to the PB1 subunit is required for viral RNA synthesis in replication of the influenza virus genome. J. Virol., 70, 6390–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perales B. and Ortin,J. (1997) The influenza A virus PB2 polymerase subunit is required for the replication of viral RNA. J. Virol., 71, 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skorko R., Summers,D.F. and Galarza,J.M. (1991) Influenza A virus in vitro transcription: roles of NS1 and NP proteins in regulating RNA synthesis. Virology, 180, 668–677. [DOI] [PubMed] [Google Scholar]
- 29.Seong B.L. and Brownlee,G.G. (1992) A new method for reconstituting influenza polymerase and RNA in vitro: A study of the promoter elements for cRNA and vRNA synthesis in vitro and viral rescue in vivo. Virology, 186, 247–260. [DOI] [PubMed] [Google Scholar]
- 30.Hay A.J., Lomniczi,B., Bellamy,A.R. and Skehel,J.J. (1977) Transcription of the influenza virus genome. Virology, 83, 337–355. [DOI] [PubMed] [Google Scholar]
- 31.Momose F., Handa,H. and Nagata,K. (1996) Identification of host factors that regulate the influenza virus RNA polymerase activity. Biochimie, 78, 1103–1108. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu K., Handa,H., Nakada,S. and Nagata,K. (1994) Regulation of influenza virus RNA polymerase activity by cellular and viral factors. Nucleic Acids Res., 22, 5047–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beaton A.R. and Krug,R.M. (1986) Transcription antitermination during influenza viral template RNA synthesis requires the nucleocapsid protein and the absence of a 5′ capped end. Proc. Natl Acad. Sci. USA, 83, 6282–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro G.I. and Krug,R.M. (1988) Influenza virus RNA replication in vitro: synthesis of viral template RNAs and virion RNAs in the absence of an added primer. J. Virol., 62, 2285–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krug R.M., Ueda,M. and Palese,P. (1975) Temperature-sensitive mutants of influenza WSN virus defective in virus-specific RNA synthesis. J. Virol., 16, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scholtissek C. (1978) The genome of the influenza virus. Curr. Top. Microbiol. Immunol., 80, 139–169. [DOI] [PubMed] [Google Scholar]
- 37.Thierry F. and Danos,O. (1982) Use of specific single stranded DNA probes cloned in M13 to study the RNA synthesis of four temperature-sensitive mutants of HK/68 influenza virus. Nucleic Acids Res., 10, 2925–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mena I., Jambrina,E., Albo,C., Perales,B., Ortin,J., Arrese,M., Vallejo,D. and Portela,A. (1999) Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J. Virol., 73, 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith G.L., Levin,J.Z., Palese,P. and Moss,B. (1987) Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology, 160, 336–345. [DOI] [PubMed] [Google Scholar]
- 40.Fuerst T.R., Niles,E.G., Studier,F.W. and Moss,B. (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl Acad. Sci. USA, 83, 8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Earle P.L., Cooper,N. and Moss,B. (1991) Protein expression. In Struhl,K. (ed.), Current Protocols in Molecular Biology. Greene Publishing and Wiley-Interscience, New York, NY, Vol. 2, pp. 16.16.11–16.16.17.
- 42.Fiering S., Northrop,J.P., Nolan,G.P., Mattila,P.S., Crabtree,G.R. and Herzenberg,L.A. (1990) Single cell assay of a transcription factor reveals a threshold in transcription activated by signals emanating from the T-cell antigen receptor. Genes Dev., 4, 1823–1834. [DOI] [PubMed] [Google Scholar]
- 43.Ohlsson H. and Edlund,T. (1986) Sequence-specific interactions of nuclear factors with the insulin gene enhancer. Cell, 45, 35–44. [DOI] [PubMed] [Google Scholar]
- 44.Shinshi H., Miwa,M., Kato,K., Noguchi,M., Matsushima,T. and Sugimura,T. (1976) A novel phosphodiesterase from cultured tobacco cells. Biochemistry, 15, 2185–2190. [DOI] [PubMed] [Google Scholar]
- 45.Luo Y., Mao,X., Deng,L., Cong,P. and Shuman,S. (1995) The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia virus capping enzyme. J. Virol., 69, 3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fodor E., Pritlove,D.C. and Brownlee,G.G. (1994) The influenza virus panhandle is involved in the initiation of transcription. J. Virol., 68, 4092–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flick R., Neumann,G., Hoffmann,E., Neumeier,E. and Hobom,G. (1996) Promoter elements in the influenza vRNA terminal structure. RNA, 2, 1046–1057. [PMC free article] [PubMed] [Google Scholar]
- 48.Poon L.L., Pritlove,D.C., Sharps,J. and Brownlee,G.G. (1998) The RNA polymerase of influenza virus, bound to the 5′ end of virion RNA, acts in cis to polyadenylate mRNA. J. Virol., 72, 8214–8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pritlove D.C., Poon,L.L., Devenish,L.J., Leahy,M.B. and Brownlee,G.G. (1999) A hairpin loop at the 5′ end of influenza A virus virion RNA is required for synthesis of poly(A)+ mRNA in vitro. J. Virol., 73, 2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honda A., Mukaigawa,J., Yokoiyama,A., Kato,A., Ueda,S., Nagata,K., Krystal,M., Nayak,D.P. and Ishihama,A. (1990) Purification and molecular structure of RNA polymerase from influenza virus A/PR8. J. Biochem., 107, 624–628. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez S. and Ortin,J. (1999) Characterization of influenza virus PB1 protein binding to viral RNA: two separate regions of the protein contribute to the interaction domain. J. Virol., 73, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi M., Tuchiya,K., Nagata,K. and Ishihama,A. (1992) Reconstitution of influenza virus RNA polymerase from three subunits expressed using recombinant baculovirus system. Virus Res., 22, 235–245. [DOI] [PubMed] [Google Scholar]
- 53.Inglis S.C., Carroll,A.R., Lamb,R.M. and Mahy,B.W.J. (1976) Polypeptides specified by the influenza virus genome. I. Evidence for eight distinct gene products specified by Fowl Plague virus. Virology, 74, 489–503. [DOI] [PubMed] [Google Scholar]
- 54.Fodor E., Pritlove,D.C. and Brownlee,G.G. (1995) Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J. Virol., 69, 4012–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shuman S. and Hurwitz,J. (1981) Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme–guanylate intermediate. Proc. Natl Acad. Sci. USA, 78, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng Q., Galarza,J.M., Shi,L. and Summers,D.F. (1996) Influenza A virus RNA-dependent RNA polymerase cleaves influenza mRNA in vitro. Virus Res., 42, 149–158. [DOI] [PubMed] [Google Scholar]
- 57.Hagen M., Tiley,L., Chung,T.D. and Krystal,M. (1995) The role of template-primer interactions in cleavage and initiation by the influenza virus polymerase. J. Gen. Virol., 76, 603–611. [DOI] [PubMed] [Google Scholar]
- 58.Hwang J.S., Yamada,K., Honda,A., Nakade,K. and Ishihama,A. (2000) Expression of functional influenza virus RNA polymerase in the methylotrophic yeast Pichia pastoris. J. Virol., 74, 4074–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Honda A., Endo,A., Mizumoto,K. and Ishihama,A. (2001) Differential roles of viral RNA and cRNA in functional modulation of the influenza virus RNA polymerase. J. Biol. Chem., 276, 31179–31185. [DOI] [PubMed] [Google Scholar]
- 60.Honda A., Ueda,K., Nagata,K. and Ishihama,A. (1988) RNA polymerase of influenza virus: role of NP in RNA chain elongation. J. Biochem. (Tokyo), 104, 1021–1026. [DOI] [PubMed] [Google Scholar]
- 61.Penn C.R. and Mahy,B.W.J. (1984) Capped mRNAs may stimulate the influenza virion polymerase by allosteric modulation. Virus Res., 1, 1–13. [DOI] [PubMed] [Google Scholar]