

Abstract
Background and Aims:
Biliary atresia is a severe inflammatory and fibrosing cholangiopathy of neonates of unknown etiology. The onset of cholestasis at birth implies a prenatal onset of liver dysfunction. Our aim was to investigate the mechanisms linked to abnormal cholangiocyte development.
Approach and Results:
We generated biliary organoids from liver biopsies of infants with biliary atresia and normal and diseased controls. Organoids emerged from biliary atresia livers and controls and grew as lumen-containing spheres with an epithelial lining of cytokeratin-19posalbuminnegSOX17neg cholangiocyte-like cells. Spheres had similar gross morphology in all three groups and expressed cholangiocyte-enriched genes. In biliary atresia, cholangiocyte-like cells lacked a basal positioning of the nucleus, expressed fewer developmental and functional markers, and displayed misorientation of cilia. They aberrantly expressed F-actin, β-catenin, and Ezrin, had low signals for the tight junction protein zonula occludens-1 (ZO-1), and displayed increased permeability as evidenced by a higher Rhodamine-123 (R123) signal inside organoids after verapamil treatment. Biliary atresia organoids had decreased expression of genes related to EGF signaling and FGF2 signaling. When treated with EGF+FGF2, biliary atresia organoids expressed differentiation (cytokeratin 7 and hepatocyte nuclear factor 1 homeobox B) and functional (somatostatin receptor 2, cystic fibrosis transmembrane conductance regulator [CFTR], aquaporin 1) markers, restored polarity with improved localization of F-actin, β-catenin and ZO-1, increased CFTR function, and decreased uptake of R123.
Conclusions:
Organoids from biliary atresia are viable and have evidence of halted epithelial development. The induction of developmental markers, improved cell-cell junction, and decreased epithelial permeability by EGF and FGF2 identifies potential strategies to promote epithelial maturation and function.
INTRODUCTION
The presence of congenital abnormalities or tissue injury at birth generally results from a genetic defect, prenatal virus infection, or toxic insult.[1] A variant of this paradigm is biliary atresia, a severe disease of multifactorial pathogenesis that targets the liver and biliary system and produces a severe phenotype of bile duct obstruction within 3 months of life.[2,3] Most affected infants have no major congenital abnormalities, transition uneventfully to extrauterine life before the full onset of symptoms, and then develop direct hyperbilirubinemia and acholic stools caused by an underlying obstruction of bile ducts that completely disrupts bile flow and drives a rapidly progressive fibrosis.[4] Searching for pathogenic mechanisms of hepatobiliary injury, we and others have used a neonatal mouse model of rotavirus-induced biliary injury that recapitulates several clinical and pathologic features of human biliary atresia.[5] In this model, mechanisms of tissue injury have been directly attributed to a transient population of hepatic erythroblasts, a lack of regulatory T-helper lymphocytes, and an unrestrained activation of the innate and adaptive immune systems in response to virus or environmental toxin(s) that target the bile duct epithelium.[5,6] Among environmental factors, individual viruses have been identified in affected infants, but there is limited reproducibility in different patient cohorts. The isolation of an environmental toxin known as biliatresone, which induces biliary atresia–like disease in lambs, has been shown to target bile duct epithelial cells in zebrafish, but it has not been directly associated with pathogenesis of the disease in humans.[7–10]
Several lines of evidence point to a prenatal onset of disease. Clinically, an analysis of serum bilirubin levels in the first postnatal days of a large cohort of live births showed that infants subsequently diagnosed with the biliary atresia had increased levels of direct bilirubin.[3] In a study investigating the expression of proteins that regulate embryonic development, liver sections had abnormal expression of Hedgehog proteins.[11] Experimentally, a defect in morphogenesis is supported by the biliary dysfunction in zebrafish lacking functional Add3a[12] and by the transcriptional shift from cholangiocytes to hepatocytes in organoids generated from progenitor cells.[13] Searching for mechanisms linked to abnormal cholangiocyte development, we derived biliary organoids directly from liver tissues, examined their developmental profiles, and performed functional assays to explore their relationship to pathogenesis of biliary atresia.
MATERIALS AND METHODS
Human subjects and study approval
The collection of patient data and liver tissue for the generation of organoids was performed according to the institutional guidelines of the Research Ethics Committees at Cincinnati Children’s Hospital Medical Center. All patients’ guardians signed the written informed consent form before collection of the specimens. A total of 29 liver-tissue specimens were collected either as surgical biopsies or explants from deceased healthy donors (n = 2; healthy; 13 and 17 years), patient with urea cycle defect (n = 1; used as a healthy control; 10 months), non-biliary atresia cholestasis (n = 6; disease controls; age at diagnosis, 4 months; transplant ages, 8 weeks to 17 years), and subjects diagnosed with biliary atresia (n = 20; age at diagnosis, 5 weeks to 3 months; transplant ages, 6 months to 17 years) (Table S1).
Generation of organoids
Organoids were generated from human liver tissues as described previously.[14] Briefly, human liver tissues were minced and washed 2 times with ice-cold basal medium consisting of Advanced DMEM/F12 (Catalog #12634; Gibco, Carlsbad, CA) supplemented with 1% Penicillin/Streptomycin (Catalog #15140122; Gibco), 1% GlutaMAX (Catalog #35050; Gibco), and 10 mM HEPES (Catalog #15630; Gibco). Minced tissue was strained through a membrane to isolate structures less than 0.1-mm size, which were mixed into Cultrex Reduced Growth Factor Basement Membrane Extract type 2 PathClear Matrix (Catalog #3533-005-02; AMSBIO LLC, Cambridge, MA). A total of 50 μl of tissue-embedded matrix was plated in each well of a 24-well plate and incubated for 10 min at 37°C to solidify the matrix. The matrix was overlaid with 500 μl isolation medium (expansion medium supplemented with 25 ng/ml recombinant human Noggin [Catalog #120–10C; PeproTech, Rocky Hill, NJ], 30% vol/vol homemade Wnt3a-conditioned medium, and 10 μM ROCK-inhibitor/Y-27632 [Catalog #SCM075; Sigma-Aldrich, St. Louis, MO]) and cultured for 5 days. Subsequently, cultures were maintained in expansion medium (basal medium supplemented with 1× B27 [Catalog #12587010; Gibco], 1× N2 supplement minus vitamin A [Catalog #17502048; Gibco], 1 mM N-acetylcysteine [Catalog #A7250; Sigma-Aldrich], 10% vol/vol homemade RSPO1-conditioned media, 10 mM Nicotinamide [Catalog #N0636; Sigma-Aldrich], 10 nM recombinant human-Gastrin [Catalog #G9145; Sigma-Aldrich], 50 ng/ml recombinant human EGF [Catalog #100–15; PeproTech], 100 ng/ml recombinant human FGF10 [Catalog #100–26; PeproTech], 25 ng/ml recombinant human HGF [Catalog #100–39; PeproTech], 10 μM Forskolin [Catalog #1099; Tocris, Minneapolis, MN], and 5 μM A83–01 [Catalog #SML0788; Sigma-Aldrich]) (Table S2). After emergence, organoids were transferred to new wells and grown to high density (near confluency), at which time they underwent mechanical dissociation by successive pipetting into small fragments and split into 4–6 new wells or cryopreserved for future use. Phase contrast images were captured at ×10 magnification with an Olympus IX71 inverted microscope using DP71 camera operated by Olympus Cell Sens standard version 1.18 software (Olympus America Inc., Center Valley, PA).
The methods summarized below are described in greater detail in the Supporting Information. To avoid the potential for confounding variables related to differences in cell function or expression of markers induced by handling of organoids, we used organoids with the same number of passages for all three groups in all studies described here.
RNA sequencing and analysis
Bulk RNA sequencing (RNA-seq) and transcriptomic analysis were performed as described previously.[15] Organoids cultured in either expansion or differentiation media were harvested into Trizol to isolate RNA, which was sequenced in Illumina Hiseq 2500 and Novaseq 6000 (San Diego, CA); 100–125 base-pair reads were trimmed and aligned with reference sequences using FASTQC, Trim Galore, and SAMtools. The gene-expression platform containing data from organoids and livers (as a tissue references) was analyzed by principal component analysis (PCA), supervised pathway mining, and ToppGene Suite to identify differentially expressed biological processes.
Immunofluorescence and transmission electron microscopy
Immunofluorescence staining of organoids was performed using protocols described previously,[16] with paraformaldehyde-fixed organoids embedded in M-1 matrix (Catalog #1310TS; Thermo Fisher Scientific, Grand Island, NY) and cryosectioned. Tissue sections were then rehydrated, blocked, stained with primary, matching secondary antibodies, and covered with a mounting solution containing DAPI. Protocols for transmission electron microscopy (TEM) are described in the Supporting Information.
Whole-mount staining
Whole-mount organoid processing and staining procedures were adapted from previously published reports.[17,18] Briefly, organoids devoid of matrix were fixed in paraformaldehyde, and precautions were taken not to rupture while processing. Whole organoids were incubated with blocking solution, primary antibodies, matching secondary antibodies, and stained with Hoechst for nuclei. Whole organoids were suspended in chamber slides with PBS and were imaged under confocal microscope.
Rhodamine 123 assay
Organoids cultured either in expansion medium or differentiation medium were extracted from matrix and pretreated with or without 10 μM verapamil (Catalog #V4629; Sigma-Aldrich) before incubation with 5 μM Rhodamine123 at 37°C for 2 h (Catalog #83702; Sigma-Aldrich). Images were captured after wash. Luminal fluorescence was measured and normalized to background using Image J software.[19] Protocol for the cystic fibrosis transmembrane conductance regulator (CFTR) assay is described in the Supporting Information.
Differentiation of organoids to mature cholangiocytes
Organoids were cultured in differentiation media containing 20 ng/ml EGF (Catalog #500-P45; PeproTech) and 5 ng/ml FGF2 (Catalog #100–18B; PeproTech) in William’s E medium (Table S2). Cultures were maintained for 14 days with media changed every other day.
Statistics
Data are shown as mean with SD from three independent experiments. Throughout the manuscript, we present the number of biological replicates (from individual liver donors) in each figure legend. For each biological replicate, we include several organoids to control for any potential variability among organoids from the same donor. The numbers of samples for individual experiments are presented with the results. The results were analyzed using GraphPad Prism version 7.00 (GraphPad Software, La Jolla, CA) by two-tailed, unpaired Student t test for two group comparisons and Mann-Whitney U test for three group comparisons. p values of <0.05 were considered statistically significant.
RESULTS
Derivation of organoids from livers with biliary atresia
To test the hypothesis that infants with biliary atresia have abnormal development of the bile duct epithelium, we cultured fresh liver biopsies from 20 infants with biliary atresia and 6 with other forms of cholestasis (intrahepatic cholestasis; used as diseased controls) in 100% matrix scaffold and media containing R-Spondin, Forskolin, FGF10, and A83–01[14,20]; biopsies from unused liver-transplant donor grafts and from a patient with urea cycle defect served as normal controls (Table S1). Daily monitoring with phase contrast microscopy revealed the emergence of budding structures in normal controls by 3 ± 1 (mean ± SD) days of culture, which grew as identifiable spheroids by 15 days (Figure 1A), whereas the emergence of spheroids was delayed for biliary atresia and diseased controls (12.5 ± 4.8 and 20.5 ± 6.1 days, respectively; p < 0.05 (Figure 1B). Because our protocols did not involve flow-sorting of cells expressing the epithelial cell adhesion molecule (EPCAM) used in other studies,[13,14] we determined whether this delay in emergence was due to paucity of EPCAMpos cells in liver tissues.[20] By immunostaining, we found easily identifiable EPCAMpos cells in the periportal region of all three groups (Figure S1). All three spheroids from normal controls underwent successive cryopreservation and passage, with the same being true for 61% of the organoids from biliary atresia (Figure 1C). Morphologically, spheroids from all three groups had similar features by phase-contrast microscopy and were formed by a layer of epithelial cells and a lumen (Figure 1D). Notably, the ability of biliary atresia organoids to form a lumen and undergo cryopreservation and successful passage differs from the outcome of organoids generated from EpCAMpos cells reported previously, in which the organoids lacked a lumen and were unable to establish cell lines.[13]
FIGURE 1.
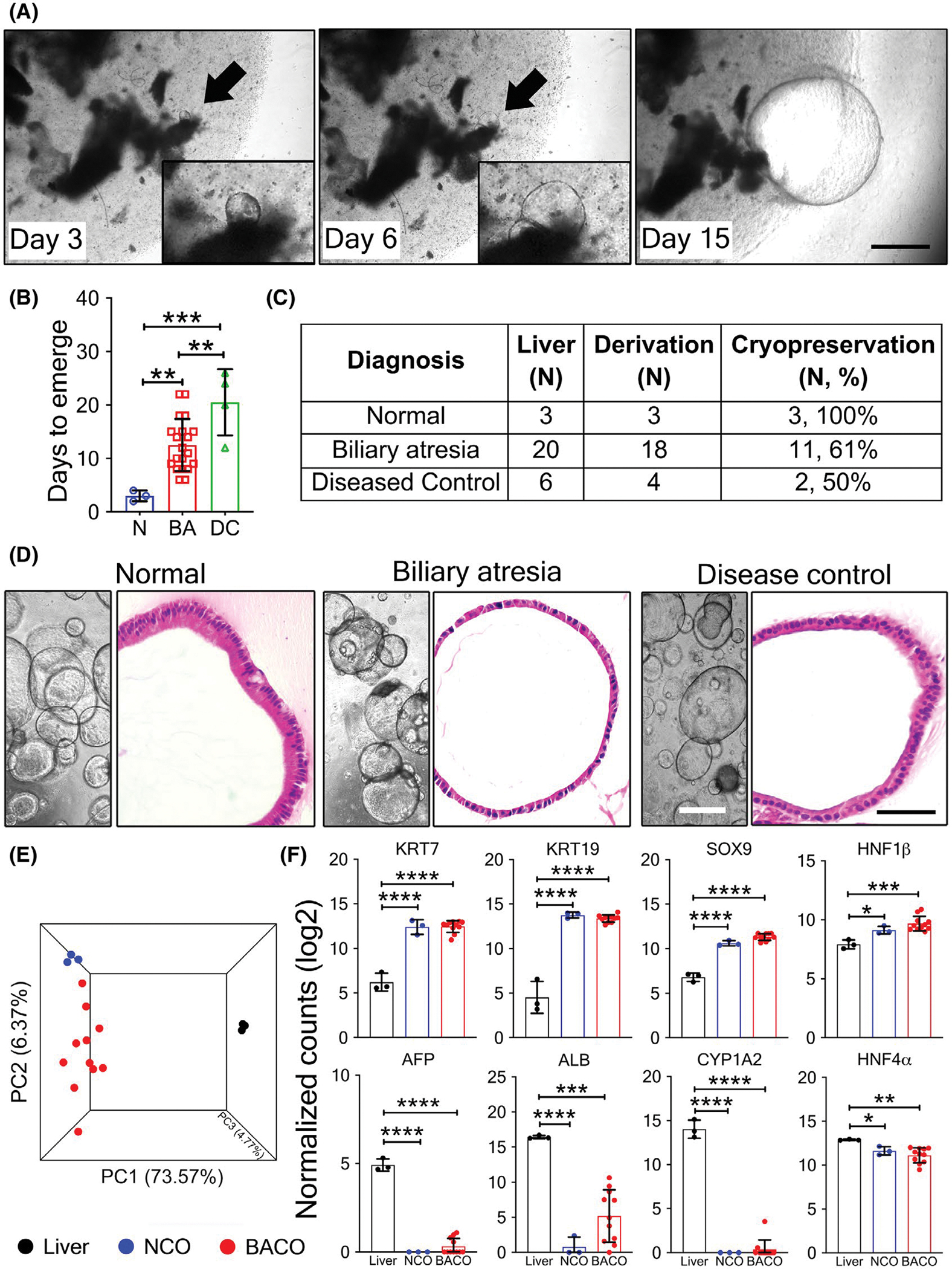
Emergence and growth of biliary organoids from human livers. (A) Phase contrast microscopy show emergence (arrows) of organoids from liver biopsies from healthy subjects at days 3, 6, and 15 of culture; insets show magnified view. (B) Number of days for organoids to emerge. (C) Number of organoids successfully derived, cryopreserved, and passaged. (D) Phase contrast images (left panels; ×10 magnification) depicting organoid growth, and hematoxylin and eosin staining (right panels; ×40 magnification) showing histological features from NCO, BACO, and DCO cholangiocyte-like organoids (scale bar, 50 μm). (E) Three-way PCA analysis of RNA-seq data comparing NCOs (n = 3), BACOs (n = 11), and liver tissues (n = 3). (F) RNA expression levels for cholangiocyte-enriched (top panel) and hepatocyte-enriched (lower panel) genes. Statistical analysis was performed using Mann-Whitney U test; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.5. AFP, alpha-fetoprotein; ALB, albumin; KRT, keratin; ns, not significant; PC, principal component; SOX9, SRY (sex determining region Y)-box 9
In initial steps to examine the cellular composition of organoids, we compared global gene-expression signatures among individual groups, using the liver as a tissue reference. PCA of RNA-seq showed a clear separation of spheroids from livers (Figures 1E and S2), with increased expressions of cholangiocyte-enriched genes and decreased expression of hepatocyte-enriched genes (Figure 1F). A list of differentially expressed genes between spheroids and liver tissues is shown in Files S1 and S2. We then applied enrichment analysis using differentially expressed genes to investigate molecular profiles and found that the most distinguishing biological processes up-regulated in the spheroids were related to cell cycle and cytoskeletal and epithelium development, whereas processes linked to metabolism were significantly down-regulated when compared with livers (Figures S3 and S4). Together, the findings showed that liver-derived spheroids are populated by cells expressing a molecular profile enriched for cholangiocyte markers, heretofore referred to as cholangiocyte-like organoids (COs).
To investigate how normal COs (NCOs) and biliary atresia COs (BACOs) relate to anatomical domains of bile ducts, we built a gene-expression platform containing RNA-seq data from our organoids and data from the recent report by Rimland et al.,[21] which is publicly available as FASTQ files for organoids derived from extrahepatic (EHBD) and intrahepatic (IHBD) bile ducts (E-MTAB-7569). The integration of the data into the platform included sequence alignment, normalization of sequencing depth using DESeq2, correction for batch differences, and functional annotations (described in the Supporting Information). Applying PCA to the curated data platform showed that NCOs clustered close to EHBD organoids, whereas some BACOs tended to cluster with EHBDs (Figure S5), and still others were physically dispersed between EHBDs and IHBDs.[21]
Expression of development and functional markers in organoids
To directly determine whether COs from subjects with biliary atresia have abnormal development of the biliary epithelium, we profiled BACO, NCO, and diseased CO (DCO) controls by immunostaining of cellular markers of development and function. All three groups expressed similar numbers of cytokeratin 19 cells and lacked albumin and SOX17 (SRY [sex determining region Y]-box 2) expression (Figure S6); in contrast, the numbers of organoid cells expressing the developmental and cholangiocyte markers cytokeratin 7 (CK7), EPCAM, and the transporters aquaporin 1 (AQP1), CFTR, and somatostatin receptor 2 (SSTR2) were significantly lower in biliary atresia when compared with normal and disease controls (Figure 2A,B). Furthermore, hepatocyte nuclear factor (HNF)-4α expression was confined only to NCOs and DCOs (Figure S6), suggesting a preserved hepatocyte/cholangiocyte progenitor potential. When considering the qualitative features of expressed proteins, subtle differences in staining patterns were seen between NCOs and DCOs and were more pronounced in BACOs (in addition to decreased number of positive cells). Altogether, the major differences in the expression patterns of cell markers in BACOs were the evidence of a potential delay in epithelial cell development and maturation in biliary atresia.
FIGURE 2.
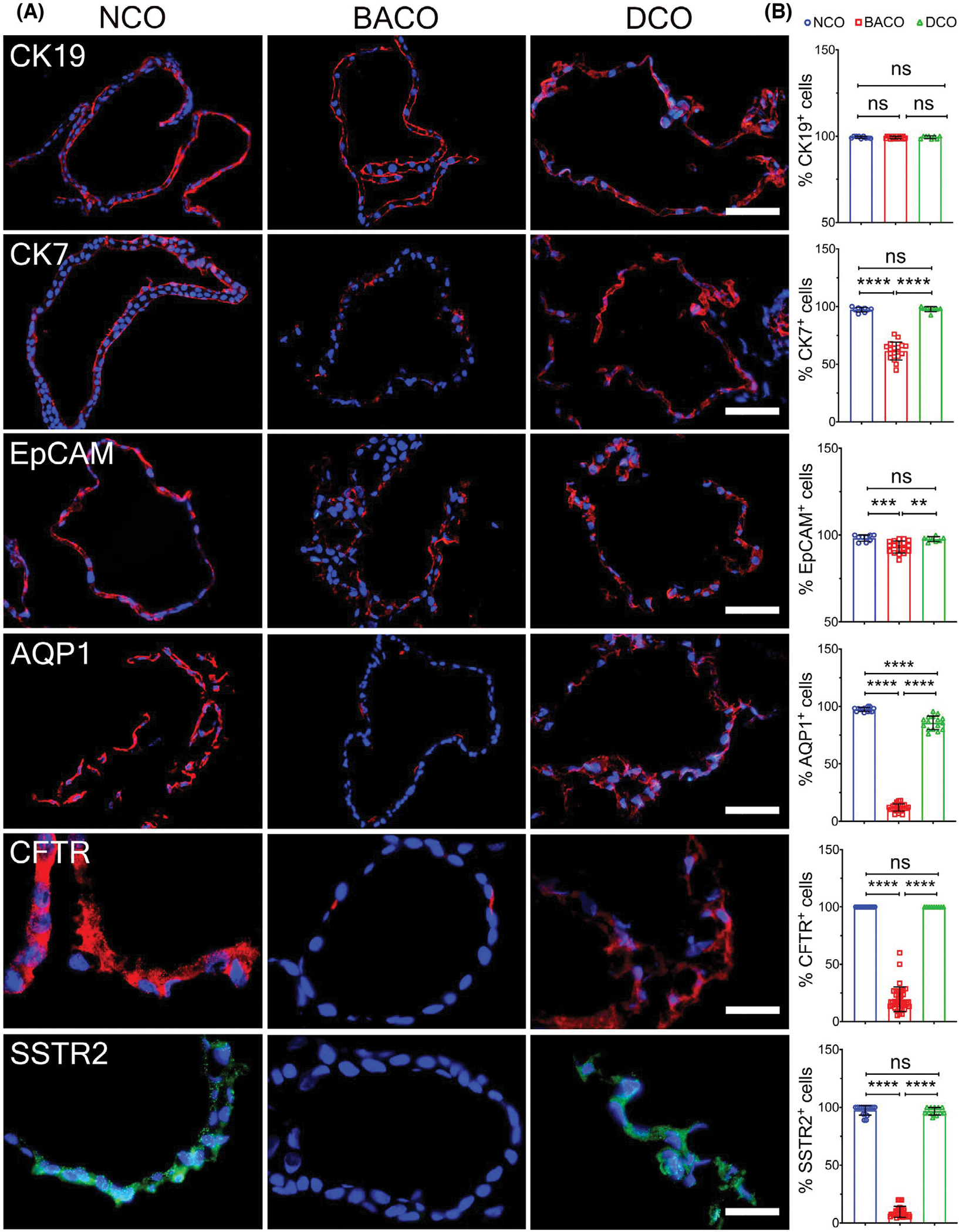
Immunophenotyping of organoids. (A) Immunofluorescence staining to detect expression of cytokeratin 19, CK7, EpCAM, AQP1, CFTR, and SSTR2 in NCO, BACO, and DCO (scale bar, 50 μm; CFTR and SSTR2 scale bar, 20 μm). (B) Graphs on the right panel show comparative quantifications of the percent of positive cells. Data are presented as mean ± SD. Statistical analysis was performed using Mann-Whitney U test; ****p < 0.0001, ***p < 0.001, and **p < 0.01. NCO, n = 3; BACO, n = 4; and DCO, n = 2. CK19, cytokeratin 19
Abnormal cell polarity in biliary atresia organoids
Searching for additional evidence of delayed cellular development in biliary atresia, we examined the cellular morphology of organoids. NCOs and DCOs had a layer of columnar cells with a basal nucleus, in contrast to the predominantly cuboidal cells with a small cytoplasm to nucleus ratio in BACOs (Figure 3A). To determine whether these features represented a defect in cell polarization, we applied whole-mount immunostaining of organoids with anti-acetylated-α-tubulin antibodies to visualize the cholangiocyte cilium.[22] Three-dimensional rendering of confocal z-stack images easily identified cilium in epithelial cells oriented toward the center/lumen of the organoid in NCOs and DCOs, whereas cilia of BACOs pointed laterally toward neighboring cells or outwardly (Figure 3B; Movie S1A,B); quantitatively, BACOs had a lower number of ciliated cells (Figure 3C; Movie S2A–C). This lower population of ciliated cells in BACOs was associated with an ectopic expression of filamentous actin (F-actin) and β-catenin, in contrast to NCOs and DCOs, which had apical expression and basolateral expression for F-actin and β-catenin, respectively (Figure 3D). Searching for further evidence of abnormal cell polarity, we determined the mRNA expression for the genes encoding the Ezrin (EZR)–Radixin (RDX)–Moesin (MSN) complex.[23] We found that EZR mRNA expression (but not RDX or MSN) was selectively decreased in BACOs (Figure 3E); at the protein level, Ezrin was confined to the basal region of BACOs instead of the prominent apical (and basolateral) expression in NCOs (Figure 3F). These findings were consistent with a decreased number of ciliated cells and loss of polarity in the organoid epithelium in biliary atresia.
FIGURE 3.
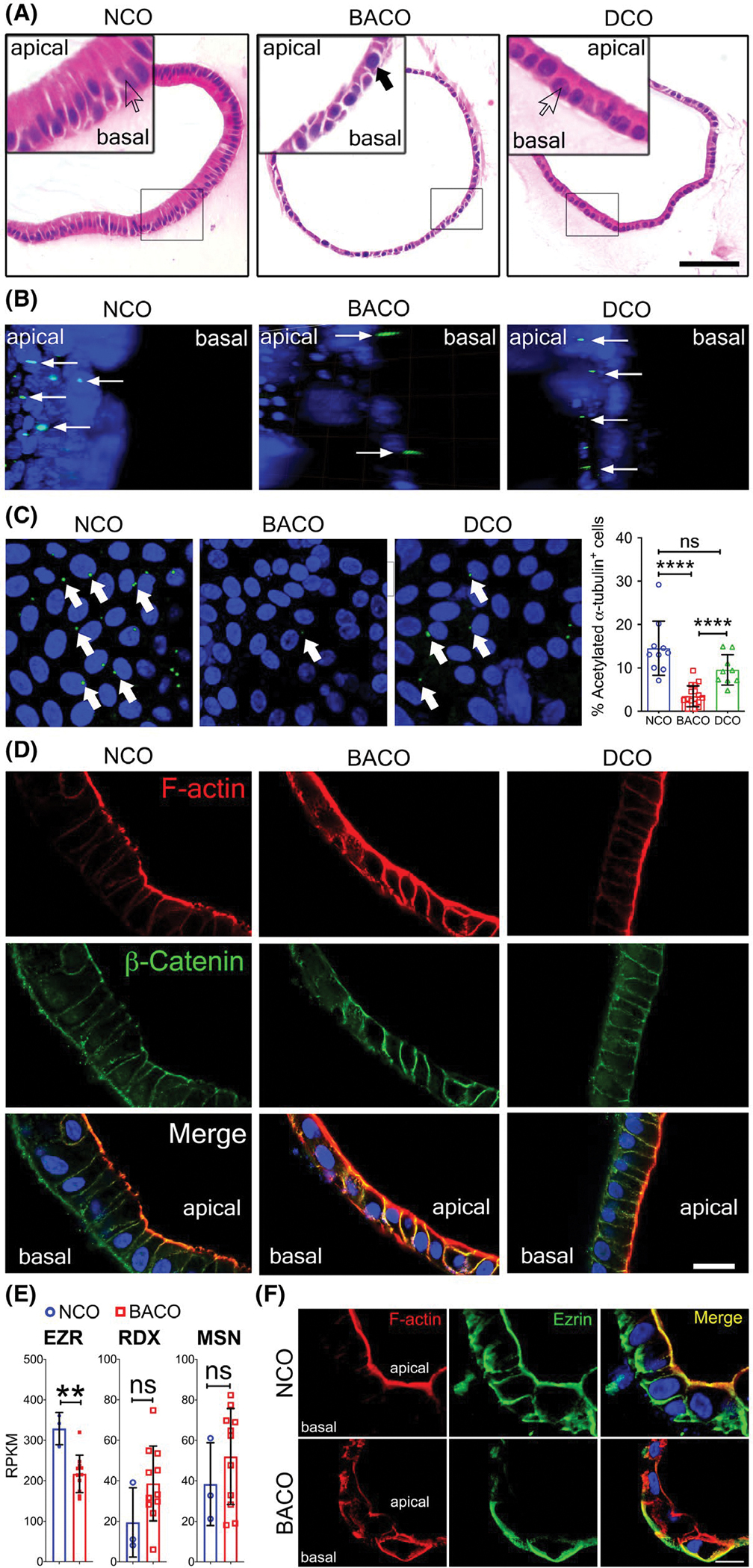
Biliary atresia organoids have abnormal cell polarity. (A) Hematoxylin and eosin staining of NCO and DCO organoids show columnar cells with basal nucleus (white arrows; high magnification insets), whereas BACO organoids show cuboidal cells with centrally located nucleus (solid black arrow; high magnification inset). Scale bar, 50 μm. (B,C) Three-dimensional surface rendering of confocal z-stacks of whole mount organoids stained for cholangiocyte cilium with acetylated-α-tubulin antibodies. In (B), cilia emerge toward the apical or luminal side in NCOs and DCOs, and toward the basal side in BACOs (thin white arrows), with computer-generated flattening to enable counting in (C), showing a decreased number of ciliated cells in BACOs (graph depicts percent of acetylated-α-tubulin+/Hoechst+ cells in organoids). Magnification, ×20. (D) Immunofluorescence confocal microscopy shows localization of filamentous-actin (F-actin) as an apical marker (red) and β-catenin as a basolateral marker (green) in NCOs and DCOs, whereas BACOs show extension of expression of both proteins. Nuclei were counterstained with Hoechst; ×60 magnification. (E) RNA expressions of EZR, RDX, and MSN. (F) Immunofluorescence staining of NCOs shows apicobasal EZR (green) signals co-localizing with F-actin (red). BACOs show loss of apical EZR expression localizing only to the basal region. Nuclei were counterstained with Hoechst. Scale bar, 20 μm. Graph data are presented as mean ± SD. Statistical analysis was performed using Mann-Whitney U test; ****p < 0.0001. NCO, n = 3; BACO, n = 4; and DCO, n = 2. RPKM, reads per kilobase of exon model per million mapped reads
Increased permeability in biliary atresia organoids
Based on the role of cell polarity in epithelial permeability, we next determined the expression of zonula occludens-1 (ZO-1), a tight junction protein located in the cytoplasmic membrane that provides a major barrier function. ZO-1 was uniformly expressed between cells of NCOs and DCOs, but it was fragmented and restricted to only 13.1 ± 2.7% of the cells in BACOs (p < 0.001; Figure 4A). Using TEM to directly visualize tight junctions, we observed a significantly smaller number of intercellular junctions per cell in BACOs (1–2 per cell) when compared with the epithelial lining of NCOs (4–5 junctions per cell; p < 0.0001; Figure 4B). Altered patterns of ZO-1 expression were also found in liver tissues from patients with biliary atresia when compared with normal donor liver tissue staining (Figure S7), raising the possibility of an increased epithelial cell permeability in biliary atresia. To examine this possibility, we incubated organoids in the presence of rhodamine 123 (R123), a cationic fluorescent substrate actively transported by multidrug resistance protein-1 (MDR1).[24] Initial measurements of fluorescence in NCOs in 30-min increments of incubation showed a progressive lumen accumulation of R123 for 2 h, a time when R123 fluorescence was 19.2 ± 7.5 fluorescence units (FU) in NCOs, 48.8 ± 9.7 FU in DCOs, and 89.6 ± 30.2 FU in BACOs (p < 0.001; Figure 4C). When a similar assay was performed in the presence of 10 μM verapamil (an MDR1 inhibitor), R123 signal was effectively prevented in NCOs and DCOs. In BACOs, the lumen accumulation of R123 was lower at 2 h than the value without verapamil, but the signal remained higher than controls at 50.7 ± 25.2 FU (Figure 4C). These data pointed to a leaky epithelium or an MDR1-independent uptake in BACOs.
FIGURE 4.
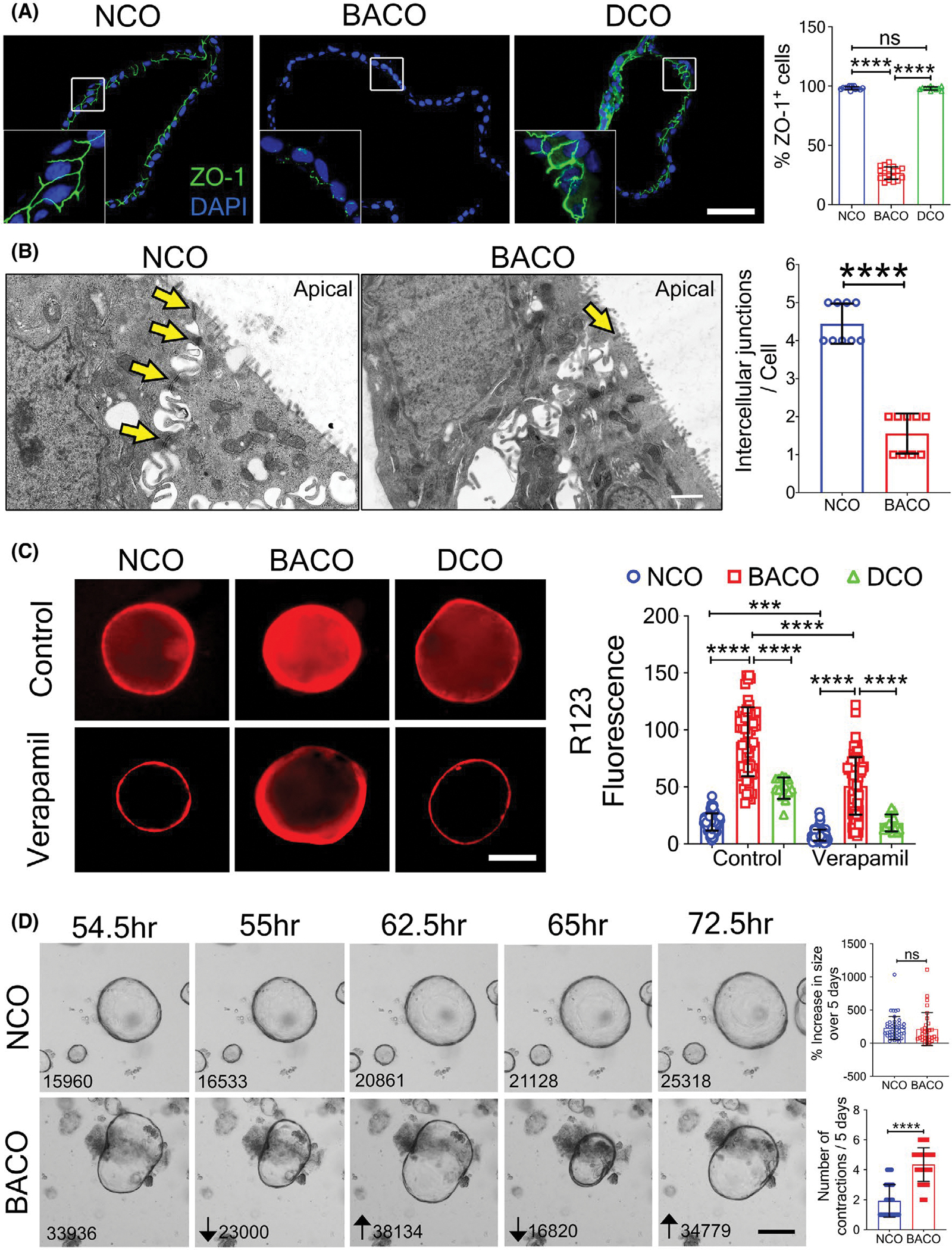
Biliary atresia organoids have increased permeability. (A) Immunofluorescence staining for ZO-1 shows normal tight junction in NCOs and DCOs, with decreased and spotty expression in BACOs (insets = higher magnification; nuclei were stained with DAPI; graph shows percentage of ZO-1+/DAPI+ cells; scale bar, 50 μm. (B) TEM imaging depicts preserved and loss of intercellular junctions (yellow arrows) in NCOs and BACOs, respectively. Graph shows number of intercellular junctions per cell (bar, 1 μm). (C) Immunofluorescent images depict uptake of R123 into the lumen of organoids without (top row) or with Verapamil (lower row) to inhibit uptake. Magnification, ×10. Graph shows quantification of luminal fluorescence corrected for background fluorescence. (D) Image frames taken 0.5 h apart from a 5-day time-lapse microscopy to monitor growth of organoids document contractions in BACOs but not in NCOs (numbers indicate area of the organoids measured in pixel2 using ImageJ software; graphs indicate the percent change in area at the end of 5 days [top] and the number of contractions [bottom] over 5 days; scale bar, 100 μm). Data are presented as mean ± SD. Statistical analysis was performed using Mann-Whitney U test or unpaired t test; ****p < 0.0001 and ***p < 0.001. NCO, n = 3; BACO, n = 4; and DCO, n = 2
Looking for further evidence of epithelial leak, we plated organoids and monitored their growth for 5 days by time-lapse microscopy in the presence of forskolin. As reported for intestinal organoids,[25] NCOs and DCOs grew uniformly over time when cultured with forskolin and had occasional contractions followed by a prompt recovery of its surface area. BACOs also grew uniformly, but they exhibited frequent contractions and a gradual return to their surface area (Figure 4D and Movie S3A,B). Altogether, these data supported an abnormal cell junction associated with a leaky epithelial lining in BACOs.
Differentiation of cholangiocyte-like cells by activation of EGF receptor/FGF signaling
Based on the underexpression of developmental markers, abnormal cell polarity, and intercellular junction in BACOs, we reasoned that the delayed epithelial development in biliary atresia may result from a suppression of molecular circuits regulating cholangiocyte differentiation. To investigate this possibility, we quantified the expression of genes functionally related to EGF receptor (EGFR) and FGF because of their reported effects on cholangiocyte specification,[26–29] polarization,[30] and formation of junctional complexes.[31] Analysis of the RNA-seq data identified 16 EGFR/FGF-related genes with lower expression levels in BACOs compared with NCOs (Figure 5A). To determine whether activation of EGFR/FGF pathways would correct the cellular defect(s), we added basic FGF2 to the organoid culture media that already contained EGF, forskolin, dexamethasone, and dickkopf WNT signaling pathway inhibitor 1, and removed HGF and ROCK inhibitor (Y27632).[28,32] After 14 days of culture in this differentiation medium, organoids maintained the similar growth/expansion pattern and external morphology (Figure 5B). Analysis of RNA-seq from BACOs cultured in the differentiated media compared with BACOs cultured in the previous media showed the highest predominance of biological processes to be enriched for genes regulating cell adhesion, matrix, and morphogenesis (Figure 5C) and down-regulation of metabolic processes that are typically associated with hepatocytes (Figure 5D). Immunostaining of NCOs, BACOs, and DCOs showed that BACO cells largely restored the expression levels for CK7, substantially increased AQP1, CFTR, and SSTR2 (well above the near absent signal shown in Figure 2), and improved ZO-1 expression to 38.7 ± 4.8% of cells (Figure 6A,B). A comparative analysis of cells stained with HNF-1β, CK7, AQP1, CFTR, SSTR2, ZO-1, and α-tubulin before and after differentiation showed increased numbers when compared with organoids grown in the previous media (Figures 6C and S8; Movie S4A–C). Using TEM to directly visualize the impact of differentiation on cellular junctions, we found a significantly improved number of intercellular junctions in BACOs, which reached the numbers seen in NCOs (Figure 6D).
FIGURE 5.
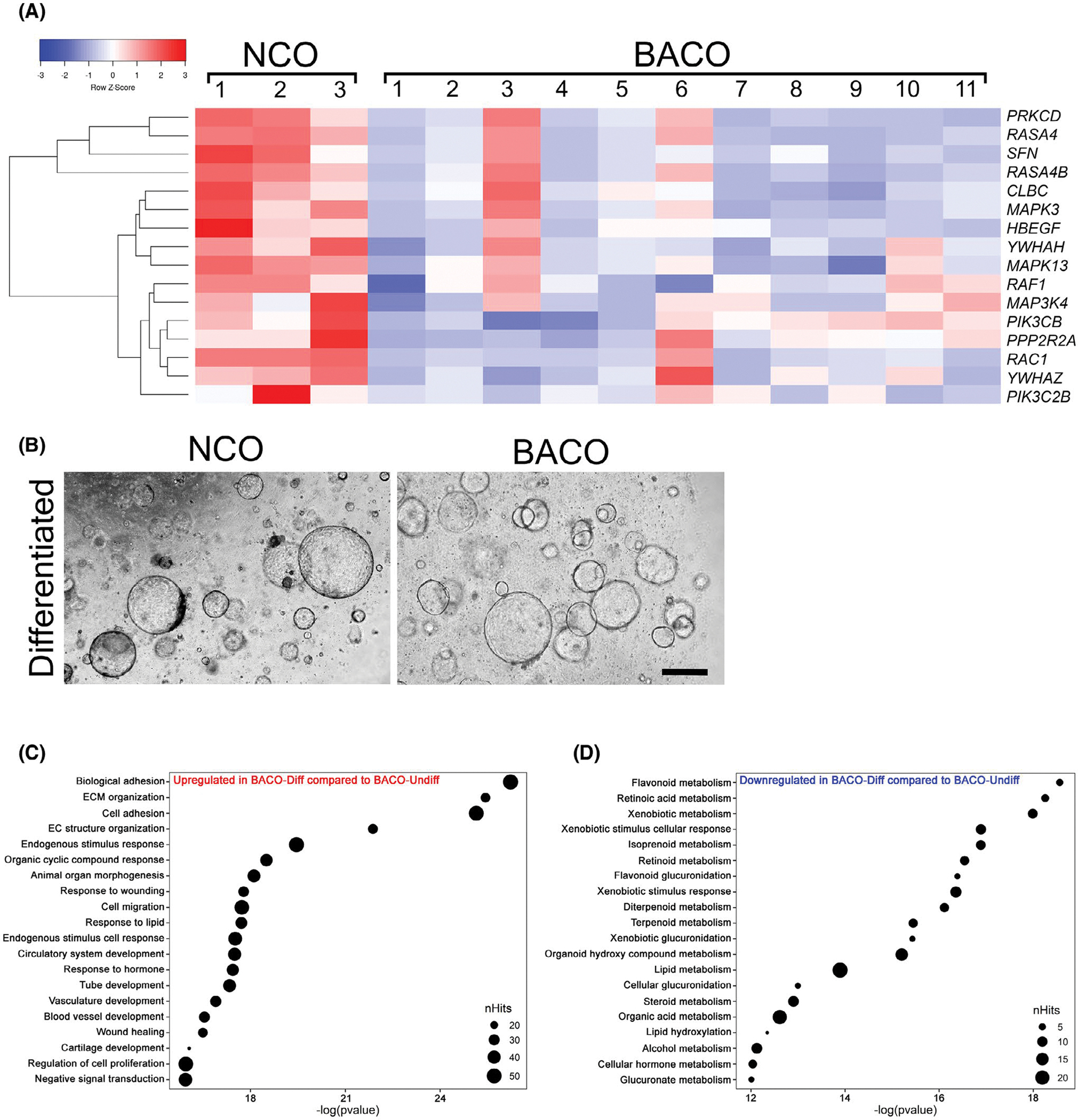
Expression and activation of EGFR/FGF signaling in organoids. (A) RNA-seq analysis of NCOs and BACOs contained a group of 16 genes functionally related to EGFR and FGF signaling pathways shown in a heatmap (p < 0.05). (B) Phase contrast images of COs cultured in differentiation medium for 14 days show similar cell morphology in NCOs and BACOs. In (C) and (D), gene ontology analysis in ToppGene using differentially expressed genes from RNA-seq data for BACOs identifies biological processes up-regulated (C) and down-regulated (D) after subjecting to differentiation protocols. NCO, n = 3; and BACO, n = 11. Abbreviations: CLBC, A-kinase anchoring protein 13; ECM, extracellular matrix; HBEGF, heparin binding EGF like growth factor; MAP3K4, mitogen-activated protein kinase kinase kinase 4; MAPK13, mitogen-activated protein kinase 13; MAPK3, mitogen-activated protein kinase 3; PIK3C2B, phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta; PIK3CB, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta; PPP2R2A, protein phosphatase 2 regulatory subunit Balpha; RAC1, Rac family small GTPase 1; RAF1, Raf-1 proto-oncogene, serine/threonine kinase; RASA4, RAS p21 protein activator 4; RASA4B, RAS p21 protein activator 4B; RKCD, protein kinase C delta; SFN, stratifin; YWHAH, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein eta; YWHAZ, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta
FIGURE 6.
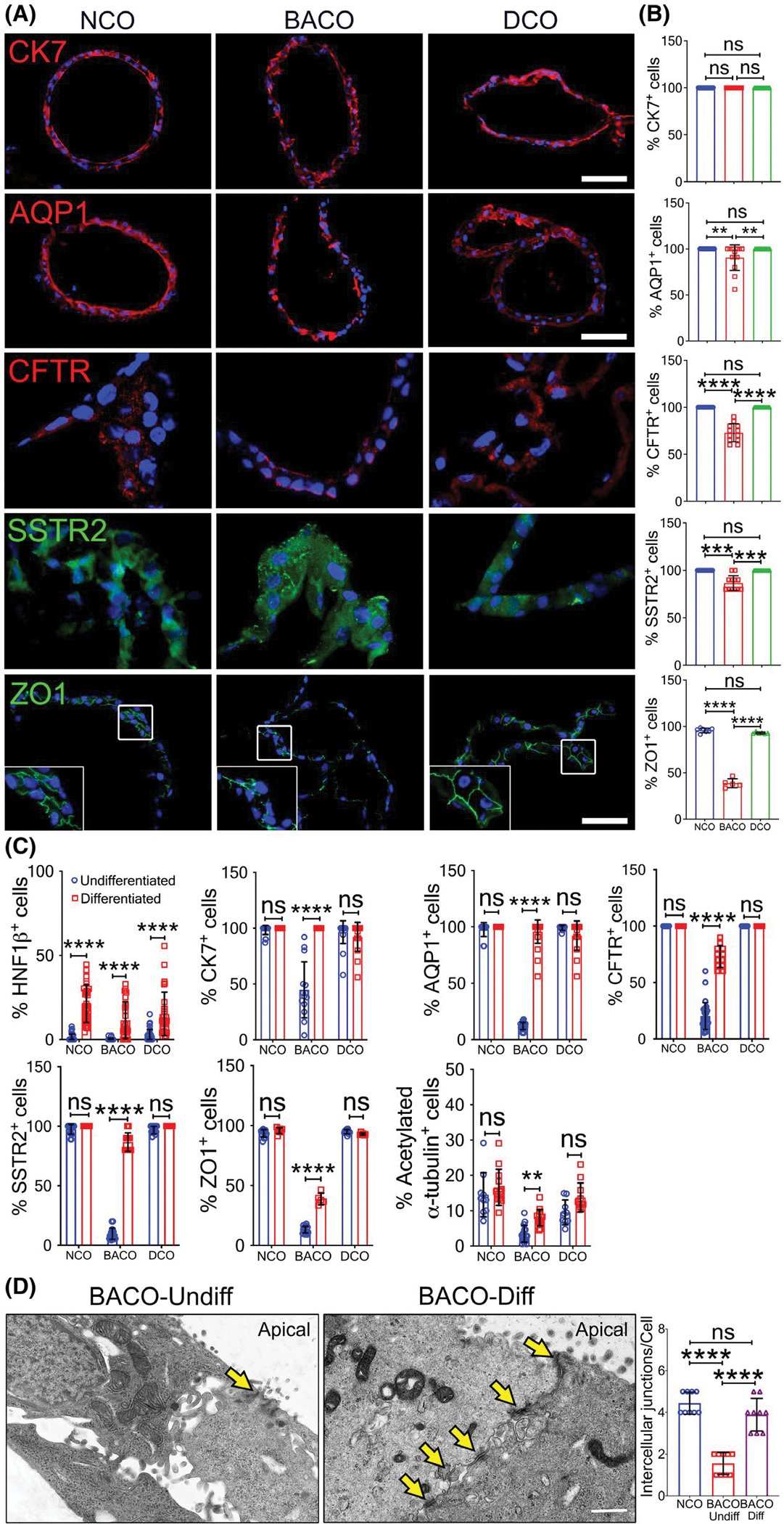
Induction of cell differentiation improves expression of developmental markers. (A) Immunofluorescence staining of CK7, AQP1, CFTR, SSTR2, and ZO1 in organoids after growth in cell differentiation conditions, with graphs in (B) showing percent of total DAPI+ cells (scale bar, 50 μm). A similar approach is shown in (C) with the inclusion of the same groups in the undifferentiation controls for the listed proteins. (D) TEM identifies occasional intercellular junction (yellow arrows) in undifferentiated BACOs and more easily identifiable junctions (yellow arrows) in BACOs cultured in differentiation medium. Graph represents number of intercellular junctions per cell (bar, 1 μm; NCOs cultured in differentiation medium are shown as controls). NCO, n = 3; BACO, n = 4; and DCO, n = 2. Data are presented as mean ± SD. Statistical analysis was performed using Mann-Whitney U test; ****p < 0.0001 and **p < 0.01. BACO-Diff, differentiated BACOs; and BACO-Undiff, undifferentiated BACOs
Finally, to investigate whether EGF-FGF activation affected epithelial cell polarization and function, we stained organoids for F-actin and β-catenin and found an improved apical and basolateral staining pattern, with only few focal areas of aberrant expression of F-actin in BACOs (Figure 7A). Functionally, we subjected NCO and BACO organoids to a CFTR assay that uses quenching of the fluorescent dye N-ethoxycarbonylmethyl-6-methoxyquinolinium bromide in response to chloride transport. In NCOs, about 65% of the fluorescence in the organoid lumen was lost within 30 min, both in the undifferentiated and differentiated organoids (Figure 7B). In the undifferentiated state, BACOs retained the fluorescence in the lumen, whereas differentiated BACOs lost about 50% of the fluorescence in the same time frame (Figure 7C), thus reflecting a gain of function after differentiation. To assess epithelial permeability, we quantified R123 fluorescence intensity of differentiated organoids in culture and found lower R123 signals in the BACOs, which suggested that the signal accumulation was effectively blocked by verapamil (Figure 7D,E).
FIGURE 7.
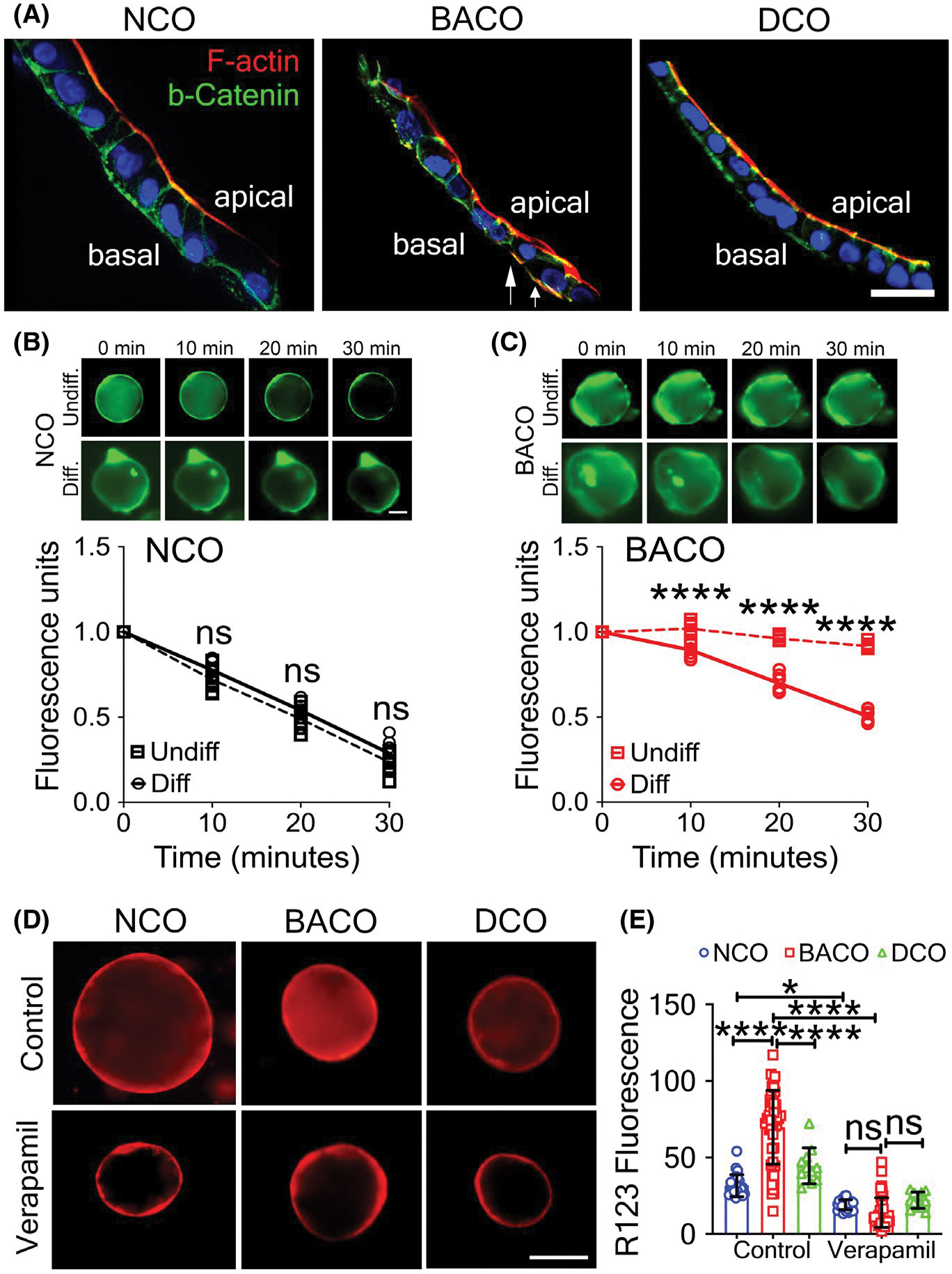
Cholangiocyte differentiation restores cell polarity and decreases epithelial permeability. (A) Immunofluorescence staining of COs identifies anatomically correct localization of F-actin and β-catenin in NCOs and DCOs, and significant correction of apicobasal polarity in BACOs following differentiation protocol, with aberrant expression limited to localized areas (white arrows; scale bar, 5 μm). (B) Measurement of CFTR activity shows similar fluorescent dye quenching by undifferentiated and differentiated NCOs. (C) BACOs cultured in differentiation medium quench the fluorescent dye by 30 min, whereas undifferentiated BACOs continue to retain the dye, indicating defective CFTR function. (D) Immunofluorescent images depict absorption and luminal uptake of the fluorescent substrate R123 into differentiated organoids from NCOs, BACOs, and DCOs without (top row) or with (lower row) verapamil to inhibit uptake (magnification, ×10). (E) Quantification of R123 luminal fluorescence of control and verapamil-treated organoids comparing within the same groups. NCO, n = 3; BACO, n = 4; and DCO, n = 2. Data are presented as mean ± SD. Statistical analysis was performed using an unpaired t test; ****p < 0.0001 and ***p < 0.001. Diff., differentiated; and Undiff., undifferentiated
DISCUSSION
Our strategy to derive organoids from liver biopsies of subjects with biliary atresia generated lumen-containing spheroids with a layer of epithelial cells displaying an expression signature of cholangiocyte-enriched genes. A detailed survey of these organoids identified cellular, molecular, and functional features that were unique to biliary atresia, such as fewer cholangiocyte-like cells expressing developmental (e.g., CK7) and functional (e.g., AQP1, CFTR) markers. They also had an abnormal pattern of expression of cell polarity proteins, had cilium that pointed laterally or outwardly, and formed an epithelial layer with anatomical and functional evidence of increased permeability. The restriction of these findings to biliary atresia (with disease controls behaving like normal controls) is in keeping with a potential role of a defective epithelial development and as chief elements among the pathogenic mechanisms of disease.
The abnormal expression of polarity proteins and the increased uptake of R123 were described previously in biliary atresia organoids,[13] with an unusual cellular aggregation and the inability to form lumen, which suggested a severe defect in epithelial development. Our findings reproduce the defect in polarity and the increase in epithelial permeability, but a detailed anatomical analysis of biliary organoids in our experiments clearly showed that spheroids had a continuous epithelial layer and a lumen. These findings support the population of biliary organoids by cholangiocyte-like cells with a less severe developmental (or maturational) defect. It is possible that the different biological outcomes are due to the use of hepatic EpCAMpos cells to generate organoids in Babu et al.[13]; such an approach requires the cells to undergo activation of molecular pathways that may not be readily available when the cells are removed from livers of children with biliary atresia. In contrast, our approach enabled the growth of organoids that emerged directly from liver tissues, thus naturally selecting for cells primed to develop into cholangiocyte-like cells. It is also possible that the more severe defect may have been present in biliary atresia organoids that emerged but could not undergo successful cryopreservation (39% of livers in our studies). Among these possibilities, we favor a biological scenario in which the bile duct epithelium in children with biliary atresia has a less severe defect in cell development that is compatible with organogenesis, but nevertheless represents an important susceptibility factor of disease. This is further supported by the ability of cholangiocyte-like cells to express markers of more differentiated cells and to form an epithelium with decreased epithelial permeability when our organoids were cultured in the presence of EGF and FGF2.
In addition to an abnormal expression of the intercellular tight junction protein ZO-1, we found other lines of evidence pointing to a significant defect in epithelial barrier function in biliary atresia organoids. Although the formation and expansion of spheroid-shaped organoids occurred similarly in biliary atresia and normal and disease controls, the monitoring of the expansion of spheroids by time-lapse microscopy showed frequent decompressions of organoids, suggesting a leakage of internal constituents, followed by an expansion of their dimension. Their ability to achieve final spheroid sizes comparable to normal and disease controls suggests that they ultimately established cell-cell contact and formed an epithelial continuum. However, the increased uptake of R123 independently of verapamil inhibition indicates that the epithelium does not build an efficient epithelial barrier function. Although speculative, it is possible that an increased permeability may allow bile acids to enter the subepithelial compartment and trigger a secondary inflammatory response that further injures the epithelial lining, thus setting in motion a series of cellular events that ultimately result in an obstruction of the bile duct lumen.
The delayed development of epithelial cells in biliary atresia organoid may depend on an intrinsic suppression of circuits regulating cholangiocyte differentiation. This is supported by the decreased expression of 16 EGF/FGF-related genes and by the improved differentiation of organoids in response to their treatment with EGF and FGF2.[32,33] When cultured in this enriched media, biliary atresia organoids increased the number of cells expressing HNF-1β, CK7, AQP-1, CFTR, SSTR2, and the number of ciliated cells. Furthermore, treatment with FGF2 and EGF partially rescued the expression of ZO-1 and improved the barrier function of the epithelium. Although the data reported herein do not directly dissect these molecular mechanisms, patient-derived organoids may prove to be powerful tools for this type of future mechanistic studies and to investigate disease pathogenesis.
In summary, the detailed molecular and functional phenotyping of organoids derived from liver biopsies revealed molecular and functional evidences of delayed epithelial development in biliary atresia. The existence of these findings in multicellular organoids completely independent from the surrounding complex cellular environment that is typical of the liver is in keeping with a primary defect in cholangiocyte maturation. These data support the proposal of a pathogenic model of disease in which a delayed development of cholangiocytes impairs the barrier function of the bile duct epithelium. The resultant increase in epithelial permeability renders the fetus or neonate susceptible to a more severe injury induced by an external (e.g., toxin, virus) or internal (e.g., hypoxemia) insult. In this setting, even a mild injury to cholangiocyte may facilitate the exposure of luminal bile acids to the subepithelial compartment and trigger an inflammatory response that expands the epithelial damage, facilitates luminal obstruction by cellular debris and immune cells, and progression to biliary atresia. The mechanistic studies focused on restoring EGF/FGF signaling point to the potential of a pharmacologic approach to promote cholangiocyte differentiation and strengthen the barrier function of the bile duct epithelium. In this regard, our ability to produce viable patient-specific organoid lines opens new opportunities to design and test therapeutic interventions to restore intercellular adhesion and block pathogenic mechanisms of bile duct injury.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Clinical Component and the Gene Analysis Core, the Integrative Morphology Core, and the Stem Cell/Organoid and Genome Editing Core of the Digestive Health Center at Cincinnati Children’s Hospital Medical Center.
Funding information
Supported by the National Institutes of Health (DK 64008, DK 83781, and DK 78392)
Abbreviations:
- AQP1
aquaporin 1
- BACO
biliary atresia cholangiocyte-like organoid
- CFTR
cystic fibrosis transmembrane conductance regulator
- CK7
cytokeratin 7
- CO
cholangiocyte-like organoid
- DCO
diseased cholangiocyte-like organoid
- EGFR
EGF receptor
- EHBD
extrahepatic bile duct
- EPCAM
epithelial cell adhesion molecule
- EZR
Ezrin
- F-actin
filamentous actin
- FU
fluorescence units
- HNF
hepatocyte nuclear factor
- MDR1
multidrug resistance 1
- MSN
Moesin
- NCO
normal cholangiocyte-like organoid
- ns
not significant
- PCA
principal component analysis
- R123
rhodhamine 123
- RDX
Radixin
- RNA-seq
RNA sequencing
- SSTR2
somatostatin receptor 2
- TEM
transmission electron microscopy
- ZO-1
zonula occludens 1
Footnotes
CONFLICT OF INTEREST
Nothing to report.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
All RNAseq data generated during this study are deposited in the GEO repository under the study ID: GSE186444. The data can be accessed at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186444.
DATA AVAILABILITY STATEMENT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors: Jorge A. Bezerra (Jorge.bezerra@cchmc.org) and Pranavkumar Shivakumar (pranav.shivakumar@cchmc.org).
REFERENCES
- 1.Hemberger M, Hanna CW, Dean W. Mechanisms of early placental development in mouse and humans. Nat Rev Genet. 2020;21:27–43. [DOI] [PubMed] [Google Scholar]
- 2.Bessho K, Bezerra JA. Biliary atresia: will blocking inflammation tame the disease? Annu Rev Med. 2011;62:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harpavat S, Garcia-Prats JA, Anaya C, Brandt ML, Lupo PJ, Finegold MJ, et al. Diagnostic yield of newborn screening for biliary atresia using direct or conjugated bilirubin measurements. JAMA. 2020;323:1141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz KB, Haber BH, Rosenthal P, Mack CL, Moore J, Bove K, et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology. 2013;58:1724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol. 2015;12:342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Shivakumar P, Kinder J, Way SS, Donnelly B, Mourya R, et al. Regulation of bile duct epithelial injury by hepatic CD71+ erythroid cells. JCI Insight. 2020;5:e135751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorent K, Gong W, Koo KA, Waisbourd-Zinman O, Karjoo S, Zhao X, et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci Transl Med. 2015;7:286ra267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X, Lorent K, Wilkins BJ, Marchione DM, Gillespie K, Waisbourd-Zinman O, et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology. 2016;64:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waisbourd-Zinman O, Koh H, Tsai S, Lavrut P-M, Dang C, Zhao X, et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology. 2016;64:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bezerra JA, Wells RG, Mack CL, Karpen SJ, Hoofnagle JH, Doo E, et al. Biliary atresia: clinical and research challenges for the twenty-first century. Hepatology. 2018;68:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omenetti A, Bass LM, Anders RA, Clemente MG, Francis H, D. Guy C, et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology. 2011;53:1246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang V, Cofer ZC, Cui S, Sapp V, Loomes KM, Matthews RP. Loss of a candidate biliary atresia susceptibility gene, add3a, causes biliary developmental defects in zebrafish. J Pediatr Gastroenterol Nutr. 2016;63:524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babu RO, Lui VCH, Chen Y, Yiu RSW, Ye Y, Niu B, et al. Beta-amyloid deposition around hepatic bile ducts is a novel pathobiological and diagnostic feature of biliary atresia. J Hepatol. 2020;73:1391–403. [DOI] [PubMed] [Google Scholar]
- 14.Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. 2016;11:1724–43. [DOI] [PubMed] [Google Scholar]
- 15.Luo Z, Shivakumar P, Mourya R, Gutta S, Bezerra JA. Gene expression signatures associated with survival times of pediatric patients with biliary atresia identify potential therapeutic agents. Gastroenterology. 2019;157:1138–52.e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33–dependent cholangiocyte proliferation. J Clin Investig. 2014;124:3241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiPaola F, Shivakumar P, Pfister J, Walters S, Sabla G, Bezerra JA. Identification of intramural epithelial networks linked to peribiliary glands that express progenitor cell markers and proliferate after injury in mice. Hepatology. 2013;58:1486–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rios AC, Clevers H. Imaging organoids: a bright future ahead. Nat Methods. 2018;15:24–6. [DOI] [PubMed] [Google Scholar]
- 19.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen M, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rimland CA, Tilson SG, Morell CM, Tomaz RA, Lu W-Y, Adams SE, et al. Regional differences in human biliary tissues and corresponding in vitro-derived organoids. Hepatology. 2021;73:247–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fouassier L, Duan C, Feranchak AP, Yun CHC, Sutherland E, Simon F, et al. Ezrin-radixin-moesin–binding phosphoprotein 50 is expressed at the apical membrane of rat liver epithelia. Hepatology. 2001;33:166–76. [DOI] [PubMed] [Google Scholar]
- 24.Wu F, Wu DI, Ren Y, Huang Y, Feng BO, Zhao N, et al. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J Hepatol. 2019;70:1145–58. [DOI] [PubMed] [Google Scholar]
- 25.Boj SF, Vonk AM, Statia M, Su J, Vries RRG, Beekman JM, et al. Forskolin-induced swelling in intestinal organoids: an in vitro assay for assessing drug response in cystic fibrosis patients. J Vis Exp. 2017;120:55159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitade M, Factor VM, Andersen JB, Tomokuni A, Kaji K, Akita H, et al. Specific fate decisions in adult hepatic progenitor cells driven by MET and EGFR signaling. Genes Dev. 2013;27:1706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33:853–61. [DOI] [PubMed] [Google Scholar]
- 28.Sampaziotis F, Cardoso de Brito M, Madrigal P, Bertero A, Saeb-Parsy K, Soares FAC, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33:845–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sampaziotis F, de Brito MC, Geti I, Bertero A, Hannan NR, Vallier L. Directed differentiation of human induced pluripotent stem cells into functional cholangiocyte-like cells. Nat Protoc. 2017;12:814–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sirica AE, Gainey TW. A new rat bile ductular epithelial cell culture model characterized by the appearance of polarized bile ducts in vitro. Hepatology. 1997;26:537–49. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Meng Z, Wang X, Zeng S, Huang W. The nuclear receptor CAR modulates alcohol-induced liver injury. Lab Invest. 2011;91:1136–45. [DOI] [PubMed] [Google Scholar]
- 32.Sampaziotis F, Justin AW, Tysoe OC, Sawiak S, Godfrey EM, Upponi SS, et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat Med. 2017;23:954–63. [DOI] [PubMed] [Google Scholar]
- 33.Tysoe OC, Justin AW, Brevini T, Chen SE, Mahbubani KT, Frank AK, et al. Isolation and propagation of primary human cholangiocyte organoids for the generation of bioengineered biliary tissue. Nat Protoc. 2019;14:1884–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors: Jorge A. Bezerra (Jorge.bezerra@cchmc.org) and Pranavkumar Shivakumar (pranav.shivakumar@cchmc.org).