

Abstract
Chimeric antigen receptor (CAR) T-cell based immunotherapy has shown its potential in treating blood cancers, and its application to solid tumors is currently being extensively investigated. For glioma brain tumors, various CAR T-cell targets include IL13Rα2, EGFRvIII, HER2, EphA2, GD2, B7-H3, and chlorotoxin. In this work, we are interested in developing a mathematical model of IL13Rα2 targeting CAR T-cells for treating glioma. We focus on extending the work of Kuznetsov et al. (1994) by considering binding of multiple CAR T-cells to a single glioma cell, and the dynamics of these multi-cellular conjugates. Our model more accurately describes experimentally observed CAR T-cell killing assay data than the models which do not consider multi-cellular conjugates. Moreover, we derive conditions in the CAR T-cell expansion rate that determines treatment success or failure. Finally, we show that our model captures distinct CAR T-cell killing dynamics from low to high antigen receptor densities in patient-derived brain tumor cells.
Keywords: Immunotherapy, Adoptive cell therapy, Chimeric antigen receptor T-cell, Mathematical Oncology, Dynamical system
1. Introduction
Adoptive cell-based immunotherapy has shown to be successful in treating patients with cancer. In particular, Chimeric Antigen Receptor T cell (CAR T-cell) therapy is one of the adoptive immunotherapies that has been successful in clinical and pre-clinical models, and has been FDA-approved since 2017 [1,2]. In this therapy, a patient or donor’s T cells are collected and genetically engineered to express a receptor specific to an antigen found on cancer cells, thus improving the ability of T-cells to eradicate the target cancer cells. Finally, these CAR T-cells are cultured to large numbers, then introduced back to the patient [3,4]. CAR T-cell therapy has shown its potential in blood cancer, and solid tumors [3,5]. However, the success of CAR T-cell therapy for solid tumors has been challenging due to difficulties in (1) trafficking CAR T-cells into solid tumors, (2) hostile tumor microenvironment that suppresses T cell activity, and (3) tumor antigen heterogeneity [5,6]. Since CAR T-cells mostly exist in bloodstream and lymphatic system, which makes CAR T-cell therapy a great weapon for hematological tumors (e.g. blood tumor cells), it may be hard for CAR T-cells to penetrate tumor tissue through the vascular endothelium. Moreover, various types of cells infiltrate solid tumors to support tumor growth and restrict the efficacy of CAR T-cell therapy [6].
One of the first mathematical models describing the interaction between immune cells and cancer cells is from Kuznetsov et al. (1994) [7], which is a dynamical system model with two populations, tumor cells and cytotoxic T cells, or T lymphocytes. The model can describe the formation of a tumor dormant state and evasion of the immune system. A subsequent model developed by Kirschner and Panetta (1998) [8] considered the cytokine interleukin-2 in addition to the dynamics between tumor cells and immune effector cells, and was able to model short-term tumor oscillations as well as long-term tumor relapses. In order to model persistent oscillations that were observed in immune systems, periodic treatment and time delay was added to the model in [9], and stability analysis of the model was done in [10]. Thereafter, the later built models were improved by adding new types of cells, such as natural killer cells, normal cells, and different kinds of cytokines [11,12]. These models not only capture tumor immune escape, but also explain multiple equilibrium phases of coexisting immune cells and cancer cells. Other than dynamical system models, spatial models that describe the spatial temporal interaction are developed as well. [13] develops a spatial temporal version of [7] using partial differential equations, [14] develops a hybrid cellular automata-partial differential equation model, and [15] develops a hybrid off-lattice agent-based and partial differential equation model.
The surge of clinical trials and the success of CAR T-cell therapy also drew a lot of interest in mathematical modeling of CAR T-cell therapy. This includes modeling CD19 CAR T-cell therapy targeting acute lymphoblastic leukemia in [16] as a dynamical system, which includes healthy B cell populations and circulating lymphocytes. Later study in [17] further showed relationships between CAR T-cell doses and diseases burden by the observed clinical data. In order to study cytokine release syndrome, which is one of the primary side effects of CAR T-cell therapy, another dynamical system of nine cytokines responding to CAR T-cell therapy was proposed in [18] as well. More recent work in [19] attempted to understand the dynamics of CAR T-cell therapy by considering not only tumor cells and CAR T-cells but also normal T cells. Meanwhile, the model introduced in [20] includes long-term memory CAR T-cells, which are produced by memory pool formation of effector CAR T-cells. The corresponding stability analysis of this model was done in [21].
Here we develop a mathematical model of glioma cells and CAR T-cells inspired by the experimental data provided in [22]. The experiments study the interaction between glioma cell lines, derived from glioblastoma patients undergoing tumor resections at City of Hope [23,24], and IL13Rα2 targeting CAR T-cells. As shown in Fig. 1, cells were co-cultured in vitro and images were taken under a light microscope over a 72 h period. The experimental images illustrate two aspects of the glioma cell killing of CAR T-cells. First, the elimination process of glioma by CAR T-cell is not immediate, and second, multiple CAR T-cells can interact with glioma cells. Such phenomena can be also observed in a close-up video in [25]. In subsequent experiments, the glioma cells and CAR T-cells are mixed at different ratios (CAR T-cell to glioma cell ratios of 1:5, 1:10, and 1:20), and glioma cells with different antigen receptor density levels (low, medium, and high) were tested. We remark that number of CAR T-cells binding to glioma cells will depend on the antigen receptor density levels since glioma cells with high antigen receptor density levels will have more capacity for CAR T-cells to bind. Throughout the course of experiment, real-time monitoring of glioma population size was performed by using xCELLigence cell analyzer system [26]. This system quantifies the glioma cell population with a dimensionless number referred to as cell-index (CI) (1 CI ≈ 104 cells [22]), where the population size is tracked every 15 min. See appendix Fig. C.1 for the full set of data.
Fig. 1.
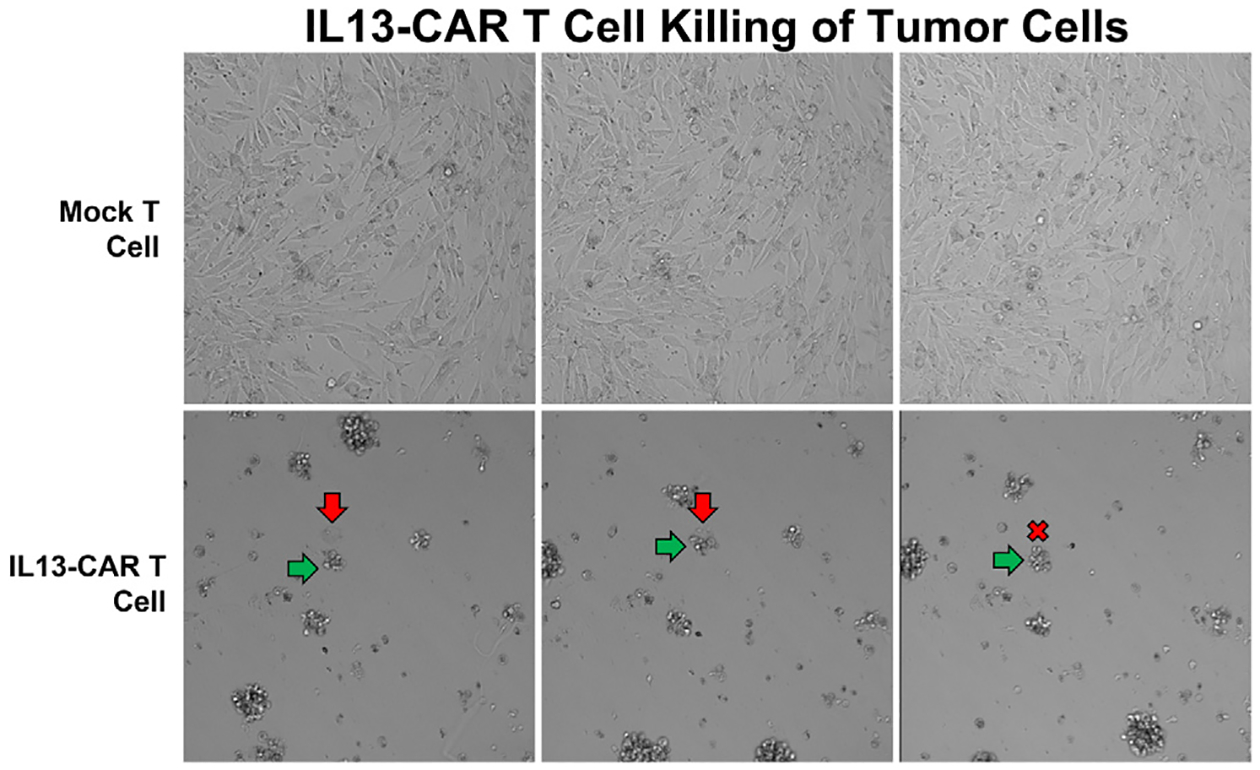
IL13-CAR T-cell killing of brain tumor cells. The first row corresponds to when glioma cells are mixed with mock-T cells, and the second row is when mixed with IL13-CAR T-cells. It shows the process of glioma cell (red arrow) being eliminated by the CAR T-cells (green arrow).
The rest of the paper is structured as follows. In Section 2, we describe the proposed model, where we assume that one glioma cell may interact with multiple CAR T-cells, and further come up with a system of ODE that describes not only the size of tumor cells and CAR T-cells, but also the size of multiple CAR T-cell binding conjugates. We denote the model with conjugates up to n CAR T-cells binding to glioma cell as the n-binding model. The stability analysis of one-binding slow reaction model is presented in Section 3, and we provide parameter conditions that guarantee either CAR T-cell treatment success or failure. In Section 4, we compare the accuracy between the slow reaction and fast reaction one-binding model, and show that the slow reaction model more accurately describes the experimental data. We also simulate the multiple binding slow reaction models from one- to five-binding conjugates, and study the hypotheses in reaction rates that captures the experimental result regarding low to high antigen receptor density levels of glioma cells. Summary of our findings and future work is discussed in Section 5.
2. Mathematical model
This section summarizes the mathematical models that we study in this work. The motivation of building these models comes from the experiments that show multiple CAR T-cells interacting with glioma cells [25] and non-instantaneous killing process that can take hours. We start by deriving the dynamical system model proposed in Kuznetsov et al. (1994) [7] that considers one CAR T-cell interacting with one glioma cell. The two versions of the model – slow and fast reaction models – are compared. Then we extend the models to consider the multi-cellular conjugate of multiple CAR T-cells and glioma cell.
2.1. One CAR T-cell bound to one glioma cell model
Among the mathematical models considering interaction of glioma cells and immune cells, one of the most recognized models is the model developed in Kuznetsov et al. (1994) [7], modeling the one to one binding of cancer and cytotoxic immune cells. The glioma cells are subject to be attacked by cytotoxic effector cells, e.g. cytotoxic T lymphocytes and natural killer cells. Here, we consider CAR T-cells instead of the general effector cells. The killing process of CAR T-cell has mainly 4 stages: binding, recognition, lethal hit, and target cell rounding [5,27]. More explicitly, tumor cells are first scanned by CAR T-cells, then antigens are recognized through proteins or glycans on the surface. After that, tumor cells are killed by CAR T-cells and cell conjugates lose adhesion and commit to cancer cells’ death. In our model, we describe the interaction between the glioma cancer cells C(t) and CAR T-cells T(t) by the kinetic scheme illustrated in Fig. 2 (top), where I1(t) is the conjugate of one CAR T-cell and one glioma cell. The kinetic parameter describes the rate that the glioma cell binds to the CAR T-cell, and is the rate that the conjugate detaches without damaging the cells. is the rate that the CAR T-cell and glioma cell interaction kills the glioma cell, and corresponds to the rate of reaction that the CAR T-cells become no longer active, for example, due to cell death or exhaustion [28]. Hence stands for the successful killing process, and and indicates that some stage in the killing process did not work properly. In addition to the interaction, the growth dynamics of the glioma cells and CAR T-cells are included in the model, which yields in the following system of differential equation.
| (1) |
Here, parameter ρ is the maximal growth rate of the glioma cells assuming logistic growth; parameter K is the maximal carrying capacity of the biological environment for the glioma cells; p is the rate that the CAR T-cells accumulate in the region due to the presence of the tumor, in other words, CAR T-cell expansion rate; g is a concentration of glioma cells that halves the maximum rate p; θ represents the death rate of the CAR T-cells. Lysed cancer cells and CAR T-cells will be formed at the end of this interaction, but since their dynamics are simply decaying, we do not include those equations in our model as in [7]. We reparameterize the equation as
| (2) |
where
The new parameter a represents the rate of net inflow of CAR T-cells from the conjugate I1, β represents the inflow rate for glioma cells, and γ stands for the net decay rate of the conjugate I1 due to either detachment or lysed reaction. We will refer to Eqs. (1) or (2) as the slow reaction model, since following the dynamics of the conjugate I1 allows the interaction to be in a comparable time scale as the other dynamics, in contrast to the assumption that will be made in the following model.
Fig. 2.
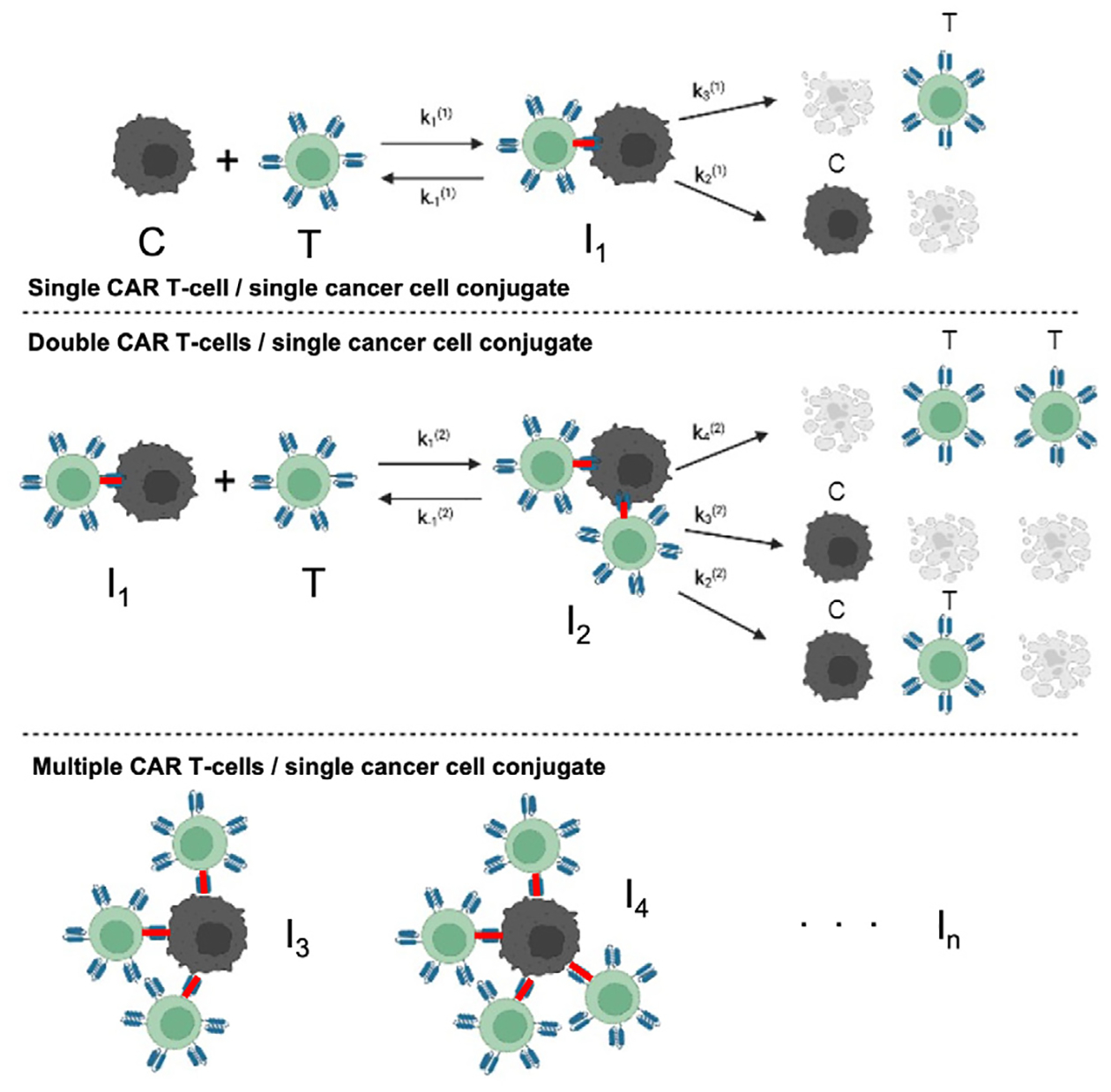
Kinetic interaction between glioma cancer cells (C) and CAR T-cells (T), assuming single CAR T-cell and single cancer cell conjugates I1 (top), double CAR T-cell and cancer cell conjugates I2 (middle), and multiple CAR T-cells and cancer cell conjugates In (bottom).
2.1.1. One-to-one binding model with fast reaction
The model proposed in [7] further reduces Eq. (1) by a separation of time scale between the dynamics of glioma cells and CAR T-cells compared to the dynamics of the conjugates I1. Assuming that the dynamics of conjugate I1 reaches equilibrium quickly, we can take İ1 = 0, and we have , that is,
Then, Eq. (2) reduces to the Kuznetsov et al. (1994) model as
| (3) |
where we define
| (4) |
We will refer to this model as the fast reaction model of one CAR T-cell binding case, as opposed to the slow reaction model in Eq. (2). The fast reaction scenario can happen, for example, when the glioma cell is mixed with a large number of CAR T-cells or the receptor density levels of glioma cells are high, which may lead to a fast elimination of glioma cells.
2.2. Two CAR T-cells bound to one glioma cell model
The following system, building up on the one-binding models in Eqs. (2) and (3), will govern the population dynamics of the scenario where up to two CAR T-cells can interact and bind to one glioma cell. As in the diagram of Fig. 2 (middle), conjugate I1 of one CAR T-cell and one glioma cell can interact with another CAR T-cell (T) and compose conjugate I2 of two CAR T-cells and one glioma cell. The kinetic interaction rates are defined similarly as before, where and will describe the binding and detachment rate of cells without damage, and (i=2,3,4) will be the rates of the interaction events. The system of equations can be written as follows.
| (5) |
This model follows the dynamics of I1 and I2 conjugates, so we will refer to this as a two-to-one or two binding slow reaction model. This model describes the scenario that if one CAR T-cell cannot kill a glioma cell immediately, then another CAR T-cell may come to react with the conjugate I1 formed by previous reaction.
2.2.1. Two-to-one binding model with fast reaction
Using a similar approach when we derived the Kuznetsov et al. (1994) model, the two-binding slow reaction system can be reduced to the following two-binding fast reaction system.
| (6) |
Note that we added the assumption that the detach rate is small, that is, , for simplicity. Derivation of model details are included in Appendix A.
2.3. Multiple CAR T-cells bound to one glioma cell model
Building up on the two-binding model, we can further extend the model to n numbers of CAR T-cells binding to one glioma cell. We denote Ij as the conjugate of j CAR T-cells and one glioma cell. The notation denotes the rate of different reactions, where the superscript (n) denotes the n-binding and the subscript i denotes the ith reaction in the n-binding reaction. The governing equations of the n-binding model that includes the conjugates I1,…, In can be written as follows.
| (7) |
where , , and . The parameters and their biological meanings are summarized in Table 1.
Table 1.
Model parameters and their biological interpretation.
| Parameter | Biological meaning |
|---|---|
| ρ | Proliferation rate of glioma cells |
| K | Carrying capacity of glioma cells |
| p | Rate of CAR T-cell expansion induced by glioma |
| θ | Death rate of CAR T-cells |
| g | Steepness coefficient of CAR T-cell expansion |
| Binding rate of CAR T-cell and conjugate In−1 | |
| Detaching rate of CAR T-cell and conjugate In | |
| Death rate of glioma cells from conjugate In | |
| Death rate of CAR T-cells from conjugate In |
In the following sections, we will test various hypotheses on the reaction rates to reproduce the phenomena of saturation of CAR T-cell therapy efficacy when the antigen receptor density of cancer increases.
3. Stability analysis
In order to better understand the dynamics of CAR T-cell therapy with the slow and fast reaction models, we present stability analysis of the one-to-one binding model.
3.1. One-to-one binding slow reaction model
In this section, we study the stability of one-binding slow reaction model (2). There are four steady states (T, C, I1) in the slow reaction model
where
and
Note that we order the points as C1 < C2, so that C1 is the state with a smaller cancer size. Among these four states, we are interested in (0, K, 0) and , which represent CAR T-cell treatment failure and success, respectively. The steady state (0, 0, 0) is always an unstable saddle point (See Appendix B), thus it is not of our interest. We aim to find the parameter conditions that yield these two states, especially focusing on the CAR T-cell expansion rate induced by the presence of cancer, p.
3.1.1. CAR T-cell treatment failure
One of the steady states is (0, K, 0) that represents the tumor reaching its maximal capacity, or in other words, the CAR T-cell treatment failure. This steady state becomes stable if
| (8) |
or using the kinetic reaction parameters,
| (9) |
This condition implies that if the engineered CAR T-cell expansion rate p is less than , the tumor reaches its maximum capacity; meanwhile, if the engineered CAR T-cells can expand enough so that , then the CAR T-cell therapy will prevent the cancer from growing to its maximum capacity. Therefore, this condition provides a minimal level of CAR T-cell expansion rate which can prevent cancer from growing to its maximal size.
Let us further examine the condition in Eqs. (8) and (9). Observe that as the reaction rate increases, or equivalently, α/γ becomes closer to 1, the right-hand side of the inequality decreases. Thus, if the killing of cancer becomes more effective, the minimum expansion rate of the CAR T-cell required to prevent treatment failure becomes smaller. It means that a CAR T-cell with a high cancer killing rate can prevent the tumor from growing to its maximum size despite a relatively low expansion rate. On the other hand, as increases, or α/γ gets closer to 0, the right-hand-side of the inequality increases. Thus if the inactivation rate of the CAR T-cells from interaction increases, CAR T-cells will need a higher expansion rate to successfully treat the tumor. As for the parameter K, which stands for the capacity of cancer cells, the right-hand-side is an increasing function with respect to K on the range of . This means that a tumor with a larger capacity will need a larger value of expansion rate p to prevent CAR T-cell treatment failure.
3.1.2. Potential CAR T-cell treatment success
Another steady state of our interest is the CAR T-cell treatment success state, . Then, the tumor is reduced to size C1 compared to reaching its maximal capacity K. For this equilibrium point to exist, the following condition needs to be satisfied,
| (10) |
or
| (11) |
This condition provides a minimum level of CAR T-cell expansion rate that makes it possible to avoid treatment failure that results in tumor growing to its maximal capacity, although the treatment success will depend on other parameters and initial CAR T-cell dosage. When is large, the term will be closer to 1, and our lower bound for p will approach . This implies that with a less effective cancer killing rate, the CAR T-cell requires a higher expansion rate to achieve potential treatment success. Also if the reaction rate has a large value, our lower bound for p will decrease to θ. It follows that the interaction requires a lower expansion rate to achieve a potential treatment success with a more effective cancer killing rate. Detailed analysis can be found in Appendix B, and further discussion on the stable nonzero CAR T-cell population is in Section 5.
3.2. One-to-one binding fast reaction model
The stability analysis of the one-to-one binding fast reaction model Eq. (3) is comparable to the slow reaction model. The analysis can be found in many literature including [7,29]. Here, we briefly summarize it to compare it with the slow reaction model. Identical to the slow reaction model, there are four possible steady states (T, C),
where
The steady state that the tumor growing to its maximum capacity, (T, C) = (0, K), becomes stable if
| (12) |
This condition is comparable to Eq. (8), which provides the condition that the CAR T-cell expansion rate prevents the cancer from growing to its maximum. The other condition related to the equilibrium point (T1, C1), the CAR T-cell therapy success case, is
| (13) |
comparable to Eq. (10). The CAR T-cell needs to expand at least this level to have chances for successful CAR T-cell treatment. We remark that the other equilibrium points (0, 0) and (T2, C2) are always saddles, so that they are not of our interest.
4. Numerical study of glioma cells and CAR T-cells interaction
Our experimental data consist of multiple doses of CAR T-cells and multiple types of glioma cells based on the antigen receptor densities. Specifically, we look into the experiment data using IL13BB CAR T-cells [22]. The data includes glioma cells measured in a unitless cell index (CI) with low, medium, and high antigen receptor densities, where the latter is likely to have a better response to CAR T-cell treatment, although not strictly better. For each density level, the experiments were initialized with different mixture ratios between CAR T-cells and glioma cells as 1:5, 1:10, and 1:20 (See Fig. C.1). The number of initial CAR T-cells are determined to be proportional to the initial glioma cell number. For instance, for a 1:5 mixture ratio, the glioma cells are mixed with 0.2C0 (CI) = 0.2C0 · 104 (cells) CAR T-cells, where C0 is the initial glioma size in CI. Using this rich dataset, we numerically study the proposed multiple CAR T-cell binding model to find a better model that describes the experimental data.
To numerically solve the models, we use the built-in ODE solver (ode23) in MATLAB, which is based on an explicit Runge–Kutta pair of Bogacki and Shampine [30,31]. The calibration of our model is done by minimizing the sum of squares norm of the error between the data and our model fit. To improve the accuracy and parameter identifiability of the calibration, we calibrate the parameters sequentially, first using the tumor growth data without treatment, then using the data with treatment. Using the no treatment data, we first calibrate the tumor growth parameter ρ with fixed K in Ċ = ρC (1−C/K). We obtain three sets of values of ρ and K given three different types of tumor considering antigen receptor densities – low, medium, and high. Afterward, we calibrate the remaining parameters for each density level while parameters ρ, K, and θ are fixed. See Tables in Appendix C for the full set of parameters. We comment that the calibrated values of CAR T-cell expansion rate p change more than a magnitude when mixed with low to high antigen density glioma, where varying levels of T-cell expansion depending on the level of presented antigens has been observed [32]. In the remaining section, we show that the slow reaction model Eq. (2) describes the data more accurately compared to the fast reaction model Eq. (3). Then, we compare different assumptions in the reaction rates to investigate the saturation of CAR T-cell treatment efficacy regarding the antigen density levels.
4.1. Comparison between fast reaction and slow reaction models
The fast reaction model, for example, Eq. (3) does not describe the dynamics of the conjugates Ij, assuming that they reach the equilibrium state immediately. However, in reality, the reaction is not always fast enough as it takes time for the CAR T-cells to detect and infiltrate the glioma cells, which is also depicted in the experimental data [22]. Therefore, we expect the slow reaction model (2) to describe the data more accurately. Our hypothesis is confirmed in Fig. 5, where we compare the accuracy of the slow reaction model Eq. (2) and the fast reaction model Eq. (3). The error is computed as the sum of squares differences between the data and the calibrated model fit. In these plots, the errors from the three sets of data with the same receptor density (low and high) and CAR T-cell ratio (1:5, 1:10, 1:20) are marked as black crosses, and the bar shows the average of the errors. We observe that the slow reaction model results in significantly smaller errors for all cases. Let us comment on each dataset more closely.
Fig. 5.
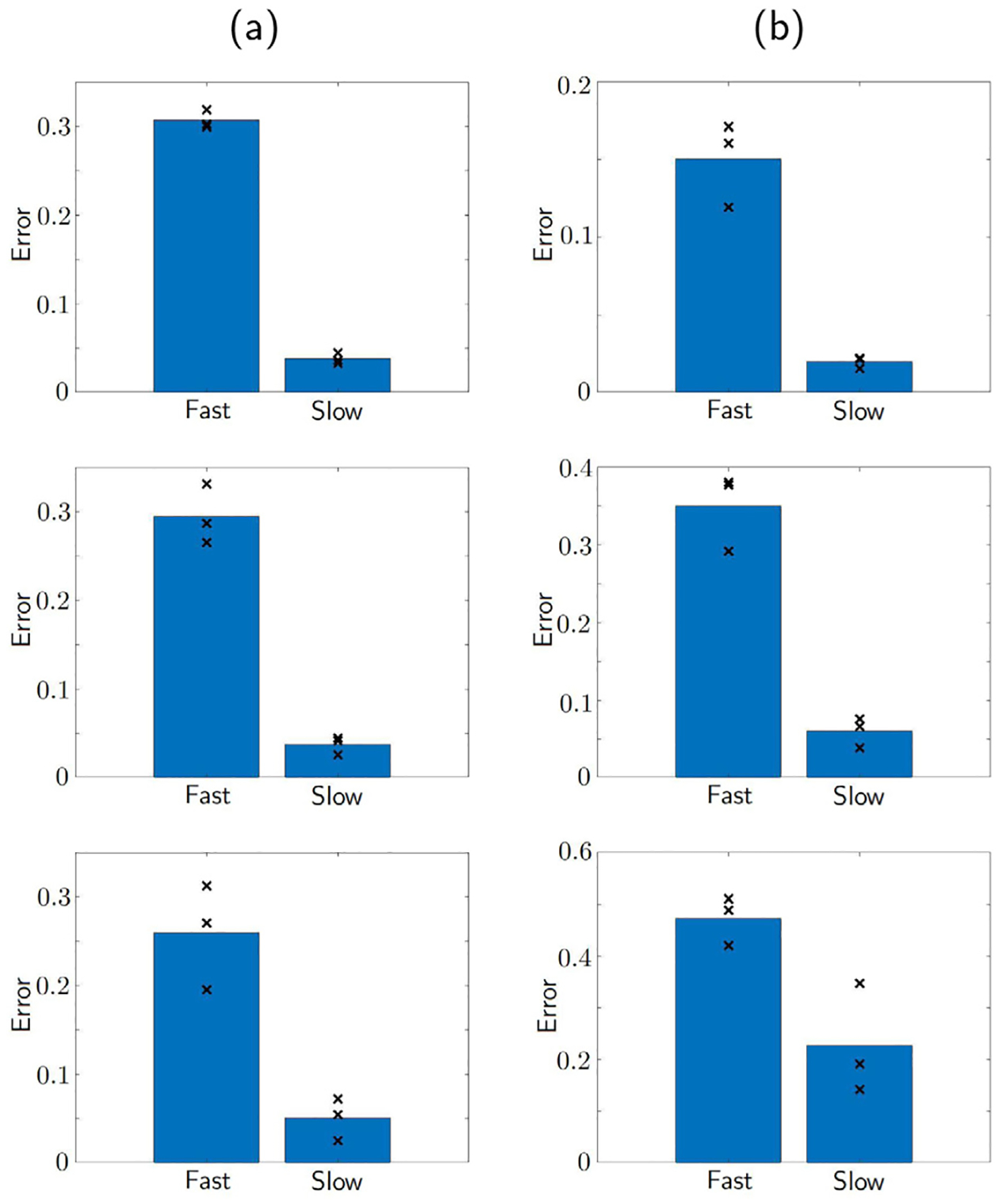
Comparison of model fit errors between the fast reaction model Eq. (3) and the slow reaction model Eq. (2). The shown bar plots are the average of the three marked errors (×) for fitting the data of low antigen receptor density glioma (a) and high antigen receptor density glioma (b), with CAR T-cell to cancer ratio 1:5 (top), 1:10 (middle), and 1:20 (bottom). It can be seen that slow reaction model has smaller errors in all cases.
Fig. 3 compares the accuracy of slow binding and fast reaction models calibrated to the low receptor density glioma data. We observe that the slow reaction model does have a better fitting than the fast reaction model. The top row shows the case of initial ratio 1:5, where we can see that the data points are closer to the calibrated model curve especially near t = 0.5 and t = 1.5 using the slow reaction model. Similarly in the second row that corresponds to the 1:10 ratio, the fitting of the slow reaction model is significantly better at capturing the saturating tail at the later time points after t = 3. The last row shows the result of initial ratio 1:20, which is the case with the smallest CAR T-cell dosage. Again, the slow reaction model better describes the non-monotonic data. In all three cases of low antigen receptor density glioma experiments, we observe that the slow reaction model is more accurate, especially when the concavity is nonzero. The reason behind this result is that the reaction speed is slower when the antigen receptor density is low, which makes the slow reaction model more appropriate than the fast reaction model.
Fig. 3.
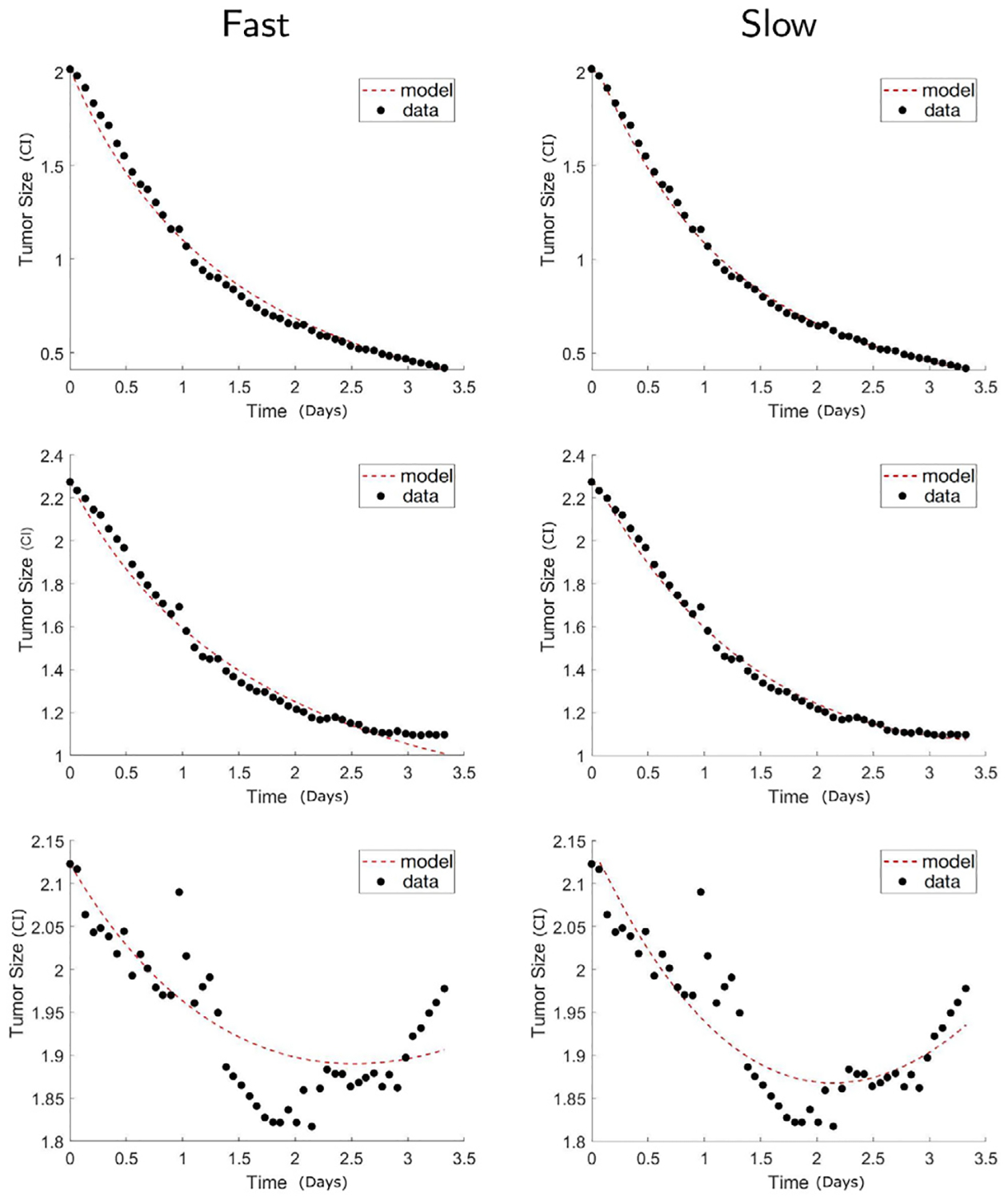
Low IL13Rα2 antigen density glioma. Comparison of the calibrated model fits between the fast reaction model Eq. (3) (Fast) and the slow reaction model Eq. (2) (Slow). The shown results are for low receptor density cancer with CAR T-cell to cancer ratio 1:5 (top), 1:10 (middle), and 1:20 (bottom). The calibrated model fits using the slow reaction model are closer to the data points, demonstrating that the slow reaction model more accurately describes the experimental data than the fast reaction model in the case of low receptor density.
On the other hand, we hypothesize that the fast reaction model could be more appropriate to fit the experiments with high antigen receptor density glioma. Since the high receptor density glioma cells have more receptors for the CAR T-cells to bind, the reactions can be more efficient. We observe that from the data plotted in Fig. 4 that the tumor size decay faster compared to the low receptor density case. This is the case especially when the glioma cells are mixed with a large number of CAR T-cells, that is, the 1:5 ratio case among our experiments. The top row of Fig. 4 shows the 1:5 ratio mixture, where the CAR T-cells eliminate the glioma cells most rapidly. The model fits using the slow and fast reaction models both look accurate, and we confirm that this is the case that the fast reaction model can accurately capture the data. No big difference can be seen between the slow and fast reaction model fits, although the error is smaller using the slow reaction model (see Fig. 5(b)). Nonetheless, in case of lower dosages of CAR T-cells, for instance, 1:20 mixture, the slow reaction model improves the accuracy by more than 75%, being able to capture the initial bump of the data. Finally, Akaike information criterion (AIC) [33,34] that quantifies the quality of model fitness is computed in Table 2. The AIC values of slow reaction model is smaller than the fast reaction model across all datasets thus confirms the better model fitness of the slow reaction model.
Fig. 4.
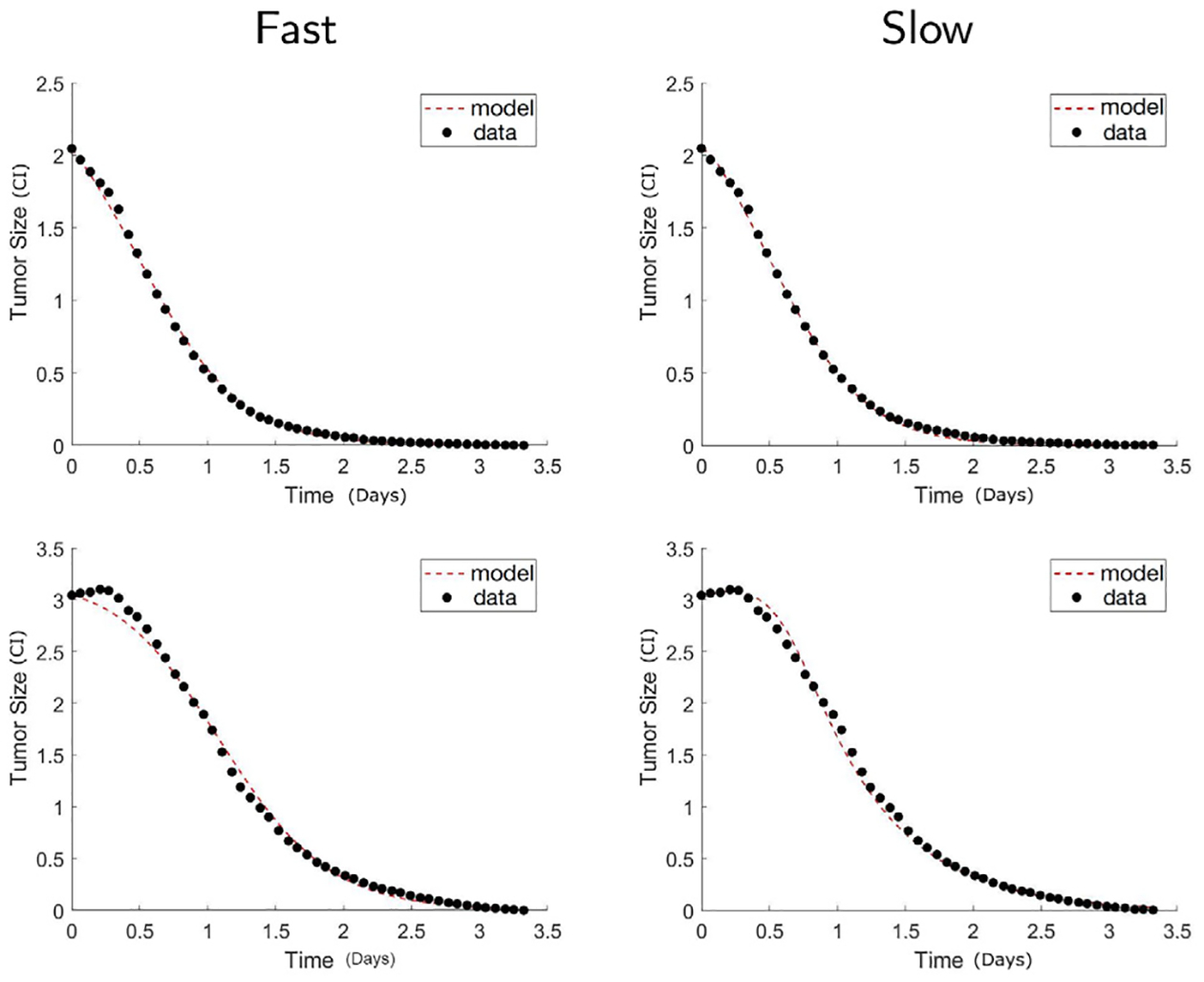
High antigen receptor density glioma. Comparison of calibrated model between the fast reaction model Eq. (3) (Fast) and the slow reaction model Eq. (2) (Slow). The shown results are for high receptor density cancer with CAR T-cell to cancer ratio 1:5 (top), 1:20 (bottom). The calibrated model fits between the two models are similarly good in the case of 1:5 ratio, that is the case of higher dosage. Since the high receptor density glioma cells with high dosage of CAR T-cells is the case that the reaction can be the most efficient, the fast reaction model is not significantly better when it comes to this case.
Table 2.
Comparison of model fitness using Akaike information criterion (AIC) [33,34] between fast reaction model Eq. (3) (Fast) and the slow reaction model Eq. (2) (Slow). Mean of AIC values among all datasets and in the parenthesis, minimal and maximal values are shown. The slow reaction model show better model fitness with lower values of AIC across all datasets.
| Low | Medium | High | |
|---|---|---|---|
| Fast | −300.0 (−337,−285) |
−346.9 (−378,−304) |
−298.1 (−385,−242) |
| Slow | −495.8 (−540,−438) |
−455.8 (−590,−340) |
−453.0 (−588,−284) |
We conclude that the slow reaction model is more appropriate to be considered in general, since the model fit is more accurate compared to the fast reaction model, and moreover, it agrees with the experimental observation that showed the reaction between the glioma cells and the CAR T-cells not being immediate. This happens especially when the glioma cells have low antigen receptor density and CAR T-cell numbers are small. In addition to the model fit comparison, we argue that the slow reaction model can either match the fast reaction model or deviate from it based on the parameter values. We remark that the slow reaction model has four interaction parameters that defines two parameters of fast reaction model, ℓ and m, presented in Eq. (4). Therefore there are multiple sets of parameters of slow reaction model that reduces to an identical fast reaction model. Fig. 6 shows such an example. The bottom figures are computed from two distinct parameter sets of the slow reaction model that yields identical ℓ and m values, ℓ = 2.785 and m = 0.0223, for the fast reaction model. Large values of indicate fast reaction, for example, when , the I1 conjugate dynamics are trivial and the slow reaction model agrees the fast reaction model. However, when , we observe the I1 conjugates number increasing and the slow reaction model show distinct result from the fast reaction model. While the fast reaction model shows the number of glioma cells declining, the slow reaction model with a different choice of changes the glioma cell dynamics from decreasing to increasing. This example shows the richer dynamics that the slow reaction model contains. However, the slow reaction model has a drawback that more uncertainty is present in the estimated parameters due to its larger number of parameters, thus interpretation of the fitted parameters needs to be done carefully, and it is our future work to study parameter identifiability in the proposed model when additional data of CAR T-cells and conjugates Ii are available.
Fig. 6.
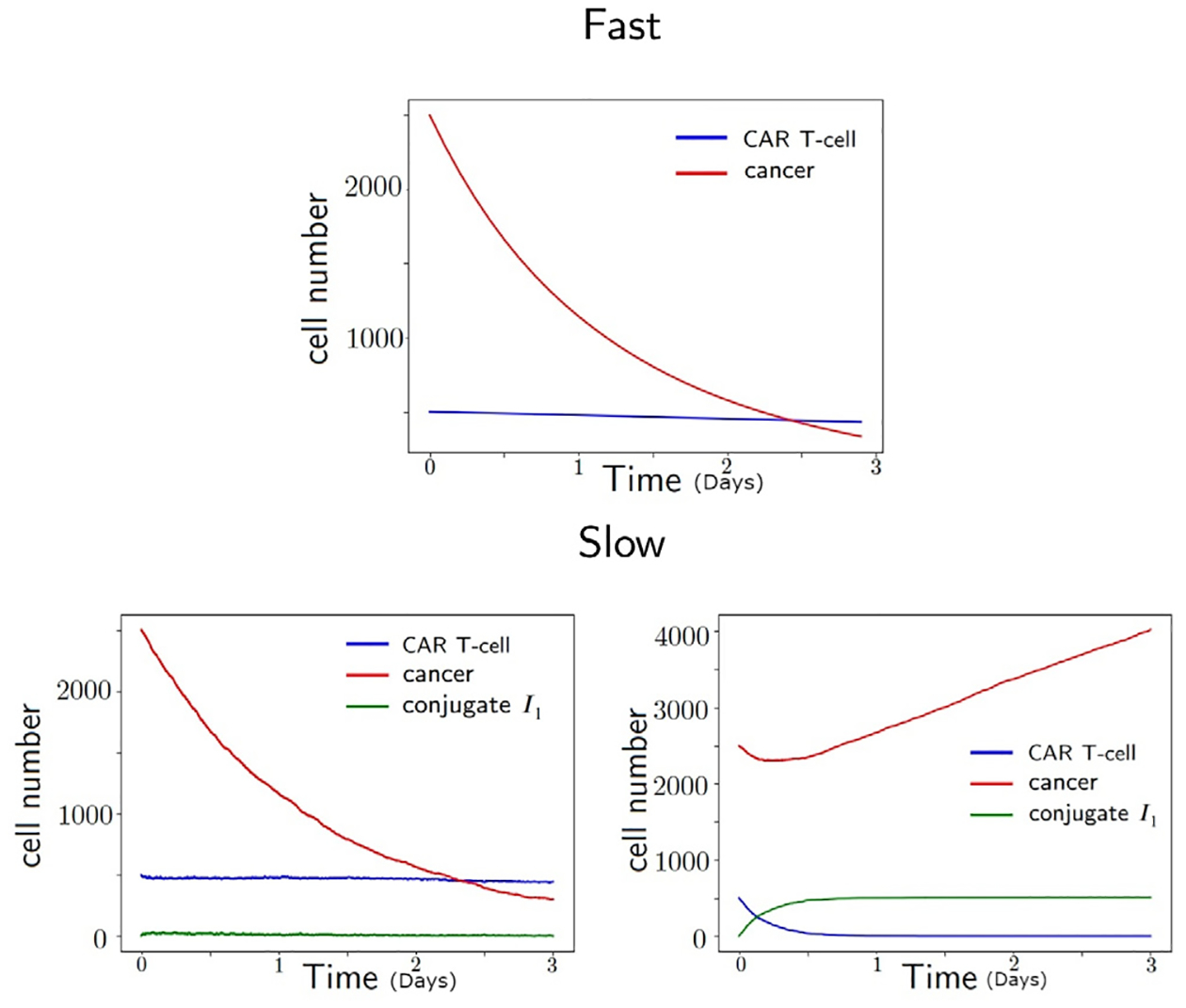
Dynamics of cancer, CAR T-cell, and conjugate I1 using two parameter sets of slow reaction model (bottom) that reduces to the same fast reaction model (top) parameter set. The unit of time is in days. When the cancer killing rate is large as (bottom, left), the dynamics of slow and fast reaction models agree. However, when the cancer killing rate is small as (bottom, right), the conjugate I1 have non-trivial dynamics and the dynamics of slow reaction model differs from the fast reaction model.
4.2. Simulation of multiple-binding slow reaction models
Having compared the slow and fast reaction models in the previous section, we now study the multiple CAR T-cells binding model Eq. (7). In particular, we focus on the experimental results of glioma cells with different levels of antigen receptor density: low, medium, and high. When mixed with the same number of CAR T-cells, glioma cells with low antigen receptor density respond less than the glioma cells with medium antigen receptor density. In other words, the decay rate of glioma cell is positively correlated with the antigen receptor density in general. However, an interesting observation was made in [22] that the effectiveness CAR T-cell therapy saturated after a certain receptor density level. As shown in Fig. 7, the high receptor density cells did not respond better than the medium receptor density glioma cells, or responded rather worse. We aim to study these phenomena using the multiple CAR T-cells binding models Eq. (7). Assuming that the antigen receptor density levels of glioma cells determines the number of CAR T-cells that can bind to a single glioma cell, we associate the one CAR T-cell binding model (n = 1) to the low density receptor glioma, and multiple CAR T-cells binding model up to the five binding (n = 5) to higher density receptor levels of glioma. One problem we face for the multiple CAR T-cells binding models is that the number of interaction parameters increases as the number of binding increases. Since we do not have data for the dynamics of any conjugates Ij for j = 1, …, n, it is difficult to estimate parameters from the data. Therefore, using the parameter set estimated for the one-binding model, we test the following two different hypotheses describing the relationships between the reaction rates for the multiple binding models with more than one binding.
Fig. 7.
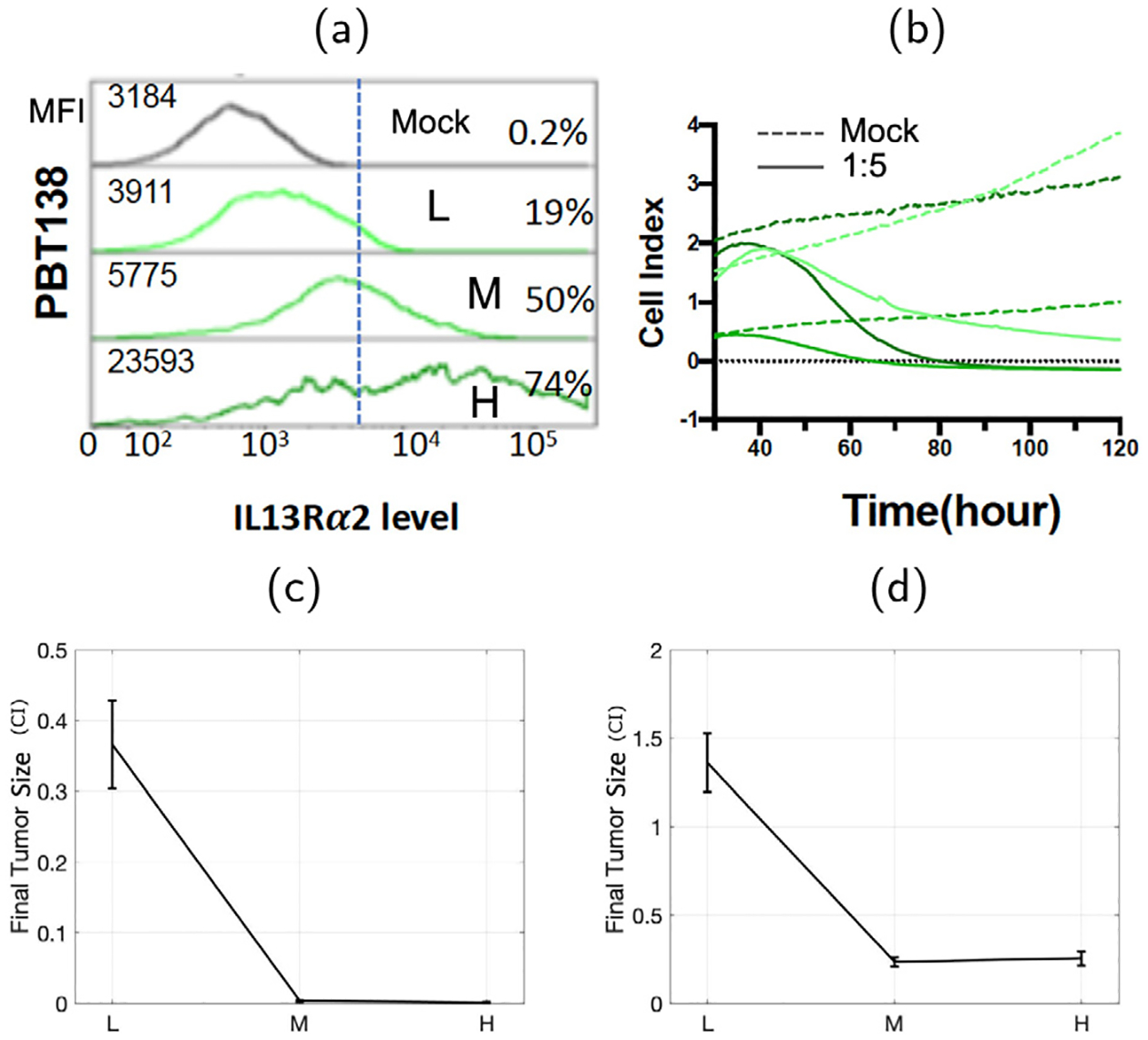
Antigen expression measured with flow cytometry (mean florescence intensity MFI, and the percentage of cells positive) for cell line PBT138 (mock, low (L), medium (M), high (H)) reported in Sahoo et al. [22] (a). Dynamics of the size of glioma cell population measured by xCELLigence (b). Glioma tumor size data (CI) after mixed with CAR T-cells in 1:5 ratio (c) and 1:10 ratio (d) in terms of antigen receptor density level of glioma cells: low (L), medium (M), and high (H) are shown as well.
Hypothesis 1: Assume that the reaction rates are uniform across conjugates Ij.
- The CAR T-cell attaches to the glioma cell or the conjugate Ij with an identical rate, i.e.,
(14) - The reactions that the glioma cell dies from the conjugate Ij have the same rates across number of bindings, i.e.,
(15) -
The reactions that glioma cells’ survive also have the same reaction rates with equal chances of CAR T-cells dying i.e,
so that .(16) Note that denotes the number of cases when j − 1 CAR T-cells die after the detachment of In, while 2n2−6n+7 denotes the number of all cases when at least one CAR T-cell dies (in other words, glioma cell survives) from the detachment of In.
Hypothesis 2: Instead of assuming that the reaction rates are uniform across the conjugates Ij, in this hypothesis, we assume that the reaction rates are non-uniform, either increasing or decreasing, and saturate after a certain number of CAR T-cells bind.
- The reaction rates of a new CAR T-cell attaching to the conjugate Ij decrease geometrically, i.e.,
for some positive integer M ≥ 1.(17) -
The glioma cells’ death rate increases geometrically as the number of bindings increases, but saturates after three CAR T-cells binding, i.e.,
for some positive integer L ≥ 1.(18) We observe that the parameter L directly leads to efficiency of cancer killing rate. With a bigger L, glioma cells’ death rate gets bigger for the first 3 bindings, which is more likely to bring a successful treatment. To estimate the parameter L directly from experiments, it can be determined by comparing the glioma cells’ death rates between n +1-binding interaction and n-binding interaction.
The reactions that glioma cells’ survive are distributed as in hypothesis 1, Eq. (16).
We compare the two hypotheses to find which assumption yields the experimental data we have considering different levels of antigen receptor densities in glioma cells. In particular, we compare the final tumor size after 3 days of CAR T-cell treatment, i.e. C (t) at t = 3. In the top row of Fig. 8, we show the results testing hypothesis 1. The results do not match what we observe in the experiments, as the final tumor size increases from one-binding to five-binding models. We presume that this is due to the relative reaction rate of cancer death decreasing as the number of reaction increases. Hence, by simply considering uniform reaction rates for one to multiple CAR T-cell conjugates as in hypothesis 1, we cannot recover the dynamics of glioma cells responding better in higher antigen receptor density levels.
Fig. 8.
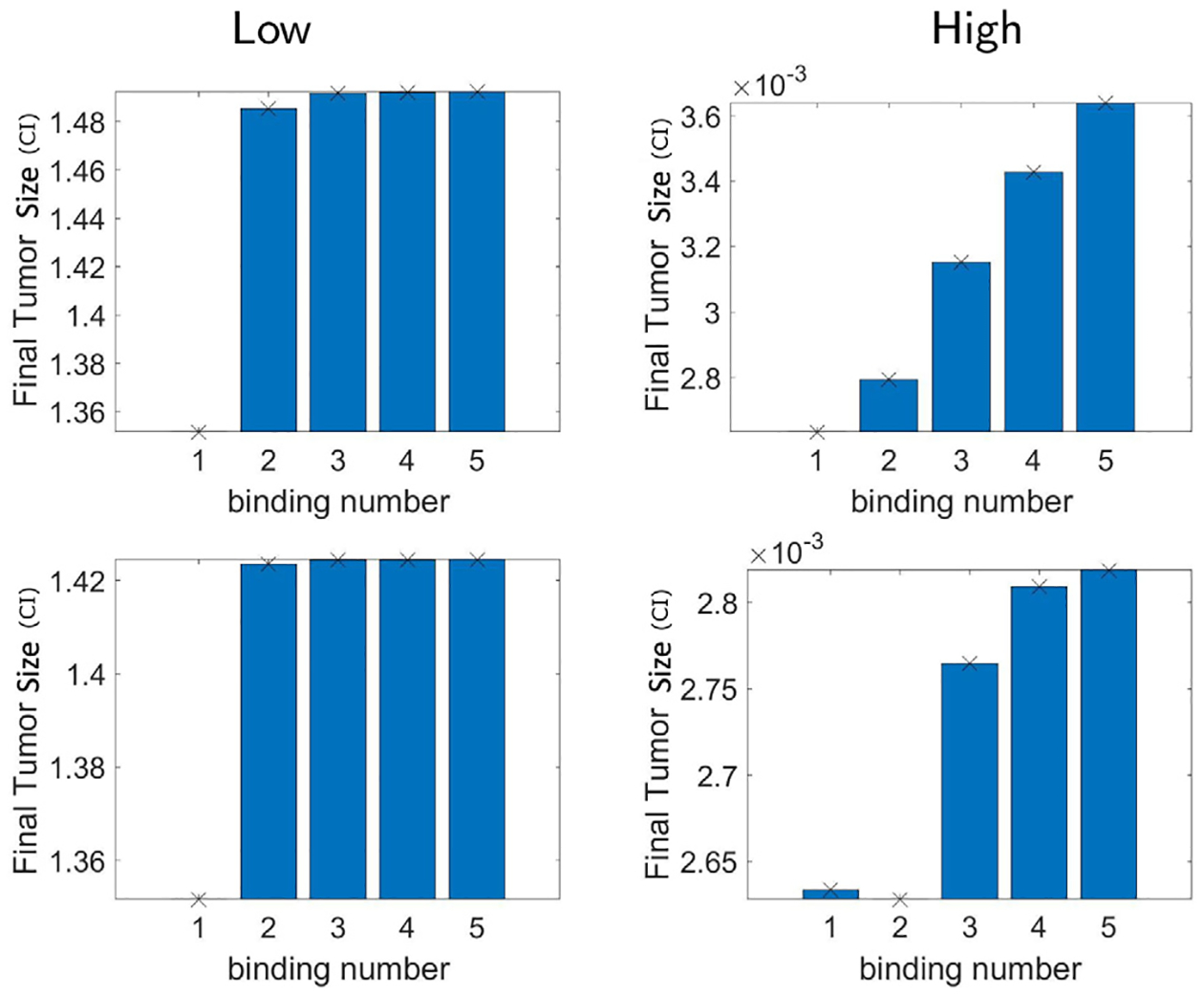
Final tumor size (CI) using the n-binding model Eq. (7) assuming up to n number of CAR T-cells binding to glioma cell forming the conjugates I1, …, In. We consider n = 1,2,…,5. The kinetic rate parameters of the multiple CAR T-cells binding model are taken by hypothesis 1 (top) and hypothesis 2 with M = 2, L = 1 (bottom). In fact, the tumor size does not decay, but rather increases as the number of binding n increases, in both low and high antigen receptor density cases.
Nevertheless, results from hypothesis 2 show distinctive outcomes from hypothesis 1. The simulation results taking different values of M and L are shown in the bottom row of Figs. 8 and 9. In particular, Fig. 9 shows the result of M = 1, L = 2, and M = 2, L = 2. It turns out that we no longer have the increase of tumor size as n increases, instead, we have a decrease for both low and high densities. We can further observe that the reaction saturates after the numbers of binding become more than three, as the final tumor size remains very small eventually. However, if we consider M = 2, L = 1, the tumor size increases as n increases, and the results again deviate from the experimental data as shown in the bottom row of Fig. 8. Thus, we conclude that hypothesis 2 on the reaction rates with L > 1 agrees with the experimental data shown in Fig. 7 (c,d) regarding the decay and saturation of tumor size with respect to increasing antigen density receptor levels. It makes sense that L is the critical parameter for this result, since it is the parameter that makes the death rate of the glioma cells increase as n increases. In addition, parameter sensitivity analysis [35] is included in Appendix D that shows larger Pearson correlation coefficient of L compared to M to the final tumor volume despite various levels of magnitude.
Fig. 9.
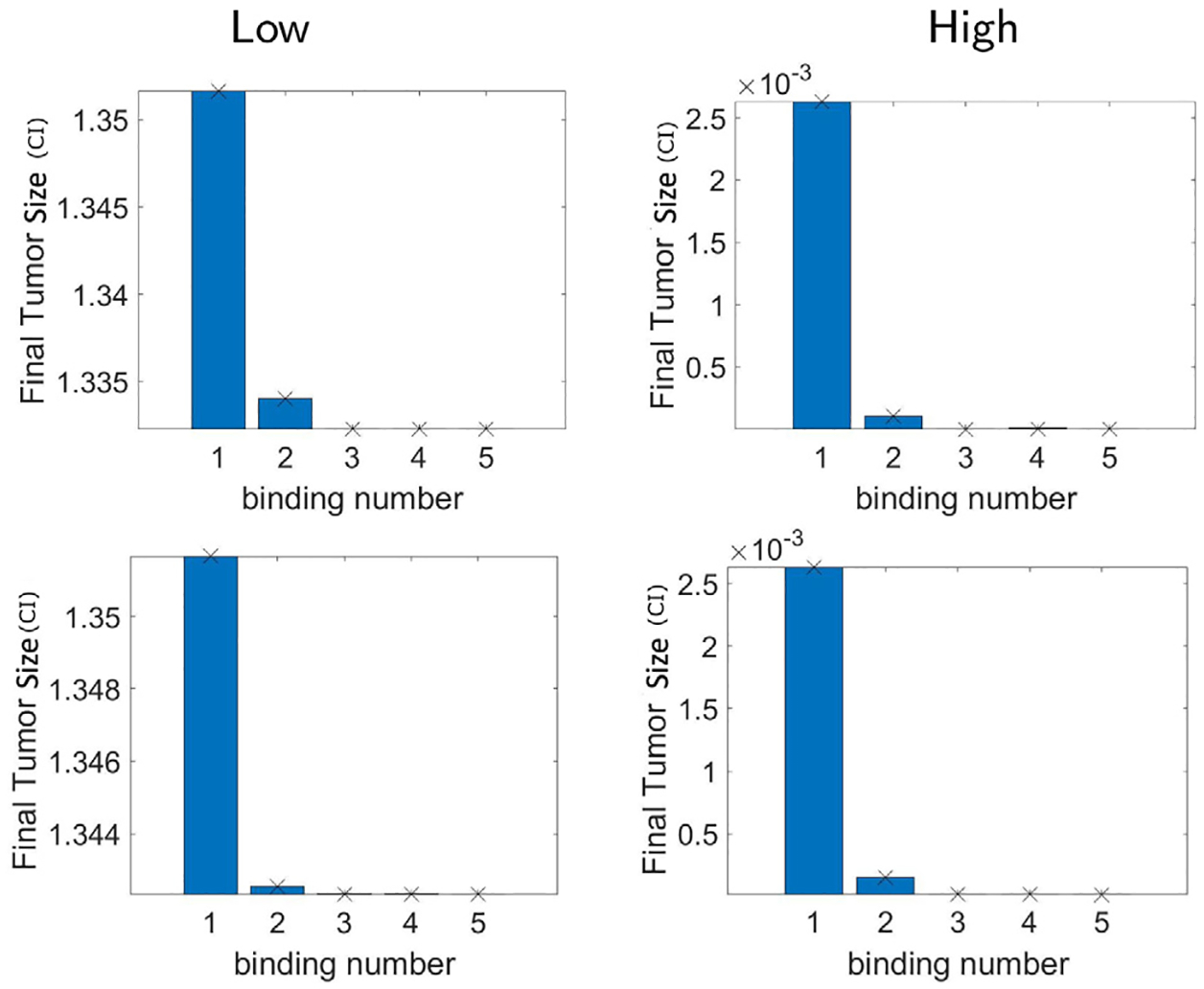
Hypothesis 2. Final tumor size (CI) using the n-binding model Eq. (7) assuming up to n number of CAR T-cells binding to glioma cell forming the conjugates I1, …, In. We consider n = 1,2,…,5. The kinetic rate parameters of the multiple CAR T-cells binding model are taken by hypothesis 2 with M = 1, L = 2 (top) and M = 2, L = 2 (bottom). Unlike what we had in hypothesis 1, there is no increase from two bindings to three bindings for all densities, and all results show saturation of final tumor size after three bindings. Thus the relationships between reaction rates that we consider in hypothesis 2 with L > 1 describe the experimental data regarding the cancer antigen density receptor level.
5. Summary and future work
Here we developed an ODE model extending [7]. While the original model considers one conjugate I1 of one glioma cell and one CAR T-cell, and assumes that the dynamics of I1 conjugate is in equilibrium, our model considers multiple conjugates Ij, j = 1, …, n with more than one CAR T-cells binding to the glioma cell, and follow their dynamics. We denote the original model as one binding fast reaction model, and our models as n binding slow reaction models.
First, we study the stability of the one-binding slow reaction ODE system, and compare it with the fast reaction model. We obtain similar equilibrium states, but in terms of the parameter of the slow reaction model. Also, we derive the conditions, especially regarding the range of the CAR T-cell expansion rate parameter p, (i) the minimum level of CAR T-cell expansion rate that provides the possibility that the CAR T-cell treatment can be successful, and (ii) the level of CAR T-cell expansion rate that guarantees the escape from reaching the maximum tumor size.
Using our model, we then compare the fast and slow reaction model numerically, and show that the slow reaction model describes the experimental data more accurately, especially when the glioma antigen receptor density is low and/or mixed with relatively small numbers of CAR T-cells. In addition, we use the one-binding to five-binding slow reaction models to simulate low to high receptor density glioma cells, and study the assumption in the reaction rates that yield desired outcome, the decay and saturation of tumor size with respect to the antigen density levels. We come up with two different hypotheses to describe their connections, where the first hypothesis considers homogeneous rates across different numbers of CAR T-cell binding conjugates. More precisely, we assume identical rates for reactions involving glioma cells dying from the conjugates Ij. We show that the second hypothesis, where we consider non-homogeneous reaction rates, in particular, increasing tumor killing rates as the number of bindings increases, reflects the reduced but saturated tumor size.
One of our future works is to calibrate the reaction rate parameters from experimental data, and verify whether our assumptions in hypothesis 2 on the reaction rates of multiple binding model is valid. Currently, our model is calibrated to the time series data of glioma cells, and additional CAR T-cell dynamics data and experiments can be used to infer the reaction rates from data. We also hope to validate the effectiveness of the slow reaction model to in vivo data where we expect the interaction between the glioma cells and CAR T-cells to be slower than our current in vitro data. Different CAR T-cell designs lead to different affinities and cytotoxicities [36–38], thus we hope to estimate and compare the kinetic reaction rates for different immunological synapse designs of CAR T-cells. In addition, parameter identifiability study and strategies to find universal parameters that can be fixed across patients are our future work as well.
A limitation of our model is that it is fitted to the in vitro data that does not include the human immune system or tumor microenvironment. We propose to study and model the interaction of glioma cells and CAR T-cells with other immune cells in the future. Modeling the interaction with endogenously produced immune cells including non CAR T-cells, natural killer cells, and antigen presenting cells that include macrophages, dendritic cells, and B cells, along with cytokines may help understand how and why CAR T-cell conjugates are created as well as the role of other immune cells in the immune system in this process. This is also related to further investigating the persisting nonzero CAR T-cell population in case of treatment success predicted by our model in the stability analysis. Such a stable CAR T-cell population has been shown to be achieved in the clinics treating blood cancer [39,40]. In particular, [41] studies the persistence of CD4+ CAR T-cell population over 10 years after therapy treating leukemia, and finds that CD8+ and CD4+ CAR T-cells are critical for elimination of leukemic cells at early stage and in long-term, respectively [42]. This indicates the importance of considering different populations of T-cells to better achieve durable remission, and this is also our future work.
Acknowledgments
Research reported in this publication was supported by the California Institute for Regenerative Medicine (CLIN2-10248, CB), the National Cancer Institute of the National Institutes of Health under grant numbers R01CA236500 (CB), P30CA033572 (S. Rosen). The content is solely the responsibility of the authors and does not necessarily represent the official views of the California Institute for Regenerative Medicine or National Institutes of Health. We want to thank Renate Starr and Jim O’Hearn for help with the manuscript and figures. This manuscript is dedicated to the memory of Xin (Cindy) Yang and her contributions on this study.
Appendix A. Computation of two-binding model with fast reaction
Our assumption in this case is that
| (19) |
| (20) |
which means that the two conjugates decompose immediately right before the other CAR T-cells come to react with the conjugates themselves. By rearranging Eq. (19), we obtain
| (21) |
and by rearranging Eq. (20), we obtain
| (22) |
where we further assume that . Now we look at Ṫ
by substituting (21) into our equation and simplify it, we obtain
| (23) |
For Ċ, by using (21) and (22), we have
which can be rewritten as
| (24) |
Therefore, (23) and (24) give our two-binding model with fast reaction.
Appendix B. Stability analysis of one-binding model with slow reaction
The equilibrium points can be computed by:
Observe that because of kCT − γI1 = 0, this ODE system have the equilibrium points stated in Section 3.1. More precisely, using the relation , we can rewrite the three dimensional ODE system into a two dimensional ODE system:
The first equation gives us
and
If we let and , these agree with (T1, C1) and (T2, C2) obtained with the fast reaction model. Therefore, again we have 4 equilibrium points
We then notice the Jacobian matrix of the ODE system Eq. (2) is
Let us examine each equilibrium points and compare them with the fast reaction model.
1. The equilibrium point (T, C, I1) = (0, 0, 0) is the tumor-free and CAR T-cellfree case. The Jacobian matrix becomes
The corresponding characteristic polynomial will be
Because this is an upper triangular matrix, the eigenvalues can be observed easily, i.e. λ1 = −θ, λ2 = ρ, λ3 = −γ. We conclude that (0, 0, 0) is an unstable saddle point.
Fig. C.1.
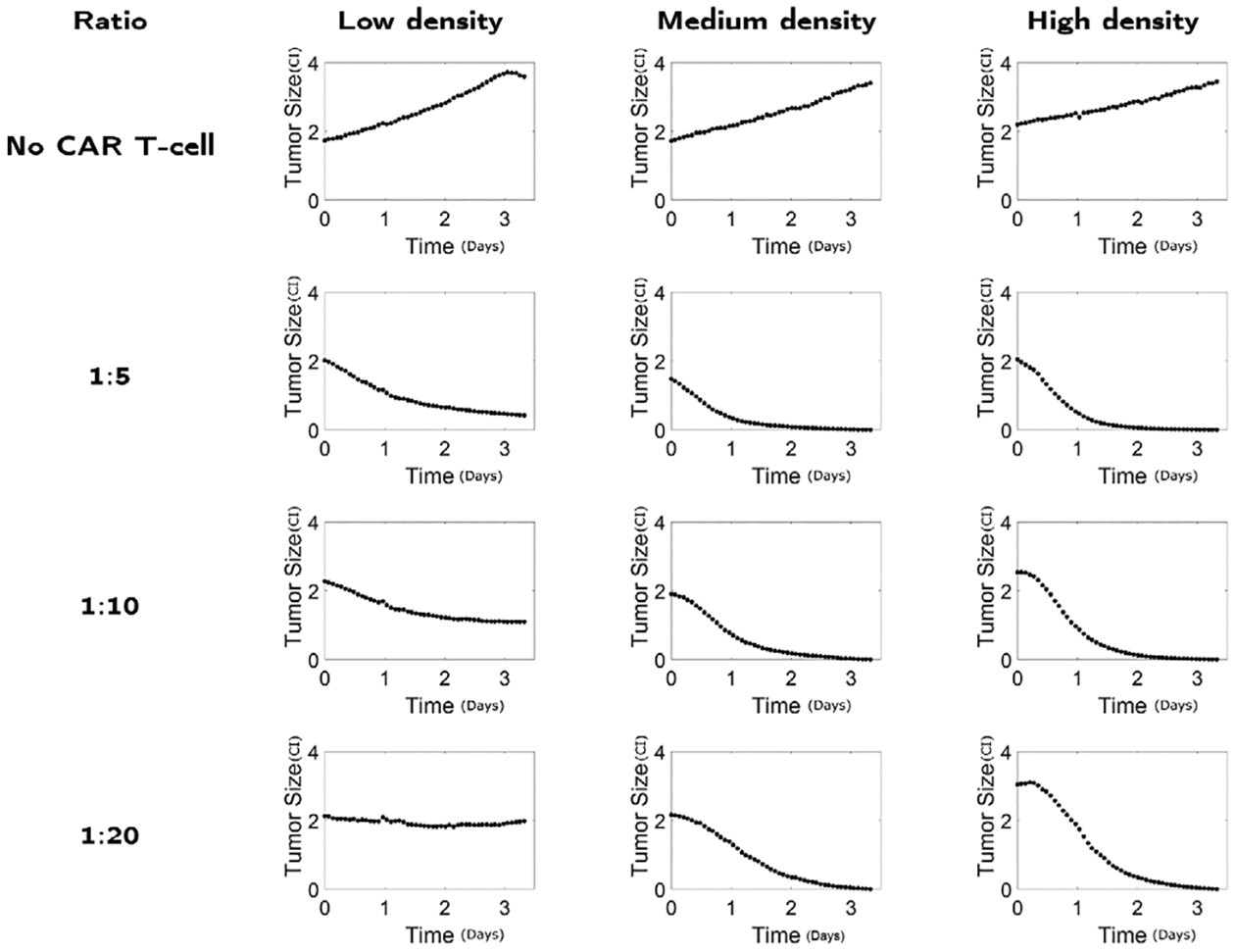
Data of tumor size (CI) dynamics in time (days) for different experimental design considering the number of mixed CAR T-cells (no CAR T-cell, 1:5, 1:10, 1:20) and glioma antigen receptor density (low, medium, high) [22].
2. The equilibrium point (T, C, I1) = (0, K, 0) is the tumor reaching the maximal capacity with no CAR T-cell surviving. The Jacobian matrix becomes
For simplicity, let , so . The corresponding characteristic polynomial will be
By setting P(λ) = 0, we obtain the first eigenvalue λ1 = −ρ, and the following quadratic equation,
This gives two additional solutions
Note that the expression inside the square root is always positive since
Therefore, these two eigenvalues are always real. For this equilibrium point to be stable, we need the eigenvalues to be negative, i.e.
or equivalently,
| (25) |
which comes from γg(K) < −Kka. If Eq. (25) is satisfied, the equilibrium point (0, K, 0) is stable. Therefore, this condition provides the minimal level of CAR T-cell expansion rate to avoid treatment failure.
3. The equilibrium points include (T1, C1) that can be denoted as the CAR T-cell therapy success case by ordering the points as 0 < C1 < C2 and T1 > T2 > 0. For these equilibrium points to exist, we can obtain the two conditions, , and , that reduces to
| (26) |
This condition provides a minimum level of CAR T-cell expansion rate that makes treatment success possible. To examine the stability, let us define . The characteristic polynomial becomes
where the coefficients are computed as following
The stability of the equilibrium points will depend on the roots of this polynomial. Although it is difficult to analyze the condition from this polynomial, we know that for the CAR T-cell therapy success case (T1, C1), and h′(C1) < 0, which provide us that (T1, C1) is either stable or a saddle.
Table C.1.
Parameters for the slow binding model Eq. (2) with low IL13Rα2 antigen density. The left column lists all the parameters that are considered in the calibration. The top row specifies different CAR T-cell ratios.
| 1:5 | 1:10 | 1:20 | |
|---|---|---|---|
| ρ (day−1) | 0.3953 | 0.3953 | 0.3953 |
| K (CI) | 6 | 6 | 6 |
| p (day−1) | 3.2430 | 8.8245 | 7.0226 |
| g (CI) | 16.8454 | 17.0687 | 10.7698 |
| 6.1317 | 14.2485 | 9.9151 | |
| 1.1658 | 0.8095 | 1.1463 | |
| 9.1820 | 8.4086 | 12.8200 | |
| 0.1017 | 0.0658 | 0.7729 | |
| θ (day−1) | 0.0412 | 0.0412 | 0.0412 |
Table C.2.
Parameters for the fast binding model Eq. (3) with low IL13Rα2 antigen density. The left column lists all the parameters that are considered in the calibration. The top row specifies different CAR T-cell ratios.
| 1:5 | 1:10 | 1:20 | |
|---|---|---|---|
| ρ (day−1) | 0.3953 | 0.3953 | 0.3953 |
| K (CI) | 6 | 6 | 6 |
| p (day−1) | 0.4351 | 5.3280 | 7.9233 |
| g (CI) | 5.2676 | 11.5747 | 11.4983 |
| m (day−1 · CI−1) | 0.1179 | 0.4640 | 0.6260 |
| ℓ (day−1 · CI−1) | 2.3818 | 3.0213 | 3.4057 |
| θ (day 1) | 0.0412 | 0.0412 | 0.0412 |
Table C.3.
Parameters for the slow binding model Eq. (2) with high IL13Rα2 antigen density. The left column lists all the parameters that are considered in the calibration. The top row specifies different CAR T-cell ratios.
| 1:5 | 1:10 | 1:20 | |
|---|---|---|---|
| ρ (day−1) | 0.2187 | 0.2187 | 0.2187 |
| K (CI) | 6 | 6 | 6 |
| p (day−1) | 18.5305 | 16.5100 | 16.6778 |
| g (CI) | 8.0816 | 1.6240 | 2.3731 |
| 3.7075 | 3.7439 | 1.6125 | |
| 4.3446 | 0.2965 | 2.0446 | |
| 13.0198 | 1.8054 | 1.8582 | |
| 0.3386 | 0.0495 | 0.7008 | |
| θ (day−1) | 0.0412 | 0.0412 | 0.0412 |
Table C.4.
Parameters for the fast binding model Eq. (3) with high IL13Rα2 antigen density. The left column lists all the parameters that are considered in the calibration. The top row specifies different CAR T-cell ratios.
| 1:5 | 1:10 | 1:20 | |
|---|---|---|---|
| ρ (day−1) | 0.2187 | 0.2187 | 0.2187 |
| K (CI) | 6 | 6 | 6 |
| p (day−1) | 10.5097 | 16.7512 | 15.2942 |
| g (CI) | 5.5172 | 11.8066 | 17.9938 |
| m (day−1 · CI−1) | 0.5691 | 0.3766 | 0.0566 |
| ℓ (day−1 · CI−1) | 1.8054 | 1.2394 | 1.4700 |
| θ (day−1) | 0.0412 | 0.0412 | 0.0412 |
Fig. D.1.
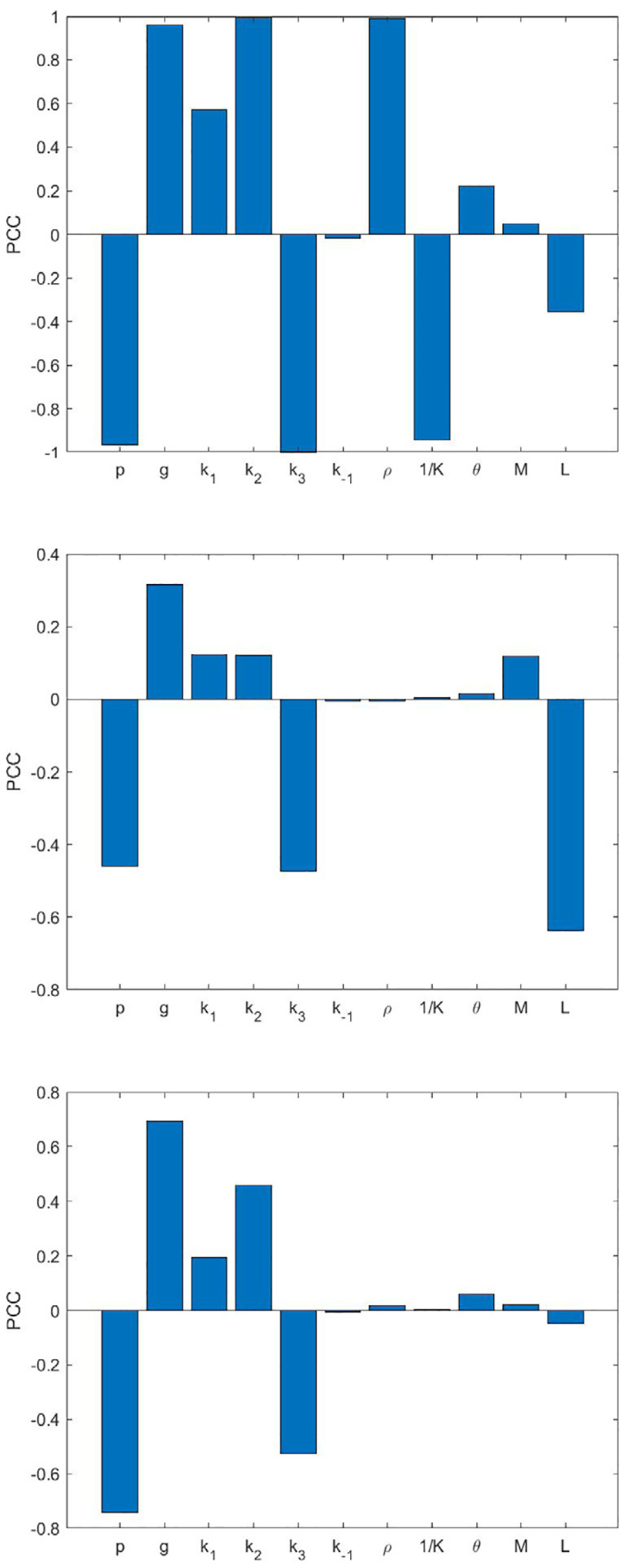
Partial correlation coefficients (PCC) of the model parameters of the five-binding model (7) to the final tumor volume. Shown cases are IL13Rα2 antigen density and CAR T-cell ratio, low and 1:10 (top), high and 1:20 (middle), and high and 1:5 (bottom).
Appendix C. Data and parameters
The glioma growth data used to calibrate the models are shown in Fig. C.1.
The calibrated parameter values of one-to-one binding slow and fast reaction models to plot Figs. 3 and 4 are provided in Tables C.1–C.4. The tumor growth rate ρ is calibrated with the tumor data without CAR T-cell treatment, while K is fixed as 6 CI ≈ 6 · 104 cells. The other parameters are calibrated with tumor data with CAR T-cell treatment while θ is fixed [7] to reduce uncertainty in parameter estimations.
Table D.1.
Partial correlation coefficients of parameters M and L in multiple binding models (7) and hypothesis 2 in section 4.2. Presented cases of densities and CAR T-cell ratios are from Figs. 8–9. Both M and L are taken to be 2. The mean of partial correlation coefficients with the range of minimums and maximums across 2 bindings to 5 bindings in parentheses are shown.
| M | L | |
|---|---|---|
| Low density, 1:10 | 0.04286 (0.03768, 0.04676) |
−0.3575 (−0.3683, −0.3488) |
| High density, 1:5 | 0.01673 (0.01322, 0.02230) |
−0.05055 (−0.05823, −0.04516) |
Appendix D. Parameter sensitivity analysis
In Fig. D.1, we list partial correlation coefficients for all parameters in the five-binding model. In Table D.1, we include the partial correlation coefficients for parameters M and L to final tumor volume.
Footnotes
CRediT authorship contribution statement
Runpeng Li: Analysis, Investigation, Visualization, Software, Writing – original draft. Prativa Sahoo: Data curation, Interpretation, Writing – review & editing. Dongrui Wang: Data curation. Qixuan Wang: Methodology, Writing – review & editing. Christine E. Brown: Data curation, Writing – review & editing. Russell C. Rockne: Data curation, Interpretation, Writing – review & editing. Heyrim Cho: Conceptualization, Methodology, Analysis, Visualization, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
I have shared the link to my code at the Attached File step.
References
- [1].Rotolo A, Caputo V, Karadimitris A. The prospects and promise of chimeric antigen receptor immunotherapy in multiple Myeloma. Br J Haematol 2016;173:350–64. [DOI] [PubMed] [Google Scholar]
- [2].Mohanty R, Chowdhury CR, Arega S, Sen P, Ganguly P, Ganguly N. CAR T cell therapy: A new era for cancer treatment (review). Oncol Rep 2019;42:2183–95. [DOI] [PubMed] [Google Scholar]
- [3].Paucek D, Baltimore D, Li G. The cellular immunotherapy revolution: Arming the immune system for precision therapy. Trends Immunol 2019;40:292–309. [DOI] [PubMed] [Google Scholar]
- [4].Hinrichs CS. Molecular pathways: Breaking the epithelial cancer barrier for chimeric antigen receptor and T-cell receptor gene therapy. Clin Cancer Res 2016;22(7):1559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018;359(6382):1361–5. [DOI] [PubMed] [Google Scholar]
- [6].Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid Tumors: Challenges and opportunities. Stem Cell Res Therapy 2021;12(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kuznetsov VA, Makalkin IA, Taylor MA, Perelson AS. Nonlinear dynamics of immunogen1c Tumors: Parameter estimation and global bifurcation analysis. Bull Math Biol 1994;56(2):295–321. [DOI] [PubMed] [Google Scholar]
- [8].Kirschner D, Panetta JC. Modeling immunotherapy of the Tumor-immune interaction. J Math Biol 1998;37:235–52. [DOI] [PubMed] [Google Scholar]
- [9].Sotolongo-Costa O, Molina LM, Perez DR, Antoranz JC, Reyes MC. Behavior of tumors under nonstationary therapy. Physica D 2003;178(3–4):242–53. [Google Scholar]
- [10].D’Onofrio A Metamodeling tumor-immune system interaction, tumor evasion and immunotherapy. Math Comput Modelling 2008;47(5–6):614–37. [Google Scholar]
- [11].De Pillis LG, Radunskaya A. The dynamics of an optimally controlled tumor model: A case study. Math Comput Modelling 2003;37(11):1221–44. [Google Scholar]
- [12].Moore H, Li NK. A mathematical model for chronic myelogenous leukemia (CML) and T cell interaction. J Theoret Biol 2004;227(4):513–23. [DOI] [PubMed] [Google Scholar]
- [13].Chaplain M, Matzavinos A. Mathematical modelling of spatio-temporal phenomena in tumour immunology. In: Tutorials in mathematical biosciences III. Springer, Berlin; 2006, p. 131–83. [Google Scholar]
- [14].Mallet DG, dePillis LG. A cellular automata model of Tumor–immune system interactions. J Theoret Biol 2006;239:334–50. [DOI] [PubMed] [Google Scholar]
- [15].Ghaffarizadeh A, Heiland R, Friedman SH, Mumenthaler SM, Macklin P. PhysiCell: An open source physics-based cell simulator for 3-D multicellular systems. PLoS Comput Biol 2018;14(2):e1005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mostolizadeh R, Afsharnezhad Z, Marciniak-Czochra A. Mathematical model of chimeric anti-gene receptor (CAR) T cell therapy with presence of cytokine. Numer Algebra Control Optim 2018;8:63–80. [Google Scholar]
- [17].Hardiansyah D, Ng CM. Pharmacology model of chimeric antigen receptor T-cell therapy. Clin Transl Sci 2019;12:343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hopkins B, Tucker M, Pan Y, Fang N, Huang Z. A model-based investigation of Cytokine storm for T-cell therapy. IFAC-PapersOnLine 2018;51:76–9. [Google Scholar]
- [19].Kimmel GJ, Locke FL, Altrock PM. The roles of T cell competition and stochastic extinction events in chimeric antigen receptor T cell therapy. Proc R Soc B 2021;288(1947):20210229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barros LRC, de Jesus Rodrigues B, Almeida RC. CAR-T cell goes on a mathematical model. J Cell Immunol 2020;2(1). [Google Scholar]
- [21].Barros LR, Paixão EA, Valli aM, Naozuka GT, Fassoni AC, Almeida RC. CARTmath—A mathematical model of CAR-T immunotherapy in preclinical studies of hematological cancers. Cancers 2021;13(12):2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sahoo P, Yang X, Abler D, Maestrini D, Adhikarla V, Frankhouser D, et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J R Soc Interface 2020;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like Tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine–redirected T cells. Clin Cancer Res 2012;18(8):2199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375(26):2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].City of Hope. CAR-T cells destroying glioblastoma cells. 2017, URL https://www.youtube.com/watch?v=8D6_xWYlLy4.
- [26].Moniri MR, Young A, Reinheimer K, Rayat J, Dai L-J, Warnock GL. Dynamic assessment of cell viability, proliferation and migration using real time cell analyzer system (RTCA). Cytotechnology 2015;67(2):379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Davenport AJ, Jenkins MR, Cross RS, Yong CS, Prince HM, Ritchie DS, et al. CAR-T cells inflict sequential killing of multiple tumor target cells. Cancer Immunol Res 2015;3(5):483–94. [DOI] [PubMed] [Google Scholar]
- [28].Lynn RC, Weber EW, Gennert D, Sotillo E, Xu P, Good Z, et al. C-jun overexpressing CAR-T cells are exhaustion-resistant and mediate enhanced antitumor activity. 2019, p. 1–33, BioRxiv. [Google Scholar]
- [29].Cho H, Wang Z, Levy D. Study of dose-dependent combination immunotherapy using engineered T cells and IL-2 in cervical cancer. J Theoret Biol 2020;505. [DOI] [PubMed] [Google Scholar]
- [30].Bogacki P, Shampine L. A 3(2) pair of Runge - Kutta formulas. Appl Math Lett 1989;2(4):321–5. [Google Scholar]
- [31].Shampine LF, Reichelt MW. The MATLAB ODE suite. SIAM J Sci Comput 1997;18(1):1–22. [Google Scholar]
- [32].Mayer A, Zhang Y, Perelson AS, Wingreen NS. Regulation of T cell expansion by antigen presentation dynamics. Proc Natl Acad Sci 2019;116(13):5914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Akaike H A new look at the statistical model identification. IEEE Trans Automat Control 1974;19(6):716–23. [Google Scholar]
- [34].Benzekry S, Lamont C, Beheshti A, Tracz A, Ebos JM, Hlatky L, et al. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol 2014;10(8):e1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Marino S, Hogue IB, Ray CJ, Kirschner DE. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theoret Biol 2008;254(1):178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJ, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate Tumour antigen escape. Nature 2019;568(7750):112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu D, Badeti S, Dotti G, Jiang J-g, Wang H, Dermody J, et al. The role of immunological synapse in predicting the efficacy of chimeric antigen receptor (CAR) immunotherapy. Cell Commun Signaling 2020;18(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu Y-H, et al. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol Therapy 2018;26(4):963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T Cells for sustained remissions in Leukemia. N Engl J Med 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell all patients. J Clin Investig 2016;126(6):2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022;602(7897):503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen AX, Derrick EB, Beavis PA, House IG. CD4+ chimeric antigen receptor T cells in for the long journey. Immunol Cell Biol 2022;100(5):304–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
I have shared the link to my code at the Attached File step.