

Abstract
Damage associated molecular patterns (DAMPs) are molecules released from dead/dying cells following toxicant and/or environmental exposures that activate the immune response through binding pattern recognition receptors (PRRs). Excessive production of DAMPs or failed clearance leads to chronic inflammation and delayed inflammation resolution. One category of DAMPs are oxidized phospholipids (oxPLs) produced upon exposure to high levels of oxidative stress, such as following ozone (O3)- induced inflammation. OxPLs are bound by multiple classes of PRRs that include scavenger receptors (SRs) such as SR class B-1 (SR-BI) and toll-like receptors (TLRs). Interactions between oxPLs and PRRs appear to regulate inflammation; however, the role of SR-BI in oxPL-induced lung inflammation has not been defined. Therefore, we hypothesize that SR-BI is critical in protecting the lung from oxPL-induced pulmonary inflammation/injury. To test this hypothesis, C57BL/6J (WT) female mice were dosed with oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidylcholine (oxPAPC) by oropharyngeal aspiration and identified increased pulmonary SR-BI expression. Following oxPAPC exposure, SR-BI deficient (SR-BI(−/−) mice exhibited increased lung pathology and inflammatory cytokine/chemokine production. Lipidomic analysis revealed that SR-BI(−/− mice had an altered pulmonary lipidome prior to and following oxPAPC exposure, which correlated with increased oxidized phosphatidylcholines (PCs). Finally, we characterized TLR4-mediated activation of NF-κB following oxPAPC exposure and discovered that SR-BI−/− mice had increased TLR4 mRNA expression in lung tissue and macrophages, increased nuclear p65, and decreased cytoplasmic IκBα. Overall, we conclude that SR-BI is required for limiting oxPAPC-induced lung pathology by maintaining lipid homeostasis, reducing oxidized PCs, and attenuating TLR4-NF-κB activation thereby preventing excessive and persistent inflammation.
Keywords: SR-BI, DAMP, TLR4, Lung, Inflammation
1. Introduction
The classical role of the immune system is to protect against invading pathogens and to detect damaged or dying cells. Damaged and/or dying cells release damage associated molecular patterns (DAMPs) that trigger inflammatory mechanisms (1). DAMPs such as oxidized phospholipids (oxPLs) are produced as a result of free radical generation during oxidative stress (2). Excess oxPL generation or a lack of oxPL clearance can result in augmented and/or persistent inflammation and tissue damage leading to various inflammatory lung diseases such as chronic obstructive pulmonary disease (COPD), asthma, idiopathic pulmonary fibrosis (IPF), and acute respiratory distress syndrome (ARDS) (3–7). Given the significant contribution of DAMPs to lung disease, it is important to understand the mechanisms by which DAMPs induce pulmonary inflammation and injury as well as how these DAMPs are cleared to prevent further tissue damage and facilitate tissue resolution.
Following DAMP generation, the innate immune system recognizes, activates, and clears DAMPs after binding to pattern recognition receptors (PRRs). PRRs known to bind DAMPs include toll-like receptors (TLRs), NOD-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), and scavenger receptors (SRs) (1, 8, 9). While TLRs and NLRs are well characterized in their role in recognizing and clearing DAMPs, SRs are understudied. There are 8 classes of SRs that are expressed either on the cell surface or intracellularly (10). SRs were initially described for their role in lipid recognition but recent research has indicated they also recognize specific pathogen associated molecular patterns (PAMPs) and DAMPs (11). Specifically, class A SRs (SR-A) have been reported to bind and facilitate TLR recognition of the DAMP high-mobility group box 1 (HMGB1) (12). In addition, another SR, known as macrophage receptor with collagenous structure (MARCO), has been shown to mediate macrophage detection and oxPL uptake as well as TLR activation (13–15). These findings demonstrate that there are more diverse roles for SRs, including participation in the innate immune response.
Recent studies have identified SR class B-I (SR-BI) as a novel PRR that recognizes PAMPs and DAMPs (16). SR-BI was first discovered to bind anionic phospholipids and was later discovered to facilitate cholesterol ester uptake from high-density lipoprotein (HDL) particles (17–21). In the airspace, SR-BI is expressed on alveolar macrophages and alveolar epithelial cells where it mediates the uptake of vitamin E (22–24). Recently, SR-BI has been reported to bind a broad array of ligands, both endogenous (oxPLs, serum amyloid A, α-1 antitrypsin) and exogenous (pathogens, lipopolysaccharide [LPS]) (25–27). Much of the current SR-BI research focuses on how SR-BI functions in cardiovascular diseases, which has been attributed to SR-BI’s ability to regulate HDL-cholesterol and low-density lipoprotein-cholesterol concentrations (28–30). Recent studies from our lab indicate that SR-BI also regulates pulmonary host defense to bacterial pneumonia (31). Furthermore, SR-BI binds and clears LPS, a well-characterized PAMP, from the lung facilitating the resolution of pulmonary inflammation (32). Given SR-BI’s expression on structural and immune cells in the airspace, its ability to clear PAMPs (LPS) and bind DAMPs such as oxPLs, we hypothesize that SR-BI influences inflammation via recognition and clearance of airspace oxPLs thereby preventing excessive and persistent lung inflammation.
Herein, we examined the role of SR-BI in oxPL-induced lung injury and inflammation. To assess oxPL-induced lung injury, we utilized oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidylcholine (oxPAPC), a class of oxPLs know to drive sterile lung injury and inflammation (33). We identified that oxPAPC oropharyngeal (o.p.) aspiration upregulates pulmonary SR-BI expression. In SR-BI deficient mice (SR-BI−/−) or following SR-BI pharmacologic inhibition, we found that the loss of SR-BI leads to increased oxPAPC driven pulmonary pathology, and pro-inflammatory cytokine and chemokine production. The increased pulmonary pathology and cyto/chemokine production correlated with increased pulmonary oxidized phosphatidylcholines (PCs) as well as alterations in pulmonary airspace lipid composition. These lipid changes were associated with increased pulmonary TLR4 expression as well as markers for increased activation of the NF-κB pathway. These results indicate that SR-BI mediates clearance of oxPLs from the airspace and maintains the pulmonary lipid composition while dampening TLR-driven NF-κB activation and limiting oxPL-induced lung injury.
2. Methods
Animals
Female wild-type (C57BL/6J, WT) and SR-BI deficient (B6;129S2-Scarb1tm1Kri/J, SR-BI−/−) mice 8 to 16 weeks of age were used for this study. SR-BI−/− mice were backcrossed >6 generations onto a C57BL/6J background using C57BL/6J that were purchased from Jackson Laboratories (Bar Harbor, ME). SR-BI−/− mice were bred in house for experiments. Previous experiments conducted using both littermate SR-BI+/+ and commercial SR-BI+/+ (WT; C57BL/6J) controls confirmed similar pulmonary responses (31). Genotypes were confirmed by tail snip DNA analysis after weaning. Only female mice were used for these studies because the cholesterol deficiency in SR-BI KO mice increases the number of females born per litter (34, 35). All experiments were performed in accordance with the Animal Welfare Act and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals after review by the Animal Care and Use Committees of East Carolina University and The Ohio State University. To ensure rigor, all experiments were repeated as two independent cohorts and experimental data were pooled.
Murine in vivo exposures
WT and SR-BI−/− mice were instilled with 200 μg/50μl/mouse of oxPAPC (Hycult Biotech, Wayne, PA) or phosphate buffered saline (PBS) via o.p. aspiration based on previous publications (36, 37). At 0, 2, 4, and 6 h following o.p. aspiration, mice were euthanized with an intraperitoneal injection of ketamine (100 mg/kg)/xylazine (10 mg/kg) mixture as previously described (38). In separate experiments, WT mice were given Blocking Lipid Transport-2 (BLT-2; ChemBridge, San Diego, CA; 250 μg/kg) or PBS by o.p. aspiration to inhibit SR-BI function as previously described (39, 40). Then, 1 h post BLT-2 exposure, mice were exposed to PBS or 200 μg/mouse oxPAPC by o.p. aspiration and were euthanized 4 h later for sample collection and analysis.
Bronchoalveolar lavage (BAL) collection and analysis
BAL was collected immediately following sacrifice. The lung lobes were lavaged 3 times with 0.9% saline solution (Braun Medical Inc., Irvine, CA). The lavage volume was based on body weight (26.25 ml per kg of body weight) as previously described (41, 42). The resulting lavage was centrifuged (460 x g, 6 min, at 4°C). Total protein was measured in BAL supernatant using the bicinchoninic acid (BCA) Protein-Assay Kit (Thermo Scientific, Hercules, CA) and albumin was measured from BAL using the Mouse Albumin ELISA kit (Immunology Consultants Laboratory, Inc.). Differential analysis of BAL cells were performed as previously described (43, 44). BAL was centrifuged to remove cells and then concentrated using a centrifugal filter (Millipore Sigma, St. Louis, MO) before measuring cytokines and chemokines using custom multiplex assays (Bio-Rad, Hercules, CA) per the manufacturer’s instructions. Cytokines and chemokines measured in BAL supernatant included: Interleukin-6 (IL-6), Interleukin-17A (IL-17A), C-C motif chemokine ligand 2 (CCL2 also known as MCP-1), C-X-C motif chemokine ligand 1 (CXCL1 also known as KC), and granulocyte-colony stimulating factor (G-CSF)
Lung histopathological scoring
The left lung lobe was fixed (10% neutral-buffered formalin for 24 h) 4 h post o.p. aspiration. The tissues were then processed, paraffin embedded, sectioned (5 μm), and stained with hematoxylin and eosin as previously described (45). Inflammation of the whole lobe was semi-quantitatively scored on 10 random sections of the lung lobe on a scale of 0 to 3 in 3 inflammation categories (periluminal infiltrates, pneumonitis, extent of injury) culminating in a maximum score of 9 for the most severe pathology, by a board-certified pathologist blinded to genotype and treatment as previously described (46). Photomicrographs were captured using an Olympus BX41 microscope with Olympus DP26 camera and CellSens Standard v1.6 software (Olympus America Inc.) (Figure 2D). Histology was performed by HistoWiz Inc. (histowiz.com) using a Standard Operating Procedure and fully automated workflow. Samples were processed, embedded in paraffin, and sectioned at 4μm. Whole slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems) (Figure 4D). Representative images of lung histology are presented in Figures 2, 4, and Supplemental Figure 3.
Figure 2. SR-BI deficiency increases susceptibility to oxPAPC-induced lung injury.
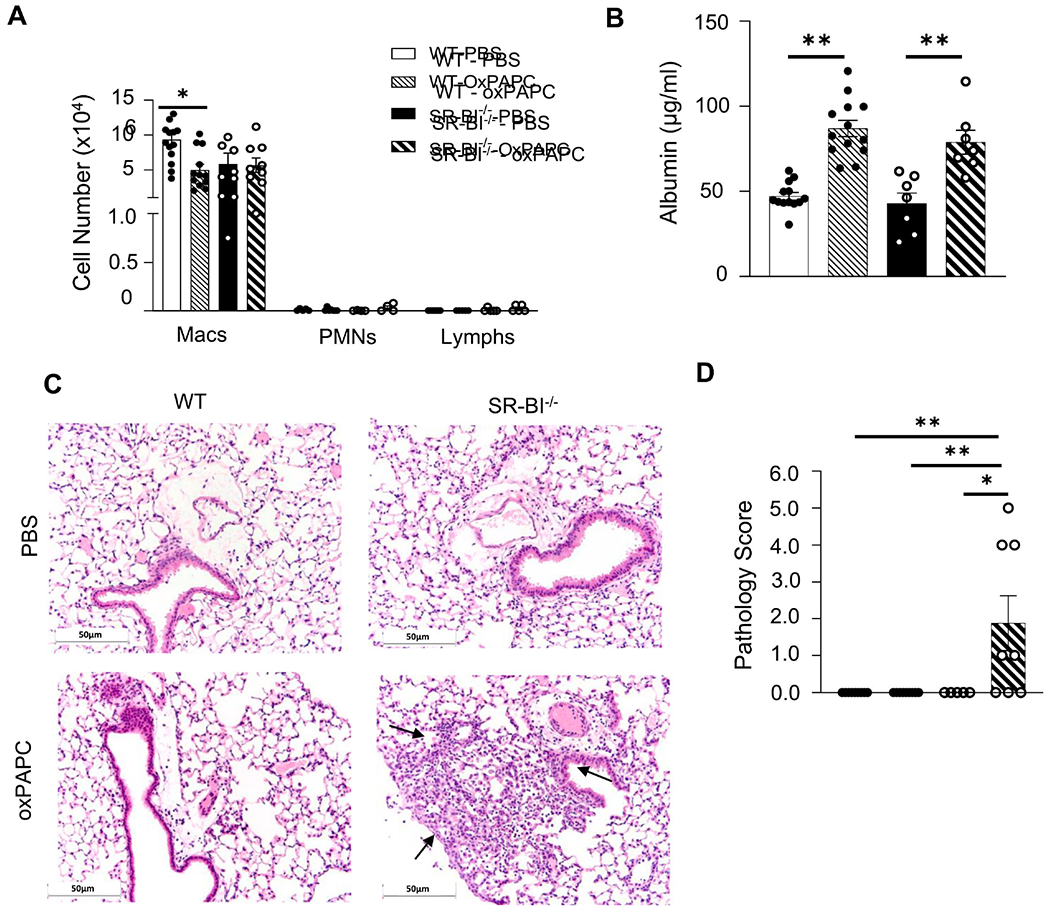
Female WT or SR-BI−/− mice were exposed to PBS or oxPAPC via oropharyngeal (o.p.) aspiration and euthanized 4 h after exposure. Bronchoalveolar lavage (BAL) was collected for (A) cell differentials, and (B) albumin analysis. Lungs were then fixed with 10% neutral-buffered formalin and stained with (C) H&E and were (D) graded by a blinded board-certified veterinary pathologist. Arrows to indicate areas of pulmonary injury. Representative images are at 20x magnification. **p<0.01, ***p<0.001, ****p<0.0001; n=5-14 per group.
Figure 4. SR-BI pharmacological inhibition increases susceptibility to oxPAPC-induced lung injury.
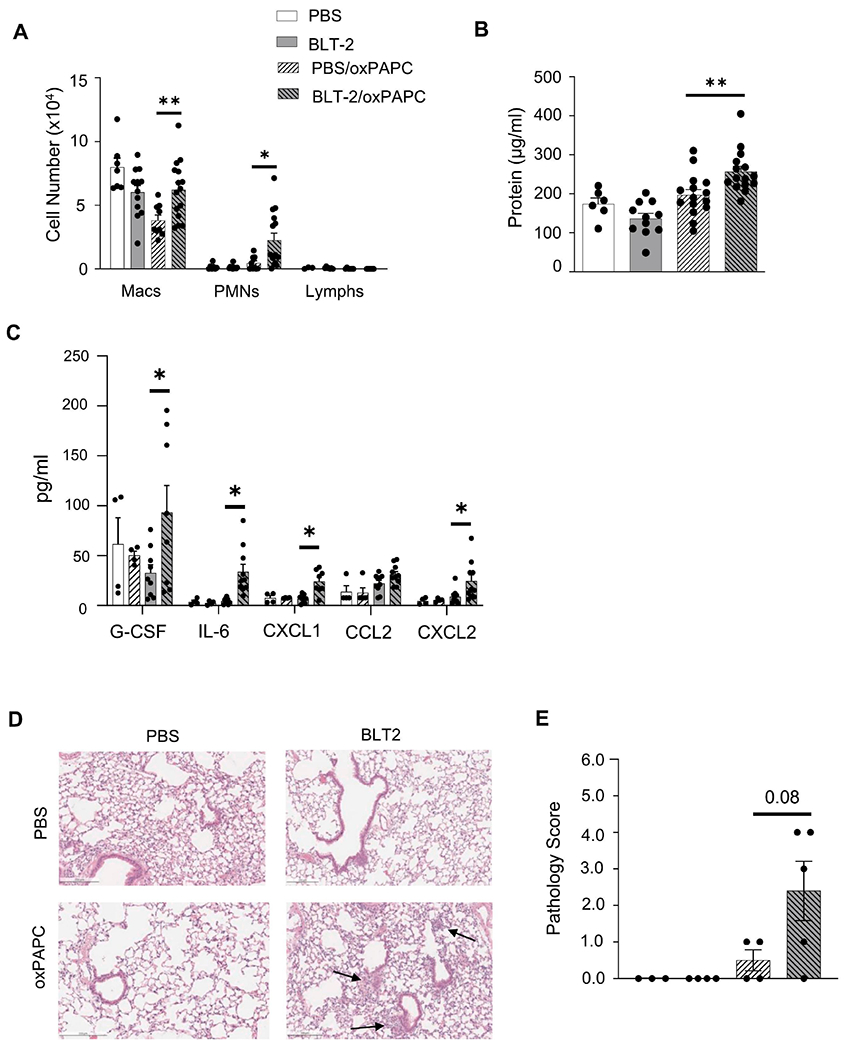
Female WT mice were exposed to PBS or BLT-2 (SR-BI inhibitor) 1 h prior to PBS or oxPAPC exposure. Then, 4 h following exposure, mice were euthanized and bronchoalveolar lavage (BAL) was collected to measure (A) cell differentials, (B) protein, and (C) cytokines/chemokines via multiplex analysis. Lungs were also fixed with 10% neutral-buffered formalin and stained for (D) H&E and were (E) graded by a blinded board-certified veterinary pathologist. Arrows to indicate areas of pulmonary injury. Representative images are at 20x magnification. *p<0.05, **p<0.01; n=3-15 per group.
BAL oxidized phospholipid (oxPL) measurements via liquid chromatography with tandem mass spectrometry (LC-MS/MS)
BAL supernatant samples from the 4 h time point were extracted with a methanol mix (300 μL) and methyl tert-butyl ether (1 mL). Equisplash® (Avanti Polar Lipids) was included as an internal standard in the methanol at 1.5 μg/mL. Samples were vortexed for 15 min then water (250 μL) and the extracts were centrifuged at 20,000 x g for 10 min. The top layer was removed, dried down, and reconstituted in 150 μL of isopropyl alcohol for analysis. Samples were analyzed with a ThermoFisher Q Exactive HF-X (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatography system. Samples were introduced via a heated electrospray source at a flow rate of 0.2 mL/min. Electrospray source conditions were set as: spray voltage 3.5 kV, sheath gas (nitrogen) 53 arbitrary unity (arb), auxiliary gas (nitrogen) 14 arb, sweep gas (nitrogen) 2 arb, nebulizer temperature 400°C, capillary temperature 300 °C, RF funnel 100 V and the mass range was set to 200-2000 m/z. Samples were analyzed using positive/negative polarity switching ionization mode with top 5 data dependent fragmentation and dynamic exclusion set to 10 seconds. All full scan measurements were recorded at a resolution setting of 60,000 and all MS2 scans were recorded at 15,000. MS2 events utilized a 1.5 Da isolation window with stepped collision energy at 25, 35 and 45 arb. OxPAPC metabolites were determined based on high resolution accurate mass (<5 ppm) and retention time as compared to the Hycult oxPAPC standard.
Separations were conducted on a Waters Acquity UPLC BEH C18 column (100 mm x 2.1 mm, 2.1 μm). Mobile phase composition was A- 60/40 acetonitrile/water with 10 mM ammonium formate and 0.1% formic acid and B- 90/10 isopropyl alcohol/acetonitrile with 10 mM ammonium formate and 0.1% formic acid. Initial liquid chromatography conditions were set to 32% B, which increased to 40% B at 1 min (held until 1.5 min) then 45% B at 4 min. This was increased to 50% B at 5 min, 60% B at 8 min, 70% B at 11 min, and 80% B at 14 min (held until 16 min). At 16 min the composition switched back to starting conditions (32% B) and was held for 4 min to re-equilibrate the column. Example raw chromatographs of standards (Supplemental Figure 1A and B) and BAL oxPLs (Supplemental Figure 2) are presented.
RNA isolation and quantitative polymerase chain reaction (qPCR)
Macrophages were isolated from BAL by pelleting cells from BAL at 460 x g for 6 min aspirating off supernatant and re-suspending in DMEM + 10% FBS and plating in a 96 well plate, one well per animal. Cells were incubated at 37°C in 5% CO2 for 1-2 hours, then the media was aspirated off and adherent cells were lysed with a guanidinium-based lysis buffer + 1% β-mercaptoethanol (Invitrogen, Carlsband, CA). RNA was isolated from isolated macrophages by PureLink RNA Mini kit (Invitrogen, Carlsband, CA). Complementary DNAs (cDNA) were generated from purified RNA using TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA). Real time PCR was performed with Taqman PCR Mix (Applied Biosystems) in the HT7900 ABI sequence Detection System (Applied Biosystems) using predesigned primers (Applied Biosystems). Fold changes in expression for mRNA quantities were calculated using Ct values and the 2−ΔΔCt method. Samples were normalized to 18S as previously described (43). See Supplemental Table 1 for primer information.
For lung tissue assessments, RNA was extracted from flash-frozen lung tissue homogenates using a Direct-zol RNA MiniPrep Plus Kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. RNA was quantified using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Rockford, IL). cDNA synthesis was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), and gene expression was measured by qPCR using the above described Taqman method or PowerUP SYBR Green Master Mix (Applied Biosystems). Fold changes in expression for mRNA quantities were calculated using Ct values and the 2−ΔΔCt method. Samples were normalized to 18s for Taqman and β-actin for SYBR Green as previously described (47). See Supplemental Table 2 for SYBR Green primer information.
Western Blot Analyses
Protein was extracted from lung tissue samples using a RIPA lysis buffer (Thermo Scientific) containing a protease inhibitor cocktail (Calbiochem, San Diego, CA), and NaF at 1M. Lysis buffer was added to each sample according to weight (50 μL/mg) and homogenized using a bead mill 4 homogenizer (Fisherbrand, Waltham, MA). Protein concentrations were quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific). 30 μg of protein was mixed with an equal volume of 2x sample buffer, loaded on a 12% SDS-PAGE gel, electrophoresed and transferred to a polyvinylidene difluoride (PVDF) membrane by a transfer system (Bio-Rad Laboratories, Inc. Des Plaines, IL). The membrane was blocked with 5% BSA in Tris-buffered saline with Tween 20 and incubated with a primary anti-rabbit polyclonal SR-BI (82 kDa) antibody (1:1000) (Novus, Littleton, CO, USA) at 4°C overnight. A goat anti-rabbit lgG (H+L) secondary antibody (1:8000) was used to incubate the membrane at room temperature for 1 h followed by incubation for 1 min with clarity western ECL substrate (Bio-Rad Laboratories, Inc.). The blot was then imaged in a molecular imager with β-actin (1:1000; 42 kDa) (catalog number sc-47778, Santa Cruz, Dallas, TX) used as an internal control. The intensity of the blot band was quantified and analyzed by Image Lab.
The preparation of cytoplasmic fractions from lung tissue followed previously published protocols (48). Nuclear-cytoplasmic fractionation was conducted using Nuclear Extract Kit (Active Motif) according to the manufacturer’s protocol. Protein levels in samples were measured using a BCA kit (Pierce, Rockford, IL). Lysates were separated 4–20% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad), transferred onto PVDF membranes, probed with the specific primary antibody for IkBa (sc371, Santa Cruz Biotechnologies) (1:1,000 dilution in PBS containing 0.1% Tween 20), and developed by enhanced chemiluminescence. Equivalent loading of the gel was determined by quantitation of protein as well as by reprobing membranes for alpha-tubulin).
p65 ELISA
For p65 detection, cytoplasmic and nuclear fractions were isolated from lung tissues using the Nuclear Extract Kit (Active Motif) according to the manufacturer’s protocol as described previously (48). Protein levels in samples were measured using a BCA kit (Pierce, Rockford, IL). Nuclear extracts were analyzed for p65 using a commercially available ELISA (Active Motif) per manufacturer’s instructions (49).
Statistical Analysis
All experiments were repeated in at least two independent experiments and data from experiments were pooled. Data are expressed as mean ± SEM. Data generated from experiments were analyzed using a one-way ANOVA (Kruskal-Wallis test; Figure 1, 5 and 7) or a two-way ANOVA followed by comparison using a Tukey’s multiple comparisons test (Figure 2 and 4), once parametric or nonparametric distribution was determined (GraphPad Prism 9.00; San Diego, CA). With comparisons of two groups an unpaired nonparametric t-test (Mann-Whitney test; Figure 3, 6, and Supplemental Figure 4) was utilized. A value of p<0.05 was considered significant.
Figure 1. SR-BI gene expression is increased in the lungs after oxPAPC exposure.
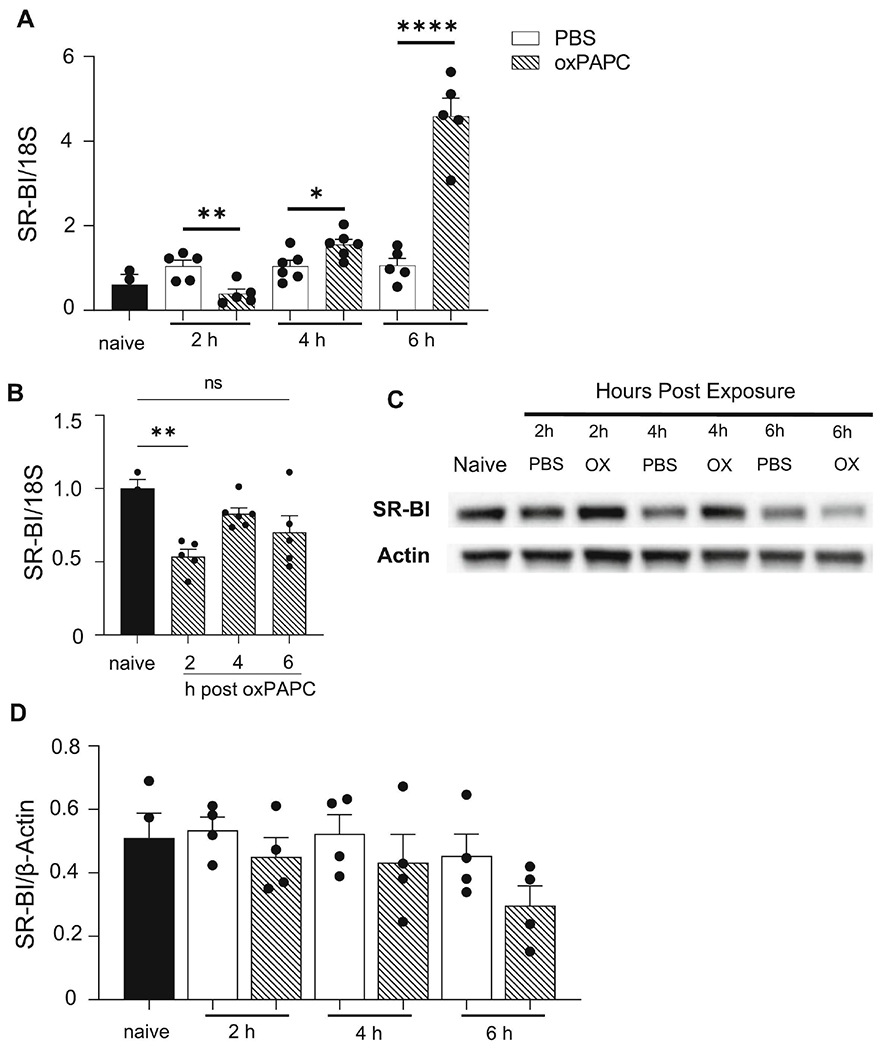
Female WT mice were unexposed (naïve), exposed to PBS, or oxPAPC via oropharyngeal (o.p.) aspiration and then euthanized 2, 4, or 6 h post- exposure. Lung tissue was collected to assess SR-BI (A) mRNA gene expression by qPCR, and (B) protein via western blot, which was quantified by densitometry and normalized to β-actin. *p<0.05, **p<0.01, ****p<0.0001; n=3-5 per group.
Figure 5. OxPLs are increased in the lungs of SR-BI−/− mice.
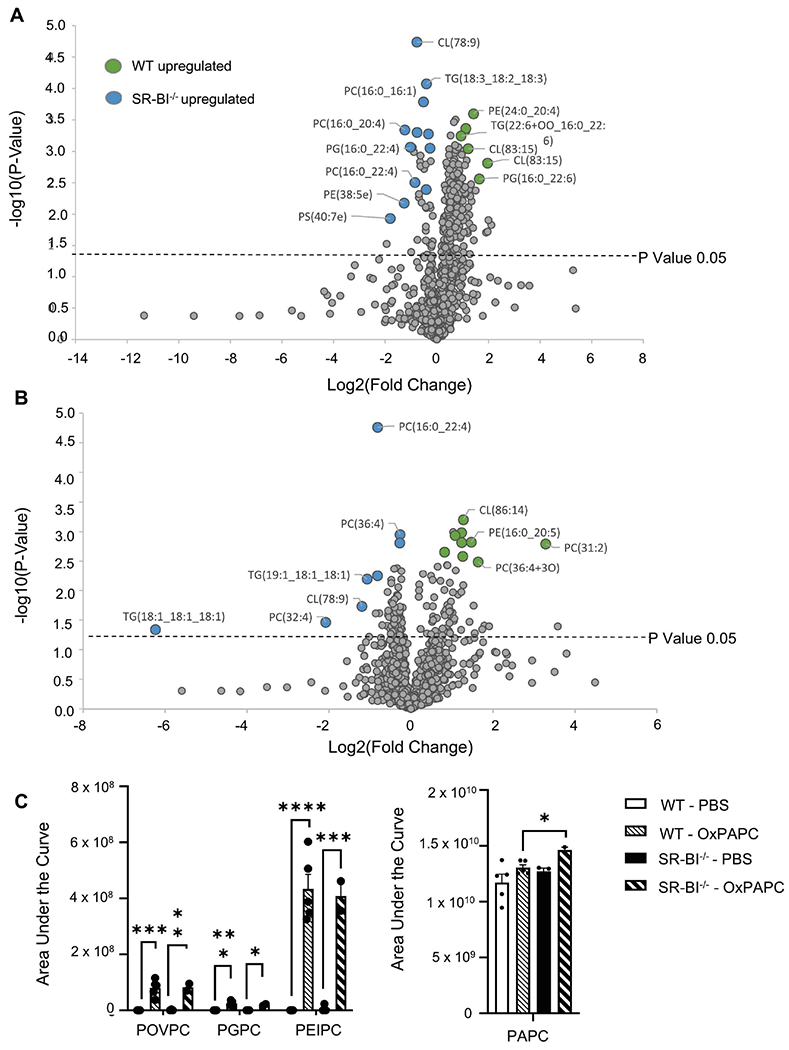
Female WT or SR-BI−/− mice were exposed to PBS or oxPAPC via oropharyngeal (o.p.) aspiration and were euthanized 4 h after exposure. Bronchoalveolar lavage (BAL) was collected for LC-MS/MS analysis. (A) Relative phospholipid concentrations were compared between WT and SR-BI−/− mice after PBS exposure and (B) after OxPAPC exposure. Increased fold change values for WT animals are shaded green while increased fold change values in SR-BI−/− mice are shaded blue and (C) the area under the curve quantitation of relative concentrations of oxidized lipid species POVPC, PGPC, PEIPC, and PAPC normalized to a deuterated PC internal standard (PC-IS). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; n=3-6 per group.
Figure 7. NF-κB activation is upregulated in SR-BI−/− mice after oxPAPC exposure.
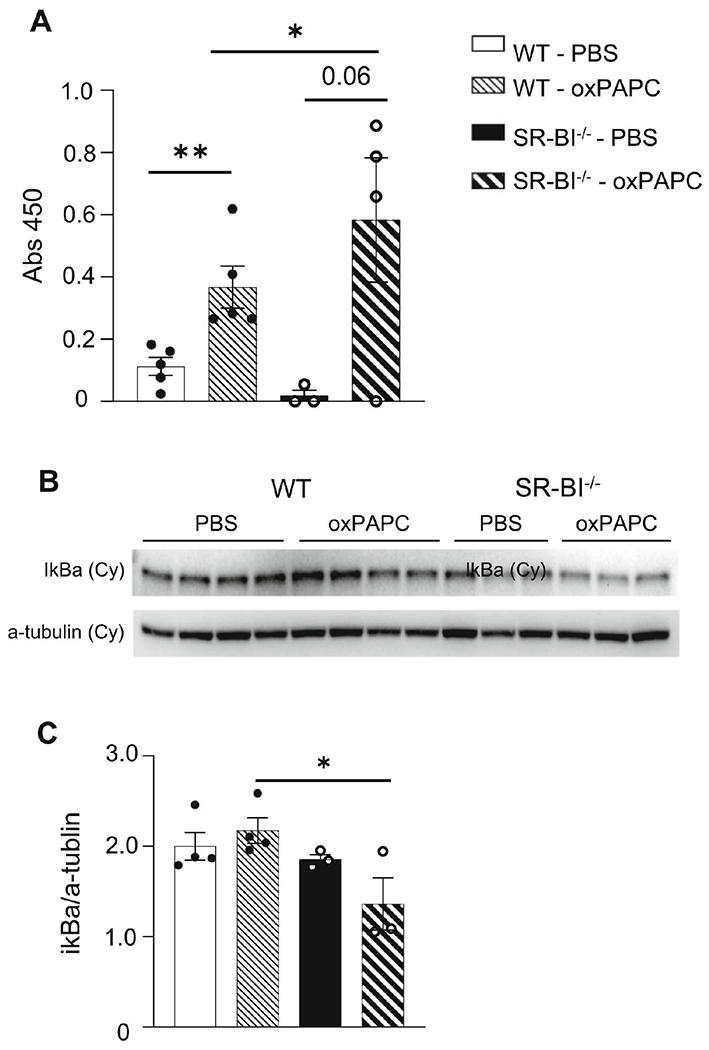
Female WT or SR-BI−/− mice were exposed to PBS or oxPAPC via oropharyngeal (o.p.) aspiration and euthanized 2h after exposure. (A) Lung tissue was collected and nuclear-cytoplasmic fractionation was conducted to measure p65 in the nuclear fraction via p65 ELISA. (B) Female WT or SR-BI−/− mice were exposed to PBS or oxPAPC via oropharyngeal (o.p.) aspiration and euthanized 4h after exposure. Lung tissue was collected and nuclear-cytoplasmic fractionation was conducted to measure cytoplasmic (cy) IκBα via western blot. (C) Densitometry of the cytoplasmic (cy) IκBα relative to α-tubulin. *p<0.05, **p<0.01; n=3-5 per group.
Figure 3. Loss of SR-BI increases cyto/chemokine production in the airspace after oxPAPC exposure.
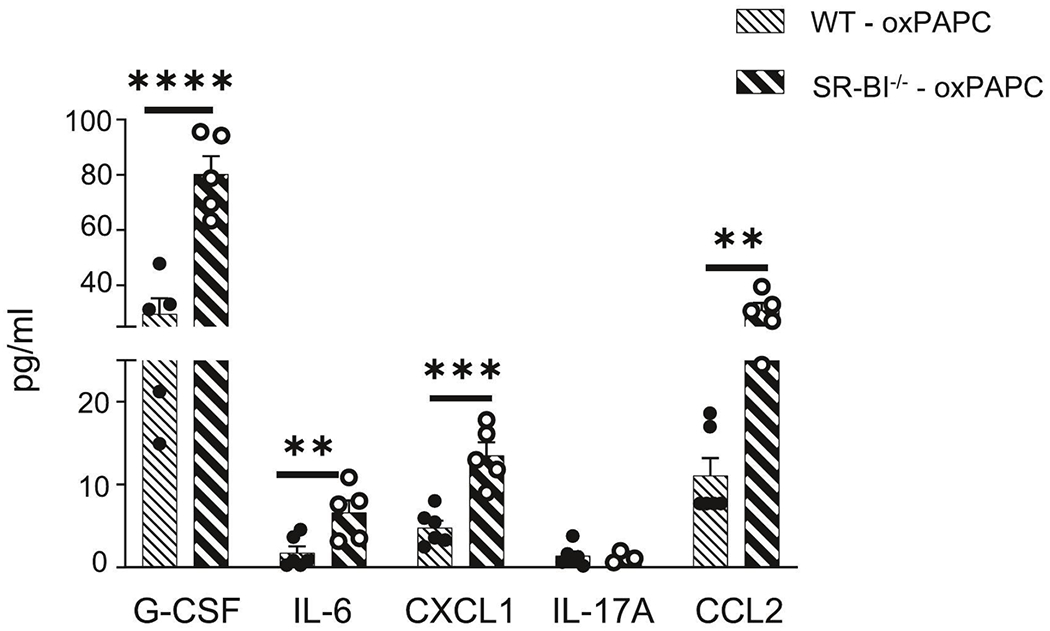
Female WT or SR-BI−/− mice were exposed to oxPAPC via oropharyngeal (o.p.) aspiration and euthanized 4 h after exposure. Bronchoalveolar lavage (BAL) was collected for multiplex analysis of G-CSF, IL-6, CXCL1, IL-17A, and CCL2. *p<0.05, **p<0.01, ***p<0.001; n=5-13 per group.
Figure 6. TLR4 expression is upregulated in SR-BI−/− mice after oxPAPC exposure.
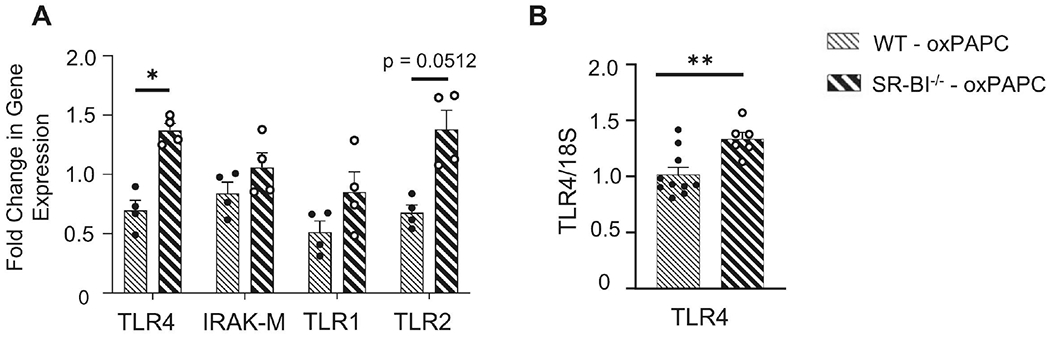
Female WT or SR-BI−/− mice were exposed to oxPAPC via oropharyngeal (o.p.) aspiration and euthanized 4 h after exposure. (A) Lung tissue was collected to analyze mRNA expression TLR4, IRAK-M, TLR1, and TLR2 by qPCR. (B) Macrophages were isolated from bronchoalveolar lavage (BAL), lysed, and mRNA expression of TLR4 was analyzed by qPCR. *p<0.05; n=4-10 per group.
3. Results
Pulmonary and airspace macrophage SR-BI mRNA expression is altered following oxPAPC induced injury
Given the role of SR-BI in pulmonary innate immune responses to PAMPs, we determined if SR-BI expression and/or production was altered during oxPL-induced acute lung injury. To induce acute lung injury, we utilized oxPAPC, an oxPLs family known to cause pulmonary inflammation and injury (33, 50). We first measured SR-BI gene expression in whole lung tissue via qPCR from naïve mice, mice challenged with PBS (vehicle control), and lungs from mice 2, 4, or 6 h following oxPAPC exposure (Fig 1A). There were no differences in pulmonary SR-BI mRNA in PBS exposed mice compared to naïve mice. OxPAPC exposure induced differential changes in SR-BI mRNA expression based on the time point following exposure. At 2 h post-exposure, SR-BI mRNA expression was decreased, whereas at 4 and 6 h post-exposure expression was increased. To examine if SR-BI mRNA expression changes were also present in airspace macrophages, qPCR was also run on macrophages isolated from lavage fluid 2, 4, or 6 h following oxPAPC exposure (Fig 1B). At 2 h post-exposure, airspace macrophages had a significant decrease in SR-BI mRNA when compared to naive controls. However, this decrease in SR-BI mRNA was not seen 4 or 6 h post-exposure. To determine if SR-BI mRNA expression changes translated to altered SR-BI protein production in the lung, western blots were performed on whole lung tissue homogenate from, naïve mice, mice exposed to the PBS control or oxPAPC and then assessed at 2, 4, or 6 h. Interestingly, despite changes in SR-BI mRNA expression, SR-BI protein levels in lung tissue were not significantly different following exposure (Fig 1C and D).
OxPAPC exposure decreases lung barrier function, which is exacerbated by SR-BI deficiency
To define the impact of SR-BI on oxPL-induced lung microvascular injury and inflammation, we challenged WT and SR-BI−/− mice to vehicle (PBS) or oxPAPC and evaluated the pulmonary inflammation and barrier permeability responses 4 h post-exposure. Following oxPAPC exposure, WT mice had decreased macrophages in the airspace (Fig 2A), while SR-BI−/− BAL macrophages were unchanged. BAL neutrophils were largely unchanged between the groups and following exposure to oxPAPC. BAL albumin was then measured to assess lung barrier function. WT and SR-BI−/− mice exposed to oxPAPC had increased BAL albumin compared to PBS exposed controls but there was no difference between the two strains (Fig 2B). Lung histopathology was assesed by a blinded board-certified veterinary pathologist. Lung pathology was unchanged in oxPAPC-exposed WT mice when compared to PBS-exposed mice; however, SR-BI−/− mice had a significant increase following oxPAPC exposure compared to PBS-exposed SR-BI−/− or WT controls (Fig 2C, D, and Supplemental Fig 3). In particular, oxPAPC exposed SR-BI−/− mice had substantial pneumonitis (including interstitial thickening) and periluminal infiltration of neutrophils, lymphocytes, and macrophages, as indicated by the arrows in Fig 2C. This pathology was primarily noted in the alveolar areas and little to no pathological changes were noted in the upper airways.
We then evaluated the production of cyto/chemokines in the airspace following oxPAPC exposure. Multiplex cytokine analysis indicated that SR-BI−/− mice had increased CXCL1 following PBS and significant increases in G-CSF, IL-6, CXCL1 and CCL2 concentrations in the BAL following oxPAPC exposure when compared to WT (Supplemental Fig 4A and Fig 3). These data support that there are augmented inflammatory responses to oxPAPC when SR-BI is deficient.
Inhibition of SR-BI with BLT-2 increases susceptibility to oxPAPC-induced lung injury
To ensure the SR-BI−/− phenotype was not the result of developmental effects, we determined if pharmacological inhibition of SR-BI in adult mice was sufficient to exacerbate oxPAPC-induced lung inflammation and injury. To block SR-BI, we dosed WT mice by o.p. aspiration with BLT-2, an SR-BI inhibitor (47), 1 h before oxPAPC dosing. BLT-2 treatment alone did not change cellular influx or lung injury after PBS exposure (Fig 4A and B). After oxPAPC exposure, BLT-2 treated mice had a significant increase in BAL neutrophils (Fig 4A), and BAL protein (Fig 4B). Multiplex cytokine analysis indicated that production of G-CSF, IL-6, CXCL1, and CXCL2 (Fig 4C) were increased in the BLT-2 treated mice compared to the PBS controls following oxPAPC treatment, indicating a similar cyto/chemokine response to SR-BI−/− mice exposed to oxPAPC. Lung pathology trended to increase (p=0.08) following BLT-2 treatment and oxPAPC exposure, although not reaching statistical significance (Fig 4D and E).
Oxidized phospholipids are increased in the lungs of SR-BI−/− mice
Given that SR-BI expression is increased after oxPAPC exposure, and removal of SR-BI leads to exacerbated pulmonary pathology, we hypothesized that SR-BI deficiency alters baseline lung lipid composition and causes defective oxPL clearance following oxPAPC exposure. We first evaluated baseline airspace lipid composition by performing untargeted lipidomics on BAL samples in PBS exposed WT and SR-B1−/− mice. We observed that SR-BI deficiency led to altered airspace lipid composition of several phosphatidylcholine (PC), triglyceride (TG), cardiolipin (CL), phosphatidylglycerol (PG), and phosphatidylethanolamine (PE) species (Supplemental Table 3 and Fig 5A). Three of the top upregulated lipids in SR-BI−/− mice included: PC(16:0-16:1), PC(16:0-20:4), and PC(16:0-22:4). Following oxPAPC exposure, PC(31:2) was significantly upregulated in the airspace of WT mice, whereas PC(16:0-22:4), PC(32:4) and PC(36:4) were increased in SR-BI−/− mice (Supplemental Table 4 and Fig 5B). These lipid species are involved in arachidonic acid and linoleic acid metabolism and are known to alter innate immune system responses (51). Furthermore, TG(18:1-18:1-18:1), a common component of glycerolipid metabolism, was increased 6-fold in the airspace of SR-BI−/− mice. Lastly, we measured airspace oxPL concentrations to define changes in oxPL clearance following exposure (Fig 5C). In both WT and SR-BI−/− mice dosed with oxPAPC, the airspace had increased concentrations of the oxidized lipids 1-palmitoyl-2-(5’-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC) and 1-palmitoyl-2-(5(6)-epoxy-9-oxo-11-hydroxy-7E,14Z-prostadienoyl)-sn-glycero-phosphocholine (PEIPC) (Fig 5C). However, WT mice did not have an increase in oxPAPC in the air space whereas SR-BI−/− mice had significantly more oxidized PC in the air space following oxPAPC exposure, potentially indicating inadequate clearance and/or additional generation of oxPAPC in the airspace of SR-BI−/− mice (Fig 5C).
Loss of SR-BI primes TLR4 activation following oxPAPC exposure
OxPLs, including oxPAPC, are known to initiate TLR4-dependent pulmonary inflammation via increased NF-κB activation (33, 52). After observing increased airspace oxPAPC in SR-BI−/− mice following oxPAPC exposure, we hypothesized that this increased oxPAPC would enhance TLR4 signaling. We first measured the pulmonary expression of TLR4 along with other TLRs (TLR1 and 2) and its negative regulator, IRAK-M, in WT and SR-BI−/− mice exposed to oxPAPC (Fig 6A). As previously reported, there was no significant increase in any of the PRRs measured in SR-BI−/− mice following PBS exposure (Supplemental Fig 4B and C) (31). TLR4 was significantly increased in the lungs of SR-BI−/− mice exposed to oxPAPC with a trend towards increased TLR2 expression (p=0.051) (Fig 6A). However, we did not observe changes in gene expression for IRAK-M or TLR1. To determine if the changes in pulmonary TLR4 expression were also noted in airspace macrophages, we isolated RNA from BAL macrophages of WT or SR-BI−/− mice and measured TLR4 expression. We observed a statistically significant increase in TLR4 expression in lung macrophages of oxPAPC exposed SR-BI−/− mice (Fig 6B).
SR-BI regulates NF-κB activity following oxPAPC exposure
To evaluate if the changes noted in TLR4 expression following oxPAPC reflected differential downstream signaling, we measured NF-κB activation in lung tissue by nuclear p65 (activated NF-κB that has translocated to the nucleus). In WT mice, oxPAPC exposure did not change the pulmonary production of cytoplasmic p65 (data not shown). However, nuclear p65 was significantly increased in WT mice after oxPAPC exposure which was further augmented in SR-BI−/− mice (Fig 7A). To evaluate if other components of the NF-κB signaling pathway were altered, cytoplasmic IκBα (NF-κB transcription factor inhibitor) was measured by western blot. In the lung tissue from oxPAPC exposed SR-BI−/− mice, there was a significant decrease in cytoplasmic IκBα (Fig 7B and C) indicating IκBα degradation and NF-κB activation.
Discussion
In the present study, we report that the PRR SR-BI is protective against oxPL-induced lung pathology by maintaining the lipid composition and reducing the amount of oxPAPC in the airspace. The altered lipid composition and increased oxPAPC in the airspace of SR-BI−/− mice was associated with increased TLR4 gene expression and NF-κB activation resulting in enhanced pro-inflammatory cytokine production. Lastly, our data indicate that airspace macrophage expression of SR-BI dampens oxPAPC-induced TLR4 mRNA expression which could result in prolonged inflammatory responses. Collectively, these data add to a growing body of literature that supports a protective role for SR-BI in dampening the pulmonary inflammatory response.
Data we present here show that oxPAPC exposure alters pulmonary and macrophage SR-BI mRNA expression but not protein production. Though the mechanism by which this occurs were not specifically elucidated in the present study, increased SR-BI mRNA expression in the lung could be a result of differential regulation of transcription factors known to drive SR-BI expression such as liver X receptor (LXR) and peroxisome proliferator-activated receptor-γ (PPAR-γ) (25, 53–55). OxPLs are known to inactivate LXR and to down-regulate lung tissue expression of PPAR-γ (25, 56); therefore, decreased SR-BI mRNA may be a consequence of altered transcription factors known to regulate expression. Alternatively, oxPAPC exposure may increase the production of other SR-BI ligands, such as HMGB1, which may upregulate SR-BI mRNA expression (55–59). Lastly, oxPLs are known to inhibit cholesterol uptake and/or disrupt cholesterol trafficking which could lead to altered SR-BI gene expression in the lung (60–62). Interestingly, increased SR-BI mRNA did not lead to a significant increase in protein production. One potential explanation is that oxPAPC-induced endoplasmic reticulum stress can result in an unfolded protein response which is an adaptive mechanism shown to inhibit SR-BI protein production (63, 64). Additionally, cell-specific regulation of SR-BI production may have been masked in whole lung protein analysis. Regardless, our data does indicate that the presence of oxPLs in the lung does modulate SR-BI gene expression indicating a potential role for SR-BI in protecting the lung from oxPL induced injury/inflammation.
OxPLs are known to induce lung injury, yet little is known about the role of SR-BI in these responses. In this study, SR-BI deficiency and/or pharmacological inhibition of SR-BI significantly exacerbated the lung pathology associated with oxPAPC exposure. This worsened lung pathology was independent of changes in BAL cell differentials and/or albumin. These data are counterintuitive to our previous examination of SR-BI in other infectious and inflammatory lung diseases where SR-BI deficiency led to an increase in pulmonary neutrophilia and BAL protein (31). The evidence for the lack of airspace neutrophilia but the observation of exacerbated pulmonary pathology could be accounted for by several considerations. Based on histopathologic evaluation, the pulmonary pathology induced by oxPAPC is heterogenous and therefore whole lung airspace sampling may exhibit volume averaging. Alternatively, it is possible that the immune cells including neutrophils and lymphocytes remained in the interstitium and did not transit to the airspace, therefore leading to evidence of pathology without significant airspace inflammation. SR-BI−/− mice have been been reported to have enhanced lymphocyte proliferation as well as enhanced numbers of T and B lymphocytes in the spleens (65). Finally, it is possible that this is due to the time course of sampling where studying delayed time points may provide greater insight into airspace inflammation. A limitation here is that the mice at later time points are more moribund, which limited assessment of later time points. Nonetheless, our data clearly demonstrate that SR-BI is essential for preventing oxPL-induced pulmonary pathology.
Prior to descriptions of pulmonary SR-BI functions, SR-BI was identified as a phospholipid transporter that extracted lipids from HDL and facilitated cellular uptake (66). Phospholipids known to be transported by SR-BI include: PC, PE, and sphingomyelin (SM). Our lipidomic analysis of the BAL of SR-BI−/− mice indicate that many PCs were increased at baseline and, following oxPAPC exposure. Increased lipid species commonly contained palmitic acid (16:0), oleic acid (16:1), linoleic acid (18:2), arachidonic acid (20:4), and eicosapentaenoic acid (20:5) fatty acids. The increase in these BAL phospholipids could be a result of failed lipid uptake or a consequence of the augmented oxPAPC induced pathology. An increased supply of fatty acids such as linoleic and arachidonic acid can promote the production of pro-inflammatory lipid mediators (prostaglandins and leukotrienes), thus driving inflammatory responses (67). Future studies will focus on how SR-BI facilitates the uptake and metabolism of lipid mediators in the lung that can influence pulmonary injury and inflammation. In addition to their altered baseline pulmonary lipidome, SR-BI−/− mice had increased oxPAPC in the BAL following oxPAPC exposure when compared to WT mice. This increase in oxPAPC could be a result of impaired clearance or an increased generation of oxPAPC. oxPLs such as oxPAPC induce lipid peroxidation (68), which likely contributed to oxidized POVPC, PGPC, and PEIPC concentrations detected in WT and SR-BI−/− mice. These oxidized lipid species can also induce apoptosis which can perpetuate the generation of additional oxPLs (69, 70). However, given that only oxPAPC was increased in the airspace of SR-BI−/− mice and not other oxPLs, it is likely the observation of increased oxPAPC was due to reduced oxPAPC clearance leading to increased lung pathology. This remains an area for future investigation.
It is known that oxPLs are ligands for SR-BI, TLR2, and TLR4, which signal via NF-κB to generate inflammation (33, 71). Our data indicated that SR-BI−/− mice dosed with oxPAPC had increased pulmonary and alveolar macrophage TLR4 mRNA expression. We speculate that this increase in TLR4 expression is driving the increased lung pathology and production of select cytokines and chemokines in SR-BI−/− mice. The increased TLR4 expression noted in SR-BI deficiency has been previously reported by our group and others and adds to a body of literature suggesting that SRs regulate TLR expression and signaling (27, 31, 72). However, it is still unclear how SR-BI and/or SRs regulate TLR expression and the proceeding downstream signaling cascade. This may be the result of several factors. First, the diminished uptake of oxPAPC by SR-BI may augment subsequent TLR signaling. We have previously reported that SR-BI is required to mediate clearance of LPS and inhibition of TLR4 signaling (31). Furthermore, SR-BI expression is increased upon administration of TLR4 ligands (73). It is possible that oxPAPC, a known TLR4 and SR-BI ligand, is both cleared from the lung by SR-BI to prevent TLR4 activation and binds to TLR4 leading to SR-BI upregulation to further facilitate TLR4 activation. Alternatively, our results may be the result of decreased suppressive effects of HDL in the lung. SR-BI−/− mice are known to have altered systemic lipid profiles and HDL isolated from SR-BI−/− mice have deficient antioxidant activities, resulting in increased oxidative stress (74, 75). It would be of interest to confirm whether these factors are involved in the protective role of SR-BI in DAMP-induced pulmonary inflammation and injury.
Despite our novel findings, we acknowledge that our study has limitations. We principally focus on pulmonary inflammation and injury at 4 h post-exposure. This is based on observing increased SR-BI gene expression in the lungs at that time point. While the expression was further increased at the 6 h time point, the mice were near or at morbidity criteria and we could not maintain sufficient animal survival to adequately analyze the inflammatory response. Additionally, we chemically inhibited SR-BI in the lung through BLT-2 to observe if SR-BI expression in the lung is responsible for mitigating oxPAPC-induced inflammation which is not cell-specific. Development of inducible cell-specific SR-BI null mice would be necessary to decipher the cellular source of SR-BI that is involved in protecting the lung from oxPAPC exposure. Lastly, we acknowledge that the route of exposure and dose of oxPAPC used to induce lung injury/inflammation is not physiologically relevant but does allow for mechanistic studies of how oxPLs can trigger the innate immune response in the lung. The use of oropharyngeal aspiration provides a direct dose to the lungs but can have differential deposition in the lung, which could be the reasoning for the lack of biological changes with some of the endpoints measured. The dose of oxPAPC used could potentially have cytotoxic effects that can contribute to the inflammation and injury responses as well as the increase in TLR4 and NF-κB activation. We measured caspase 3 activation following oxPAPC exposure and did not note any increases (data not shown) however cell death by other mechanisms besides apoptosis could contribute to this phenotype. Future investigation into the ability of oxPAPC in this model and other models where differential doses are used will be required to further understand the pathway through which oxPAPC induces lung inflammation and pathology.
In conclusion, these data provide evidence that SR-BI mitigates the pro-inflammatory effects of oxPAPC by decreasing TLR4 driven activation of NF-κB and attenuating lung pathology and production of cytokines and chemokines. Given that genetic alterations in SR-BI have been identified in the human population, this could contribute to the persistence and severity of pulmonary pathology following lung injury where there is elevated production of DAMPs (76, 77). Overall, these data indicate that augmenting SR-BI and identifying individuals with known loss-of-function mutations may be a potential strategy to decrease the negative health effects of DAMP driven pulmonary diseases (78, 79).
Supplementary Material
These studies demonstrate that scavenger receptor BI (SR-BI) limits the lung pathology and inflammation following the generation of the danger associated molecular pattern oxidized phospholipids (oxPL). These findings indicate that if SR-BI is dysfunctional, there is a lack of oxPL clearance, dysfunctional lipid metabolism, and increased toll-like receptor 4 signaling in the lung leading to increased lung pathologies.
6. Acknowledgments
We would like to acknowledge the Comparative Pathology & Mouse Phenotyping Shared Resource (CPMPSR) at Ohio State University for their help with lung pathology.
5. Funding
R01ES028829 (K.M.G.), R01ES027574 (R.M.T.), R01ES031253 (S.H.), P30 ES025128, and P30 CA016058.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Author Dr. Debra Tokarz is employed by Experimental Pathology Laboratories, Inc. Author Dr. Katelyn Dunigan-Russell is currently employed by AmplifyBio. but completed the majority of her contributions (concept and design, acquisition of data, analysis and interpretation) during her postdoctoral scholar position at Ohio State University. Author Dr. Brita Kilburg-Basnyat is currently employed by Arcus Biosciences, Inc. but completed the majority of her contributions (concept and design, acquisition of data, analysis and interpretation) during her postdoctoral scholar position at East Carolina University. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
7 References
- 1.Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112. [DOI] [PubMed] [Google Scholar]
- 2.Stamenkovic A, Pierce GN, Ravandi A. Oxidized lipids: not just another brick in the wall (1). Can J Physiol Pharmacol. 2019;97(6):473–85. [DOI] [PubMed] [Google Scholar]
- 3.Minagawa S, Yoshida M, Araya J, Hara H, Imai H, Kuwano K. Regulated Necrosis in Pulmonary Disease. A Focus on Necroptosis and Ferroptosis. Am J Respir Cell Mol Biol. 2020;62(5):554–62. [DOI] [PubMed] [Google Scholar]
- 4.Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017;3:17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, et al. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154(2 Pt 1):477–83. [DOI] [PubMed] [Google Scholar]
- 6.Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M, et al. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J. 2019;286(3):507–22. [DOI] [PubMed] [Google Scholar]
- 7.Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215–26. [DOI] [PubMed] [Google Scholar]
- 8.Cao X Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16(1):35–50. [DOI] [PubMed] [Google Scholar]
- 9.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J Immunol. 2017;198(10):3775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alquraini A, El Khoury J. Scavenger receptors. Curr Biol. 2020;30(14):R790–R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komai K, Shichita T, Ito M, Kanamori M, Chikuma S, Yoshimura A. Role of scavenger receptors as damage-associated molecular pattern receptors in Toll-like receptor activation. Int Immunol. 2017;29(2):59–70. [DOI] [PubMed] [Google Scholar]
- 13.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277(41):38517–23. [DOI] [PubMed] [Google Scholar]
- 14.Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, et al. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest. 2007;117(3):757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kissick HT, Dunn LK, Ghosh S, Nechama M, Kobzik L, Arredouani MS. The scavenger receptor MARCO modulates TLR-induced responses in dendritic cells. PLoS One. 2014;9(8):e104148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao D, Ashraf MZ, Kar NS, Lin D, Sayre LM, Podrez EA. Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class B scavenger receptors CD36 and SR-BI. J Biol Chem. 2010;285(7):4447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linton MF, Tao H, Linton EF, Yancey PG. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol Metab. 2017;28(6):461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115(10):2870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krieger M, Kozarsky K. Influence of the HDL receptor SR-BI on atherosclerosis. Curr Opin Lipidol. 1999;10(6):491–7. [DOI] [PubMed] [Google Scholar]
- 20.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98(4):984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigotti A, Acton SL, Krieger M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J Biol Chem. 1995;270(27):16221–4. [DOI] [PubMed] [Google Scholar]
- 22.Santander N, Lizama C, Parga MJ, Quiroz A, Perez D, Echeverria G, et al. Deficient Vitamin E Uptake During Development Impairs Neural Tube Closure in Mice Lacking Lipoprotein Receptor SR-BI. Sci Rep. 2017;7(1):5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valacchi G, Vasu VT, Yokohama W, Corbacho AM, Phung A, Lim Y, et al. Lung vitamin E transport processes are affected by both age and environmental oxidants in mice. Toxicol Appl Pharmacol. 2007;222(2):227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolleck I, Witt W, Wissel H, Sinha P, Rustow B. HDL and vitamin E in plasma and the expression of SR-BI on lung cells during rat perinatal development. Lung. 2000;178(4):191–200. [DOI] [PubMed] [Google Scholar]
- 25.Shen WJ, Asthana S, Kraemer FB, Azhar S. Scavenger receptor B type 1: expression, molecular regulation, and cholesterol transport function. J Lipid Res. 2018;59(7):1114–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280(4):2954–61. [DOI] [PubMed] [Google Scholar]
- 27.Cai L, Wang Z, Meyer JM, Ji A, van der Westhuyzen DR. Macrophage SR-BI regulates LPS-induced pro-inflammatory signaling in mice and isolated macrophages. J Lipid Res. 2012;53(8):1472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahebi R, Hassanian SM, Ghayour-Mobarhan M, Farrokhi E, Rezayi M, Samadi S, et al. Scavenger receptor Class B type I as a potential risk stratification biomarker and therapeutic target in cardiovascular disease. J Cell Physiol. 2019;234(10):16925–32. [DOI] [PubMed] [Google Scholar]
- 29.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90(3):270–6. [DOI] [PubMed] [Google Scholar]
- 30.Ueda Y, Gong E, Royer L, Cooper PN, Francone OL, Rubin EM. Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J Biol Chem. 2000;275(27):20368–73. [DOI] [PubMed] [Google Scholar]
- 31.Gowdy KM, Madenspacher JH, Azzam KM, Gabor KA, Janardhan KS, Aloor JJ, et al. Key role for scavenger receptor B-I in the integrative physiology of host defense during bacterial pneumonia. Mucosal Immunol. 2015;8(3):559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baranova IN, Bocharov AV, Vishnyakova TG, Chen Z, Birukova AA, Ke Y, et al. Class B Scavenger Receptors BI and BII Protect against LPS-Induced Acute Lung Injury in Mice by Mediating LPS. Infect Immun. 2021;89(10):e0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miettinen HE, Rayburn H, Krieger M. Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J Clin Invest. 2001;108(11):1717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santander NG, Contreras-Duarte S, Awad MF, Lizama C, Passalacqua I, Rigotti A, et al. Developmental abnormalities in mouse embryos lacking the HDL receptor SR-BI. Hum Mol Genet. 2013;22(6):1086–96. [DOI] [PubMed] [Google Scholar]
- 36.Aldossari AA, Shannahan JH, Podila R, Brown JM. Scavenger receptor B1 facilitates macrophage uptake of silver nanoparticles and cellular activation. Journal of Nanoparticle Research. 2015;17(7):313. [Google Scholar]
- 37.Thimmulappa RK, Gang X, Kim JH, Sussan TE, Witztum JL, Biswal S. Oxidized phospholipids impair pulmonary antibacterial defenses: evidence in mice exposed to cigarette smoke. Biochem Biophys Res Commun. 2012;426(2):253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tighe RM, Birukova A, Yaeger MJ, Reece SW, Gowdy KM. Euthanasia- and Lavage-mediated Effects on Bronchoalveolar Measures of Lung Injury and Inflammation. Am J Respir Cell Mol Biol. 2018;59(2):257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shannahan JH, Podila R, Aldossari AA, Emerson H, Powell BA, Ke PC, et al. Formation of a protein corona on silver nanoparticles mediates cellular toxicity via scavenger receptors. Toxicol Sci. 2015;143(1):136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aldossari AA, Shannahan JH, Podila R, Brown JM Scavenger receptor B1 facilitates macrophage uptake of silver nanoparticles and cellular activation. Journal of Nanoparticle Research. 2015;17, Article number: 313. [Google Scholar]
- 41.Wang X, Katwa P, Podila R, Chen P, Ke PC, Rao AM, et al. Multi-walled carbon nanotube instillation impairs pulmonary function in C57BL/6 mice. Part Fibre Toxicol. 2011;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Draper DW, Madenspacher JH, Dixon D, King DH, Remaley AT, Fessler MB. ATP-binding cassette transporter G1 deficiency dysregulates host defense in the lung. Am J Respir Crit Care Med. 2010;182(3):404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilburg-Basnyat B, Reece SW, Crouch MJ, Luo B, Boone AD, Yaeger M, et al. Specialized Pro-Resolving Lipid Mediators Regulate Ozone-Induced Pulmonary and Systemic Inflammation. Toxicol Sci. 2018;163(2):466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Hoecke L, Job ER, Saelens X, Roose K. Bronchoalveolar Lavage of Murine Lungs to Analyze Inflammatory Cell Infiltration. J Vis Exp. 2017(123). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Messenger ZJ, Hall JR, Jima DD, House JS, Tam HW, Tokarz DA, et al. C/EBPbeta deletion in oncogenic Ras skin tumors is a synthetic lethal event. Cell Death Dis. 2018;9(11):1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaeger MJ, Reece SW, Kilburg-Basnyat B, Hodge MX, Pal A, Dunigan-Russell K, et al. Sex Differences in Pulmonary Eicosanoids and Specialized Pro-Resolving Mediators in Response to Ozone Exposure. Toxicol Sci. 2021;183(1):170–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reader BF, Sethuraman S, Hay BR, Thomas Becket RV, Karpurapu M, Chung S, et al. IRAK-M Regulates Monocyte Trafficking to the Lungs in Response to Bleomycin Challenge. J Immunol. 2020;204(10):2661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung KF, Adcock IM. Precision medicine for the discovery of treatable mechanisms in severe asthma. Allergy. 2019;74(9):1649–59. [DOI] [PubMed] [Google Scholar]
- 49.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, et al. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 2010;11(6):493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Birukova AA, Starosta V, Tian X, Higginbotham K, Koroniak L, Berliner JA, et al. Fragmented oxidation products define barrier disruptive endothelial cell response to OxPAPC. Transl Res. 2013;161(6):495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radzikowska U, Rinaldi AO, Celebi Sozener Z, Karaguzel D, Wojcik M, Cypryk K, et al. The Influence of Dietary Fatty Acids on Immune Responses. Nutrients. 2019;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walton KA, Hsieh X, Gharavi N, Wang S, Wang G, Yeh M, et al. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278(32):29661–6. [DOI] [PubMed] [Google Scholar]
- 53.Speen AM, Kim HH, Bauer RN, Meyer M, Gowdy KM, Fessler MB, et al. Ozone-derived Oxysterols Affect Liver X Receptor (LXR) Signaling: A POTENTIAL ROLE FOR LIPID-PROTEIN ADDUCTS. J Biol Chem. 2016;291(48):25192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez D, Sandhoff TW, McLean MP. Steroidogenic factor-1 mediates cyclic 3′,5′-adenosine monophosphate regulation of the high density lipoprotein receptor. Endocrinology. 1999;140(7):3034–44. [DOI] [PubMed] [Google Scholar]
- 55.Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL, Hobbs HH. Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenic factor 1. J Biol Chem. 1997;272(52):33068–76. [DOI] [PubMed] [Google Scholar]
- 56.Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta. 2016;1860(12):2771–81. [DOI] [PubMed] [Google Scholar]
- 57.Lockett AD, Petrusca DN, Justice MJ, Poirier C, Serban KA, Rush NI, et al. Scavenger receptor class B, type I-mediated uptake of A1AT by pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2015;309(4):L425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol. 2011;187(9):4686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gillotte-Taylor K, Boullier A, Witztum JL, Steinberg D, Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J Lipid Res. 2001;42(9):1474–82. [PubMed] [Google Scholar]
- 60.Fessler MB. A New Frontier in Immunometabolism. Cholesterol in Lung Health and Disease. Ann Am Thorac Soc. 2017;14(Supplement_5):S399–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai C, Yao X, Keeran KJ, Zywicke GJ, Qu X, Yu ZX, et al. Apolipoprotein A-I attenuates ovalbumin-induced neutrophilic airway inflammation via a granulocyte colony-stimulating factor-dependent mechanism. Am J Respir Cell Mol Biol. 2012;47(2):186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashraf MZ, Kar NS, Chen X, Choi J, Salomon RG, Febbraio M, et al. Specific oxidized phospholipids inhibit scavenger receptor bi-mediated selective uptake of cholesteryl esters. J Biol Chem. 2008;283(16):10408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eberhart T, Eigner K, Filik Y, Fruhwurth S, Stangl H, Rohrl C. The unfolded protein response is a negative regulator of scavenger receptor class B, type I (SR-BI) expression. Biochem Biophys Res Commun. 2016;479(3):557–62. [DOI] [PubMed] [Google Scholar]
- 64.Mozzini C, Fratta Pasini A, Garbin U, Stranieri C, Pasini A, Vallerio P, et al. Increased endoplasmic reticulum stress and Nrf2 repression in peripheral blood mononuclear cells of patients with stable coronary artery disease. Free Radic Biol Med. 2014;68:178–85. [DOI] [PubMed] [Google Scholar]
- 65.Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, et al. Deficiency of scavenger receptor BI leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Urban S, Zieseniss S, Werder M, Hauser H, Budzinski R, Engelmann B. Scavenger receptor BI transfers major lipoprotein-associated phospholipids into the cells. J Biol Chem. 2000;275(43):33409–15. [DOI] [PubMed] [Google Scholar]
- 67.Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14(10):461–9. [DOI] [PubMed] [Google Scholar]
- 68.Pascoe CD, Roy N, Turner-Brannen E, Schultz A, Vaghasiya J, Ravandi A, et al. Oxidized phosphatidylcholines induce multiple functional defects in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2021;321(4):L703–L17. [DOI] [PubMed] [Google Scholar]
- 69.Fruhwirth GO, Moumtzi A, Loidl A, Ingolic E, Hermetter A. The oxidized phospholipids POVPC and PGPC inhibit growth and induce apoptosis in vascular smooth muscle cells. Biochim Biophys Acta. 2006;1761(9):1060–9. [DOI] [PubMed] [Google Scholar]
- 70.Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. 2018;558(7709):301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S, et al. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. 2011;51(10):1903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, et al. Scavenger Receptor BI Protects against Septic Death through Its Role in Modulating Inflammatory Response. J Biol Chem. 2009;284(30):19826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taront S, Dieudonne A, Blanchard S, Jeannin P, Lassalle P, Delneste Y, et al. Implication of scavenger receptors in the interactions between diesel exhaust particles and immature or mature dendritic cells. Part Fibre Toxicol. 2009;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK, et al. Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol. 2007;27(11):2413–9. [DOI] [PubMed] [Google Scholar]
- 75.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94(23):12610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Helgadottir A, Sulem P, Thorgeirsson G, Gretarsdottir S, Thorleifsson G, Jensson BO, et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur Heart J. 2018;39(23):2172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vickers KC, Rodriguez A. Human scavenger receptor class B type I variants, lipid traits, and cardiovascular disease. Circ Cardiovasc Genet. 2014;7(6):735–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351(6278):1166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, et al. Genetic variant of the scavenger receptor BI in humans. N Engl J Med. 2011;364(2):136–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.