

Abstract
Introduction:
Traumatic brain injuries (TBIs) impact the breadth of society and remain without any approved pharmacological treatments. Despite successful Phase II clinical trials, the failure of many Phase III clinical trials may be explained by insufficient drug targeting and retention, preventing the proper attainment of an observable dosage threshold. To address this challenge, nanoparticles can be functionalized to protect pharmacological payloads, improve targeted drug delivery to sites of injury, and can be combined with supportive scaffolding to improve secondary outcomes.
Areas covered:
This review briefly covers the pathophysiology of TBIs and their subtypes, the current pre-clinical and clinical management strategies, explores the common models of focal, diffuse, and mixed traumatic brain injury employed in experimental animals, and surveys the existing literature on nanoparticles developed to treat TBIs.
Expert opinion:
Nanoparticles are well suited to improve secondary outcomes as their multifunctionality and customizability enhance their potential for efficient targeted delivery, payload protection, increased brain penetration, low off-target toxicity, and biocompatibility in both acute and chronic timescales.
Keywords: TBI, nanoparticles, drug delivery, neural engineering
1. Introduction
Traumatic brain injuries (TBIs) are a leading cause of death and disability worldwide [1]. Without accounting for TBI cases that are left untreated, seen in outpatient care, and in federal facilities [2], there are still 69 million people who reportedly experience TBIs each year globally [3]. The incidence of TBIs spans all ages and affects particularly racial and ethnic minorities [4], military and public service members [5], people experiencing houselessness [6], and victims of assault and motor vehicle crashes [7]. TBIs in extreme conditions like in space travel will also require attention in the future. In a recent longitudinal study, blood samples from cosmonauts who spent, on average, 169 days on the International Space Station, saw significant elevations of neurofilament light chain, a biomarker of TBI and other neurodegenerative disease [8, 9].
TBIs constitute both closed head injuries (CHI) and, less common, penetrating injuries. CHIs are caused by blunt trauma, rapid acceleration, or deceleration forces [10] whereas in penetrating injuries, an object perforates the skull, breaches the meninges, and injures the brain [11]. To categorize the severity of TBIs and triage patients, the Glasgow Coma Scale (GCS), a physiologic measure of injury for all types of acute medical and trauma patients, is often employed in the clinical setting [12]. Typically, GCS scores between 13-15 are classified as mild TBI, 9-12 for moderate TBI, and 3-8 for severe TBI. Though useful, the GCS is weakly correlated with survival and functional outcomes [13, 14], thus, physicians also use imaging tools such as computerized tomography (CT) scans [15, 16] and magnetic resonance imaging (MRI) to determine injury severity [17]. Evidence has shown that employing additional anatomical measurements in combination with the GCS can improve the correlation with Glasgow Outcome Scale Extended (GOS-E) scores, a common tool used to assess global disability and recovery after a TBI [18]. For example, combining the GCS with the Injury Severity Score (ISS) and the Abbreviated Injury Score (AIS) has shown to improve the correlation with GOS-E scores, R=.335, p<.001 and R=.275, p<.001 respectively [19]. Other studies point to measuring a combination of biomarkers of injury to improve outcome correlation, including neuroendocrine hormones, micro-RNA, and brain-specific proteins [20].
TBIs begin with a primary injury as a result of the immediate impact. This event is not treatable and can be characterized by intra- and extraparenchymal hemorrhage, focal contusions, cerebral edema, and focal and/or diffuse axonal injury (DAI), due to the biomechanical shearing, tearing, or stretching of white matter tracts [21]. Concurrently initiated with the primary injury, the secondary injury sequelae can evolve over minutes to days and even months [22], providing a window for intervention. During this period, cellular and neurochemical mechanisms can result in an inflammatory response characterized by ischemia, hypotension, hypoxia, increased intracranial pressure (ICP), decreased cerebral perfusion pressure, and edema [21]. Glutamate excitotoxicity, the generation of reactive oxygen species (ROS), mitochondrial dysfunction, DAI, apoptosis, and the eventual death of neuronal, endothelial, and glial cells may also occur [23, 24]. These delayed deleterious processes can lead to acute and chronic sensorimotor [25, 26] and cognitive impairments [27-29].
Clinical management to reduce secondary damage and improve the prognosis of motor and cognitive functioning remains a challenge as no pharmacologic agents that have succeeded in Phase III clinical trials to treat TBIs. For instance, progesterone initially showed promise in pre-clinical studies by reducing inflammatory cytokines [30], stimulating neurogenesis, and synaptogenesis [31], and enhancing functional recovery of sensory, cognitive, and motor tasks [32-34]. Phase II clinical trials even proved moderately successful, but two Phase III trials, ProTECT III in the USA and SyNAPSe in China, failed to meet their primary outcome of improving scores on the GOS-E [35]. Though there may have been flaws in the experimental design [36], it is noteworthy that the mode of drug delivery and location of administration were problematic for efficient targeted treatment and determining an effective dosage. As another example, dexanabinol also failed to see a difference in GOS-E score at 6 months, despite being able to show improved ICP in Phase II trials [37]. Researchers of these trials noted that the distribution of drug volume may have varied in patients, necessitating a more targeted delivery mechanism.
2. Pathophysiology of TBI
The pathophysiology of a TBI is biphasic, beginning with the primary injury, a direct consequence of the mechanophysical forces responsible for the insult. This phase can not be treated and only preventative measures can be taken to avoid the incident [38]. Upon immediate impact, the resulting damage can be focally concentrated, diffuse, or a combination of both. Focal brain injuries stem from collision forces acting on the skull, resulting in epidural, subdural, and intraparenchymal hematomas, compression, and contusion of the underlying brain tissue at either the site of impact (coup), opposite the site of impact (contrecoup) or both [39]. Such physical changes restrict blood flow and often produce necrotic areas near the site(s) of impact. Diffuse injuries occur when rapid acceleration-deceleration or rotational forces shift the brain inside the skull causing dynamic shear, tension, and compressive forces that impact the brain. These processes elicit DAIs predominantly in subcortical and deep white matter tissue such as the brain stem and corpus callosum [40] and result in traumatic axonal damage, diffuse vascular injury, hypoxic-ischemic injury, and edema [41].
The secondary injury phase represents the body’s attempt to limit the extent of damage, repair consequences of the primary injury, and restore the brain’s structural and functional integrity. The physical, cellular, and biochemical processes that occur during the primary injury progress into secondary injury mechanisms on a time scale of hours to years, resulting in cerebral swelling, herniation phenomena, and neurodegeneration, which can lead to cognitive and motor impairment [42]. This secondary phase reveals the only window for pharmacological intervention to reduce injury progression and improve outcome severity. For example, delayed Wallerian degeneration, a phenomenon where severed axons maintain independent function for weeks after the initial impact event, has revealed a special window for TBI treatment and has been a topic of interest in studies aimed to improve functional recovery [43, 44].
The blood-brain barrier (BBB) is a selectively permeable microvasculature system that shields the brain from toxic substances in the blood, provides nutrients to brain tissue, and relinquishes harmful substances in the brain to the bloodstream. At the moment of impact, the BBB is often compromised, triggering the downregulation of the platelet-derived growth factor (PDGF)-B/ PDGF receptor-β signaling pathway, impairing pericyte-endothelium interactions and resulting in pericyte loss near the BBB, further compromising the BBB’s integrity (see Figure 1) [45]. This dysfunction of BBB dynamics is followed by an increased permeability to water, marked by a significant increase in aquaporin4 (AQP4) expression and possibly edema [45]. Also contributing to brain swelling, a ruptured BBB can encourage vasogenic edema, where protein-rich fluid from circulating blood enters the brain’s interstitial fluid [46]. Cytotoxic edema can also form, where the dysregulation of homeostatic ion channels and pumps disrupt the ionic gradient leading to intracellular swelling of astrocytes and glial cells. Both cytotoxic and vasogenic edema can elevate ICP, worsening functional outcome [47].
Figure 1.

BBB-level cellular and molecular response after a TBI. Leukocytes including neutrophils, macrophages, mast cells, eosinophils, natural killer cells, and dendritic cells transmigrate between and through endothelial cells, as well as through breaks in the BBB following the gradient of DAMPs released into the parenchyma after a TBI. Concurrently, PDGF-B/PDGF receptor-β expression is downregulated resulting in impaired pericyte-endothelium interactions evidenced by a decreased in the expression of gap junction proteins like Connexin-43, adherent junction proteins like N-cadherin, which connect endothelium and pericytes, and tight junction proteins, increasing BBB permeability. Vasogenic edema can also be seen by the increased expression of AQP4 towards the astrocyte end-feet surrounding the perivascular region. Created with BioRender.com.
Evidence has shown that prolonged neuroinflammation recruits macrophages, activates microglia, and promotes astrogliosis [48]. In response to the injury microenvironment, microglial polarization allows for a phenotypic response to surrounding biochemical cues which alter microglia function accordingly. Infiltrating microglia/macrophages can assume either two main terminally defined states: M1, which is pro-inflammatory/neurotoxic, or M2, which is anti-inflammatory/neuroprotective [49]. Research has shown that early after a TBI, both M1- and M2-like phenotypic markers are expressed followed by a transient up-regulation of the M2-like phenotype, which is soon replaced by a predominant M1-like phenotype associated with neurodegeneration up to 7 days post-injury [50]. During astrogliosis, activated astrocytes undergo hypertrophy and intermingle their processes with oligodendrocytes, meningeal cells, microglia, and fibroblasts to develop scar-like structures in an attempt to limit the injured area. Such mechanisms curtail the potential for axonal regeneration [51], though, some investigators believe that glial scarring provides perineuronal nets for both synaptic maturation and plasticity [52].
Synchronous with the processes described, a TBI insult also signals the release of damage-associated molecular patterns (DAMPs) as a response to cellular stress from injured tissue and are recognized by macrophages and pattern recognition receptors such as toll-like receptors and inflammasomes of the innate immune system [53]. DAMPs trigger inflammatory responses through multiple pathways which create a positive feedback loop of DAMPs production and inflammation (see Figure 2). A pathway of popular research interest, the nuclear factor-κB (NF-κB) inflammatory pathway, is a central mediator in pro-inflammatory gene induction [54] and plays a large role in edema [55] and neuronal apoptosis [56].
Figure 2.

Brain tissue-level response to TBI. After the BBB is compromised, DAMPs are released from injured cells and are recognized by pattern recognition receptors. In response to the DAMPs signals, an inflammatory response is elicited by leukocytes, microglia, and astrocytes which release pro-inflammatory molecules in a positive feedback loop. Prolonged inflammation promotes astrogliosis resulting in a scar-like formation around the injury perimeter and a reversal of glutamate uptake mechanisms which can contribute to persistent activation of N-methyl-d-aspartate acid (NMDA), α-amino-3-hydroxy-5-methylisoxazole propionic acid (AMPA) receptors, and voltage-gated ion channels, resulting in a lethal influx of calcium and subsequent neuronal excitotoxicity. Macrophages, microglia, and other cell types also respond to DAMPs signals by activating pro-inflammatory pathways such as the NF-κB inflammatory pathway which contributes to edema and neuronal apoptosis. The pro-inflammatory mechanisms and molecules described, along with glutamate accumulation from sheared neurons, impaired reuptake mechanisms, and neighboring glutamate-induced aggravated release from pre-synaptic terminals, exacerbate excitotoxic mechanisms, reduce ATP production leading to mitochondrial dysfunction, increase oxidative stress, and eventually cell death, triggering the further release of DAMPs. Created with BioRender.com.
DAMPs also guide the extravasation of activated leukocytes into the brain parenchyma which, in concert with microglia and astrocytes, produce ROS and inflammatory molecules including cytokines such as IL-1β, IL-6, and TNF-α, and chemokines such as MIP-α, MCP-1, and IL-8, recruiting more leukocytes to the injury site [57, 58]. These inflammatory biomolecules that follow leukocyte invasion encourage the demyelination and degradation of axonal cytoskeletons, resulting in axonal swelling and the accumulation of transport proteins at the synaptic terminals, compromising neuronal activity [59, 60]. Neurotransmitters can also accumulate in the synaptic space due to release from sheared neurons, glutamate-induced aggravated release from pre-synaptic terminals, and impaired reuptake mechanisms of the ischemic brain and activate both ionotropic and metabotropic receptors on the post-synaptic membranes fostering an influx of calcium ions, inducing excitotoxicity [61]. Furthermore, the activation of mitogen-activated protein kinases (MAPK), protein phosphatases, ROS, nitric oxide, calcineurin, calpain, and caspases follow, which induce apoptosis and increase ROS-mediated lipid peroxidation [62]. These processes impair mitochondrial function by depolarizing the membrane, reducing ATP production, and increasing oxidative stress, further exacerbating the secondary injury [61, 62].
3. Current management of TBI
Management of TBIs includes preventative measures to avoid TBIs, pre-hospital strategies, and clinical interventions to manage injuries. Primary prevention aims to circumvent brain trauma events by transforming social behavior through an array of mechanisms from public policy reforms [63], such as revising speed limits [64], improving road engineering [65], and enforcing helmet use [66], to altering cultural practices, such as creating programs to address alcohol abuse [67] and teaching athletes proper tackling techniques while playing rugby [68]. Secondary prevention focuses on minimizing the biological injury sequelae [63]. These efforts include addressing systemic issues through blood-pressure management, choice of fluid for resuscitation, temperature management, ICP management, monitoring cerebral oxygenation, and improving ventilation techniques [63]. As mentioned, clinical strategies to reduce secondary injury progression from a subcellular approach have not resulted in any successful Phase III clinical trials. Lastly, tertiary prevention, which is now often seen as a part of secondary prevention, aims to maximize the patient’s functional abilities and restore their quality of daily life after injury [63]. Such efforts include neuro-rehabilitation and understanding the relationships between imaging, function, and the underlying pathology to administer the most effective treatment for all injury types. The Centers for Disease Control and Prevention has recently published guidelines on tertiary prevention techniques for mild TBI in children which include symptoms scales, proper sleep methods, and assistance with the time given to complete assignments, all of which can aid in recovery [69].
The clinical management of TBIs aims to categorize the severity of the injury, relieve cerebral swelling and mass effect through medical and surgical strategies, and provide treatment to improve cognitive and functional outcomes. After an impact, patients may experience physical symptoms such as headache, fatigue, anxiety, light sensitivity, and confusion for mild TBIs. With moderate to severe TBIs, patients can experience seizures, loss of consciousness, and significant cognitive, motor, and sensory deficits alongside the previously mentioned symptoms [70]. Clinicians commonly use the GCS or extended GCS [71] which measures impaired consciousness based on the eye-opening, motor, and verbal responsiveness to categorize injury extent as either mild, moderate, or severe and predict prognosis. As mentioned the correlation between outcome and GCS is weak therefore, clinicians often employ various imaging techniques to improve the accuracy of the prognosis [15-17].
Once injury severity is determined, clinicians focus on relieving the cerebral mass effect, a phenomenon in which a focal lesion, such as a blood clot or a contusion, compresses the surrounding brain tissue [72]. Understanding of the mass effect is, in part, driven by the Monro-Kellie doctrine, a hypothesis that posits that the sum of the volumes of brain tissue, cerebrospinal fluid, and intracranial blood is constant [73]. Therefore, alternations in one will elicit opposite fluctuations in one or both of the remaining two variables. This behavior, in combination with principles of cerebral blood flow, accounts for many of the pathophysiological changes that occur after TBI. Depending on the extent of the acute focal mass effect, brain swelling, and increased ICP, physicians commonly administer mannitol and preferably hypertonic saline, since evidence has shown that this osmotic diuretic may increase pressure in the skull with excessive administration and present detrimental effects on mortality when compared to the latter [74]. In cases where herniation persists, surgeons are prompted to perform a decompressive craniectomy (DC) to relieve focal mass effect or high ICP. Early DC reduces mortality in patients with TBI but the clinical outcome compared to the standard medical management remains the same and determining the most optimal time frame for performing DCs still requires further investigation [75]. Unfortunately, there are still no proven treatment strategies to relieve the progressive secondary injury effects of TBI.
4. Modeling TBI
To investigate TBI treatment strategies and therapeutics, researchers are faced with the challenge of choosing an appropriate model organism and injury model to investigate the heterogeneous nature of TBIs in vivo. While larger mammals and non-human primates phylogenetically closer to humans may more accurately reflect the human biological response to TBIs, rodent models are often preferred due to their accessibility, comparatively lower cost, ease of handling, cheap post-surgical care, and existent standardized outcome measurements. For instance, to measure motor function in rodents there are many assessments including the rotarod test [76], beam walking [76], and cylinder test [77]. To assess cognition, there are also varied experimental apparatuses such as the Morris Water Maze [78] and Y maze [79]. Comparatively, large animals lack such a span of outcome measurements and models of delivering TBIs, though investigations are underway to bridge this gap [80, 81].
When choosing an appropriate TBI model, researchers must consider the type of injury that is to be modeled. There is no established guideline to measure injury severity in experimental TBI thus, investigators must tune mechanical parameters such as impact speed, depth, and dwell time [82]. Reasearchers can also note neurological changes, such as righting time [83], physiological changes, like weight loss [84], histological changes, including infarct volume and neuronal loss [85], and observe motor and cognitive performance [76-79]. It should be noted that there are limitations to using animal models. For example, although severe TBIs in rodents often reveal extensive tissue degeneration and motor dysfunction, these animals rarely mimic the state of coma often seen in diffuse TBI patients [86-88]. Furthermore, many TBI models require anesthesia prior to injury which can confer cerebral metabolic and physiological effects [89]. For instance, isoflurane, a commonly inhaled anesthetic used during surgical procedures, has been evidenced to attenuate functional deficits and elicit some neuroprotective action on the injured brain [90, 91]. Propofol, an anesthetic used in the clinic, has been associated with poor outcomes in rat focal TBI models [92]. For these reasons, it is important to have a sham animal group when designing TBI studies.
Broadly, animal models of TBI are classified as either mainly focal, diffuse, or mixed injuries. Mixed injuries represent a combination of focal and diffuse injuries, reflecting falls or sports-related injuries in humans. Details of commonly employed animal models to administer TBIs are discussed in Table 1. Ideally, when designing an experiment, the model should be tunable (commensurate with the desired injury severity), quantifiable, reproducible, and have some clinically relevant correlation. Common methods to mimic a focal brain injury include weight drop injury models (WDI), the open skull controlled cortical impact (CCI) model, and penetrating ballistic-like brain injury models. Diffuse injuries are commonly modeled using blast-wave models and mixed injuries can be modeled by some WDI, acceleration-deceleration models, and fluid percussion injuries. Though much of the current literature describes most injury models as being developed to represent a binary classification of TBI, mainly focal or diffuse, a large percentage of TBIs are of the mixed type. A recent MRI study revealed both focal lesions and diffuse injuries in 50% of patients studied with moderate and severe TBI [93].
Table 1.
Common traumatic brain injury models. Created with BioRender.com.
| Model | Setup | Examples of Injury Model | Advantages | Disadvantages |
|---|---|---|---|---|
| Weight Drop Injury Models |
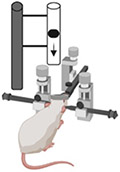
|
Feeney WDI model Focal injury Shohami WDI model Focal injury Marmarou WDI model Mainly diffuse injury |
|
|
| Controlled Cortical Impact Models |
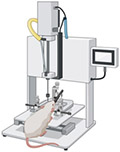
|
Focal injury |
|
|
| Blast-like Injury Models |
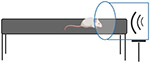
|
Penetrating ballistic injury model Mainly focal injury Parks Tube model Diffuse/mixed injury Clemedson Tube model Diffuse/mixed injury |
|
|
| Fluid Percussion Injury Models |
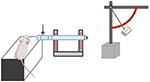
|
Midline FPI model Diffuse injury Lateral FPI model Mixed injury model |
|
|
In general, WDI models take advantage of the gravitational force elicited by a free-falling weight to generate the desired injury severity. The Feeney weight-drop model represents a focal injury model and generates reproducible graded cortical contusions using a 40 cm stainless steel tube attached to a circular footplate. A craniotomy is performed and the footplate is positioned above the exposed dura. The weight is then released, dislodging the footplate and impacting the region of interest [94]. This impact results in a cortical contusion characterized by edema [95], hemorrhaging [96], and damage to the BBB [97]. The subsequent inflammatory processes [94, 96] which activate microglia, astrocytes, the complement system [98], as well as the infiltration of neutrophils and macrophages, are commensurate with the weight falling height and consequentially, the injury severity [99]. The Shohami WDI model is a similar model except that the focal injury is created over one side of an intact skull using a flat silicone tip fixed at the end of an impacting rod [100]. The animal in both models is often fixed to a hard surface to minimize the dissipation of energy and avoid diffuse injury.
This practice is in contrast to the Marmarou weight drop model, often employed to mimic motor vehicle injuries and induce diffuse axonal injuries, where a rotational acceleration follows a linear acceleration injury [88, 101]. In this model, the animal is strapped to a spongiose material, such as a foam-covered platform, which serves to decelerate the head after impact [102]. A segmented brass weight then free-falls through a Plexiglass tube striking a metal disk affixed to its skull which allows for widespread axonal damage while reducing the chances of cranial fracture and the appearance of focal injuries. In a graded manner, dependent upon the release height of the weight, characteristics of diffuse injury are apparent such as bilateral hemorrhaging, cell loss, DAI, activated microglia, and astrogliosis. While these WDI models are similar to the mechanism of human TBI, can undergo neuroscoring post-injury using the Neurological Severity Score (NSS) [103], and are easy, cheap, and convenient, these models have high mortality rates due to apnea and skull fractures, can sometimes produce rebound injuries, often have varied impact velocities, and thus present inaccuracies with brain tissue deformation and injury severity.
A more reproducible TBI model is the CCI model which generates a mainly focal TBI and has been used in many animals including rodents [82, 104-106], swine [107-110], and non-human primates [111]. A craniectomy is first performed, leaving the dura intact over the targeted area. Using an electromagnetic or pneumatic impactor, investigators can precisely control quantifiable mechanical parameters such as the velocity of impact, dwell time, and cortical depth penetration to achieve the desired severity of injury [104, 106]. While there is no consensus regarding the behavioral or histological criteria that constitute mild, moderate, or severe injuries as seen in patients, research is actively trying to standardize the CCI model injury severity. Recently, one study recommends injury severity to be determined by a multifactorial approach including the extent of tissue loss, NSS, and cognitive deficits in addition to the surgical parameters previously mentioned [82].
Similar to humans with injury-induced epilepsy, rodents with CCI-induced TBIs may experience post-traumatic seizure activity [112]. Furthermore, the CCI model may inflict cytotoxic and vasogenic brain edema, often seen in the clinical human pathophysiology of TBI [46, 105]. Dependent upon the severity, the neuropathology of rodent CCIs often includes a cortical contusion proximal to the impact site, subdural hematoma, BBB disruption, hypoperfusion, cavitation, and neurodegeneration. Upregulated inflammatory cascades lead to excitotoxicity, neuronal cell death, astrogliosis, microglial activation, axonal damage, and cortical spreading depressions [96, 107]. Weaknesses of this model include the lack of brainstem deformation resulting in minimal mortality, as well as the lack of post-injury neuroscoring though, implementation of an NSS could improve this. Other focal injury models such as cryogenic injury models [113-115] and penetrating injury models, the only one in use today is the balloon inflation technique [116], can mimic some aspects of human TBI pathology but their clinical relevance is limited. For instance, in the cryogenic models, the focal traumas often lack the contrecoup injury and DAI often associated with human cerebral injuries.
Blast-like injury models, apart from open field designs, represent another tunable and highly reproducible set of TBI models. The penetrating ballistic brain injury model represents the focal damage shrapnel from an explosion can cause where a cavity forms corresponding to the penetration location [117]. Most other blast injury models reflect mechanisms of diffuse and mixed injuries. Blast tube models employ the detonation of an explosive to simulate a shock wave and blast wind without reflected shock fronts from the ground and other surfaces. When designing these experiments researchers must consider accommodations for the model organism, intended placement of the organisms within or away from the tubes/subject standoff, and plan for specialized testing locations and personnel training for the safe use of explosives. Common models include the Parks tube, often used in swine models[118, 119], and the Clemedson tube[120] which can be used to study blast injury in rats [121, 122]. Shock-wave primary blast injury models are similar except that they use compressed gas instead of explosives, though the physics of gas-driven shock waves may differ from explosive shock waves and the resulting pathology may not accurately reflect that of the human condition [123].
Fluid percussion injury (FPI) models represent another mixed injury model where both cerebral and focal concussions are produced, similar to sports-related injuries such as boxing. A pendulum first strikes a piston at the end of a tube filled with fluid and the fluid pressure impulse rapidly injects the exposed dura directly through a Luer-Lock surgically implanted onto the intact dural surface exposed via craniotomy which is usually made either centrally [124], over the sagittal suture midway between the bregma and lambda, or laterally over the parietal cortex [125]. The implant is connected to an FPI device fluid impulse outlet and fluid pressure detector which accurately measures the impact pressure. Injury severity is determined by adjusting the force of the fluid pressure pulse. Weaknesses of this model include the need for a craniectomy and like WDI and other models, FPI models demonstrate a high mortality rate due to apnea and lack immediate post-injury neuroscoring.
5. Nanoparticles address the challenges of treating TBI
The failure of many Phase III clinical trials [35-37, 126, 127] has revealed to investigators that to successfully improve TBI outcomes, novel pharmacological interventions must efficiently deliver therapeutics to the targeted injured area(s) and be retained in the brain long enough to confer its therapeutic effect while avoiding off-target toxicity. Successful therapies will also need to specifically address the dysfunction of the neurovascular unit (NVU) during a TBI, which includes the cellular and extracellular components of the brain and its vasculature such as neurons, perivascular astrocytes, microglia, pericytes, endothelial cells, and the basement membrane [128]. Drugs that can mediate the immune trauma response without needing to penetrate the cerebral tissue, such as a free radical scavenger, would not necessarily require NVU targeting, but those which must cross the BBB, such as ion channel blockers, calcium channel antagonists, trophic factors, and cell-specific targets should focus on increasing delivery efficiency when designing therapies [127]. Addressing these challenges will lower the barriers to establishing dosage thresholds, or the minimum amount of drug below which a biological effect does not occur, and enhance the potential of meeting the set endpoints of clinical trials.
NP-based drug delivery systems have the potential to meet the requirements that can increase the efficiency and efficacy of TBI therapies. Their customizable size, stealthy chemistry, and multifunctionality allow NPs to be optimized for stable targeted drug delivery systemically and to the brain [129, 130]. Modifications can also include functionalizing NPs with target-appropriate charges. To illustrate, in peptide-modified NPs that are delivered directly to the brain, positively charged peptides show restricted distribution compared to neutral, zwitterionic, or negatively charged particles, while the opposite is true for off-target organs [131]. Depending on their engineering, NPs can themselves elicit some therapeutic benefit in addition to shielding pharmacological payloads during targeted delivery.
The materials used in developing NP therapeutics are chosen based on some permutation of their ability to be functionalized with immune-evading, therapeutic, or targeting ligands, their capacity to protect their payload in the injury microenvironment, their chemistry to foster optimal timing for drug release, their improved biocompatibility, and low off-target accumulation and toxicity [130]. Poly(ethylene)glycol (PEG), a polyether compound, is regularly conjugated to NP polypeptides to prevent aggregation, improve solubility, and increase half-life in the circulatory system [132]. The PEG coating minimizes opsonization by avoiding plasma protein adsorption via steric hindrance and shield charging, thereby abating interactions with phagocytotic cells and avoiding clearance [133, 134]. Poly(lactic -co-glycolic acid) (PLGA) and Tween® 80, an amphiphilic non-ionic polysorbate (PS) surfactant, are also commonly employed in NP synthesis to improve biodegradability and biocompatibility as well as foster delivery across the BBB through low-density lipoprotein-mediated endocytosis, respectively [135, 136]. Incorporation of metals, such as gold [137, 138] and cerium[139, 140], are also used when synthesizing NPs to target injured parenchyma since their native properties can be utilized for their enhanced drug release chemistry, enzymatic actions, and anti-inflammatory capabilities.
Across the literature, investigations which harness this multifunctional diversity and potential for developing NP therapeutics to treat TBIs center around different strategies to improve the secondary injury sequelae. Similar therapeutic applications of NPs have been achieved through employing different nanomaterials. Therefore, here, NPs are discussed based on their shared function or pharmacological effect. In general, the NPs studied to treat TBI, and their respective payloads, function to elicit some combination of immunomodulatory, hemostatic, neuroprotective, anti-oxidative, anti-inflammatory, and stem-cell integration assistance through either modified surface chemistry, peptide functionalizations on the NPs, encapsulating and delivering gene therapies, pharmaceutical agents, neuroprotective substances, or growth factors. A summary of the most recent NPs investigated to treat TBIs are categorized by their primary function listed in Table 2.
Table 2.
List of NPs developed to treat TBIs categorized by function.
| Targeted Effect | Relevant NPs Developed |
Payload | Target | Reference |
|---|---|---|---|---|
| Immunomodulation | Immunomodulatory NP (PLGA-COOH) | N/A | MARCO+ hMos | [142] |
| Leuko NPs (Leukocyte-Based Biomimetic NP) | N/A | Activated endothelial cells through CD11b | [143] | |
| Interleukin-4 protein-loaded- liposome | IL-4 | Peroxisome proliferator-activated receptor gamma | [144] | |
| shCCL20-CCR6 nanodendriplexes | shRNA (CCL20 or CCR6) | Chemokine (C-C motif) ligand 20 via receptor chemokine receptor 6 | [145] | |
| p5RHH+miR-146a NPs | miR-146a | Nuclear factor-kappaB inflammatory modulators TRAF6 and IRAK1 | [146] | |
| Hemostasis | PLGA-PLL-PEG-GRGDS NPs | Dexamethasone Or Poly(acrylic) acid (PAA) |
Glycoprotein IIb/IIIa receptors & Glucocorticoid receptor | [150] |
| Antithrombin mechanisms | [152] | |||
| Polyurethane NPs in Gelatin (Spongostan™) | Polyurethane NPs | M1/M2 mRNA expression / Nuclear factor-kappaB pathway | [153] | |
| Neuroprotection | Neuron-targeted nanocomplex NP (TP-RVG NP) | Caspase 3 siRNA | Receptor for rabies virus (e.g. nicotinic acetylcholine receptor) on neurons and caspase 3 expression | [155] |
| Tau siRNA-loaded-PS 80 (High Density) (PS 80 (H)-NPs) | Tau siRNA |
PS 80: Endogenous apolipoproteins such as lipoprotein receptor-related protein 1 to promote BBB penetration Tau expression: Pathology linked to neurodegeneration and brain dysfunction in TBI |
[135] | |
| NP-wrapped siRNAs-Fyn/siRNA-c-Src | Fyn and c-Src siRNA | Fyn & cSrC messenger RNA (to inhibit ROCK) | [157] | |
| Poly(lactide-co-glycolide)-graft-polyethylenimine | RhoA siRNA or Rolipram |
RhoA expression | [158] | |
| Restore cAMP | [159] | |||
| Cerebrolysin-loaded PLGA NPs | Cerebrolysin | Injured neuronal tissue | [160, 161] | |
| BDNF-loaded-PLGA NPs coated with poloxamer 188 (NP-BDNF-PX) | BDNF | Penetrate CNS to deliver BDNF | [164] | |
| Tat-NR2B9c-loaded Activatable protein nanoparticle (TN-APNPs) *conjugated with CAQK or CCAQK for TBI | NR2B9c | Disrupt interaction between NMDARs & PSD-95 | [166, 167] | |
| Angiopep-2 functionalized and manganese doped eumelanin NPs (ANG-MnEMNPs-Cur/AMEC) | Curcumin |
Angiopep-2; low-density lipoprotein receptor-related protein-1 for transcytosis across BBB Eumelanin: Reactive oxygen species Curcumin: Inflammatory cytokines |
[171] | |
| Curcumin-loaded niosome NPs (CM-NPs) | Curcumin | TLR4/NF-κB and inflammatory cytokines | [172] | |
| Curcumin-loaded AmyloLipid nanovesicles (ALNs) | Curcumin | Amyloid-β-protein, β-secretase, acetylcholinesterase, and anti-inflammatory cytokines such as IL-4 and IL-10 | [173] | |
| Nanocoffee particles | N/A | Amyloid-β-protein, β- & γ-secretase Poly adenosine diphosphate-ribose polymerase (PARP), p-Erk, Erk2, phosphorylated GSK3 | [175] | |
| Nerve Growth Factor (β-NGF) bound to Poly(n-butyl-2-cyanoacrylate) (PBCA) NPs | β-NGF | Mitogen-activated protein kinase kinase (MAPKK) | [176] | |
| Recombinant human erythropoietin-loaded Tween 80 modified albumin NPs (rh-EPO-Tw-ABNPs) | rh-EPO | AQP4 and pro-inflammatory processes (e.g. reactive astroglia) | [177] | |
| Anti-inflammatory/Anti-oxidative action | Thioether core-cross-linked NP | Sulfoxides and sulfones via thioether cores | Hydrogen peroxide & Superoxide | [179, 180] |
| Cerium Oxide NP (CeONPs) | N/A | Radical species (e.g. superoxide, hydroxyl, and nitroxyl radicals) | [139] | |
| Au24Cd1, Au24Cu1 Clusters | N/A | HER, OER, O2, H2O2 | [137] | |
| Gold nanoclusters - dihydrolipoic acid (DHLA-AuNCs) | DHLA | Reactive oxygen species, NF-κB pathway | [182] | |
| Redox-active nitroxide radical- containing NPs (RNP) | 4-amino-2,2,6,6-Tetramethylpiperide-1-oxyl | Reactive oxygen species | [181] | |
| Sinomenine conjugated to hydroxyl-terminated generation-4 Poly(amidoamine) dendrimer) (D-Sino) | Sinomenine | NF-κB nuclear translocation | [183] | |
| Triphenyl-phosphonium-Dendrimer-N-acetyl cysteine (TPP-D-NAC) | NAC | Reactive oxygen species, glutathione | [184] | |
| Glyburide-Loaded Betulinic Acid NP | Glyburide |
Betulinic Acid NPs: cross BBB via CB1-mediated transcytosis Glyburide: antagonist to the sulfonylurea receptor -transient receptor potential melastatin 4 channel |
[185, 186] | |
| Nimodipine (CL-PPS/Np) NPs Surface material : lecithin, CAQK-DSPE-PEG2000, and DSPE-PEG2000 Core material: poly(propylene sulfide)60 (PPS60) |
Nimodipine |
PPS60 : Reactive oxygen species Nimodipine: Ca2+ channels |
[168] | |
| Poly(ethylene)glycol conjugated hydrophilic carbon clusters (PEG-HCCs) | N/A | Reactive oxygen species (e.g. superoxide anions & hydroxyl radicals) | [188] | |
| Surfactant poloxamer 188-N-acetylcysteine-loaded poly(lactic-co-glycolic acid) (P188-NAC-loaded PLGA) NPs | NAC |
P-selectin glycoprotein ligand-1: E-selectin, P-selectin, L-selectin P188 & NAC: Reactive oxygen species |
[189] | |
| Stem cell therapy | Gold NP-Bax inhibiting pentapeptide-linker-modified(gene) (Au-NP-Ku70) | Ku70 Plasmid | Mesenchymal stem cell to secrete BDNF | [138] |
| Neurogenin-2-loaded poly(β-amino ester)-based NPs | Neurogenin-2 | Induce differentiation of hNSCs to neurons | [190] | |
| Stromal-cell derived factor-1 loaded Polydopamine NPs (PDA@SDF-1α) | SDF-1α | Receptor CXCR4/Guide stem-cell homing/ proliferate neuroblast | [191, 192] |
5.1. Immunomodulatory NPs
Immunomodulatory NPs include those nanoscale technologies which alter the immune system in a manner that improves TBI outcome. Investigators in this field have focused on designing NPs that mitigate upstream inflammatory players and sequester inflammation both in the vasculature and around the injured parenchyma sometimes solely through surface modifications, omitting a payload [141].
Immunomodulatory nanoparticles (IMPs), 500nm-diameter NPs made from Food and Drug Administration (FDA)-approved biodegradable biopolymer carboxylated PLGA, are shown to improve TBI in the acute phase by binding to macrophage receptors with collagenous structure (MARCO) positive hematogenous monocytes (hMos) [142]. These monocytes are then sequestered to the spleen (see Figure 3A for possible translation in humans), unable to home sites of inflammation evidenced by an overall reduction in infiltrating immune cells and glial scarring, mitigation of the inflammatory status of infiltrating cells exemplified by microglia shifting to a more M2-like profile, improved visual and motor function, and reduced edema in both CCI and WDI models [142]. Even at the chronic time point of 10 weeks, CCI animals saw a significant 44.7% reduction in lesion volume compared to the vehicle-treated animals.
Figure 3.

Nanoparticles can accumulate in the brain and peripheral organs to improve secondary injury outcomes. (A) Immunomodulatory NPs attenuate peripheral inflammation by accumulating in the spleen, liver, kidneys, and lungs to block inflammatory processes and render immune cells unable to home sites of inflammation. (B) Neuromodulatory nanoparticles enter the brain to elicit, hemostatic, immunomodulatory, and neuroprotective functions as well as deliver targeted therapeutics into the injured parenchyma. Depending on its chemistry, size, and charge NPs can enter the injured brain through breaks in the BBB, mechanisms of transcytosis such as cannabinoid receptor 1-mediated transcytosis and low-density lipoprotein receptor mediated transcytosis (not shown), and to a lesser extent paracellular transport which allows permeation of selective substances regulated by tight junction proteins between endothelia of the BBB. Created with BioRender.com.
Biomimetic nanoparticles can also be employed as they provide the stealth of evading clearance by the mononuclear phagocyte system (MPS), reduce elimination by Kupffer cells in the liver, and glomerular filtration in the kidneys, while still being able to maintain the delivery capabilities of a synthetic NP [141]. Leukocyte-based biomimetic NPs (Leuko NPs) are <200 nm and are fabricated using liposomes and membrane proteins extracted from cultivated leukocytes. These NPs target activated endothelia through CD11b targeting and evade MPS uptake through CD45 present on their surface [143]. Using the In Vivo Imaging System, Leuko NPs were shown to accumulate in the brain through paracellular diffusion and breaks in the BBB (see Figure 3B) as well as in peripheral organs such as the liver, spleen, kidneys, and lungs where no abnormal or pathological morphology was found as a result of this treatment. A significant increase in Iba-1 positive cells 24 hours post-injury was observed and can be linked to an increase in M2 microglia, decreased lesion size, and a reduction in F4/80 positive cells which are usually highly expressed on infiltrating macrophages. On a systemic level, Leuko NPs also attenuated peripheral inflammation as those organs which saw NP accumulation are speculated to aid in blocking the inflammatory response, thereby contributing to the reduction in infiltrating macrophages observed (see Figure 3A) [143]. In line with immunomodulating NPs, interleukin-4 protein-loaded liposomes were evidenced to facilitate oligodendrocyte precursor cell differentiation and oligodendrogenesis through peroxisome proliferation-activated receptor-γ signaling, thereby reducing oxidative stress, promoting mitochondrial function, and improving sensorimotor neurological recovery [144].
Also in the arsenal to modulate the immune response are NPs which deliver gene therapies. Short hairpin RNA-loaded shCCL20-CCR6 nanodedriplexes, made of polyamidoamine, target chemokine (C-C motif) ligand 20 via receptor chemokine receptor 6 to attenuate inflammation after repeated TBI [145]. This reduction of the inflammatory microenvironment improves the efficacy of human mesenchymal stem cell transplantation, resulting in reduced pro-inflammatory cytokine IL-6, neurodegeneration, microgliosis, and astrogliosis, and increased brain-derived neurotrophic factor (BDNF) expression in the cerebral cortex indicating possible neurogenesis in mice. MicroRNAs (miRNAs), small non-coding RNA molecules that regulate post-transcriptional gene expression, have also been employed in gene therapy strategies toward thwarting the deleterious immune response post-TBI. Upon observing that certain miRNAs in the mitochondria, compared to the cytosol, display a compartmental re-distribution post-injury, one study developed melittin-derived cell-permeable peptide (p5RHH)+miR-146a NPs which have shown to significantly reduce the expression of tumor necrosis factor (TNF) receptor-associated factor 6 and interleukin-1 receptor-associated kinase 1, two important modulators of the NF-κB pro-inflammatory pathway after injury [146]. Future studies in this field must continue to consider how inflammatory cells and immunological signaling regulate the post-TBI recovery mechanisms, keeping in mind the timing of pro- and anti-inflammatory processes when developing immunomodulatory NPs [147].
5.2. Hemostatic NPs
Though all TBIs are susceptible to hemorrhaging, severe blast-induced injuries can especially elicit internal bleeding in multiple organs. Immediate intervention is among the most effective solutions to minimizing mortality in cases of severe polytrauma yet, the only available treatments are pressure dressings and absorbent materials which are only effective for superficially exposed wounds and are unable to ameliorate internal injuries. To find more viable solutions to reduce the chance of coagulopathy and mortality in severe trauma, many investigators are developing hemostatic NPs which aim to stop bleeding and reduce the progression of inflammatory processes. Commonly functionalized to hemostatic NPs, the synthetic peptide Gly-Arg-Gly-Asp-Ser (GRGDS) contains the amino acid sequence Arg-Gly-Asp (RGD) which mimics a recognition site in the interaction between extracellular matrix molecules and cell membrane receptors. When platelets are activated, they expose glycoprotein IIb/IIa receptor. RGD can block the receptors of these glycoproteins, inhibiting fibrinogen binding and modulating hemostasis through anti-thrombotic actions [148, 149].
In one study 500 nm PLGA-poly(ε-cbz-L-lysine)-PEG-GRGDS NPs (hNPs) were synthesized to encapsulate dexamethasone (hDNPs), an anti-inflammatory glucocorticoid loaded at a final concentration of 22 ± 1μg of dexamethasone/ mg of NP, to stop bleeding and mitigate pro-inflammatory processes in a blast TBI model [150]. Results revealed an elevation in survival with these NPs regardless of whether dexamethasone is encapsulated or not. Interestingly, hDNPs were able to confer a decrease in apoptosis and BBB disruption in the amygdala which correlated with a significant reduction in anxiety-like behavior when tested in an open-field tracking system. Importantly, hNPs significantly mitigated hemorrhaging and hDNPs conferred a significant reduction in astrogliosis, a restoration of microglia levels, an alleviation of vascular endothelial growth factor expression, which at high levels can indicate BBB permeability, and increased expression of SM1–71, a recognized antibody against rat endothelial barrier antigen that indicates the functionality of the BBB [150].
In an earlier study, PLGA-poly(ε-cbz-L-lysine)-PEG-GRGDS NPs encapsulated poly(acrylic acid) (PAA) [151], a flocculating agent implicated as an anticoagulant due to its antithrombin-activating properties [152]. Results from these hemostatic NP studies showed a significant increase to 95% survival following a 20-psi blast compared to 60% in the control and there were no complications or toxicity observed in the experimental groups. In a more recent study, carboxyl-functionalized biodegradable polyurethane (PU) NPs have been delivered with Spongostan™ as a hemostatic agent and has shown that PU NP-contained gelatin attenuated brain edema, suppressed expression of M1 biomarkers such as IL-1β, elevated M2 biomarkers, reduced the activation of inflammatory cells near the implant site, and increased BDNF by almost 3-folds [153].
5.3. Neuroprotective NPs
Neuroprotectants are those agents which restrict injury to the brain parenchyma and attempt to salvage or regenerate neuronal cell structure and function [154]. Though in general, NPs which elicit some immunomodulatory, hemostatic, anti-inflammatory, and anti-oxidant effects can result in neuroprotection downstream, discussed here are those NPs that contain agents in which their primary function is to elicit a direct neuroprotective or neuronal-related interaction.
Recently, many studies have utilized NP encapsulation of short interfering or silencing RNA (siRNA) to deliver gene therapies across the BBB and improve neuronal pathology post-TBI, since their efficiency and specificity avoid off-target effects that can be seen with some small-molecule drugs. Neuron-targeted nanocomplex (TP-RVG) NPs were developed to encapsulate siRNA against caspase 3, a lysosomal enzyme involved in apoptosis. Researchers synthesized this NP using a tandem peptide consisting of a targeting peptide found in the rabies virus, RVG, and intracellular trafficking molecule transportan (TP) [155]. Data revealed that after treatment with these TP-RVG NPs 5 minutes post-TBI, there was a significant accumulation of NPs in neurons. By day 3 there was an ~80% decrease in caspase 3 in the injured hemisphere, with no knockdown observed in the contralateral hemisphere. These results show the potential for TV-RVG NPs to target injured neurons and deliver siRNA to reduce neuronal apoptosis. Another group utilized 60 nm high-density Tau siRNA-loaded-PLGA-PS 80-coated NPs (PS 80 (H)-NPs) to penetrate an intact BBB and accumulate in the brain at a level 3-folds higher than conventional PEGylated NPs in mice with WDI modeled TBI [135]. Tau pathology is strongly linked to TBI-caused neurodegeneration and brain dysfunction [156]. This study was the first to show that PS 80 (H)-NPs could reduce tau expression in cultured primary neural cells and also achieved 40-50% tau silencing in TBI mice regardless of being administered within or outside the window of the physically breached BBB.
In a recent study, NP-wrapped siRNA-Fyn and siRNA-c-Src were shown to knockdown Fyn and c-Src messenger RNA, members of the Src family kinases activated by transmembrane receptors implicated in pathways leading to the release of toxic molecules post-TBI [157]. Researchers in this study support that Src kinases activate neurotoxic downstream signaling including the Rho-associated protein kinase (ROCK) pathway which suppresses central nervous system regeneration, leading to neuronal loss and cognitive decline post-injury. Data from this study revealed that combined NP-wrapped siRNA-Fyn and siRNA-c-Src delivery demonstrated significantly reduced CA2/3 NeuN+ neuronal cell loss in the hippocampus post-TBI and improved cognitive function 12-16 days post-injury. Another group recently looked to target the RhoA/Rock pathway using poly (lactide-co-glycolide)-graft-polyethylenimine (PgP) as a NP to encapsulate and deliver siRNA targeting RhoA (siRhoA) [158]. Data showed that in rats with CCI modeled TBIs, the PgP/siRhoA NPs significantly reduced RhoA expression, lesion volume, neuroinflammation, apoptosis, and increased neuronal survival. This team recently used PgP NPs to deliver Rolipram which restored cyclic adenosine monophosphate (cAMP) levels in the injured brain close to the sham level and reduced brain lesion volume, neuroinflammation, and apoptosis at 7 days post-TBI [159].
The use of peptides in NPs systems have also been a popular target of neuroprotective studies on improving TBI outcome. A couple of studies have explored the use of NPs for sustained release of cerebrolysin (CBL), a neuroprotective agent characterized by a peptide mixture able to ameliorate TBI symptomology with factors including BDNF, glial cell line-derived neurotrophic factor, nerve growth factor, and ciliary neurotrophic factor [160, 161]. CBL-loaded PLGA NPs, ranging 250-300nm, showed superior neuroprotective effects following a CHI compared to free CBL [161]. BDNF, in particular, is known to regulate neuronal plasticity, neuronal growth, proliferation, cell survival, and long-term memory but its short half-life and low BBB permeability create a barrier to effective therapeutic delivery [162, 163]. BDNF-loaded-PLGA NPs coated in poloxamer 188 have also circumvented this obstacle by demonstrating that they can increase BNDF levels in both sham and WDI mice in both ispsi- and contralateral hemispheres and also markedly improved NSS [164].
Another peptide, NR2B9c, has also been studied for its neuroprotective properties. N-methyl-d-aspartate receptor (NMDAR)-dependent excitotoxicity, induced by postsynaptic density protein-95 (PSD-95) binding, is the primary mechanism of neuronal injury following ischemic injury [165]. Activatable protein nanoparticles (APNPs) are made of 3 independent polypeptides with enzyme-responsive sequences located between therapeutic peptides. APNPs home to target tissues and when activated by proteases rich in the injury microenvironment, release their payload. In one study Tat-NR2B9c, a post-synaptic PSD-95 inhibitor, was encapsulated with APNPs conjugated with PEG (TN-APNPs). When TN-APNPs were delivered to the injured ischemic brain, there was a significant reduction in infarct size, which was on average 22.6% larger than the contralateral side (compared with 58.8% and 38.7% in rats treated with PBS and free Tat-NR2B9c, respectively), and significant improvement in neurological function [166]. Tat-NR2B9c colocalized with PSD-95 only in the ipsilateral hemisphere of the ischemic injury, suggesting that the improved biological outcomes are due to the release of Tat-NR2B9c from TN-APNPs. In a more recent study, researchers enhanced the efficiency of TN-APNPs to deliver Tat-NR2B9c to TBI brain tissue by conjugating the NPs with peptides CAQK, which has a high affinity for extracellular matrix at the site of the injured brain, and CCAQK which contains an extra cysteine since the N-terminal cysteine could be important for the targeting effect of CAQK. In mice that underwent CCI and were treated with both peptide conjugated TN-APNPs, both showed improved brain penetrability and CCAQK-TN-APNPs demonstrated higher brain targeting efficiency, reduced injury size, and improved psychological outcomes [167]. CAQK has been implicated in many other TBI NP studies [168, 169] including the use of CAQK-conjugated silver NPs for targeting chondroitin sulfate proteoglycans-rich scars on mature oligodendrocytes of the injured region which would address a major barrier to regeneration [170].
Neuroprotectants can also be derived from natural and unconventional sources. For instance, curcumin, the principal secondary metabolite of turmeric or Curcuma longa, has been explored as a neuroprotectant in many studies [171-173]. Angiopep-2 functionalized and manganese-doped eumelanin NPs encapsulate curcumin (AMEC) as a neuroprotectant to influence anti-oxidation and anti-neuroinflammation in TBI parenchyma [171]. According to results from this study, after CCI in mice and subsequent intravenous administration of AMEC, these specialized ~160 nm nanoparticles can penetrate the brain through angiopep-2 binding low-density lipoprotein receptor-related protein-1 on the BBB enhancing drug accumulation, reduce inflammation and oxidative stress as exemplified by a lower M1/M2 ratio through ROS scavenging, and promote neuronal regeneration and functional recovery through the synergistic effects of eumelanin and curcumin [171]. In another study, curcumin-loaded niosome NPs were orally administered, after transplantation of human neural stem/progenitor cells, for 10 days. This combinatorial treatment showed a significant improvement in brain edema after TBI, increased locomotor activity, a decrease in astrogliosis, and an overall decrease in TLR4-, NF-κB, and TNR-α-positive cells [172]. Another recent study evaluated curcumin-loaded AmyloLipid nanovesicles (ALNs) administered intranasally and showed evidence for its potential to increase its bioavailability to treat CNS disorders. ALNs are NPs based on a cross-linked starch enveloping a solid lipid core which liquifies at body temperature exposing the payload [173]. Multi-lamellar vesicle NPs have also shown great potential as a carrier for therapeutic agents to the injured brain considering they are non-toxic, biocompatible, biodegradable, can cross the BBB, and maintain stability in the blood [174].
Nanocoffee NPs have also been investigated to exploit the neuroprotective potential of caffeine to prevent neuronal death. Caffeine can increase protein kinase A, suppressing β-secretase by reducing the Raf-1/ NF-κB inflammatory pathway, and reducing γ-secretase, through lowering GDK-3α, which decreases brain amyloid-beta, a marker of neurodegenerative disease [175]. Data from CHI animals treated intraperitoneally with nanocoffee NPs showed an enhancing effect on recognition memory, reduced anxiety, displayed an increase in T-type spines associated with learning, mitigated the increase in poly(ADP-ribose) polymerase (PARP) expressions seen after TBI which has been linked to microglia activation, and demonstrated a decrease in Erk and other MAPK signaling pathway members which mediate secondary injury [175]. Another NP encapsulating a neuroprotectant is poly(n-butyl-2-cyanoacrylate) (PBCA) coated in Tween® 80 which can cross the BBB via low-density lipoprotein receptor-mediated transcytosis and was recently studied for its potential to delivery β-nerve growth factor (β-NGF), a neurotrophin essential to maintaining the survival and normal functioning of neurons [176]. After a TBI, intravenous administration of PBCA- β-NGF-Tween® 80 NPs revealed a dramatic elevation in β-NGF concentration within 1 day which kept increasing significantly to day 7 compared to treatment with free β-NGF. The NP-treated group saw reduced mortality in vivo and increased neurite regeneration in vitro in PC12 cells through PBCA-delivered β-NGF enhanced MAPK kinase activity [176].
Recombinant human erythropoietin (rh-EPO) has shown pre-clinical evidence in protecting nerves, resisting apoptosis, promoting vascular regeneration, and reducing cerebral edema [177]. In efforts to increase neuroprotectant rh-EPO penetrability and accumulation in the brain, rh-EPO loaded Tween® 80 modified albumin nanoparticles (~438 nm) administered after TBI was shown to be non-toxic and significantly enhanced the distribution of rh-EPO (5000 IU/kg), reduced the expression of activated astroglia, and recovered AQP4 levels, relieving brain edema. As demonstrated the efficacy of neuroprotectants has been greatly augmented by the use of NP encapsulation technology for efficient targeted delivery. Relevant to previously failed Phase III clinical trials, progesterone has been studied for efficient encapsulation using Flash NanoPrecipitation to produce 300 nm progesterone-loaded polymeric NPs [178]. Work continues in this field to more effectively harness the neuroprotective potential of progesterone and other agents to improve secondary outcomes of TBI.
5.4. Anti-inflammatory and anti-oxidative NPs
The pro-inflammatory and oxidative pathways described in the Pathophysiology of TBI section contribute to the neurodegenerative and other deleterious effects exacerbated by positive feedback loops, such as those prompted by DAMPs, in secondary injury processes. Described here are those NPs which have been employed in brain injury studies to directly confer either anti-oxidative, anti-inflammatory, or both, effect(s).
Thioether core-crosslinked NPs (NP1) were shown to enter the brain parenchyma through a break in the BBB to scavenge and inactivate ROS such as hydrogen peroxide and superoxide [179]. NP1, made from PS 80, is evidenced to reduce ROS in the ipsilateral hemisphere 1-day post-CCI and reduce acrolein, a product of lipid peroxidation, up to 1-month post-CCI [180]. Furthermore, NP1 conferred a significant reduction in activated microglia in the contralateral CA1 region and neuron-astrocyte-microglia triad formation in the contralateral CA1, ipsilateral CA2/3, and bilateral DG region 1 month post-CCI [180].
Many studies including the development of cerium oxide nanoparticles (CeONPs) [139] and redox-reactive nitroxide radical containing NPs [181] have also elicited antioxidative effects that resulted in improved TBI outcomes. The crystal lattice of cerium oxide has multiple valence states that confer its high redox capacity. After FPI, the higher concentration and dosing paradigm of CeONPs were shown to increase neuronal survival, preserve endogenous calcium anti-oxidant activity, reduce oxidative stress in part through improved superoxide dismutase activity, and restored reduced glutathione (GSH)/oxidized glutathione (GSSG) ratios [139]. Interestingly the shape of cerium oxide nanoparticles affects its efficiency. Ceria nanorods, ~130nm, were shown to exhibit better antioxidant activity and cytotoxicity than Ceria nanospheres, ~3.5 nm, due to nanorods displaying more high-energy surfaces with more active sites [140]. Other metal-containing NPs developed to treat TBIs include Au24Cd1 and Au24Cu1 clusters [137] which utilize the enzymatic activity of gold while taking advantage of its negligible toxicity and high renal clearance, ability to safely target hydrogen evolution reactions (HER), oxygen evolution reactions (OER), and reduce O2. Gold nanoclusters have also been explored with dihydrolipoic acid (DHLA) functionalization, an anti-oxidant with demonstrated neuroprotective potential that suppresses pro-inflammatory processes by inducing M2 polarization in macrophage/microglia associated with a reduction of NF-κB signaling, a decrease in ROS, and improved cell survival [182]. This study also highlighted the role of autophagy as a potential target for immunomodulation in regenerative medicine.
Other studies that focus on reducing oxidative and inflammatory processes include redox-active nitroxide radical-containing NPs which encapsulate antioxidant 4-amino-2,2,6,6-Tetramethylpiperidine-1-oxyl to scavenge free radicals and improve lesion volume and cognitive behavior while greatly lowering the toxicity of its payload [181]. In another study using sinomenine, an anti-inflammatory/anti-oxidant drug, conjugated to hydroxyl-terminated generation-4 poly(amidoamine) dendrimers (D-Sino), these NPs were able to target activated microglia/macrophages without conjugation of a targeting ligand [183]. D-Sino NPs were synthesized using enzyme-sensitive covalent linkages via a highly efficient and robust copper (I) catalyzed alkyne-azide click reaction to attach sinomenine to dendrimers. Results demonstrate that D-Sino NPs specifically target activated microglia/macrophages via endocytotic activity and can inhibit NF-κB, a key regulator of atherosclerotic pathogenesis, proving its potential in suppressing acute inflammation post-TBI [183]. Another dendrimer NP, Triphenyl-phosphonium-Dendrimer-N-acetyl cysteine encapsulated NAC (TPP-D-NAC), an antioxidant and anti-inflammatory agent, for mitochondrial targeting under oxidative stress. Results showed that TTP-D-NAC specifically co-localized with mitochondria in activated microglia after systemic administration, confirming promise in BBB penetrability and targeting injured glia [184].
Betulinic acid (BA) NPs derived from the herb E. ulmoides have been shown to penetrate the BBB and elicit an antioxidative effect, improving functional recovery after a middle cerebral artery occlusion injury. BA NPs can cross the BBB via cannabinoid receptor 1-mediated transcytosis (see Figure 3B) [185]. Recently, researchers converted BA to betulinic amine (BAM). BAM accelerates drug release in the acidic conditions of the ischemic environment after a stroke and when surface conjugated with AMD3100, which interacts with CXCR4 abundant in ischemic tissue, enhances brain penetration [186]. In this study, investigators delivered NA1, a peptide designed to protect neurons against NMDA receptor-mediated excitotoxicity. These NA1-AMD3100-BAM NPs significantly increased survival, reduced infarct volumes by 69.8%, and enhanced neurological scores [186]. Acidic conditions also characterize injured tissue post-TBI [187], thus acid-responsive NPs such as these which can cross the BBB and quickly deliver a payload would be advantageous to treating TBI in the acute phase. There are many other ROS scavengers considered in NP therapeutics for TBI treatment including nimodipine [168], PEG conjugated hydrophilic carbon clusters [188], and surfactant poloxamer 188- N-acetylcysteine-loaded PLGA NPs [189].
5.5. NPs enhance stem-cell integration for improved TBI outcome
In the past decade, nanotherapeutics began to focus on enhancing stem cell transplantation and recruitment to repair the secondary injury-induced lesion cavity and promote tissue regeneration usually through some NP-complex [138, 145, 172, 190-192]. Mesenchymal stem cells (MSCs) are of special interest to cell therapy approaches as their autologous origins evade any immune response, display inherent pluripotency, and demonstrate reparative properties at sites of injury [193, 194]. In an in-vitro study, gold NPs surface-modified with a zwitterionic pentapeptide designed from Bax inhibiting peptide (Ku70) to enhance cellular uptake and a linearized expression vector to induce expression of BDNF in rat-derived MSCs have shown promise in engineering for efficient transplantation of MSCs post-TBI in the future [138]. This technique demonstrated higher transfection efficiency than more complicated viral gene transfer technology as MSCs exposed to a single transfection by the gold NP-gene construct showed ~80% cell transfection and exhibited successive expression of BDNF/mCherry fusion [138].
In vivo, transfected human fetal tissue-derived neural stem cells (hNSCs) with neurogenin-2-loaded poly(β-amino ester)-based NPs produced a larger number of neuronal cells compared to non-transfected cells. Delivery of these hNSCs with a hyaluronic acid hydrogel promoted vascular formation at the lesion site at 4 weeks following injection in a CCI injury model [190]. In a different study, another hydrogel model was investigated using imidazole groups-modified gelatin methacrylate (GelMA-imid) loaded with polydopamine NPs used as a carrier for stromal-derived factor-1 (SDF-1α). Human amniotic mesenchymal stromal cells (hAMSCs) were injected into the damaged area of cryogenic TBI in rats and SDF-1α promoted the migration of hAMSCs and their differentiation into nerve cells [191]. More recently, SDF-1α-loaded NPs were shown to slowly release SDF-1α and induce NSC/neuroblasts to migrate to injured areas and potentially improve the lesion volume [192]. Though the work in this field is few and in the early stages, results show promise in recovering lesion volume through efficient NP complexing.
6. Conclusion
The multifunctionality and customizability of NPs make this versatile technology best suited to address the challenges that have previously impeded potential therapeutics from meeting their clinical endpoints. In this review, we have presented an overview of the major pathways that constitute the complex pathophysiology of the continuum of focal to diffuse TBIs, described the current pre-clinical and clinical management of TBI, discussed the logistical considerations and clinical relevance of popular animal models of TBI, and reported on the existing literature centered on the use of NPs for TBI treatment. NP technology shows promise in safely and specifically delivering pharmacological payloads across the BBB while potentially eliciting therapeutic benefits through surface chemistry and functionalizations, improving secondary outcomes.
7. Expert opinion
The prospect of an approved TBI therapy is contingent upon the specific delivery of a therapeutic which confers minimal off-target effects while maintaining a predictable release profile. TBIs present on a spectrum of focal to diffuse types and therapeutics must be able to penetrate both the intact and compromised BBB not only to address this injury continuum but also to be used in both acute and chronic therapies. Nanoparticle technology is the best fit to meet these demands as their modifiable chemistry, size, charge, and customizable multifunctionality harness the potential to foster payload protection and improve therapeutic efficacy, avoiding off-target toxicity and biodegradation. NPs can also complex with other materials like hydrogels to improve therapies to regain structural and cognitive functioning of TBI patients, in combination with existing post-injury clinical treatment strategies. To facilitate the translation of the use of NPs to treat TBIs, NPs should be simple to formulate, stable at clinically relevant temperatures, and easily stored and transported for use in both acute and long-term treatment.
Though in general, research in this field is in the nascent phase of clinical translation, its potential to address multiple facets of downstream injury cascades is noteworthy. Stem cell therapies alone have shown great promise in treating TBI [195-199], and their efficacy can be improved by NP complexing as demonstrated in section 5.5. For instance, in the 2021 STEMTRA Phase II trial, intracranial implantation of allogenic modified bone marrow-derived mesenchymal stromal cells in patients with chronic motor deficits secondary to TBI showed significant improvement from baseline Fugl-Meyer Motor Scale scores at 6 months but failed to meet the secondary efficacy endpoint at statistically significant levels [200].
Future studies should investigate the effects of combining multiple NP therapeutics on TBI outcomes, consider how the dosage volume and frequency can be optimized for human applications, and study how practical these approaches are in a clinical setting or even pre-hospital environments, like in the case where, for example, hemostatic NPs could potentially improve acute survival on-site. Investigators must also be diligent about measuring off-target NP accumulation in other organs, measuring for toxicity, and having quantifiable evidence that the NPs improve physiological, functional, and cognitive outcomes. Future studies should also focus on designing more assessments in these areas for animals phylogenetically closer to humans as well as continue to improve the injury model designs to better simulate human TBI outcomes. Bearing in mind these considerations, NP technology has the capacity to greatly improve the chances of achieving an approved therapeutic for TBI treatment.
Article highlights.
The pathophysiology of TBIs is biphasic and pharmacological intervention is only possible in the secondary injury phase. Determining prognosis can be complex and is accomplished through a combination of interpreting imaging data such as MRI and CT scans in addition to employing the Glasgow Coma Scale and assessing other motor and cognitive symptomology.
Many Phase III clinical trials fail mainly due to poor targeting efficiency, experimental design, and low retention of the drug in the injured brain.
When using animal models to understand the pathophysiology and cognitive and functional outcomes of TBI, researchers must choose between models that elicit either focal, diffuse, or mixed injuries, tune the parameters of the injury setup, when possible, to reflect the target injury severity, and use sham animal groups to account for cerebral metabolic and physiological effects that are a result of the surgical environment.
Nanoparticles can increase the efficiency and efficacy of therapeutics as their customizable size, stealthy chemistry, and multifunctionality allow NPs to cross the BBB, target the injured brain, and protect and deliver its pharmacological payload all while avoiding off-target effects.
Nanoparticles designed to treat TBIs broadly include NPs with hemostatic, immunomodulating, anti-inflammatory, anti-oxidative, and gene-editing properties to enhance trauma recovery.
Funding
This work was supported by NIH Grant NS110721 (Jiangbing Zhou), the Ford Foundation Predoctoral Fellowship (Farrah S. Mohammed), and the National Science Foundation Graduate Research Fellowship Program (Farrah S. Mohammed).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- 1.Estimates, W.G.H. The top 10 causes of death 2020 [cited 2022 May 2, 2022]; Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- 2.Frieden TR, Houry D, and Baldwin G, REPORT TO CONGRESS Traumatic Brain Injury In the United States: Epidemiology and Rehabilitation. 2015, Centers for Disease Control and Prevention; National Center for Injury Prevention and Contro; Division of Unintentional injury Prevention; p. 1–59. [Google Scholar]
- 3.Dewan MC, et al. , Estimating the global incidence of traumatic brain injury. J Neurosurg, 2018: p. 1–18. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty J, et al. , Traumatic Brain Injury-Related Deaths by Race/Ethnicity, Sex, Intent, and Mechanism of Injury - United States, 2000-2017. MMWR Morb Mortal Wkly Rep, 2019. 68(46): p. 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swanson TM, et al. , Traumatic Brain Injury Incidence, Clinical Overview, and Policies in the US Military Health System Since 2000. Public Health Reports 2017. 132(2): p. 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stubbs JL, et al. , Traumatic brain injury in homeless and marginally housed individuals: a systematic review and meta-analysis. Lancet Public Health, 2020. 5(1): p. e19–e32. [DOI] [PubMed] [Google Scholar]
- 7.Cassidy JD, Boyle E, and Carroll LJ, Population-based, inception cohort study of the incidence, course, and prognosis of mild traumatic brain injury after motor vehicle collisions. Arch Phys Med Rehabil, 2014. 95(3 Suppl): p. S278–85. [DOI] [PubMed] [Google Scholar]
- 8.Zu Eulenburg P, et al. , Changes in Blood Biomarkers of Brain Injury and Degeneration Following Long-Duration Spaceflight. JAMA Neurol, 2021. 78(12): p. 1525–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao W, et al. , Neurofilament light chain level in traumatic brain injury: A system review and meta-analysis. Medicine (Baltimore), 2020. 99(38): p. e22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodnar CN, et al. , A Systematic Review of Closed Head Injury Models of Mild Traumatic Brain Injury in Mice and Rats. J Neurotrauma, 2019. 36(11): p. 1683–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Temple N, et al. , Neuroimaging in adult penetrating brain injury: a guide for radiographers. J Med Radiat Sci, 2015. 62(2): p. 122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teasdale G, et al. , The Glasgow Coma Scale at 40 years: standing the test of time. Lancet Neurology 2014. 13(8): p. 844–54. [DOI] [PubMed] [Google Scholar]
- 13.Demetriades D, et al. , Mortality prediction of head Abbreviated Injury Score and Glasgow Coma Scale: analysis of 7,764 head injuries. J Am Coll Surg, 2004. 199(2): p. 216–22. [DOI] [PubMed] [Google Scholar]
- 14.Lannoo E, et al. , Early predictors of mortality and morbidity after severe closed head injury. J Neurotrauma, 2000. 17(5): p. 403–14. [DOI] [PubMed] [Google Scholar]
- 15.Maas AIR, et al. , Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery 2005. 57(6): p. 1173–82. [DOI] [PubMed] [Google Scholar]
- 16.Maas AI, et al. , Prognostic value of computerized tomography scan characteristics in traumatic brain injury: results from the IMPACT study. J Neurotrauma, 2007. 24(2): p. 303–14. [DOI] [PubMed] [Google Scholar]
- 17.Aldossary NM, Kotb MA, and Kamal AM, Predictive value of early MRI findings on neurocognitive and psychiatric outcomes in patients with severe traumatic brain injury. J Affect Disord, 2019. 243: p. 1–7. [DOI] [PubMed] [Google Scholar]
- 18.Wilson L, et al. , A Manual for the Glasgow Outcome Scale-Extended Interview. J Neurotrauma, 2021. 38(17): p. 2435–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foreman BP, et al. , Usefulness of the abbreviated injury score and the injury severity score in comparison to the Glasgow Coma Scale in predicting outcome after traumatic brain injury. J Trauma, 2007. 62(4): p. 946–50. [DOI] [PubMed] [Google Scholar]
- 20.Wang KKW, et al. , Systems biomarkers as acute diagnostics and chronic monitoring tools for traumatic brain injury. Proceedings SPIE 8723, Sensing Technologies for Global Health, Military Medicine, and Environmental Monitoring III, 87230O, 2013. [Google Scholar]
- 21.Jarrahi A, et al. , Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines, 2020. 8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loane DJ and Faden AI, Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci, 2010. 31(12): p. 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis AE, Mechanisms of traumatic brain injury: biomechanical, structural and cellular considerations. Crit Care Nurs Q, 2000. 23(3): p. 1–13. [DOI] [PubMed] [Google Scholar]
- 24.Kaur P and Sharma S, Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr Neuropharmacol, 2018. 16(8): p. 1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galea OA, et al. , Sensorimotor and Physiological Indicators of Impairment in Mild Traumatic Brain Injury: A Meta-Analysis. Neurorehabil Neural Repair, 2018. 32(2): p. 115–128. [DOI] [PubMed] [Google Scholar]
- 26.Caeyenberghs K, et al. , Correlations between white matter integrity and motor function in traumatic brain injury patients. Neurorehabil Neural Repair, 2011. 25(6): p. 492–502. [DOI] [PubMed] [Google Scholar]
- 27.Peltz CB, et al. , Blood biomarkers of traumatic brain injury and cognitive impairment in older veterans. Neurology, 2020. 95(9): p. e1126–e1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorgoraptis N, et al. , Cognitive impairment and health-related quality of life following traumatic brain injury. NeuroRehabilitation, 2019. 44(3): p. 321–331. [DOI] [PubMed] [Google Scholar]
- 29.de Freitas Cardoso MG, et al. , Cognitive Impairment Following Acute Mild Traumatic Brain Injury. Front Neurol, 2019. 10: p. 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei B, et al. , Anti-inflammatory effects of progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS One, 2014. 9(7): p. e103969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, et al. , Progesterone influences postischemic synaptogenesis in the CA1 region of the hippocampus in rats. Synapse, 2011. 65(9): p. 880–91. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Ovejero D, et al. , Progesterone reduces secondary damage, preserves white matter, and improves locomotor outcome after spinal cord contusion. J Neurotrauma, 2014. 31(9): p. 857–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si D, et al. , Progesterone treatment improves cognitive outcome following experimental traumatic brain injury in rats. Neurosci Lett, 2013. 553: p. 18–23. [DOI] [PubMed] [Google Scholar]
- 34.Amirkhosravi L, et al. , Improved spatial memory, neurobehavioral outcomes, and neuroprotective effect after progesterone administration in ovariectomized rats with traumatic brain injury: Role of RU486 progesterone receptor antagonist. Iran J Basic Med Sci, 2021. 24(3): p. 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howard RB, Sayeed I, and Stein DG, Suboptimal Dosing Parameters as Possible Factors in the Negative Phase III Clinical Trials of Progesterone for Traumatic Brain Injury. Journal of Neurotrauma 2016. 34(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein DG, Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Injury 2015. 29(11): p. 1259–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maas AIR, et al. , Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurology, 2006. 5(1). [DOI] [PubMed] [Google Scholar]
- 38.Park E, Bell JD, and Baker AJ, Traumatic brain injury: Can the consequences be stopped? CMAJ 2008. 178(9): p. 1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andriessen TM, Jacobs B, and Vos PE, Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med, 2010. 14(10): p. 2381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huisman TAGM, et al. , Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol, 2004. 25(3): p. 370–6. [PMC free article] [PubMed] [Google Scholar]
- 41.Smith DH, Meaney DF, and Shull WH, Diffuse axonal injury in head trauma. J Head Trauma Rehabil, 2003. 18(4): p. 307–16. [DOI] [PubMed] [Google Scholar]
- 42.Kim JJ and Gean AD, Imaging for the diagnosis and management of traumatic brain injury. Neurotherapeutics, 2011. 8(1): p. 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koliatsos V and Alexandris A, Wallerian degeneration as a therapeutic target in traumatic brain injury. Current Opinion in Neurology 2020. 32(6): p. 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziogas NK and Koliatsos VE, Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms with High-Resolution Anatomical Methods. J Neurosci, 2018. 38(16): p. 4031–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhowmick S, et al. , Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp Neurol, 2019. 317: p. 260–270. [DOI] [PubMed] [Google Scholar]
- 46.Thal C, and W S. Neuhaus, The Blood–Brain Barrier as a Target in Traumatic Brain Injury Treatment. Archives of Medical Research, 2014. 45(8): p. 698–710. [DOI] [PubMed] [Google Scholar]
- 47.Rungta RL, et al. , The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell, 2015. 161(3): p. 610–621. [DOI] [PubMed] [Google Scholar]
- 48.Bye N, et al. , Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Experimental Neurology 2007. 204(1): p. 220–233. [DOI] [PubMed] [Google Scholar]
- 49.Alam A, et al. , Cellular infiltration in traumatic brain injury. J Neuroinflammation, 2020. 17(1): p. 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar A, et al. , Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J Neurotrauma, 2016. 33(19): p. 1732–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fawcett JW and Asher RA, The glial scar and central nervous system repair. Brain Res Bull, 1999. 49(6): p. 377–91. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, et al. , Portrait of glial scar in neurological diseases. Int J Immunopathol Pharmacol, 2018. 31: p. 2058738418801406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roh JS and Sohn DH, Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw, 2018. 18(4): p. e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu T, et al. , NF-kappaB signaling in inflammation. Signal Transduct Target Ther, 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang ZR, et al. , Regulating effect of activated NF-kappaB on edema induced by traumatic brain injury of rats. Asian Pac J Trop Med, 2016. 9(3): p. 274–7. [DOI] [PubMed] [Google Scholar]
- 56.Mettang M, et al. , IKK2/NF-kappaB signaling protects neurons after traumatic brain injury. FASEB J, 2018. 32(4): p. 1916–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonald B, et al. , Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science, 2010. 330(6002): p. 362–6. [DOI] [PubMed] [Google Scholar]
- 58.Gong T, et al. , DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol, 2020. 20(2): p. 95–112. [DOI] [PubMed] [Google Scholar]
- 59.Povlishock JT, Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol, 1992. 2(1): p. 1–12. [PubMed] [Google Scholar]
- 60.Lotocki G, et al. , Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J Neurotrauma, 2009. 26(7): p. 1123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yi JH and Hazell AS, Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int, 2006. 48(5): p. 394–403. [DOI] [PubMed] [Google Scholar]
- 62.Ansari MA, Roberts KN, and Scheff SW, A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma, 2008. 25(5): p. 513–26. [DOI] [PubMed] [Google Scholar]
- 63.Park E, Bell JD, and Baker AJ, Traumatic brain injury: can the consequences be stopped? CMAJ, 2008. 178(9): p. 1163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng P, et al. , Trends in traumatic brain injury mortality in China, 2006-2013: A population-based longitudinal study. PLoS Med, 2017. 14(7): p. e1002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shekhar C, et al. , An epidemiological study of traumatic brain injury cases in a trauma centre of New Delhi (India). J Emerg Trauma Shock, 2015. 8(3): p. 131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Du RY, et al. , Primary prevention of road traffic accident–related traumatic brain injuries in younger populations: a systematic review of helmet legislation. Journal of Neurosurgery 2020. 25(4). [DOI] [PubMed] [Google Scholar]
- 67.Bombardier CH, Rimmele CT, and Zintel H, The magnitude and correlates of alcohol and drug use before traumatic brain injury. Archives of Physical Medicine and Rehabilitation, 2002. 83(12): p. 1765–1773. [DOI] [PubMed] [Google Scholar]
- 68.Hendricks S and Lambert M, Tackling in Rugby: Coaching Strategies for Effective Technique and Injury Prevention. International Journal of Sports Science & Coaching, 2010. 5(1): p. 117–135. [Google Scholar]
- 69.Lumba-Brown A, et al. , Centers for Disease Control and Prevention Guideline on the Diagnosis and Management of Mild Traumatic Brain Injury Among Children. JAMA Pediatr, 2018. 172(11): p. e182853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prevention, C.f.D.C.a. TBI: Symptoms of traumatic brain injury. 2019. [cited 2022 May25]; Available from: https://www.cdc.gov/traumaticbraininjury/concussion/symptoms.html.
- 71.Lu J, et al. , A method for reducing misclassification in the extended Glasgow Outcome Score. J Neurotrauma, 2010. 27(5): p. 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rush B, Mass Effect Encyclopedia of Clinical Neuropsychology 2011. [Google Scholar]
- 73.Mokri B, The Monro-Kellie hypothesis: applications in CSF volume depletion. Neurology, 2001. 56(12): p. 1746–8. [DOI] [PubMed] [Google Scholar]
- 74.Wakai A, et al. , Mannitol for acute traumatic brain injury. Cochrane Database Syst Rev, 2013(8): p. CD001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fatima N, et al. , The Role of Decompressive Craniectomy in Traumatic Brain Injury: A Systematic Review and Meta-analysis. Asian J Neurosurg, 2019. 14(2): p. 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Curzon P, et al. , The Behavioral Assessment of Sensorimotor Processes in the Mouse: Acoustic Startle, Sensory Gating, Locomotor Activity, Rotarod, and Beam Walking, in Methods of Behavior Analysis in Neuroscience, nd and Buccafusco JJ, Editors. 2009: Boca Raton (FL). [PubMed] [Google Scholar]
- 77.Wang X, et al. , Neuroprotective effect of Bax-inhibiting peptide on neonatal brain injury. Stroke, 2010. 41(9): p. 2050–5. [DOI] [PubMed] [Google Scholar]
- 78.Tucker LB, Velosky AG, and McCabe JT, Applications of the Morris water maze in translational traumatic brain injury research. Neurosci Biobehav Rev, 2018. 88: p. 187–200. [DOI] [PubMed] [Google Scholar]
- 79.Zou H, et al. , Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil Neural Repair, 2013. 27(9): p. 878–88. [DOI] [PubMed] [Google Scholar]
- 80.Kim K, et al. , Evaluation of cognitive function in adult rhesus monkeys using the finger maze test. Applied Animal Behavior Science 2020. 224. [Google Scholar]
- 81.Vink R, Large animal models of traumatic brain injury. J Neurosci Res, 2018. 96(4): p. 527–535. [DOI] [PubMed] [Google Scholar]
- 82.Siebold L, Obenaus A, and Goyal R, Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol, 2018. 310: p. 48–57. [DOI] [PubMed] [Google Scholar]
- 83.Grin'kina NM, et al. , Righting Reflex Predicts Long-Term Histological and Behavioral Outcomes in a Closed Head Model of Traumatic Brain Injury. PLoS One, 2016. 11(9): p. e0161053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Crenn P, et al. , Changes in weight after traumatic brain injury in adult patients: a longitudinal study. Clin Nutr, 2014. 33(2): p. 348–53. [DOI] [PubMed] [Google Scholar]
- 85.Kwon SK, et al. , Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front Neurol, 2011. 2: p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith DH, et al. , Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg, 2000. 93(2): p. 315–22. [DOI] [PubMed] [Google Scholar]
- 87.Meaney DF, et al. , Modification of the cortical impact model to produce axonal injury in the rat cerebral cortex. J Neurotrauma, 1994. 11(5): p. 599–612. [DOI] [PubMed] [Google Scholar]
- 88.Foda MA and Marmarou A, A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg, 1994. 80(2): p. 301–13. [DOI] [PubMed] [Google Scholar]
- 89.Kannan G, Kambhampati SP, and Kudchadkar SR, Effect of anesthetics on microglial activation and nanoparticle uptake: Implications for drug delivery in traumatic brain injury. J Control Release, 2017. 263: p. 192–199. [DOI] [PubMed] [Google Scholar]
- 90.Statler KD, et al. , Isoflurane exerts neuroprotective actions at or near the time of severe traumatic brain injury. Brain Res, 2006. 1076(1): p. 216–24. [DOI] [PubMed] [Google Scholar]
- 91.Bickler PE, Buck LT, and Hansen BM, Effects of isoflurane and hypothermia on glutamate receptor-mediated calcium influx in brain slices. Anesthesiology, 1994. 81(6): p. 1461–9. [DOI] [PubMed] [Google Scholar]
- 92.Statler KD, et al. , The simple model versus the super model: translating experimental traumatic brain injury research to the bedside. J Neurotrauma, 2001. 18(11): p. 1195–206. [DOI] [PubMed] [Google Scholar]
- 93.Skandsen T, et al. , Prevalence and impact of diffuse axonal injury in patients with moderate and severe head injury: a cohort study of early magnetic resonance imaging findings and 1-year outcome. J Neurosurg, 2010. 113(3): p. 556–63. [DOI] [PubMed] [Google Scholar]
- 94.Feeney DM, et al. , Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res, 1981. 211(1): p. 67–77. [DOI] [PubMed] [Google Scholar]
- 95.Dail WG, et al. , Responses to cortical injury: II. Widespread depression of the activity of an enzyme in cortex remote from a focal injury. Brain Res, 1981. 211(1): p. 79–89. [DOI] [PubMed] [Google Scholar]
- 96.Morales DM, et al. , Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience, 2005. 136(4): p. 971–89. [DOI] [PubMed] [Google Scholar]
- 97.Li H, et al. , Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res, 2016. 1642: p. 70–78. [DOI] [PubMed] [Google Scholar]
- 98.Bellander BM, et al. , Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J Neurosurg, 1996. 85(3): p. 468–75. [DOI] [PubMed] [Google Scholar]
- 99.Holmin S, et al. , Delayed cytokine expression in rat brain following experimental contusion. J Neurosurg, 1997. 86(3): p. 493–504. [DOI] [PubMed] [Google Scholar]
- 100.Flierl MA, et al. , Mouse closed head injury model induced by a weight-drop device. Nat Protoc, 2009. 4(9): p. 1328–37. [DOI] [PubMed] [Google Scholar]
- 101.Marmarou A, et al. , A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg, 1994. 80(2): p. 291–300. [DOI] [PubMed] [Google Scholar]
- 102.Maruichi K, et al. , Graded model of diffuse axonal injury for studying head injury-induced cognitive dysfunction in rats. Neuropathology, 2009. 29(2): p. 132–9. [DOI] [PubMed] [Google Scholar]
- 103.Khalin I, et al. , A mouse model of weight-drop closed head injury: emphasis on cognitive and neurological deficiency. Neural Regen Res, 2016. 11(4): p. 630–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dixon CE, et al. , A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods, 1991. 39(3): p. 253–62. [DOI] [PubMed] [Google Scholar]
- 105.Elliott MB, Jallo JJ, and Tuma RF, An investigation of cerebral edema and injury volume assessments for controlled cortical impact injury. J Neurosci Methods, 2008. 168(2): p. 320–4. [DOI] [PubMed] [Google Scholar]
- 106.Smith DH, et al. , A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma, 1995. 12(2): p. 169–78. [DOI] [PubMed] [Google Scholar]
- 107.Manley GT, et al. , Controlled cortical impact in swine: pathophysiology and biomechanics. J Neurotrauma, 2006. 23(2): p. 128–39. [DOI] [PubMed] [Google Scholar]
- 108.Shin SS, et al. , Transcriptional Profiling in a Novel Swine Model of Traumatic Brain Injury. Neurotrauma Rep, 2022. 3(1): p. 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hawryluk GW, et al. , Brain tissue oxygen tension and its response to physiological manipulations: influence of distance from injury site in a swine model of traumatic brain injury. J Neurosurg, 2016. 125(5): p. 1217–1228. [DOI] [PubMed] [Google Scholar]
- 110.Korley FK, et al. , Valproic Acid Treatment Decreases Serum Glial Fibrillary Acidic Protein and Neurofilament Light Chain Levels in Swine Subjected to Traumatic Brain Injury. J Neurotrauma, 2018. 35(10): p. 1185–1191. [DOI] [PubMed] [Google Scholar]
- 111.King C, et al. , Brain temperature profiles during epidural cooling with the ChillerPad in a monkey model of traumatic brain injury. J Neurotrauma, 2010. 27(10): p. 1895–903. [DOI] [PubMed] [Google Scholar]
- 112.Kelly KM, et al. , Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res, 2015. 117: p. 104–16. [DOI] [PubMed] [Google Scholar]
- 113.Pappius HM, Local cerebral glucose utilization in thermally traumatized rat brain. Ann Neurol, 1981. 9(5): p. 484–91. [DOI] [PubMed] [Google Scholar]
- 114.Raslan F, et al. , Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J Cereb Blood Flow Metab, 2010. 30(8): p. 1477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eriskat J, et al. , Correlation of lesion volume and brain swelling from a focal brain trauma. Acta Neurochir Suppl, 2003. 86: p. 265–6. [DOI] [PubMed] [Google Scholar]
- 116.Williams AJ, et al. , Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma, 2005. 22(2): p. 313–31. [DOI] [PubMed] [Google Scholar]
- 117.Williams AJ, et al. , Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation, 2007. 4: p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bauman RA, et al. , An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma, 2009. 26(6): p. 841–60. [DOI] [PubMed] [Google Scholar]
- 119.de Lanerolle NC, et al. , Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol, 2011. 70(11): p. 1046–57. [DOI] [PubMed] [Google Scholar]
- 120.Clemedson CJ and Criborn CO, A detonation chamber for physiological blast research. J Aviat Med, 1955. 26(5): p. 373–81. [PubMed] [Google Scholar]
- 121.Saljo A, et al. , Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J Neurotrauma, 2000. 17(8): p. 719–26. [DOI] [PubMed] [Google Scholar]
- 122.Skotak M, et al. , Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma, 2013. 30(13): p. 1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kumar R and Nedungadi A, Using Gas-Driven Shock Tubes to Produce Blast Wave Signatures. Frontiers Neurology 2020. 11(90). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rowe RK, et al. , Midline (central) fluid percussion model of traumatic brain injury in pediatric and adolescent rats. J Neurosurg Pediatr, 2018. 22(1): p. 22–30. [DOI] [PubMed] [Google Scholar]
- 125.McIntosh TK, et al. , Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience, 1989. 28(1): p. 233–44. [DOI] [PubMed] [Google Scholar]
- 126.Zafonte R, et al. , The citicoline brain injury treatment (COBRIT) trial: design and methods. J Neurotrauma, 2009. 26(12): p. 2207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Doppenberg EM, Choi SC, and Bullock R, Clinical trials in traumatic brain injury: lessons for the future. J Neurosurg Anesthesiol, 2004. 16(1): p. 87–94. [DOI] [PubMed] [Google Scholar]
- 128.Lok J, et al. , Targeting the neurovascular unit in brain trauma. CNS Neurosci Ther, 2015. 21(4): p. 304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ma J, et al. , Targeted Drug Delivery to Stroke via Chemotactic Recruitment of Nanoparticles Coated with Membrane of Engineered Neural Stem Cells. Small, 2019. 15(35): p. e1902011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kyriakides TR, et al. , Biocompatibility of nanomaterials and their immunological properties. Biomedical Materials 2021. 16(4). ** Review provides a comprehensive survey of the major classes of nanomaterials, their applications, biocompatibility, and their topical and systemic interaction with the immune system.
- 131.Waggoner LE, et al. , Pharmacokinetic Analysis of Peptide-Modified Nanoparticles with Engineered Physicochemical Properties in a Mouse Model of Traumatic Brain Injury. AAPS J, 2021. 23(5): p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tobio M, et al. , The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf B Biointerfaces, 2000. 18(3-4): p. 315–323. [DOI] [PubMed] [Google Scholar]
- 133.Torchilin VP, et al. , Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta, 1994. 1195(1): p. 11–20. [DOI] [PubMed] [Google Scholar]
- 134.Levchenko TS, et al. , Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int J Pharm, 2002. 240(1-2): p. 95–102. [DOI] [PubMed] [Google Scholar]
- 135. Li W, et al. , BBB pathophysiology-independent delivery of siRNA in traumatic brain injury. Sci Adv, 2021. 7(1). * The first reported example of BBB pathophysiology-independent delivery of siRNA-loaded NPs for TBI treatment.
- 136.Alyautdin RN, et al. , Delivery of loperamide across the blood-brain barrier with polysorbate 80-coated polybutylcyanoacrylate nanoparticles. Pharm Res, 1997. 14(3): p. 325–8. [DOI] [PubMed] [Google Scholar]
- 137.Zhang Y, et al. , Catalytically active gold clusters with atomic precision for noninvasive early intervention of neurotrauma. J Nanobiotechnology, 2021. 19(1): p. 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Muroski ME, et al. , A gold nanoparticle pentapeptide: gene fusion to induce therapeutic gene expression in mesenchymal stem cells. J Am Chem Soc, 2014. 136(42): p. 14763–71. [DOI] [PubMed] [Google Scholar]
- 139.Bailey ZS, et al. , Cerium Oxide Nanoparticles Improve Outcome after In Vitro and In Vivo Mild Traumatic Brain Injury. J Neurotrauma, 2020. 37(12): p. 1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Youn DH, et al. , Shape effect of cerium oxide nanoparticles on mild traumatic brain injury. Sci Rep, 2021. 11(1): p. 15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sushnitha M, et al. , Cell Membrane-Based Biomimetic Nanoparticles and the Immune System: Immunomodulatory Interactions to Therapeutic Applications. Front Bioeng Biotechnol, 2020. 8: p. 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sharma S, et al. , Intravenous Immunomodulatory Nanoparticle Treatment for Traumatic Brain Injury. Ann Neurol, 2020. 87(3): p. 442–455. * A novel investigation of using immunomodulatory NPs to specifically treat TBI, in both focal and diffuse models, to ablate a subset of hematogenous monocytes and quell systemic and local sources of inflammation.
- 143.Zinger A, et al. , Biomimetic Nanoparticles as a Theranostic Tool for Traumatic Brain Injury. Adv Funct Mater, 2021. 31(30): p. 2100722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pu H, et al. , Interleukin-4 improves white matter integrity and functional recovery after murine traumatic brain injury via oligodendroglial PPARgamma. J Cereb Blood Flow Metab, 2021. 41(3): p. 511–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mayilsamy K, et al. , Treatment with shCCL20-CCR6 nanodendriplexes and human mesenchymal stem cell therapy improves pathology in mice with repeated traumatic brain injury. Nanomedicine, 2020. 29: p. 102247. [DOI] [PubMed] [Google Scholar]
- 146.Wang WX, et al. , Temporal changes in inflammatory mitochondria-enriched microRNAs following traumatic brain injury and effects of miR-146a nanoparticle delivery. Neural Regen Res, 2021. 16(3): p. 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McKee CA and Lukens JR, Emerging Roles for the Immune System in Traumatic Brain Injury. Front Immunol, 2016. 7: p. 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Beer JH, Springer KT, and Coller BS, Immobilized Arg-Gly-Asp (RGD) peptides of varying lengths as structural probes of the platelet glycoprotein IIb/IIIa receptor. Blood, 1992. 79(1): p. 117–28. [PubMed] [Google Scholar]
- 149.Brown AC, Lavik E, and Stabenfeldt SE, Biomimetic Strategies To Treat Traumatic Brain Injury by Leveraging Fibrinogen. Bioconjug Chem, 2019. 30(7): p. 1951–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hubbard WB, et al. , Hemostatic nanoparticles increase survival, mitigate neuropathology and alleviate anxiety in a rodent blast trauma model. Sci Rep, 2018. 8(1): p. 10622. * The first study to provide evidence that subacute outcomes of blast injury can improve following treatment with hemostatic nanoparticles and hemostatic nanoparticles encapsulating dexamethasone.
- 151.Lashof-Sullivan MM, et al. , Intravenously administered nanoparticles increase survival following blast trauma. Proc Natl Acad Sci U S A, 2014. 111(28): p. 10293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Monien BH, Cheang KI, and Desai UR, Mechanism of poly(acrylic acid) acceleration of antithrombin inhibition of thrombin: implications for the design of novel heparin mimics. J Med Chem, 2005. 48(16): p. 5360–8. [DOI] [PubMed] [Google Scholar]
- 153.Huang AP, et al. , An anti-inflammatory gelatin hemostatic agent with biodegradable polyurethane nanoparticles for vulnerable brain tissue. Mater Sci Eng C Mater Biol Appl, 2021. 121: p. 111799. [DOI] [PubMed] [Google Scholar]
- 154.Vajda FJ, Neuroprotection and neurodegenerative disease. J Clin Neurosci, 2002. 9(1): p. 4–8. [DOI] [PubMed] [Google Scholar]
- 155.Kwon EJ, et al. , Neuron-Targeted Nanoparticle for siRNA Delivery to Traumatic Brain Injuries. ACS Nano, 2016. 10(8): p. 7926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Edwards G 3rd, Moreno-Gonzalez I, and Soto C, Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem Biophys Res Commun, 2017. 483(4): p. 1137–1142. [DOI] [PubMed] [Google Scholar]
- 157.Ye Z, et al. , Combined Inhibition of Fyn and c-Src Protects Hippocampal Neurons and Improves Spatial Memory via ROCK after Traumatic Brain Injury. J Neurotrauma, 2022. 39(7-8): p. 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Macks C, Jeong D, and Lee JS, Local delivery of RhoA siRNA by PgP nanocarrier reduces inflammatory response and improves neuronal cell survival in a rat TBI model. Nanomedicine, 2021. 32: p. 102343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Macks C, Jeong D, and Lee JS, Therapeutic efficacy of rolipram delivered by PgP nanocarrier on secondary injury and motor function in a rat TBI model. Nanomedicine (Lond), 2022. 17(7): p. 431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ruozi B, et al. , PLGA Nanoparticles Loaded Cerebrolysin: Studies on Their Preparation and Investigation of the Effect of Storage and Serum Stability with Reference to Traumatic Brain Injury. Mol Neurobiol, 2015. 52(2): p. 899–912. [DOI] [PubMed] [Google Scholar]
- 161.Ruozi B, et al. , Poly (D,L-lactide-co-glycolide) nanoparticles loaded with cerebrolysin display neuroprotective activity in a rat model of concussive head injury. CNS Neurol Disord Drug Targets, 2014. 13(8): p. 1475–82. [DOI] [PubMed] [Google Scholar]
- 162.Wurzelmann M, Romeika J, and Sun D, Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen Res, 2017. 12(1): p. 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Obermeyer JM, et al. , Local Delivery of Brain-Derived Neurotrophic Factor Enables Behavioral Recovery and Tissue Repair in Stroke-Injured Rats. Tissue Eng Part A, 2019. 25(15-16): p. 1175–1187. [DOI] [PubMed] [Google Scholar]
- 164. Khalin I, et al. , Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv, 2016. 23(9): p. 3520–3528. * In vivo study which for the first time demonstrates that IV injection of poloxamer 188 coated PLGA NPs with BDNF increased BDNF levels in the CNS to improve neurological and cognitive outcomes in animals with CHI TBIs.
- 165.Aarts M, et al. , Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science, 2002. 298(5594): p. 846–50. [DOI] [PubMed] [Google Scholar]
- 166.Yu X, et al. , Activatable Protein Nanoparticles for Targeted Delivery of Therapeutic Peptides. Adv Mater, 2018. 30(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wu P, et al. , Targeted delivery of polypeptide nanoparticle for treatment of traumatic brain injury. Int J Nanomedicine, 2019. 14: p. 4059–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Han Z, et al. , A Novel Targeted Nanoparticle for Traumatic Brain Injury Treatment: Combined Effect of ROS Depletion and Calcium Overload Inhibition. Adv Healthc Mater, 2022. 11(11): p. e2102256. [DOI] [PubMed] [Google Scholar]
- 169.Bharadwaj VN, et al. , Nanoparticle-Based Therapeutics for Brain Injury. Adv Healthc Mater, 2018. 7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mann AP, et al. , A peptide for targeted, systemic delivery of imaging and therapeutic compounds into acute brain injuries. Nat Commun, 2016. 7: p. 11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sun D, et al. , Intrinsically Bioactive Manganese-Eumelanin Nanocomposites Mediated Antioxidation and Anti-Neuroinflammation for Targeted Theranostics of Traumatic Brain Injury. Adv Healthc Mater, 2022: p. e2200517. [DOI] [PubMed] [Google Scholar]
- 172.Narouiepour A, et al. , Neural stem cell therapy in conjunction with curcumin loaded in niosomal nanoparticles enhanced recovery from traumatic brain injury. Sci Rep, 2022. 12(1): p. 3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sintov AC, AmyloLipid Nanovesicles: A self-assembled lipid-modified starch hybrid system constructed for direct nose-to-brain delivery of curcumin. Int J Pharm, 2020. 588: p. 119725. [DOI] [PubMed] [Google Scholar]
- 174.Whitener R, et al. , Localization of Multi-Lamellar Vesicle Nanoparticles to Injured Brain Tissue in a Controlled Cortical Impact Injury Model of Traumatic Brain Injury in Rodents. Neurotrauma Rep, 2022. 3(1): p. 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ratliff WA, et al. , Behavior, protein, and dendritic changes after model traumatic brain injury and treatment with nanocoffee particles. BMC Neurosci, 2019. 20(1): p. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang Y, Jia F, and Lin Y, Poly(butyl cyanoacrylate) nanoparticles-deliveredbeta-nerve growth factor promotes the neurite outgrowth and reduces the mortality in the rat after traumatic brain injury. Nanotechnology, 2022. 33(13). [DOI] [PubMed] [Google Scholar]
- 177.Grasso G, et al. , The role of erythropoietin in neuroprotection: therapeutic perspectives. Drug News Perspect, 2007. 20(5): p. 315–20. [DOI] [PubMed] [Google Scholar]
- 178.Figueroa CE, et al. , Highly loaded nanoparticulate formulation of progesterone for emergency traumatic brain injury treatment. Ther Deliv, 2012. 3(11): p. 1269–79. [DOI] [PubMed] [Google Scholar]
- 179.Yoo D, et al. , Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano, 2017. 11(9): p. 8600–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tarudji AW, et al. , Antioxidant thioether core-crosslinked nanoparticles prevent the bilateral spread of secondary injury to protect spatial learning and memory in a controlled cortical impact mouse model of traumatic brain injury. Biomaterials, 2021. 272: p. 120766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Takahashi T, et al. , Novel neuroprotection using antioxidant nanoparticles in a mouse model of head trauma. J Trauma Acute Care Surg, 2020. 88(5): p. 677–685. [DOI] [PubMed] [Google Scholar]
- 182.Xiao L, et al. , Dihydrolipoic Acid-Gold Nanoclusters Regulate Microglial Polarization and Have the Potential To Alter Neurogenesis. Nano Lett, 2020. 20(1): p. 478–495. [DOI] [PubMed] [Google Scholar]
- 183.Sharma R, et al. , Dendrimer mediated targeted delivery of sinomenine for the treatment of acute neuroinflammation in traumatic brain injury. J Control Release, 2020. 323: p. 361–375. [DOI] [PubMed] [Google Scholar]
- 184.Sharma A, et al. , Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics, 2018. 8(20): p. 5529–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Deng G, et al. , Anti-edema and antioxidant combination therapy for ischemic stroke via glyburide-loaded betulinic acid nanoparticles. Theranostics, 2019. 9(23): p. 6991–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang S, et al. , Brain-targeting, acid-responsive antioxidant nanoparticles for stroke treatment and drug delivery. Bioact Mater, 2022. 16: p. 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Clausen T, et al. , Cerebral acid-base homeostasis after severe traumatic brain injury. J Neurosurg, 2005. 103(4): p. 597–607. [DOI] [PubMed] [Google Scholar]
- 188.Mendoza K, et al. , Functional and Structural Improvement with a Catalytic Carbon Nano-Antioxidant in Experimental Traumatic Brain Injury Complicated by Hypotension and Resuscitation. J Neurotrauma, 2019. 36(13): p. 2139–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Inyang E, et al. , Engineering Delivery of Nonbiologics Using Poly(lactic-co-glycolic acid) Nanoparticles for Repair of Disrupted Brain Endothelium. ACS Omega, 2020. 5(24): p. 14730–14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li X, et al. , Nanoparticle-mediated transcriptional modification enhances neuronal differentiation of human neural stem cells following transplantation in rat brain. Biomaterials, 2016. 84: p. 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zheng Y, et al. , Neuro-regenerative imidazole-functionalized GelMA hydrogel loaded with hAMSC and SDF-1alpha promote stem cell differentiation and repair focal brain injury. Bioact Mater, 2021. 6(3): p. 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhu Y, Wang Y, and Lu Z, Injection of Stromal Cell-Derived Factor-1 (SDF-1) Nanoparticles After Traumatic Brain Injury Stimulates Recruitment of Neural Stem Cells. J Biomed Nanotechnol, 2022. 18(2): p. 498–503. [DOI] [PubMed] [Google Scholar]
- 193.Yi T and Song SU, Immunomodulatory properties of mesenchymal stem cells and their therapeutic applications. Arch Pharm Res, 2012. 35(2): p. 213–21. [DOI] [PubMed] [Google Scholar]
- 194.Sokolova IB, et al. , Mesenchymal stem cells restore orientation and exploratory behavior of rats after brain injury. Bull Exp Biol Med, 2011. 151(1): p. 130–2. [DOI] [PubMed] [Google Scholar]
- 195.Gao J, et al. , Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol, 2006. 201(2): p. 281–92. [DOI] [PubMed] [Google Scholar]
- 196.Wei ZZ, et al. , Intracranial Transplantation of Hypoxia-Preconditioned iPSC-Derived Neural Progenitor Cells Alleviates Neuropsychiatric Defects After Traumatic Brain Injury in Juvenile Rats. Cell Transplant, 2016. 25(5): p. 797–809. [DOI] [PubMed] [Google Scholar]
- 197.Dunkerson J, et al. , Combining enriched environment and induced pluripotent stem cell therapy results in improved cognitive and motor function following traumatic brain injury. Restor Neurol Neurosci, 2014. 32(5): p. 675–87. [DOI] [PubMed] [Google Scholar]
- 198.Zhang R, et al. , Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation, 2013. 10: p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang Y, et al. , Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem Int, 2017. 111: p. 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kawabori M, et al. , Cell Therapy for Chronic TBI: Interim Analysis of the Randomized Controlled STEMTRA Trial. Neurology, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]