


Abstract
Mig.MthI from Methanobacterium thermoautotrophicum and MutY of Escherichia coli are both DNA mismatch glycosylases of the ‘helix-hairpin-helix’ (HhH) superfamily of DNA repair glycosylases; the former excises thymine from T/G, the latter adenine from A/G mismatches. The structure of MutY, in complex with its low molecular weight product, adenine, has previously been determined by X-ray crystallography. Surprisingly, the set of amino acid residues of MutY that are crucial for adenine recognition is largely conserved in Mig.MthI. Here we show that replacing two amino acid residues in the (modeled) thymine binding site of Mig.MthI (Leu187 to Gln and Ala50 to Val) changes substrate discrimination between T/G and A/G by a factor of 117 in favor of the latter (from 56-fold slower to 2.1-fold faster). The Ala to Val exchange also affects T/G versus U/G selectivity. The data allow a plausible model of thymine binding and of catalytic mechanism of Mig.MthI to be constructed, the key feature of which is a bidentate hydrogen bridge of a protonated glutamate end group (number 42) with thymine centers NH-3 and O-4, with proton transfer to the exocyclic oxygen atom neutralizing the negative charge that builds up in the pyrimidine ring system as the glycosidic bond is broken in a heterolytic fashion. The results also offer an explanation for why so many different substrate specificities are realized within the HhH superfamily of DNA repair glycosylases, and they widen the scope of these enzymes as practical tools.
INTRODUCTION
Life depends on the faithful maintenance of genetic information for long periods of time and many generations which, in turn, requires highly accurate copying mechanisms and equally efficient repair of chemical DNA damage that occurs between two rounds of replication. Water, to give just one example, is the major constituent of all metabolically active cells and at the same time is a fairly agressive chemical. Hydrolytic DNA damage is estimated to occur at a rate of several 104 events per day in every human cell, among them deamination of DNA cytosine residues comprising several hundred events (1). Unrepaired, each resulting DNA uracil residue will direct the incorporation of 2′-deoxy-adenosine in the next round of replication—with the eventual net result of a C/G to T/A transition mutation in half of the cell progeny.
Base excision repair (BER) is the most frequently used and most diversely evolved mechanism to counteract the mutagenic potential of hydrolytic and other DNA damage. BER is initiated by a DNA glycosylase specialized to remove a particular kind of damaged nucleobase from DNA (uracil in the example sketched above) by catalyzing the hydrolysis of its glycosidic bond. Subsequent steps are (i) strand scission at the base-free DNA site, (ii) enzymatic trimming of the incision point to form a small gap, (iii) DNA synthesis to fill the gap and (iv) ligation (2).
Damage-specific DNA repair glycosylase activities are realized in a number of different protein structural families (for instance see ref. 3); among them, the ‘helix-hairpin-helix’ (HhH) superfamily is of particular interest in that its members are represented in all phylogenetic domains and act on products of a wide variety of DNA-damaging processes such as hydrolytic deamination of cytosine and 5-methyl-cytosine residues (4,5), adenine alkylation (6,7), guanine 8-hydroxylation (8–11) and various forms of saturation of the pyrimidine 5,6-double bond (12,13). Structurally, HhH DNA repair glycosylases are characterized e.g. by the presence of a HhH motif and—in most cases—a [4Fe-4S] cluster (4,8,11,13); a common mechanistic feature is the flipping of the damaged base from the continuous stack inside the DNA double helix into a base-specific binding pocket provided by the enzyme (6,7,9,11,13). This property is shared with glycosylases of other structural families, such as human UDG (14) or Escherichia coli MUG (15).
Previously, we have described DNA mismatch glycosylase Mig.MthI from Methanobacterium thermoautotrophicum as a member of the HhH superfamily that removes the thymine base from T/G mismatches and, hence, is able to initiate repair that protects the genome of this thermophilic archaeon against the mutagenic effect of hydrolytic deamination of DNA 5-meC residues (4). This assignment as the primary function of the enzyme is endorsed by the fact that the Mig.MthI gene is located in immediate vicinity to one that encodes a DNA cytosine-5-methyltransferase (16).
With the study presented here, we address questions as to the structural basis of substrate specificity and catalytic mechanism of Mig.MthI and show that two amino acid substitutions are sufficient to change its substrate preference from T/G to A/G, the preferred substrate of MutY, another class of HhH DNA repair glycosylases (10).
MATERIALS AND METHODS
Bacterial strains
DH5-α: F–, ϕ80-dlacZΔM15, endA1, recA1, hsdR1 (rk–mk+), supE44, thi-1, gyrA96 (Na1r), relA1, Δ(lacZYA-argF) U169 (17). BL21: F–, ompT hsd, SB (rB–mB–), an E.coli B-strain (18). Its BL21 (DE3) pLysS derivative: F–, ompT hsd, SB (rB–mB–) gal dcm (λDE3) pLysS (CmR), was used for transformation. pET21d was from Novagen (Madison, WI). pET21d-mig containing mig gene was from Jens-Peter Horst (Georg-August-Universität Göttingen, Germany).
Enzymes
Pfu Turbo DNA Polymerase was purchased from Stratagene (La Jolla, CA), restriction enzymes from New England Biolabs (Beverly, MA) and MBI Fermentas (Vilnius, Lithuania). Reagents were of analytical grade and supplied from Merck (Darmstadt, Germany) or Sigma (St Louis, MO).
Synthetic oligonucleotides and heteroduplex construction
All 2′-deoxyribo-oligonucleotides were purchased from Metabion GmbH (Martinsried, Germany) (sequences read from left to right in 5′ to 3′ direction). LQ187 (31mer), GTGCAGGGACTTTAACCAAGGTTTAATGGAC; AV050 (31mer), CTACTTCGCAGGACAACTGTGGGGCATGTTA; YUP1 (18mer), CAAGACCCGTTTAGAGGC; YLO1 (18mer), ATGGTGCATGCAAGGAGA. Oligonucleotides LQ187 and AV050 were used for directed mutagenesis (altered codons underlined), YUP1 and YLO1 were used for gene amplification by PCR. The following oligonucleotides, for use in multiple substrate kinetics (see below), were of ‘HPLC-purified’ quality. 35-G, CTGCGACAGATTAAGGGCCTCGGAGATAAGCCAAG; 40-T, F-GGGTACTTGGCTTATCTCCGAGGTCCTTAATCTGTCGCAG; 40-U, F-GGGTACTTGGCTTATCTCCGAGGUCCTTAATCTGTCGCAG; 45-A, F-GGCTTGGGTACTTGGCTTATCTCCGAGGACCTTAATCTGTCGCAG; 50-T, F-GTGGCGGCTTGGGTACTTGGCTTATCTCCGAGGTCCTTAATCTGTCGCAG; 50-U, F-GTGGCGGCTTGGGTACTTGGCTTATCTCCGAGGUCCTTAATCTGTCGCAG. Numbers in the names of oligonucleotides pertain to chain length, letters to residues that are mismatched in duplexes (respective residues are underlined in corresponding sequences); F, a fluorescein moiety. 35-G was used as the universal lower strand in heteroduplex duplex constructions (compare below).
Construction of genes encoding Mig.MthI (A50V), Mig.MthI (L187Q) and Mig.MthI (A50V/L187Q)
A structural gene for the Mig.MthI (A50V) mutant was constructed by applying the ‘megaprimer’ mutagenesis technique (19) to the mig gene present in plasmid pET21d. PCR reactions were carried out with 2.5 U Pfu DNA polymerase and 20 pmol of each dNTP in 50 µl 20 mM Tris–HCl pH 8.8, 2 mM MgSO4, 10 mM KCl, 10 mM (NH4)2SO4, 0.1% Triton X-100, 0.1 mg/ml nuclease-free bovine serum albumin, 1.5% DMSO. Settings for PCR (30 cycles) were as follows: 95°C for 60 s, 55°C for 80 s and 72°C for 80 s. The megaprimer was prepared by PCR starting with 3 ng mig-containing pET21d, 20 pmol primer YUP1 and 20 pmol primer AV050 (introduces the desired mutation). The second PCR reaction was performed under identical conditions except that for five cycles a 10 µl aliquot of the product of the first reaction was used as the sole primer, then 20 pmol of primer YLO1 were added for the remaining cycles. The DNA product was cleaved with restriction endonucleases NcoI and XhoI and purified by electrophoresis through low melting point agarose gel. The resulting fragment was inserted by T4 DNA ligase reaction between the corresponding restriction sites of plasmid pET21d. Ligation products were used to transform E.coli strain DH5-α. Correct clones were identified by DNA sequence analysis of the entire mig gene. Plasmid DNA from a correct clone was isolated and used to transform protein production host strain BL21 (DE3) pLysS. Likewise, a gene coding for Mig.MthI (A50V/L187Q) was constructed by an analogous procedure using the following starting materials: (i) structural gene encoding single mutant Mig.MthI (A50V); (ii) mutagenic primer LQ187; and (iii) the same flanking primers (YUP1 and YLO1) as in the first round of directed mutagenesis. Insertion of the resulting DNA fragment into the expression plasmid, identification and verification of correct clones, and transfer of recombinant plasmid into the protein production host were as above. Finally, a gene coding for Mig.MthI (L187Q) was prepared as follows. pET21d vectors harboring genes for Mig.MthI and Mig.MthI (A50V/L187Q) were separately cleaved with restriction endonuclease BsaI to yield, in each case, two fragments of 1631 and 4398 bp (BsaI cleaves within the bla gene of the vector and in between codons 50 and 187 of the mig gene). The shorter fragment of the plasmid carrying the wild-type gene and the longer fragment of the plasmid carrying the double mutant were isolated by preparative agarose gel electrophoresis and joined by DNA ligation. Transformation etc. were as described above.
Production and purification of Mig.MthI, Mig.MthI (A50V), Mig.MthI (L187Q) and Mig.MthI (A50V/L187Q)
For the production of Mig.MthI, Mig.MthI (A50V), Mig.MthI (L187Q) and Mig.MthI (A50V/L187Q), BL21 (DE3) pLysS cells, transformed by the corresponding plasmid (compare above) were grown in 1 l dYT medium containing 50 µg/ml ampicillin and 34 µg/ml chloramphenicol at 37°C to an OD600 = 0.6–0.8. IPTG was added to a final concentration of 1 mM. After 3 h of further growth, cells were harvested by centrifugation and resuspended in 25 mM HEPES–KOH, pH 7.6, 0.5 M NaCl, 5 mM β-mercaptoethanol. After cell rupture in a French pressure cell (138 Mpa) and sonification, debris was sedimented by centrifugation (25 000 g for 20 min; Sorvall SS34). The supernatant was loaded onto a Chelating Sepharose Fast Flow (Amersham Biosciences, Buckinghamshire, UK) column charged with nickel. Enzyme was eluted with a stepwise gradient of imidazole (0, 30, 60, 80, 90, 100, 300, 500 and 1000 mM). Fractions containing highly enriched Mig.MthI or derivatives, easily recognized by their light brown color, were collected, dialyzed against 20 mM HEPES–KOH, pH 7.6, 5 mM β-mercaptoethanol, and loaded onto a 7.8 ml heparin column (POROS 20; PerSeptive Biosystems Inc., Framingham, MA). Enzymes were eluted with a linear gradient of sodium chloride (0–1 M). Fractions of essentially homogeneous proteins were pooled and an equal volume of glycerol was added before storing aliquots at –70°C. Yields were as follows: Mig.MthI, 10 mg; Mig.MthI (A50V), 2.48 mg; Mig.MthI (L187Q), 0.8 mg; Mig.MthI (A50V/L187Q), 0.16 mg.
Heteroduplex construction
40-T, 45-A and 50-U (set 1), or 40-U, 45-A and 50-T (set 2) (5 pmol each) were mixed with 75 pmol of 35-G in 100 µl 1× SSC (150 mM NaCl, 15 mM trisodium citrate). The mixtures were incubated in a programmable thermocycler as follows: 90°C for 15 s, 80°C for 3 min, 50°C for 15 min and 20°C for 15 min, and subsequently diluted 1:5 with water.
Multiple substrate kinetics
A volume of 192 µl of 50 mM Tris–HCl pH 9.0, 25 mM (NH4)2SO4 containing 48 µl of the diluted hybridization mixture (see above) was pre-incubated at 50°C for ∼10 min. A 16 µl aliquot was removed and 4 µl of 25 mM Tris–HCl pH 7.5, 5 mM β-mercaptoethanol and 50% glycerol were added. This sample was used as the 0 time point. To the remainder, 44 µl enzyme (110 pmol, in storage buffer or diluted as necessary with 25 mM Tris–HCl pH 7.5, 5 mM β-mercaptoethanol, 50% glycerol; concentration determined by UV-spectroscopy with an ɛ280 = 32 525 M–1 cm–1) were added. Aliquots of 20 µl were removed at indicated time points and mixed with 2 µl 1 M NaOH (final concentration 91 mM). The samples were kept on ice until the end of the time course and then incubated at 95°C for 7–10 min for strand cleavage at AP sites. After cooling to room temperature, 10 µl 95% formamide, 20 mM EDTA and 3 mg/ml dextran blue were added and samples were stored if necessary at –70°C. To an 11% polyacrylamide gel (acrylamide/bisacrylamide 30:1), containing 7 M urea and 1.2× TBE (see below), 7 µl of each sample were applied. The gel was run in a Pharmacia Automated Laser Fluoresence sequencer (A.L.F.) at 2 W laser power. Running buffer was 1× TBE (89 mM Tris, 89 mM boric acid, 2.5 mM EDTA). Peak areas were determined using the Pharmacia ‘Fragment Manager’ program.
Molecular modeling
Molecular modeling was carried out using ‘Insight 2000’ software installed on a Silicon Graphics ‘Octane’ workstation.
RESULTS
Sequence alignment and consequences
Within the HhH superfamily of DNA repair glycosylases, members of the Mig and of the MutY families are special in that they process true mismatches, i.e. oppositions of natural DNA constituents that do not conform to the Watson–Crick base pairing rules. Mig.MthI acts against hydrolytic deamination of 5-meC residues and excises the thymine base from T/G mismatches; in addition it processes U/G oppositions and—with much lower efficiency—G/G, A/G and T/C mismatches (see below and ref. 4). DNA adenine glycosylase MutY has a role in preventing mutations arising from oxidative attack on DNA guanine residues; it removes adenine from A/8-hydroxy-G (‘8-oxoG’) oppositions and also from A/G mismatches (10). Interestingly, E.coli MutY (termed MutY.Eco in the remainder of the text) has weak but measurable activity towards T/G mismatches (Y.N.Fondufe-Mittendorf, unpublished), a situation inverse to the one found with Mig.MthI (compare above). The 221 amino acid residues of Mig.MthI align to the N-terminal 216 amino acid residues of MutY.Eco with 29% amino acid identity and 48% similarity (Fig. 1).
Figure 1.

Amino acid alignment of Mig.MthI (NC_001336) (4), MutY.Eco (M59471) (10) and Endonuclease III (J02857) (12); GenBank accession numbers are given in parentheses. The ClustalW algorithm (24) was used for alignment. The C-terminal 134 residues of MutY are not represented. MutY residues that contact the mismatched adenine base (11) and which are conserved in Mig.MthI are indicated by filled circles. Asterisks indicate MutY amino acid residues involved in adenine binding that are not conserved in Mig.MthI. The four cystein residues contributing the sulfur part to the [4Fe-4S] cluster are highlighted by filled triangles. The exclamation mark points out the aspartic acid residue involved in acid/base catalysis (11). Shading highlights sequence positions with amino acid residues conserved between at least two enzymes.
The three-dimensional structure of MutY.Eco, in complex with its low molecular weight product, adenine, was described previously (11). In the co-crystal, adenine is bound to a deep and tightly tailored pocket, characterized by a network of multiple amino acid–amino acid and amino acid–nucleobase hydrogen bridges and van der Waals contacts (Fig. 2). Several amino acid residues shown earlier by genetic methods to be involved in enzymatic catalysis are part of that pocket which was, therefore, concluded to be identical with the active site of the enzyme. Consequently, the most critical of the amino acid residues constituting the three-dimensional ensemble illustrated in Figure 2 have been termed ‘key determinants of adenine specificity’ of MutY.Eco (11). Together with other structural features of the enzyme, the near complete engulfment of the base by amino acid side chains strongly suggests initiation of repair to proceed by flipping of the mispaired adenine residue from the interior of the continuous base stack of the DNA double helix to its periphery with insertion into the active site of the enzyme, followed by hydrolytic cleavage of the glycosidic bond (11). This mechanism is in accord with that of several other DNA repair glycosylases already characterized structurally; among them AlkA (6), Ogg 1 (9) Endo III (13), hUDG (14) and E.coli MUG (15).
Figure 2.

Stereo-pair illustration of selectivity-determining and catalytically relevant contacts between adenine and MutY.Eco (according to ref. 11 with modifications). The unique pattern of hydrogen bridges established by Glu37 and Gln182 qualifies them as key determinants of adenine specificity. Acid/base catalysis is exerted by Asp138 and Glu37 (11).
By combining the structural information provided by Guan et al. (11) with the sequence alignment shown in Figure 1 it seemed possible, therefore, to locate in the Mig.MthI sequence the ‘key determinants of thymine specificity’ and to model its thymine binding pocket by grafting the corresponding amino acid residues onto the experimentally determined main chain fold of MutY.Eco. Alignment revealed a surprising degree of similarity—six out of seven MutY.Eco amino acid residues identified by Guan et al. as immediately surrounding the flipped adenine (11) (see Figs 1 and 2) are conserved in Mig.MthI, the only difference being the replacement of residue 182 of MutY.Eco (glutamine) by leucine at the corresponding position of Mig.MthI (187; see Fig. 1, conserved base-contacting residues are marked by filled circles). This raises the intriguing possibility that the difference in substrate selectivity of the two enzymes may be determined, to a large extent, by a single amino acid residue, and that the active site of MutY.Eco may resemble very closely that of Mig.MthI. The inherent assumption of a conserved main chain fold can be tested experimentally. If correct, it follows that upon replacing residue Leu187 of Mig.MthI by glutamine, base recognition should be shifted from T/G towards A/G.
Characterization of Mig.MthI (L187Q)
A structural gene coding for Mig.MthI derivative Mig.MthI (L187Q) was constructed as described in Materials and Methods. Wild-type and mutant enzymes were produced by heterologous gene expression in E.coli and purified by column chromatography analogously as illustrated in Figure 3 for another derivative described in detail below.
Figure 3.
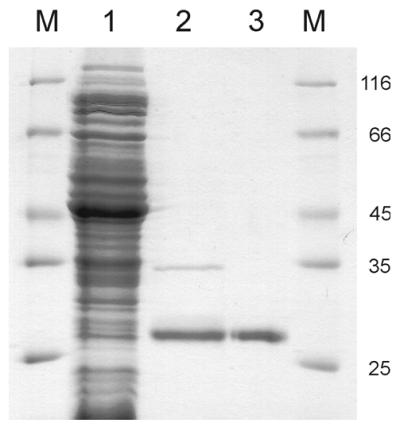
Purification of Mig.MthI (A50V/L187Q). Pattern of electrophoresis through 20% polyacrylamide–SDS gel (Coomassie blue stain). M, marker proteins (relative molecular masses indicated in the right-hand margin as multiples of 1000); lane 1, crude cell extract (flow-through fraction of chromatography on Ni2+ Chelating Sepharose); lane 2, pooled fractions eluting at 100 and 300 mM imidazole; lane 3, protein eluting from POROS heparin column at ∼600 mM NaCl (continuous gradient from 0 to 1000 mM).
Substrate selectivity of Mig.MthI (L187Q) was compared with that of the wild-type enzyme by multiple substrate kinetics (20,21). Heteroduplex DNA substrates were prepared from synthetic oligonucleotides by hybridization. Oligonucleotide 35-G served as the lower DNA strand in all substrate duplexes; it is 35 monomer units long and provides the G residue for the mismatch. For construction of the A/G substrate, oligonucleotide 45-A was hybridized to 35-G. The two oligonucleotides are complementary with the exception of an internal A/G mismatch and a single-stranded protrusion of 10 nt at the 5′-terminus of 45-A that ends in a covalently attached fluorescein moiety. Likewise, T/G and U/G substrates were prepared from 35-G plus 50-T, and 40-U, respectively. Within that set of substrates, cleavage of the upper strand at the mismatched position leads to fluorescent products of 23, 28 and 33 nt, respectively. All six molecular species are readily resolved by gel electrophoresis (compare in Fig. 4). For correction of any effect that mere length of the 5′-protruding ends might have on reaction kinetics (see below), a second set of substrates was constructed in which the lengths of upper strands carrying either T or U were reversed (40-T and 50-U).
Figure 4.

Multiple substrate kinetics: single set of fluorescence tracings. Substrate mixture was 50-T, 45-A and 40-U, the enzyme applied was MigMthI (A50V/L187Q). Time elapsed in gel electrophoresis increases from left to right. Aliquots were taken from the reaction mixture at time points indicated for each tracing. Substrate peaks are labeled in uppercase letters, peaks of corresponding products in lowercase letters. Upon processing, the data set shown gave rise to Figure 5, lower right panel.
Individual substrate sets were incubated with enzyme and strand scission following removal of the mismatched nucleobase from the fluorescently labeled strand was monitored by quantitative gel electrophoresis as decribed earlier (4,21,22) and is documented in Figures 4 and 5. All experiments were independently carried out three times. Peaks were integrated and the progress of the reaction plotted as the percentage of substrate remaining over time; curves were fitted to single exponential decay (Fig. 5 shows one series of experiments with set 1). Relative rate constants were separately derived from individual data sets, then averaged. Finally, mean values of triplicates of substrate set 1 were averaged with those of set 2 (compare in Table 1). The entire study rests on 24 data sets of the kind shown in Figure 4.
Figure 5.

Multiple substrate kinetics: four single sets of processed data derived from substrate mixture 50-T, 45-A and 40-U (ordinate scale is in percentage of substrate remaining). Substrates and graphic symbols are correlated as follows: filled circles, A/G; filled triangles, T/G; open squares, U/G.
Table 1. Relative rate constants obtained from multiple substrate kinetics.
| 40T/50U | 40U/50T | Average | ||||
|---|---|---|---|---|---|---|
| A/G : T/G | U/G : T/G | A/G : T/G | U/G : T/G | A/G : T/G | U/G : T/G | |
| WT | 0.019 ± 0.003 | 1.34 ± 0.09 | 0.017 ± 0.001 | 1.20 ± 0.05 | 0.018 | 1.27 |
| L187Q | 0.42 ± 0.12 | 2.12 ± 0.08 | 0.49 ± 0.03 | 1.83 ± 0.04 | 0.46 | 1.98 |
| A50V | 0.13 ± 0.01 | 2.96 ± 0.32 | 0.15 ± 0.01 | 2.92 ± 0.38 | 0.14 | 2.94 |
| A50V/L187Q | 2.22 ± 0.34 | 1.97 ± 0.38 | 1.94 ± 0.16 | 1.74 ± 0.20 | 2.08 | 1.85 |
Values are given as averages of three independent measurements with standard deviations.
Wild-type Mig.MthI, as the point of reference, exhibited the following properties of substrate selectivity (Table 1). Compared with T/G mismatch, A/G was processed 56 times slower, U/G 1.27 times faster. This confirms and quantitatively specifies our earlier description of substrate selectivity of Mig.MthI (4).
Since no provisions had been made to genetically deplete the E.coli strain used for enzyme production of any of its endogenous uracil glycosylases, potential contributions of these to the observed U/G processing activities must be considered. The strongest argument against such contaminating activities is as follows. Mutant Mig.MthI (Y126K) was prepared and purified exactly as all other Mig.MthI derivatives used in this study. In accord with published data (23), we found Mig.MthI (Y126K) to be an AP-lyase devoid of T/G glycosylase acitivity; in addition, and relevant to the problem of possible contamination, no trace of U/G glycosylase activity was detected either (Y.N.Fondufe-Mittendorf, unpublished).
Mutant enzyme Mig.MthI (L187Q) processes A/G mismatches only 2.2-fold more slowly than T/G mismatches which, compared with wild-type MigMthI, corresponds to a shift in substrate selectivity by a factor of ∼25. Inspection of the reaction time courses (Fig. 5) reveals that this shift has two components: acceleration of the A/G reaction on one hand, and slowing down that of T/G and U/G on the other. At the same time, the preference of U/G over T/G is increased by a factor of ∼1.5.
It should be kept in mind that the reaction proceeds in at least three steps, (i) enzyme/substrate association, (ii) substrate isomerization to the flipped-out form and (iii) cleavage of the glycosidic bond, and that multiple substrate kinetics yield selectivities as relative second-order rate constants for the global process. All experiments were done with a large excess of enzyme, i.e. under bona fide substrate saturation conditions; rates would hence be expected to be governed by the isomerization equilibria and by the rate constants (kcat) of the irreversible cleavage reaction. For comparison of T/G and U/G, it seems reasonable to assume identical or very similar kcat values, in which case the different rates would directly reflect the corresponding substrate isomerization equilibria. When comparing T/G and U/G as a group with A/G, deconvolution of the observed selectivity into contributions of kcat and of pre-equlibrium is less straightforward and must remain tentative until additional experiments.
In summary, the expectation was borne out that replacement of Leu187 of Mig.MthI would result in increased proficiency of the enzyme to process A/G mismatches. This endorsed the validity of the starting model and provided encouragement to search for additional amino acid exchanges that could possibly shift the substrate selectivity of the enzyme still further.
Properties of Mig.MthI (A50V) and of Mig.MthI (A50V/L187Q)
In order to identify additional ‘key determinants’ of base recognition in both MutY.Eco and Mig.MthI, the question of which amino acid side chains are in close contact to adenine in the co-crystal with MutY.Eco, and at the same time different in Mig.MthI, was re-examined. Val45 of MutY.Eco approaches C-2 of adenine with a minimal distance of ∼4.2 Å (Fig. 6); the corresponding position in Mig.MthI (number 50) is occupied by an alanine residue.
Figure 6.

Stereo-pair illustration of hydrophobic environment of adenine, bound to the active site of MutY.Eco (according to ref. 11). Adenine is represented as space-filling model. Red, Leu40; magenta, Trp24; orange, Leu22; green, Arg19; yellow, Val45.
Mutant enzyme Mig.MthI (A50V) was constructed and prepared as described above and characterized with respect to its substrate selectivity (Fig. 5 and Table 1). Compared with wild-type Mig.MthI, the A/G versus T/G selectivity is shifted by a factor of ∼7.7 (from 0.018 to 0.14 relative rate constant). The absolute rate with which A/G is processed by Mig.MthI (A50V) is roughly comparable with that observed with Mig.MthI (L187Q), and the lower shift in selectivity is due to a much less pronounced slowing effect on T/G processing. With a relative rate constant of 2.94, the preference for U/G over T/G rises 2.3-fold, compared with the wild-type enzyme.
Finally, the two amino acid replacements were combined in double mutant Mig.MthI (A50V/L187Q). At a factor of 117, the change of substrate selectivity (A/G versus T/G) from wild-type to double mutant is smaller than the product of the two corresponding values measured for the individual single mutants, yet large enough to make Mig.MthI (A50V/L187Q) act twice as fast on A/G than on T/G and, thus, to qualitatively invert the substrate preference.
DISCUSSION
Modeling thymine recognition and mechanism of catalysis by Mig.MthI
The binding of an adenine base by MutY.Eco can be briefly summarized as follows (compare Figs 2 and 6) (11). Two hydrophobic side chains (Leu40 and Met185) sandwich the heterocyclic ring in between them and, thus, define a plane within which the base can move. Within that plane, adenine is fixed in one particular location by two bidentated hydrogen bridges involving residues Glu37 and Gln182; this pattern commits the active site to the selective binding of adenine (11). Val45, together with Arg19, forms the rim of the pocket near position C-2 of adenine; C-8 points towards the opening of the binding site. With this position of the base, Asp138 is poised to present an activated water molecule in a favorable orientation for nucleophilic attack on the glycosidic carbon center of the sugar moiety, while Glu37—considered to be protonated—delivers a proton to N-7, stabilizing negative ring charge that builds up during the nucleophilic replacement reaction (11).
In Mig.MthI, the same ensemble of amino acid residues is present with the exceptions of Gln182 being replaced by Leu187 and Val45 by Ala50. Below, we offer a model of how Mig.MthI selectively binds thymine. The model rests on the kinetic data documented above, and on the assumption that both the catalytic mechanism and the spatial arrangement of amino acid side chains in the active site are approximately conserved between the two enzymes.
The postulate of conserved catalytic mechanism implies that the location of N-1 of thymine and the direction of the glycosidic bond emerging from it should be arranged in close accord with the corresponding features in the MutY.Eco/adenine complex. This being kept fixed, the thymine base has only one degree of motional freedom left, its rotation around the glycosidic bond. Along the full circle of rotation, Leu45 and Met190 of Mig.MthI allow only two positions for the base to adopt, one corresponding to that occupied by adenine in its complex with MutY.Eco, the other related to it by a rotational angle of 180°.
The fact that replacement of Ala50 of Mig.MthI by Val increases discrimination between thymine and uracil in favor of the latter can be tenatively interpreted as a consequence of steric hindrance, i.e. narrowing of the binding pocket on that side where the C-5 methyl group of thymine is bound. This favors the orientation of thymine in which it is rotated by 180° relative to adenine in complex with MutY.Eco (assuming that both bases start their flipping motion from an ‘anti’ conformation of the respective nucleotide residue).
Gratifyingly, this arrangement brings Glu42 of Mig.MthI (equivalent to residue 37 of MutY.Eco; compare Figs 2 and 7) into play in a plausible fashion. It can form a bidentate hydrogen bridge to thymine centers O-4 and NH-3 and, thus, contribute to both base recognition and catalysis—again by neutralizing negative charge as it builds up during the reaction (note that the two oxygen atoms of the Glu42 carboxylic acid end group swap their hydrogen donor/acceptor properties). In this orientation, Leu187 could make hydrophobic contacts near carbon center C-5 (plus attached methyl group in the case of thymine) and Ala50 could close the pocket near C-6.
Figure 7.
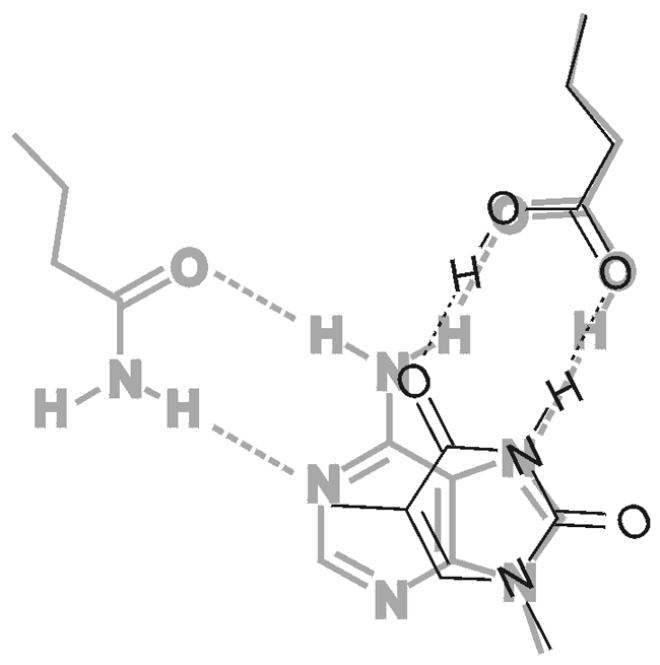
Model of thymine, bound to acitve site of Mig.MthI (narrow black lines), superimposed onto adenine, bound to active site of MutY.Eco (wide gray lines). The amino acid side chain on the top right is Glu37 in the case of MutY.Eco and Glu42 for Mig.MthI. The amino acid side chain at the top left is Gln182, which corresponds to Leu187 in Mig.MthI (also compare with Fig. 2).
As an alternative, thymine binding with formation of an analogous bidentate hydrogen bridge of Glu42 to centers O-2 and NH-3 cannot be ruled out but would require major structural adjustments of side chains making up the base binding site (model not shown). Mechanistically, the two variants of the model are very closely related; O-2 and O-4 are equally well suited for neutralizing negative ring charge by accepting a proton, and proton transfer to O-2 of the pyrimidine ring has already been implied as part of the catalytic mechanism in the case of human uracil DNA glycosylase (14).
A remaining problem concerns thymine (uracil) versus cytosine discrimination. Clearly, Glu42 could make a bidentate hydrogen bridge to cytosine and the transfer of a proton to N-3 could facilitate nucleophilic displacement of the base from the sugar. On the other hand, no excision of cytosine by Mig.MthI has been observed with any C/X opposition, where X is a natural nucleobase (4). One could argue that the different C/X mismatches are excluded on the basis of selectivity added by the nature of the ‘widowed’ base X and C/G because of its high stability as a base pair that might prevent base flipping altogether. This does not explain, however, why T/C and U/C are minor substrates of Mig.MthI, whereas C/C is not (4). Clarification of this point requires additional experiments.
In summary, our model of thymine recognition and of catalysis by Mig.MthI takes into account experimental data of reaction kinetics; it explains why a true key determinant of adenine recognition by MutY.Eco (Glu37) is nevertheless conserved in Mig.MthI (Glu42); and, in particular, the model is in agreement with known features of catalytic mechanisms of related enzymes. Eventually, the detailed mode of action of Mig.MthI needs to be investigated by direct structure analysis. As long as such data are not available, our model may serve as a surrogate; testable predictions can be derived from it and it can, thus, form the rational basis for planning further protein engineering studies.
Evolution of substrate selectivity within the HhH superfamily of repair glycosylases
As already pointed out in the Introduction, the HhH superfamily of DNA repair glycosylases comprises numerous members representing a wide variety of substrate specificities. With the present study we have shown that as few as two amino acid replacements can suffice to switch from one substrate selectivity to another. If this surprisingly small distance in sequence space also holds for the separation of other substrate selectivities within the HhH superfamily, it could explain the strikingly flexible use evolution has found for this structural scaffold. It also means that within the superfamily, association of any given enzyme with a certain substrate recognition specificity needs not necessarily be deep-rooted in an evolutionary sense. This should be kept in mind as a caveat in efforts to functionally annotate pertinent open reading frames in genomic DNA sequences.
Consequences for practical applications
Enzymes, such as Mig.MthI, that are capable of processing DNA base/base mismatches in a selective fashion are interesting tools in practical molecular genetics. Such use would greatly benefit from the availability of an entire palette of enzymes that are similar in general properties, yet able to specifically and individually address a variety of different base/base mismatches. Here we have documented a first step towards establishing such a collection.
Acknowledgments
ACKNOWLEDGEMENT
We thank Ralph Krätzner for advice with molecular modeling.
REFERENCES
- 1.Bernstein C. and Bernstein,H. (1991) Aging, Sex and DNA Repair. Academic Press Inc., San Diego, CA.
- 2.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 3.Pearl L.H. (2000) Structure and function in the uracil-DNA glycosylase superfamily. Mutat. Res., 460, 165–181. [DOI] [PubMed] [Google Scholar]
- 4.Horst J.-P. and Fritz,H.-J. (1996) Counteracting the mutagenic effect of hydrolytic deamination of DNA 5-methylcytosine residues at high temperature: DNA mismatch N-glycosylase Mig.Mth of the thermophilic archaeon Methanobacterium thermoautotrophicum THF. EMBO J., 15, 5459–5469. [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H., Fitz-Gibbon,S., Marcotte,E.M., Tai,J.H., Hyman,E.C. and Miller,J.H. (2000) Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Bacteriol., 182, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollis T., Ichikawa,Y. and Ellenberger,T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase Escherichia coli AlkA. EMBO J., 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollis T., Lau,A. and Ellenberger,T. (2000) Structural studies of human alkyladenine glycosylase and E. coli 3-methyladenine glycosylase. Mutat. Res., 460, 201–210. [DOI] [PubMed] [Google Scholar]
- 8.Nash H.M., Bruner,S.D., Schärer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 9.Bruner S.D., Norman,D.P.G. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- 10.Au K.G., Clark,S., Miller,J.H. and Modrich,P. (1989) Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc. Natl Acad. Sci. USA, 86, 8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guan Y., Manuel,R.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,R.S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- 12.Asahara H., Wistort,P.M., Bank,J.F., Bakerian,R.H. and Cunningham,R.P. (1989) Purification and characterization of Escherichia coli endonuclease III from the cloned nth gene. Biochemistry, 28, 4444–4449. [DOI] [PubMed] [Google Scholar]
- 13.Thayer M.M., Ahern,H., Xing,D., Cunningham,R.P. and Tainer,J.A. (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J., 14, 4108–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupphaug G., Mol,C.D., Kavli,B., Arvai,A.S., Krokan,H.E. and Tainer,J.A. (1996) A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature, 384, 87–92. [DOI] [PubMed] [Google Scholar]
- 15.Barrett T.E., Schärer,O.D., Savva,R., Brown,T., Jiricny,J., Verdine,G.L. and Pearl,L.H. (1999) Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J., 18, 6599–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nölling J. and de Vos,W.M. (1992) Characterization of the archaeal plasmid-encoded type II restriction-modification system MthT1 from Methanobacterium thermoformicicum THF: homology to the bacterial NgoPII system from Neisseria gonorrhoeae. J. Bacteriol., 174, 5719–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D. (1983) Studies on transformation of Escherichia coli with plasmid. J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- 18.Studier F.W. and Moffat,B.A. (1986) Use of bacteriophage T7 RNA-polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol., 189, 113–130. [DOI] [PubMed] [Google Scholar]
- 19.Barik S. and Galinski,M.S. (1991) Megaprimer method of PCR: increased template concentration improves yield. Biotechniques, 10, 489–490. [PubMed] [Google Scholar]
- 20.Schellenberger V., Siegel,R.A. and Rutter,W.J. (1993) Analysis of enzyme specificity by multiple substrate kinetics. Biochemistry, 32, 4344–4348. [DOI] [PubMed] [Google Scholar]
- 21.Gläsner W., Merkl,R., Schellenberger,V. and Fritz,H.-J. (1995) Substrate preferences of Vsr DNA mismatch endonuclease and their consequences for the evolution of the Escherichia coli K-12 genome. J. Mol. Biol., 245, 1–7. [DOI] [PubMed] [Google Scholar]
- 22.Gläsner W., Merkl,R., Schmidt,S., Cech,D. and Fritz,H.-J. (1992) Fast quantitative assay of sequence-specific endonuclease activity based on DNA sequencer technology. Biol. Chem. Hoppe Seyler, 373, 1223–1225. [DOI] [PubMed] [Google Scholar]
- 23.Begley T.J. and Cunningham,R.P. (1999) Methanobacterium thermoformicicum thymine DNA mismatch glycosylase: conversion of an N-glycosylase to an AP lyase. Protein Eng., 12, 333–340. [DOI] [PubMed] [Google Scholar]
- 24.Higgins D.G., Thompson,J.D. and Gibson,T.J. (1996) Using CLUSTAL for multiple sequence alignments. Methods Enzymol., 266, 383–402. [DOI] [PubMed] [Google Scholar]