


Abstract
We have performed rolling-circle amplification (RCA) reactions on three DNA templates that differ distinctly in their topology: an unlinked DNA circle, a linked DNA circle within a pseudorotaxane-type structure and a linked DNA circle within a catenane. In the linked templates, the single-stranded circle (dubbed earring probe) is threaded, with the aid of two peptide nucleic acid openers, between the two strands of double-stranded DNA (dsDNA). We have found that the RCA efficiency of amplification was essentially unaffected when the linked templates were employed. By showing that the DNA catenane remains intact after RCA reactions, we prove that certain DNA polymerases can carry out the replicative synthesis under topological constraints allowing detection of several hundred copies of a dsDNA marker without DNA denaturation. Our finding may have practical implications in the area of DNA diagnostics.
INTRODUCTION
Isothermal DNA amplification methods are widely used in basic and applied research (1). Among those methods, the rolling-circle amplification (RCA), also known as rolling-circle replication, has recently attracted significant attention (2–15). In RCA reactions, small single-stranded DNA (ssDNA) circles serve as templates for DNA polymerases (or sometimes RNA polymerases) generating numerous concatemerized copies of the circle. When a single primer complementary to the circle is employed, the accumulation of product proceeds in most cases linearly over time. An exponential (geometric) amplification via a so-called hyperbranched RCA (HRCA), also known as ramification or cascade RCA, is achieved by using a second primer with a sequence identical to a part of the DNA circle (7,9,10,14,15). The RCA technology is promising for molecular diagnostic and pharmacogenomic use because of its simplicity, high sensitivity, large multiplex potential, immunity to false positives/cross-contamination and easy compatibility with other detection/imaging techniques (7,12,13). Several applicable formats of the RCA technique have been described (7,11–13), and other assays may be envisioned, either employing preformed circular probes or using in situ assembly of DNA circles.
Circularized oligonucleotide templates, termed padlock probes, which target ssDNA were first reported by Nilsson et al. in 1994 (16). Since then, they have been successfully employed to analyze single-nucleotide polymorphisms (SNPs) of human genomic DNA (17,18). Recently, ourselves and others have developed approaches to selectively label double-stranded DNA (dsDNA) with circularized oligonucleotides (19–24). Ryan and Kool (19) and Escudé, Hélène and co-workers (20,24) employed triplex-directed assemblies, in which the obtained padlock-like circular probes surround both strands of the DNA double helix. We used a pair of pyrimidine peptide nucleic acid clamps, bis-PNAs (25–27), that sequence-specifically invade the DNA duplex (28–30) at closely located sites, thus providing access to the displaced DNA strand for probe hybridization (31,32). In such a way, a ssDNA circle truly topologically linked to the targeted DNA can be assembled by a post-hybridization closure procedure (21). In the final structure, a segment of the ssDNA circle, dubbed earring probe, appears to be threaded between the two strands of dsDNA. The PNA-assisted ‘topological’ labeling of dsDNA by earring probes exhibits a very high selectivity of oligonucleotide circularization: only a small part of all incorrect targets, namely those containing mismatches very close to circularization point and, therefore, virtually incapable of ligation, will be exposed by PNA openers (22).
With the exception of the earring probe, circular probes assembled on either ssDNA or dsDNA may slip away from their target sequence during post-assembly manipulations (16,19,20). Therefore, the target site cannot be located with precision better than a few hundred nucleotides. In contrast, the earring probe remains at the site of its formation on the double helix as long as the participating dsDNA macromolecule maintains its duplex conformation (21). As a result, the topological DNA labeling has a potential for not only highly selective, but also highly localized detection of designated DNA sites.
For DNA diagnostics, it is extremely attractive to combine circular oligonucleotide probes with RCA because such a combination can additionally provide high sensitivity (33,34). Up to now, however, conflicting data regarding the feasibility of the RCA on padlock probes that are pseudo-topologically linked to the ssDNA target have been reported. Lizardi et al. (7), Thomas et al. (10) and Zhang et al. (15) observed HRCA reactions with such probes hybridized to long ssDNA. While none of them addressed the issue of topological constraints experimentally, two of those research groups considered it unlikely that the circular probe slides off the lengthy ssDNA target (10,15). Banér et al. (8), however, were able to detect the RCA when such probes were tagged to short oligonucleotides, but not to longer ssDNA targets. Banér et al. therefore concluded that obstacles imposed by inter-strand topology prevent the RCA and that this reaction may proceed efficiently only if the circular probes are released from the ssDNA target (8,33,34). As the question of whether the RCA is inhibited by topological constraints is crucial for potential applications of circular DNA probes, it needs to be clarified.
To resolve the controversy, we performed in the present study the RCA reaction on earring probes. Considering the high degree of amplification and its possible non-linearity, especially in the geometric HRCA format (7), even a tiny portion of DNA minicircles, which escaped from earring complexes during RCA reactions, might give a strong signal. To exclude this possibility, we prepared, along with a free circular oligonucleotide and that linked to the regular linear dsDNA fragment (DNA pseudorotaxane in Fig. 1), a new topological construct, a catenated complex (see DNA catenane in Fig. 1) of an oligonucleotide ring with a large DNA dumbbell (dbDNA; 35). Evidently, the earring-like circular probe could not slip away from the catenane as long as the dbDNA remained intact. This provided us with an opportunity to verify, by determining the dbDNA integrity, whether the RCA reaction we observed occurred in the topologically linked setting. We found the RCA reaction to effectively proceed on linked DNA circles as templates, when certain DNA polymerases were used. Kinetic analysis revealed no substantial differences in the RCA product formation with the different templates when the same amounts of input molecules were used. As a result, the efficiency of amplification for the topologically linked DNA constructs was essentially not decreased as compared with the efficiency of amplification for free DNA circles.
Figure 1.

Three topologically different templates used in this study for RCA. Shown in blue is a primer (and the corresponding RCA product) complementary to the circular oligonucleotide probe (red), which is the same in all cases. bis-PNA openers that locally expose duplex DNA (gray) are shown in green; in all other schematics they are omitted to solely demonstrate the topology. For simplicity, only the single-primed RCA is shown.
MATERIALS AND METHODS
PNAs and oligonucleotides
Bis-PNAs, HLys2-TCTC2TC2-(eg1)3-J2TJ2TJT-LysNH2 and HLys2-TJTJ2T2J-(eg1)3-CT2C2TCT-LysNH2, were purchased from PE Biosystems (Framingham, MA). Oligodeoxynucleotides for assembly of dbDNA, 5′-CCAGTGAATTCGAGCTCGGTACCCGGGGATCGTCTCCTCCAGTAGCTTCCTCTGAT-GGTCTAGAGTCGACCTGCAGCATGCAAGCTTGGTTT-TTCCAAG-3′ and 5′-CTTGCATGCTGCAGGTCGACTCTAGACCATCAGAGGAAGCTACTGGAGGAGACGATC-CCCGGGTACCGAGCTCGAATTCACTGGCCGTCTTTTTGACGG-3′ (HIV-1 nef gene target sequences for binding PNAs and the circularizable probe are underlined and italicized, respectively), were obtained from Integrated DNA Technologies (Coralville, IA). All other oligonucleotides were purchased from MWG-Biotechnology (Hight Point, NC) and contain the following sequences: 5′-CTGGAGGAGATTTTGTGGTATCGATTCGTCTCTTAGAGGAAGCTA-3′ (circularizable oligonucleotide LO-45), 5′-GATCGTCTCCTCCAGTAGCTTCCTC (splint oligonucleotide Spl-25), 5′-GACGAATCGATACCAC-3′ (primer 1), 5′-GAGACGAATCGATACCACAA-3′ (primer 2) and 5′-GAGGAAGCTACTGGA-GGAGA-3′ (primer 3).
Preparation of a free ssDNA circle
The synthetic strategy is shown in Figure 2A. 5′-Phosphorylated, circularizable oligonucleotide LO-45 (28 pmol) was incubated with 50 pmol of splint oligonucleotide Spl-25 for 30 min at ambient temperature in 395 µl buffer containing 40 mM Tris–HCl (pH 7.8 at 25°C), 10 mM MgCl2, 10 mM DTT and 1.5 mM ATP. The mixture was then cooled to 16°C, and 5 µl of T4 DNA ligase (30 U/µl) was added. After incubation overnight at 16°C, the reaction mixture was extracted by phenol/chloroform/isoamylalcohol (25:24:1) and chloroform/isoamylalcohol (24:1), ethanol precipitated, and redissolved in TE buffer (10 mM Tris–HCl, pH 7.4 at 25°C, 0.1 mM EDTA). The analysis of the resulting sample on a denaturing polyacrylamide gel showed a 70% yield of circularized oligonucleotide. The remaining linear precursor and the splint oligonucleotide were subsequently removed by treatment of the sample with exonuclease VII, an exonuclease digesting DNA from either 5′- or 3′-ends. The sample was then desalted by gel filtration on a Sephadex G-50 spin column, extracted by phenol/chloroform/isoamylalcohol (25:24:1) and chloroform/isoamylalcohol (24:1), ethanol precipitated, and redissolved in TE buffer. The extraction and precipitation procedure was then repeated to completely remove exonuclease. As shown in Figure 2B, pure ssDNA circle was thus obtained. The concentration of the circular oligonucleotide was determined with an estimated error of 30–40% in a denaturing (8 M urea) polyacrylamide gel in comparison with known quantities of marker oligonucleotides.
Figure 2.

Preparation of a free ssDNA circle. (A) Schematics of ssDNA circularization using a splint oligonucleotide. (B) Analysis of the 45 nt ssDNA circle by denaturing polyacrylamide gel electrophoresis. Lane M, 50 nt size marker; lanes 1 and 2, splint and non-circularized oligonucleotides, respectively; lane 3, reaction mixture containing ssDNA circle after enzymatic ligation; lane 4, purified ssDNA circle after degradation of non-circularized oligonucleotides by exonuclease VII.
Assembly of closed and nicked dbDNAs
The preparation of the large dbDNA featuring a 94 bp duplex stem closed at both termini by short single-stranded loops was performed as recently described in detail (35). Briefly, two 5′-phosphorylated 99mer oligonucleotides (see above for their sequences) forming a 5 nt hairpin-like loop at their 3′-ends were annealed and ligated. Closed, double-ligated dbDNA thus obtained was purified from undesired by-products containing gaps or nicks by pre-treatment with DNA polymerase I in the presence of dATP, dGTP, dTTP and biotin-dCTP, and subsequent biomagnetic separation using streptavidin-coated microbeads. During these procedures, non-closed species were first biotinylated via nick translation and/or primer extension reactions and then selectively captured on microbeads, yielding essentially 100% pure covalently closed dbDNA (see Fig. 3, lane 3). As a control sample, a nicked, monoligated dbDNA was prepared by annealing and ligation of the same 99mer oligonucleotides, only one of which was now 5′-phosphorylated.
Figure 3.
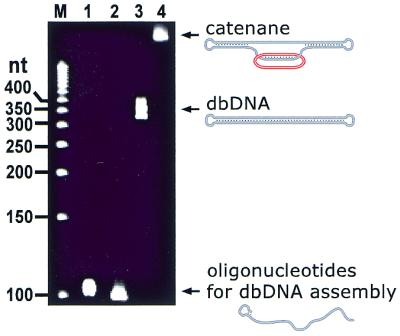
Analysis of gel-purified catenane by denaturing polyacrylamide gel electrophoresis. Lane M, 50 nt size marker; lanes 1 and 2, initial oligonucleotides used in the dbDNA preparation; lane 3, purified dbDNA; lane 4, purified catenane. Note that the dbDNA band is somewhat broad, which may reflect a high stability of the secondary structure of this closed DNA construct against denaturing conditions: during the electrophoresis, the dbDNA is not completely single stranded as its stem-forming segment may transiently restore the duplex form thus causing a smear.
Assembly of DNA pseudorotaxane and DNA catenane
The PNA-assisted assembly of pseudorotaxane was performed as recently described (21,22). Briefly, a 350 bp dsDNA fragment of recombinant plasmid carrying in the middle a part of the HIV-1 nef gene with the binding/hybridization sites for PNA openers and the circularized probe was incubated with a pair of PNAs, followed by addition of 5′-phosphorylated circularizable oligonucleotide LO-45 and its closing by ligation. The yield of pseudorotaxane was close to 90%. The complex was then treated with exonuclease VII and purified as described for the preparation of the free ssDNA circle. A similar procedure was employed to prepare the catenane, except that now the target DNA was taken in the form of pure dbDNA with the same centrally located HIV-1 target (35). The catenane was obtained in 50–60% yield and purified on a preparative, 10% native polyacrylamide gel. Its purity was confirmed by denaturing polyacrylamide gel electrophoresis demonstrating a single band with lower mobility than dbDNA (see Fig. 3, lanes 3 and 4). The concentration of pseudorotaxane and catenane in stock solutions were estimated with 50–65% accuracy by comparative electrophoretic analysis using aliquots of a commercial low-mass DNA ladder.
RCA reactions
Single-primed RCA. The desired amount of one of the three templates was preincubated with 1 µM primer 1 for 30 min at 37°C in 10 µl of buffer (20 mM MgCl2, 50 mM NaCl, 40 mM Tris–HCl, pH 7.2). Then, 5.7 µl of H2O, 1.0 µl of 100 mM DTT, 0.8 µl of a mixture containing all four dNTPs (25 mM each), 0.5 µl of SSB protein (2.2 mg/ml) and 2.0 µl of Sequenase 2.0 (1.6 U/µl) were added and the reaction mixture was incubated for 3.5 h at 37°C. To analyze the post-RCA catenane integrity, the samples were gel-filtrated on a Sephadex G-50 spin column, extracted by phenol/chloroform/isoamylalcohol (25:24:1) and chloroform/isoamylalcohol (24:1), ethanol precipitated and redissolved in TE buffer. In kinetic experiments, the RCA reactions were stopped at different incubation times by the addition of 0.5 M EDTA (pH 8.0) to a final concentration of 100 mM, followed by removal of unused primer and dNTPs by gel filtration (Sephadex G-50). The samples were then ethanol precipitated and redissolved in 250 µl of H2O. Absorbance measurements were conducted in a semi-micro quartz cuvette on a Hitachi U-3000 spectrophotometer.
Double-primed RCA. The desired amount of one of the three templates was incubated at 62°C for 1.5 h in a 35 µl reaction volume containing 20 mM Tris–HCl (pH 8.8 at 25°C), 2.5 mM MgCl2, 10 mM KCl, 10 mM (NH4)2SO4, 0.1% Triton X-100, 5% v/v DMSO, 500 µM each of the four dNTPs, 1.33 µg T4 gene-32 protein, 1 µM each of primers 2 and 3, and 10 U of Vent exo– DNA polymerase.
Gel electrophoresis. Electrophoretic analysis of RCA products was performed without their purification in 2% agarose gels filled with TAE buffer (40 mM Tris–acetate, 1 mM EDTA, pH 8.0). Polyacrylamide gel electrophoresis was performed in TBE buffer (90 mM Tris–borate, 2 mM EDTA, pH 8.0). Denaturing polyacrylamide gels (10 or 20%) contained 8 M urea and were run at 60°C. Immediately before analysis, an equal volume of denaturing loading buffer containing 95% formamide and 20 mM EDTA was added to the samples. Gels were stained by either ethidium bromide (agarose) or SYBR Green II (polyacrylamide) and visualized by transillumination at 302 nm. Quantification of bands was performed using a CCD camera and digital imaging system/image analysis software.
RESULTS AND DISCUSSION
Figure 1 shows the earring-type complexes we assembled to study the RCA reaction under topological constraints. We performed a comparative analysis of the RCA efficiency for the same circularized oligonucleotide put into three topologically different situations: a circular probe in the free state and probes linked to target DNA duplexes with either open termini (DNA pseudorotaxane) or closed termini (DNA catenane). Figures 2 and 3 demonstrate the purity of free ssDNA circle and the DNA catenane, respectively (see Materials and Methods for details). All templates were devoid of the linear precursor of the circular probe, as the RCA templates were either treated with exonuclease VII, an exonuclease digesting ssDNA from either 5′- or 3′-ends, or gel-purified before use. Thus, possible artefacts resulting from linear target isothermal multimerization and amplification (14) were excluded.
Within the linked complexes, the 21 nt hybridized part of the circular label makes two turns around the displaced DNA target strand entailing, most probably, the second-order linkage with the linking number Lk = 2 (to simplify the schematics, we show in Fig. 1 the situation for the case of the first-order link Lk = 1). The intact circular probe can therefore escape from the catenated complex only in case the dbDNA is broken. This makes it possible to check, by quantitatively analyzing the post-RCA dbDNA integrity, whether the RCA reaction takes place in the truly topologically constrained situation (see below).
We performed amplification experiments using serial dilutions of either template for both single- and double-primed RCA formats. For the single-primed RCA, a 16mer primer complementary to the linker section of the circular probe was employed to prime Sequenase 2.0 DNA polymerase at 37°C. The double-primed RCA reaction (HRCA) was performed by Vent exo– DNA polymerase at 62°C with the use of a pair of 20mer primers. Figure 4 shows that a significant amount of RCA product was formed when the initial input of DNA templates exceeded a certain quantity. As expected (2,4,7,10,14,15), the RCA products generated by one primer were characterized by a wide, essentially continuous distribution over length and could not be resolved on a gel yielding a broad smear, while distinct, ladder-type bands, which corresponded to linear, concatemeric copies of the circular template, were observed in the double-primed format. The maximal degree of amplification, as assessed by UV spectroscopy, was about 105 for the single-primed RCA format and close to 1010 when two primers were employed in the RCA reaction.
Figure 4.

Analysis of RCA products by non-denaturing agarose gel electrophoresis. Single-primed [pseudo-linear RCA (A–C)] and double-primed [geometric, HRCA (D–F)] RCA reactions were performed on the three topologically different constructs: free circular oligodeoxynucleotide (A and D), DNA pseudorotaxane (B and E) and DNA catenane (C and F). A conservative site from the HIV-1 nef gene served as a target for the PNA-assisted labeling of duplex DNA with a circular probe. The RCA products were generated starting from different initial inputs of any of the three constructs (given in number of molecules above each lane). Lanes M correspond to a 100 bp size marker except in (D), in which a 50 bp marker was employed.
We observed that topological constraints did not significantly decrease the efficiency of amplification: the sensitivity of both RCA formats was not >5-fold lower for the linked ssDNA circles as compared with the free circle. As a result, the HRCA reaction started at zeptomolar amounts of DNA templates allowing isothermal detection down to several hundred copies of the DNA marker in the duplex form. No amplification was observed for targets carrying single-nucleotide substitutions at various positions along the region (data not shown), thus demonstrating that our approach is promising for SNP analyses, which can now be performed without DNA denaturation. Potentially, our earring-based approach should provide such kind of analyses with much higher selectivity than the conventional padlock-based technique (see Introduction).
The reason for the observed concentration threshold remains unclear. Triplicate experiments performed with each template in both RCA formats resulted in the same threshold levels as shown in Figure 4 with only one exception (one HRCA experiment with the pseudorotaxane resulted in a 2-fold higher sensitivity, i.e. RCA product was observed with ≥600 input molecules). Importantly, this effect is not inherent to the linked templates as it was also observed with the free ssDNA circle. Note in this connection that our 45 nt circular probe is rather small compared with the 70 nt or longer padlock probes commonly used by others for the RCA reaction (7–10,14,15). In fact, Frieden et al. (6) found that Sequenase did not accept ssDNA circles shorter than 30 nt as templates for RCA. In contrast, they observed long amplification products on three short circular templates with the Klenow fragment of E.coli DNA polymerase I, while this enzyme was RCA-inactive on a somewhat larger circle. Thus, further studies using circular probes of different sizes (and various sequences) are needed to find the reason for the threshold for RCA. It should be emphasized that this effect is not critical for our present study investigating the feasibility of the RCA reaction under topological constraints, although understanding and overcoming the concentration threshold is extremely important for further RCA applications.
Besides the gel-electrophoretic analysis shown in Figure 4, the RCA products were also characterized by restriction enzyme cleavage and sequencing (data not shown). It was found that the majority of products corresponded to duplex DNA containing regular multiple repeats of the circular probe sequence along with randomly shuffled fragments of that sequence and its complement. While dsDNA products are expected for HRCA, the RCA reaction performed with a single primer typically results in single-stranded products. However, other researchers also observed dsDNA amplicons while performing the RCA with only one primer (2,11).
One may argue that the ∼5-fold decrease in the RCA sensitivity we observed for the linked DNA templates as compared with free ssDNA circles may be the result of slippage and subsequent amplification of a significant percentage (∼20%) of prior linked circular probes under the RCA reaction conditions, while the remaining majority of linked ssDNA circles leads to poor or even no amplification. In case of the catenated template, such an escape of the circular probe could only happen via a nick generated within the dumbbell during the amplification reaction. To check this possibility, we examined the DNA catenane by gel electrophoresis before and after the RCA reaction next to samples containing nicked dbDNA, which yielded a faster migrating band when resolved in a denaturing gel. Figure 5 shows that the integrity of the catenane (lane 1) is not affected by the RCA conditions in the presence of all components except dGTP (lane 2). Similarly, no nicked dbDNA was observed when the RCA reaction was performed on circular probes within the catenane (lane 3). As assessed by quantitative analysis using serial dilutions of the nicked dbDNA (lanes 4–9), at least 97% of the catenane remained intact during the RCA reaction. As it is thus expected, no free ssDNA circle could be observed in the samples of the catenane under RCA conditions (lanes 2 and 3; note that the sensitivity to trace this species is 4–5-fold lower than monitoring a potential nicked dbDNA).
Figure 5.

Assessment of the catenane integrity under RCA conditions by denaturing polyacrylamide gel electrophoresis. Three aliquots of 0.7 pmol purified catenane (see lane 4 in Fig. 3) were incubated in the Sequenase reaction buffer for 4 h without any other components for the RCA reaction (lane 1); with all components required for this reaction, including Sequenase (lane 3; note that the catenane and the resulting RCA products overlap with each other); or with all RCA components except dGTP (lane 2; the primer could be extended by 13 nt before dGTP was required). Lanes 4–9 correspond to a serial dilution of nicked dbDNA: 0.4 pmol (lane 4); 0.2 pmol (lane 5); 0.1 pmol (lane 6); 0.04 pmol (lane 7); 0.02 pmol (lane 8) and 0.01 pmol (lane 9; undetected). Note that the treatment of nicked dbDNA with Sequenase in the presence of all four dNTPs did not change the migration of this species. Lane M, 50 nt size marker.
Therefore, a slightly lower RCA sensitivity for the DNA catenane (and the DNA pseudorotaxane) versus the free circular oligonucleotide template could not be attributed to escaped ssDNA circles. It might be caused by additional steric hindrances imposed by PNA2–DNA triplexes to the RCA reaction (see Fig. 1). Note that we did not make any efforts to remove PNA openers after assembly of the earring probe. Therefore, two bulky PNA2–DNA triplexes could remain nearby to the earring probe/primer-extension point, at least at 37°C, thus imposing additional steric hindrances on the RCA reaction. Remarkably, the presence of PNA openers did not substantially compromise the efficiency of the RCA reaction on earring probes (see Fig. 4). In light of the control experiments in Figure 5, a very moderate decrease in the degree of amplification rules out the possibility that the RCA reaction detected in case of catenane and pseudorotaxane constructs actually occurred on a small fraction (≤3%) of minicircles escaped from the target during the RCA reaction.
This conclusion is supported by our kinetic experiments. The time courses of RCA product accumulation were determined for free and topologically linked ssDNA circles at two amounts of input template molecules differing by a factor of 30. With the free ssDNA circle, a significant difference in kinetics was observed with these different inputs (Fig. 6, blue curves). At the higher amount of DNA circle (6 fmol or ∼3.6 × 109 molecules), a rapid increase in product formation was observed between 40 and 70 min (dark blue curve). At the 30-fold lower amount of template, the accumulation of RCA product occurred mainly between 60 and 90 min (light blue curve). The acquired final amounts of RCA product were independent of the amount of input molecules, similar to previously reported data for HRCA (14). Linking the circular template to the dbDNA, significantly slower kinetics than those with the free ssDNA circle should be expected for the same number of input molecules, if the observed amplification products for the topologically linked template resulted from the slippage and subsequent amplification of a small fraction of free ssDNA circles (alternatively, slower kinetics could also be the result of an overall slower polymerization rate on the topologically linked template). However, we did not observe any substantial difference in the time courses of RCA product accumulation with the topologically linked ssDNA circle (Fig. 6, green curves) in comparison with the free ssDNA circle at the same number of input molecules (compare dark and light colored curves). Thus, the kinetic data uphold the finding that the RCA reaction can be performed in the topologically linked setting.
Figure 6.

Kinetic analysis of single-primed RCA. (A) Representative example showing the absorbance profiles for RCA product obtained at 30, 40, 50, 60, 67, 80 and 100 min (curves 1–7) using 3.6 × 109 input molecules of catenane. (B) Accumulation of RCA products with free ssDNA circle (blue curves) or catenane (green curves). The numbers of input molecules were 3.6 × 109 (dark colors) or 1.2 × 108 (light colors). The graph shows mean ± standard deviation values (n = 3).
Taken together, all of our data are in agreement with the conclusion that our topological constructs impose no serious hindrance on the RCA reaction. Our findings agree with the data by three research groups (7,10,15), who observed the RCA reaction on padlock probes hybridized to long ssDNA, but disagree with results by Banér et al. (8). This disagreement could be explained, at least partially, by different efficiencies of various DNA polymerases in performing the RCA reaction under topological constraints. We found Sequenase and Vent exo– DNA polymerase to be very efficient in the RCA reaction on earring probes. Both enzymes were reported to be able to perform HRCA on padlock probes linked to long ssDNA (7), as did the large fragment of Bst DNA polymerase (7,10,15). While we observed that Klenow exo– DNA polymerase was RCA-active on earring probes, it yielded a lesser degree of amplification than Sequenase (not shown). We also found that φ29 DNA polymerase did not work on earring probes in the single-primed RCA format, although this enzyme was very effective in performing linear (but not hyperbranched) RCA on padlock probes (7). All the data indicate that while performing the RCA reaction on topologically constrained DNA circles one has to be careful in choosing the appropriate DNA polymerase. According to our data and previous findings (7,10,14,15), Sequenase, Vent exo– DNA polymerase and Bst DNA polymerase (large fragment) are the enzymes of choice.
CONCLUSION
We have demonstrated that the RCA reaction can be performed by some DNA polymerases under true topological constraints. This resolves a major controversy in the field of the RCA reaction. Although many features of this unusual biomolecular reaction require further studies before the phenomenon will be fully understood, the obtained results are very promising with respect to developing new isothermal amplification techniques for DNA diagnostics. Using the HRCA format, we were able to detect as low as several hundred copies (zeptomolar amounts) of a dsDNA marker in model reactions without prior DNA denaturation. Our PNA-assisted approach to topologically link a circular, earring-like probe to dsDNA should provide with a higher sequence specificity than the conventional padlock-based techniques as only a small part of all incorrect targets, namely those containing mismatches very close to circularization point and, therefore, virtually incapable of ligation (22), will be exposed by PNA openers. Besides, this assay has one more substantial advantage: the probe movement along the target is restricted, thus keeping the probe at a precise position on the DNA double helix. As a result, the topological labeling of DNA duplexes with earring probes combined with RCA has the potential for a highly selective, sensitive and localized detection of designated DNA sites.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Irina V. Lavrentyeva-Smolina for technical assistance. This work was supported by NIH grant GM59173.
REFERENCES
- 1.Schweitzer B. and Kingsmore,S. (2001) Combining nucleic acid amplification and detection. Curr. Opin. Biotechnol., 12, 21–27. [DOI] [PubMed] [Google Scholar]
- 2.Fire A. and Xu,S.-Q. (1995) Rolling replication of short DNA circles. Proc. Natl Acad. Sci. USA, 92, 4641–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daubendiek S.L., Ryan,K. and Kool,E.T. (1995) Rolling circle RNA synthesis: circular oligonucleotides as efficient substrates for T7 RNA polymerase. J. Am. Chem. Soc., 117, 7818–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D., Daubendiek,S.L., Zillman,M.A., Ryan,K. and Kool,E.T. (1996) Rolling circle DNA synthesis: small circular oligonucleotides as efficient templates for DNA polymerases. J. Am. Chem. Soc., 118, 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daubendiek S.L. and Kool,E.T. (1997) Generation of catalytic RNAs by rolling transcription of synthetic DNA nanocircles. Nat. Biotechnol., 15, 273–277. [DOI] [PubMed] [Google Scholar]
- 6.Frieden M., Pedroso,E. and Kool,E.T. (1999) Tightening the belt on polymerases: evaluating the physical constraints on enzyme substrate size. Angew. Chem. Int. Ed., 38, 3654–3657. [PubMed] [Google Scholar]
- 7.Lizardi P.M., Huang,X., Zhu,Z., Bray-Ward,P., Thomas,D.C. and Ward,D.C. (1998) Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nature Genet., 19, 225–232. [DOI] [PubMed] [Google Scholar]
- 8.Banér J., Nilsson,M., Mendel-Hartvig,M. and Landegren,U. (1998) Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res., 26, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang D.Y., Brandwein,M., Hsuih,T.C.H. and Li,H. (1998) Amplification of target-specific, ligation-dependent circular probe. Gene, 211, 277–285. [DOI] [PubMed] [Google Scholar]
- 10.Thomas D.C., Nardone,G.A. and Randall,S.K. (1999) Amplification of padlock probes for DNA diagnostics by cascade rolling circle amplification or the polymerase chain reaction. Arch. Pathol. Lab. Med., 123, 1170–1176. [DOI] [PubMed] [Google Scholar]
- 11.Sabanayagam C.R., Berkey,C., Lavi,U., Cantor,C.R. and Smith,C.L. (1999) Molecular DNA switches and DNA chips. In Ferrari,M. (ed.), Micro- and Nanofabricated Structures and Devices for Biomedical and Environmental Applications II. Proceedings SPIE (The International Society for Optical Engineering), Vol. 3606, pp. 90–97.
- 12.Schweitzer B., Wiltshire,S., Lambert,J., O’Malley,S., Kukanskis,K., Zhu,Z., Kingsmore,S.F., Lizardi,P.M. and Ward,D.C. (2000) Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc. Natl Acad. Sci. USA, 97, 10113–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong X., Lizardi,P.M., Huang,X., Bray-Ward,P.L. and Ward,D.C. (2001) Visualization of oligonucleotide probes and point mutations in interphase nuclei and DNA fibers using rolling circle DNA amplification. Proc. Natl Acad. Sci. USA, 98, 3940–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafner G.J., Yang,I.C., Wolter,L.C., Stafford,M.R. and Giffard,P.M. (2001) Isothermal amplification and multimerization of DNA by Bst DNA polymerase. Biotechniques, 30, 852–867. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D.Y., Zhang,W., Li,X. and Konomi,Y. (2001) Detection of rare DNA targets by isothermal ramification amplification. Gene, 274, 209–216. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson M., Malmgren,H., Samiotaki,M., Kwiatkowski,M., Chowdhary,B.P. and Landegren,U. (1994) Padlock probes: circularizing oligonucleotides for localized DNA detection. Science, 265, 2085–2088. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson M., Krejci,K., Koch,J., Kwiatkowski,M., Gustavsson,P. and Landegren,U. (1997) Padlock probes reveal single-nucleotide differences, parent of origin and in situ distribution of centromeric sequences in human chromosomes 13 and 21. Nature Genet., 16, 252–255. [DOI] [PubMed] [Google Scholar]
- 18.Antson D.-O., Isaksson,A. and Nilsson,M. (2000) PCR-generated padlock probes detect single nucleotide variation in genomic DNA. Nucleic Acids Res., 28, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan K. and Kool,E.T. (1998) Triplex-directed self-assembly of an artificial sliding clamp on duplex DNA. Chem. Biol., 5, 59–67. [DOI] [PubMed] [Google Scholar]
- 20.Escudé C., Garestier,T. and Hélène,C. (1999) Padlock oligonucleotides for duplex DNA based on sequence-specific triple helix formation. Proc. Natl Acad. Sci. USA, 96, 10603–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn H., Demidov,V.V. and Frank-Kamenetskii,M.D. (1999) Topological links between duplex DNA and a circular DNA single strand. Angew. Chem. Int. Ed., 38, 1446–1449. [DOI] [PubMed] [Google Scholar]
- 22.Kuhn H., Demidov,V.V. and Frank-Kamenetskii,M.D. (2000) An earring for the double helix: assembly of topological links comprising duplex DNA and a circular oligodeoxynucleotide. J. Biomol. Struct. Dyn., S2, 221–225. [DOI] [PubMed] [Google Scholar]
- 23.Demidov V.V., Kuhn,H., Lavrentyeva-Smolina,I.V. and Frank-Kamenetskii,M.D. (2001) Peptide nucleic acid-assisted topological labeling of duplex DNA. Methods, 23, 123–131. [DOI] [PubMed] [Google Scholar]
- 24.Roulon T., Hélène,C. and Escudé,C. (2001) A ligand-modulated padlock oligonucleotide for supercoiled plasmids. Angew. Chem. Int. Ed., 40, 1523–1526. [DOI] [PubMed] [Google Scholar]
- 25.Egholm M., Christensen,L., Dueholm,K.L., Buchardt,O., Coull,J. and Nielsen,P.E. (1995) Efficient pH-independent sequence-specific DNA-binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res., 23, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffith M.C., Risen,L.M., Greig,M.J., Lesnik,E.A., Sprankle,K.G., Griffey,R.H., Kiely,J.S. and Freier,S.M. (1995) Single and bis peptide nucleic acids as triplexing agents – binding and stoichiometry. J. Am. Chem. Soc., 117, 831–832. [Google Scholar]
- 27.Demidov V.V., Yavnilovich,M.V., Belotserkovskii,B.P., Frank-Kamenetskii,M.D. and Nielsen,P.E. (1995) Kinetics and mechanism of polyamide (‘peptide’) nucleic acid binding to duplex DNA. Proc. Natl Acad. Sci. USA, 92, 2637–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuhn H., Demidov,V.V., Frank-Kamenetskii,M.D. and Nielsen,P.E. (1998) Kinetic sequence discrimination of cationic bis-PNAs upon targeting of double-stranded DNA. Nucleic Acids Res., 26, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn H., Demidov,V.V., Nielsen,P.E. and Frank-Kamenetskii,M.D. (1999) An experimental study of mechanism and specificity of peptide nucleic acid (PNA) binding to duplex DNA. J. Mol. Biol., 286, 1337–1345. [DOI] [PubMed] [Google Scholar]
- 30.Demidov V.V. and Frank-Kamenetskii,M.D. (2001) Sequence-specific targeting of duplex DNA by peptide nucleic acids via triplex strand invasion. Methods, 23, 108–122. [DOI] [PubMed] [Google Scholar]
- 31.Demidov V.V. (2001) PD-loop technology: PNA openers at work. Exp. Rev. Mol. Diagn., 1, 343–351. [DOI] [PubMed] [Google Scholar]
- 32.Bukanov N.O., Demidov,V.V., Nielsen,P.E. and Frank-Kamenetskii,M.D. (1998) PD-loop: a complex of duplex DNA with an oligonucleotide. Proc. Natl Acad. Sci. USA, 95, 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaksson A. and Landegren,U. (1999) Accessing genomic information: alternatives to PCR. Curr. Opin. Biotechnol., 10, 11–15. [DOI] [PubMed] [Google Scholar]
- 34.Banér J., Nilsson,M., Isaksson,A., Mendel-Hartvig,M., Antson,D.-O. and Landegren,U. (2001) More keys to padlock probes: mechanisms for high-throughput nucleic acid analysis. Curr. Opin. Biotechnol., 12, 11–15. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn H., Frank-Kamenetskii,M.D. and Demidov,V.V. (2001) High-purity preparation of a large DNA dumbbell. Antisense Nucleic Acid Drug Dev., 11, 149–153. [DOI] [PubMed] [Google Scholar]