

Abstract
While nitro and amino alkenes are common in pharmaceuticals, pesticides, and munitions, their environmental fates are not well known. Ozone is a ubiquitous atmospheric oxidant for alkenes, but the synergistic effects of nitrogen-containing groups on the reactions have not been measured. The kinetics and products of ozonolysis of a series of model compounds with different combinations of these functional groups have been measured in the condensed phase using stopped-flow and mass spectrometry methods. Rate constants span about six orders of magnitude with activation energies ranging from 4.3 to 28.2 kJ mol−1. Vinyl nitro groups substantially decrease the reactivity, while amino groups have the opposite effect. The site of the initial ozone attack is highly structure dependent, consistent with local ionization energy calculations. The reaction of the neonicotinoid pesticide nitenpyram, which forms toxic N-nitroso compounds, was consistent with model compounds, confirming the utility of model compounds for assessing environmental fates of these emerging contaminants.
Ozonolysis of model nitrogen-containing alkenes shows a wide range of reactivity and formation of toxic products.
INTRODUCTION
The American Chemical Society Chemical Abstracts Service now has more than 150 million unique compounds in its registry, while ~15,000 new compounds and biological sequences are being registered daily (1). The introduction of even a very small fraction of these into the environment presents enormous challenges in terms of assessing their transport, fates, and impacts. Emerging contaminants (EC), also known as contaminants of emerging concern (2), are naturally occurring or synthetic chemicals and materials described as those “that are not commonly monitored in the environment, but which have the potential to reach into the environment and produce known or suspected adverse ecological and (or) human health effects” (3). Many of these are multifunctional compounds for which there is a paucity of data on their reactions relevant to their potential environmental fates. Given the rapid development of a large number of new compounds, defining all of the reactions of each compound is not practical. Therefore, understanding the molecular properties that determine their reactivity, particularly in species that have multiple functional groups that can act separately or in concert, is essential for developing a predictive capability.
Compounds containing amino, nitro (─NO2), and carbon-carbon double bond (C═C) groups are a very important class of EC that are widely used in pesticides, pharmaceuticals, and insensitive munitions (4–7). Several examples are shown in Fig. 1A. The alkene group on most of these compounds is expected to be highly reactive toward atmospheric oxidants.
Fig. 1. Structures of some EC and model compounds studied.

Structures of (A) some common pharmaceuticals, pesticides, and munitions illustrating different combinations of nitro, amino, and alkene groups in these compounds and (B) selected model compounds. MW, molecular weight.
Ozone is ubiquitous in air, with concentrations from tens to hundreds of parts per billion (ppb) in highly polluted environments (8). Although the kinetics and products of alkene-O3 reactions in the condensed phase (9–13) and gas phase (8, 14, 15) have been studied extensively, reports on ozonolysis of alkenes with nitro and/or amino groups are not as plentiful. In addition, aliphatic amines in their neutral forms react rapidly with O3 as a result of the lone electron pair on the nitrogen atom that is susceptible to electrophilic attack, with second-order room temperature rate constants in solution in the range of 103 to 107 M−1 s−1 (16, 17). Whether reactions of these functional groups are additive, or whether there are synergistic effects, remains to be explored.
A previous study of the ozonolysis of solid nitenpyram (NPM), a neonicotinoid pesticide, showed that the reaction was fast and formed some unique non-oxygen–containing products (18). To explore further the ozonolysis of alkenes with ─NO2 and/or amino groups with the goal of elucidating the impacts of these groups singly or in combination on the reactivity, ozonolysis of the five model compounds and NPM shown in Fig. 1 was carried out. To avoid the simultaneous generation and reaction of OH radicals from the ozone interaction with water, the studies were carried out in the nonpolar solvent CCl4. These particular model compounds have high vapor pressures (Table 1) and can therefore exist in the gas or condensed phases. In the environment, condensed phases include water as well as low-polarity surface organic layers on natural waters (19–21), waxy plant surfaces, organic coatings on the built environment and soils (22, 23), and airborne organic particles that can be mainly hydrocarbons (24). Aerosol phase separation also can form a nonpolar organic shell with a polar inorganic core (25–27). Note that ozone is also more soluble in organics than in water, with the ratio of equilibrium concentrations in organics to that in water being ~10 (28).
Table 1. Rate constants (k) at 298 K, preexponential factors (A), and activation energies (Ea) for the reactions of ozone with model compounds and selected alkenes in CCl4.
| Compound | Structure | Vapor pressure (Pa)* | k(CCl4) ± 1σ (M−1 s−1) | A ± 1σ (M−1 s−1) | (Ea) ± 1σ (kJ mol−1) | References |
|---|---|---|---|---|---|---|
| Nitro-alkenes | ||||||
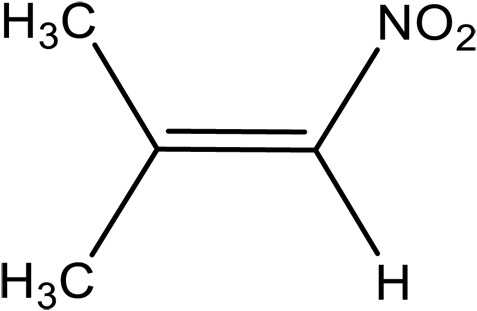
| 2-Methyl-1-nitroprop-1-ene (NTP) |
|
1.1 × 103 | 73 ± 4 | 28.2 ± 0.3 | This work | |
| 4-Methyl-4-nitro-1-pentene (MNP) |
|
4.7 × 102 | (1.4 ± 0.3) × 104 | 17.8 ± 0.5 | This work | |
| Amino-alkenes | ||||||
| E-N,N-dimethyl-1-propenylamine (DPA) |
|
2.0 × 104 | >3 × 107† | – | – | This work |
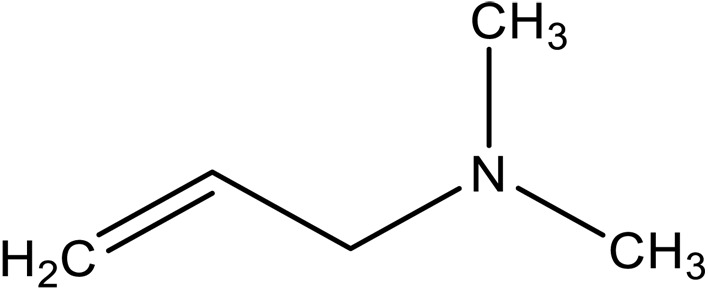
| N,N-dimethylallylamine (DMAA) |
|
2.4 × 104 | (1.1 ± 0.1) × 106 | 4.3 ± 0.4 | This work | |
| Nitro-enamines | ||||||
| 1-Dimethylamino-2-nitroethylene (DMNE) |
|
5.3 × 10−3 | (6.3 ± 0.4) × 105 | 12.9 ± 1.1 | This work | |
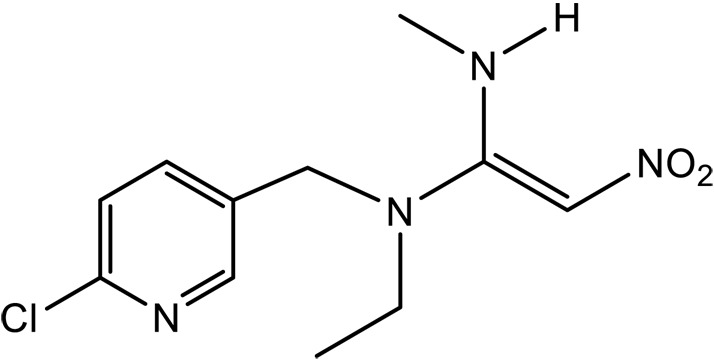
| Nitenpyram (NPM) |
|
1.1 × 10–9‡ | (1.5 ± 0.1) × 105 | 11.1 ± 0.8 | This work | |
| Reference compounds for comparison§ | ||||||
| 2-Methyl-propene |
|
– | 9.7 × 104 | – | – | (11) |
| 4,4-Dimethyl-1-pentene |
|
– | 5.4 × 104 | 3.2 × 106 | 9.6 | (12) |
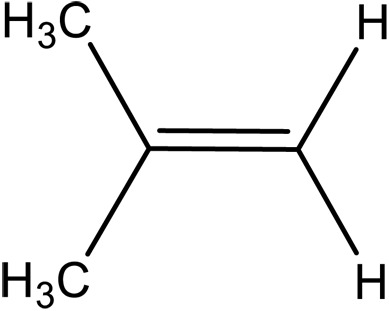
| E-4-Methyl-2-pentene |
|
– | 2.6 × 105¶ | 2.5 × 107 | 11.3 | (12) |
| 2-Methyl-l-chloro-l-propene |
|
2.1 × 104 | 8.2 × 103 | 5.0 × 106 | 15.5 | (13) |
| Trimethylamine |
|
– | 4.1 ± 0.8 × 106# | – | – | (33) |








*Saturation vapor pressures at 25°C of liquid NTP, MNP, DPA, and DMAA were calculated according to Moller formula (83). DMNE (solid) vapor pressure was reported to be 5.3 × 10−3 Pa (53).
†At 278 K.
‡From EPA CompTox Chemicals Dashboard.
§All of the reference compounds are gases at 298 K, except 2-methyl-1-chloro-1-propene.
¶Calculated from A and Ea.
#Neutral form in water.
Typical EC such as those shown in Fig. 1A have very low vapor pressures (≤10−5 Pa) (29) and, hence, are expected to partition only into the condensed phase. Some EC would partition mostly into the nonpolar organic phase, while some would prefer water or both (table S1). Thus, the reactions in the nonpolar and unreactive CCl4 solvent are directly relevant to reactions of the EC in organics, especially those of low polarity, in the environment. The use of this solvent also allowed the reactions of those compounds, such as 1-dimethylamino-2-nitroethylene (DMNE) and E-N,N-dimethyl-1-propenylamine (DPA) shown in Fig. 1B, which hydrolyze in water to be studied as part of the series. However, note that the EC in Fig. 1A do not undergo rapid hydrolysis under most environmental conditions (30–32).
The specific goals were to elucidate kinetics structure-reactivity relationships, products, and mechanisms in the condensed phase, particularly the initial sites of attack when ─NO2 and/or amino groups are present in different combinations and at different positions relative to the C═C. Thus, these data not only form the foundation for assessing their environmental degradation but also provide guidance for the development of alternatives with fewer environmental impacts.
RESULTS
Kinetics studies
Table 1 summarizes the measured room temperature rate constants for the model compounds and NPM in CCl4 and, for comparison, those previously reported for some structurally similar alkenes (11–13). One exception is trimethylamine, which was carried out in water (33). The rate constants for nitro-alkenes [2-methyl-1-nitroprop-1-ene (NTP) and 4-methyl-4-nitro-1-pentene (MNP)] and nitro-enamines (DMNE and NPM) were derived from pseudo–first-order kinetics (Fig. 2A), while those for amino-alkenes [DPA and N,N-dimethylallylamine (DMAA)] were too fast to be obtained by this method. Instead, rate constants for DMAA and DPA were obtained by finding the best fit to the experimental data through numerical integration of the rate equations using the Kintecus program (Fig. 2B) (34). Although all of the model compounds are alkenes, which are known to react with O3, their rate constants span about six orders of magnitude from NTP (73 M−1 s−1) to DPA (>3 × 107 M−1 s−1) because of the strong influence of the ─NO2 and amino groups. This is in contrast to the simple alkenes where the rate constants vary by less than a factor of three.
Fig. 2. Examples of typical kinetics data.

(A) A typical first-order kinetics plot of ozone decay for MNP ozonolysis in CCl4 solution (closed black circles) and its linear regression line (red line), and (B) an example of experimentally measured decay of O3 from the DMAA - O3 reaction in CCl4 solution using the stopped-flow apparatus (closed black circles) and the best-fit from Kintecus based on second-order kinetics (red line).
The data in Table 1 show that replacing a hydrogen in 2-methyl propene with an ─NO2 group (NTP) lowers the room temperature rate constant by about three orders of magnitude. Moving the ─NO2 group two carbons away from the terminal double bond (MNP) has a far less dramatic effect but still reduces the rate constant by a factor of approximately four compared to the structurally similar 4,4-dimethyl-1-pentene.
If an amino group is directly attached to the C═C (DPA), then the reaction is so fast that only a lower limit of 3 × 107 M−1 s−1 at 278 K could be obtained (Table 1). The lone pair of electrons on the amine nitrogen can be delocalized into an adjacent π alkene system (35), increasing the electron density and, hence, rate constant of electrophilic attack by ozone. As discussed below, the products show that there is a direct reaction of ozone with the tertiary amine and the C═C. The presence of an amino group in DMAA that is one carbon away from the double bond results in a room temperature rate constant (1.1 × 106 M−1 s−1) that is at least an order of magnitude smaller than for DPA. Note that the room temperature rate constants for reactions of ozone with the neutral forms of trimethylamine and triethylamine in water have been reported to be 4.1 × 106 and 2.1 × 106 M−1 s−1, respectively (33, 36), similar to that for DMAA in CCl4.
Most interesting is DMNE, with both an ─NO2 group and an amino group attached to the opposite sides of the double bond. Here, the rate constant is about a factor of two larger than for E-4-methyl-2-pentene. This is also about four orders of magnitude faster than NTP, indicating that the simultaneous presence of an amino group on the C═C more than overcomes the suppression due to the ─NO2 group.
Figure 3 shows Arrhenius plots of ln k as a function of T−1, from which the activation energies and preexponential factors (Table 1) are obtained. Activation energies for the reactions of structurally similar alkenes are typically ~10 kJ mol−1 (12, 13). Substitution of ─NO2 on the vinyl carbon (NTP) markedly increases the activation energy to 28.2 kJ mol−1, while the preexponential factor is similar to those for simple alkenes (Table 1). DMAA and MNP are both terminal alkenes, with the substituents (the amino or nitro groups, respectively) one or two carbons removed from the double bond. The activation energy for a similar alkene, 4,4-dimethyl-1-pentene, is 9.6 kJ mol−1. The larger Ea for MNP (17.8 kJ mol−1) reflects a large impact of the ─NO2 group on the double bond reactivity even when displaced from the double bond. The unexpected result is for DMAA, where the amino group is one carbon displaced from the double bond. Compared to 4,4-dimethyl-1-pentene, an alkene of similar structure, the activation energy is lowered by about a factor of 2, and the room temperature rate constant is increased by a factor of 20. This suggests that attack of O3 at the amine nitrogen is important, as, e.g., in DMAA and DPA, which is supported by the products discussed below.
Fig. 3. Arrhenius plots of the temperature dependence of the ozonolysis reactions.

Arrhenius plots for (A) NTP, (B) MNP, (C) DMAA, (D) DMNE, and (E) NPM ozonolysis in CCl4 solution. Error bars (1σ) for specific experiments correspond to the SD for replicate experiments, and errors on the Ea and A are from linear regression.
The neonicotinoid NPM also has the nitro-enamine structure similar to DMNE, but with an additional amino group attached to the double bond. The rate constant for NPM ozonolysis might therefore be expected to be larger. While the activation energies of DMNE and NPM are similar, the room temperature rate constant for NPM ozonolysis is smaller than that for DMNE ozonolysis (see Table 1). This is likely due to steric hindrance from the bulky amino groups around the double bond, as reflected in the smaller pre-exponential factor for NPM. Steric hindrance in ozone-alkene reactions has been reported in a number of studies (11, 12).
The ozonolysis of alkenes generally proceeds through an electrophilic addition of O3 to the C═C. Electron-donating groups increase the rate constants, while electron-withdrawing groups have the opposite effect (9, 37). The ─NO2 group is strongly electron withdrawing because of both the inductive and resonance effects (6). The room temperature rate constant for 2-methyl-1-chloro-1-propene, where ─Cl is also electron-withdrawing, is only one order of magnitude lower than 2-methylpropene but two orders of magnitude larger, with an activation energy about half, than that of NTP (Table 1). Taft constants σ* relate the relative rate constants to the relative strengths of the inductive effects for a specific type of reaction: log k/k0 = σ* ρ* (38, 39). The value of ρ* (which is a measure of the susceptibility to polar effects) is generally believed to be the same for two similar reactions (38, 39), while the values of σ* for ─NO2 and ─Cl are 0.78 and 0.2, respectively (40, 41). The more positive σ* value is indicative of greater electron-withdrawing power of the group, consistent with the lower rate constant for NTP. In DMNE and NPM, where there is a “push-pull” effect due to the combination of an amino and ─NO2 groups on the double bond, the presence of the amino groups has a marked effect, halving the activation energy compared to NTP and increasing the room temperature rate constant by four orders of magnitude. This supports the relevance of DMNE as a model compound for NPM.
Reaction products and mechanisms
Ozone can attack either the C═C, the amine nitrogen, or both. To understand the factors driving the reactivity of the model compounds in terms of these possibilities, a search for products was also carried out. The expected products vary noticeably in terms of structure, size, and volatility, and loss of some products to the gas phase or reaction with solvents used for mass spectrometry (MS) analysis may occur. As a result, obtaining quantitative yields for all of the ozonolysis products was not possible. However, using a combination of Fourier transform infrared (FTIR) and several different MS methods, the major products could be identified, and through the development of reasonable mechanisms, the initial sites of attack were identified. These are summarized in Table 2.
Table 2. Summary of ozonolysis products and the associated initial sites of O3 reaction.
| Compound | Analytical methods | Major products | Main site(s) of attack |
|---|---|---|---|
| NTP
|
FTIR | Secondary ozonide | C═C |
| MNP
|
FTIR | Aldehydes (HCHO) | C═C |
| DPA
|
FTIR and MS | Dimethylformamide and E-N,N-dimethyl-1-propenylamine N-oxide | C═C and ─N─ |
| DMAA
|
FTIR and MS | Dimethylallylamine N-oxideand N-allylmethylamine | ─N─* |
| DMNE
|
FTIR, MS, and IC | Dimethylformamide, HCOOH, dimethylamine, N-nitrosodimethylamine, and nitrite ion (in water extracts) | C═C |
| NPM
|
MS and IC | N-Nitroso and ketone productsand nitrite ion (in water extracts) | C═C |





*Small amounts of carbonyl compounds including DMF observed by FTIR.
2-Methyl-1-nitroprop-1-ene
Figure S1 shows the IR spectrum of NTP before reaction and the difference spectrum after reaction. Ozone-alkene reactions generally proceed by electrophilic addition of ozone to the double bond to form a primary ozonide (POZ), which decomposes into a carbonyl compound and a Criegee intermediate. As shown in fig. S2A, these fragments can recombine to form a secondary ozonide (SOZ) (42–49) if they are held in close proximity, for example, in a solvent cage. The product peaks at 1579 and 1226 cm−1 are characteristic of NO2 asymmetric and symmetric stretches, and the peak at 1125 cm−1 is due to a C─O absorption (fig. S1) (50). These together suggested an ─NO2 containing SOZ (fig. S2A). Because SOZ are peroxides, they are thermally labile and would be expected to decompose on heating (51, 52). To test this, the reacted solution was heated for 30 min at 55° to 65°C. Figure S1 also shows the change in the reacted spectrum after heating. The peaks at 1579, 1226, and 1125 cm−1 decrease, indicating that the product is thermally sensitive. NTP was the only compound for which there was evidence of SOZ formation. After heating, a number of peaks arise in the C═O stretching region, although the products responsible were not specifically identified. Studies of products of the gas phase ozonolysis (53) are mainly small compounds such as formaldehyde and acetone, which, if formed here, may volatilize and hence not be detected.
4-Methyl-4-nitro-1-pentene
Detection of products from the MNP ozonolysis using MS was not successful, suggesting their lowionization efficiencies or that they were too volatile or unstable to be identified. An IR peak at 1733 cm−1 in the difference FTIR spectrum of the reacted MNP sample (fig. S3) indicated the formation of an aldehyde (50). Although HCHO was an expected product (fig. S2B) from ozone attack at the double bond, its characteristic C─H stretch at 2782 cm−1 (fig. S4) was not apparent in the IR spectra. Given the very small absorbances (fig. S3), this second peak may be below the detection limit. Alternatively, given the volatility of HCHO, it may not remain in solution, so this aldehyde peak may be due to the proposed C(CH3)2(NO2)CH2CHO product (fig. S2B).
E-N,N-Dimethyl-1-propenylamine
DPA has two potential reactive sites for attack by O3, the C═C and the amine nitrogen. The IR spectrum of DPA reaction products (fig. S5) shows peaks at 1687, 1383, and 1086 cm−1, which are characteristic of N,N-dimethylformamide (DMF; fig. S4). The yield, ΔDMF/ΔO3, was estimated to be 0.17 ± 0.02 based on calibration with authentic DMF. Because DPA hydrolyzes readily, direct analysis in real-time MS (DART-MS) was adopted, and mass spectra of DPA before and after reaction are shown in fig. S6 (A and B). Peaks due to both DMF [mass/charge ratio (m/z) 74] and an N-oxide, N,N-dimethyl-1-propenylamine N-oxide (m/z 102), were also detected and identified by Orbitrap high-resolution MS (HRMS; Orbitrap) using heated electrospray ionization (HESI) coupled to ultrahigh-pressure liquid chromatography (UHPLC) and tandem MS (MS/MS) analysis (figs. S6, C and D, and S7 and table S2). DMF is expected from attack of ozone at C═C, while the N-oxide is from the reaction with the amine (Fig. 4A). Hence, product formation indicates that the reaction occurs competitively at both sites. Both DMF and DPA N-oxide were also detected in the unreacted DPA solution (fig. S6), which may be due to the reaction of DPA with small amounts of ozone that are always present in ambient air at ~10 to 40 ppb (8). Dimethylamine (DMA) (m/z 46) is expected to form from the reaction of O3 with the C═C; however, the signal intensities of m/z 46 were similar in unreacted and reacted solutions (fig. S6).
Fig. 4. Proposed ozonolysis mechanisms for model compounds.

Reaction mechanisms for (A) DPA, (B) DMAA, and (C) DMNE ozonolysis in CCl4. Red-colored compounds were identified with yields shown in brackets, while the blue-colored compounds were identified but not quantified.
N,N-Dimethylallylamine
Similar to DPA, DMAA also has two potential reaction sites with O3. Figure S8 shows the ESI-MS mass spectra of DMAA using direct infusion after reaction with ozone in CCl4. (Note that the [M + H]+ peak for DMAA is very large compared to the products necessitating a break in the axis; the peak at m/z 87 is the 13C isotope of DMAA.) The product formulae were determined by Orbitrap HRMS analysis with a mass tolerance of 3 ppm (table S2), while their chemical structures were further identified by MS/MS analysis (fig. S9). Peaks due to dimethylallylamine N-oxide (m/z 102 from [M + H]+ and its dimer [2M + H]+ at m/z 203) and N-allylmethylamine ([M + H]+ at m/z 72) were the most intense. N-Oxides have often been detected as major products during ozonation of tertiary amines (16, 54, 55). Small peaks corresponding to carbonyl compounds (50) observed in the FTIR spectrum (fig. S10) could be formed from the reactions of ozone with amine (56) or the C═C. However, it is clear that the ozone is attacking the amine nitrogen preferentially compared to the double bond (Fig. 4B) when the amine nitrogen is not attached to the vinyl carbon.
1-Dimethylamino-2-nitroethylene
This compound contains all three functional groups, C═C, amine, and ─NO2. The difference IR spectrum after reaction (fig. S11) shows small peaks at 1687, 1383, and 1086 cm−1, characteristic of DMF (fig. S4). Absorptions at 1753 and 1723 cm−1 were identified as formic acid by comparison to authentic standards (fig. S4). A peak at 2336 cm−1 was seen as well (see inset fig. S11) and identified as CO2 by comparison to a spectrum taken by bubbling pure CO2 into CCl4 (fig. S4). Figure S12 shows the ESI-MS mass spectrum of DMNE after reaction with ozone. A peak at m/z 74 from DMF is evident, consistent with the IR spectrum. DMA (m/z 46) was also seen as a major product. Unexpectedly, N-nitrosodimethylamine (NDMA) at m/z 75 was identified using UHPLC-Orbitrap MS with an authentic standard. This is of particular importance because more than 90% of N-nitroso compounds have been shown to be carcinogenic in a wide variety of animals (57–59). In the case of DMNE, quantification of many of the products was possible either from the IR spectrum or from the liquid chromatography–mass spectroscopy (LC-MS) using authentic compounds for calibration. The yields, defined as Δ [product]/Δ [DMNE], were as follows: DMF, 0.34 ± 0.02 (FTIR); DMA, 0.23 ± 0.04 (UPLC-TQD-MS); NDMA, 0.43 ± 0.05 (UHPLC-Orbitrap HRMS); and formic acid, 0.63 ± 0.04 (FTIR).
Figure 4C shows a proposed reaction scheme for DMNE ozonolysis that accounts for the observed products, which are consistent with attack mainly on the C═C. This may reflect the strong electron-withdrawing capacity of the ─NO2 group and the strong electron-donating capacity of the amino group, which increases the electron density at the C═C, making the C═C more reactive toward ozonolysis as a result of this push-pull effect. Reactions between nitrous acid (HONO) and secondary (60) or tertiary amines (61) are proposed to form the highly carcinogenic N-nitrosamine, where HONO is formed from the decomposition of nitroformaldehyde (62, 63) and the •OOCH(•)NO2 Criegee intermediate (Fig. 4C) (18). While HONO could not be directly detected, its production was indicated by measurement of nitrite ions after reaction using ion chromatography (IC) of water extracts of the CCl4 solution (fig. S13). The product yield of formic acid is larger than that of NDMA, suggesting an additional formation from other unidentified pathways.
Nitenpyram
NPM is a commercial product that also contains these three functional groups found in DMNE. Products identified by HESI-Orbitrap HRMS and MS/MS analysis (figs. S14 and S15) indicate ozone attack on the C═C, consistent with DMNE ozonolysis. As shown in the reaction scheme in fig. S16, the product with m/z 228 was proposed to be produced by the direct decomposition of the POZ, while the most intense peak at m/z 240 is from the rearrangement of the POZ. The peak at m/z 258 was attributed to a water adduct of m/z 240. The pair of ions have the same retention time, and m/z 258 has a greater intensity in LC analysis because of the presence of water in the mobile phase (fig. S17). Notably, the m/z 212 formed from the decomposition of Criegee intermediate and m/z 433 proposed from the reaction of Criegee intermediate with NPM do not contain oxygen and were also observed in ozonolysis of solid NPM at the air-solid interface (18). As was the case for DMNE, N-nitroso compounds at m/z 200 (fig. S15A) and m/z 300 (fig. S15E) were also detected for NPM ozonolysis, likely formed from similar reaction pathways involving HONO. The presence of nitrite ion in the reacted samples was confirmed by IC. Nitrous acid was also observed during NPM ozonolysis at the air-solid interface (18). Again, DMNE is seen to be useful as a model compound for the neonicotinoid pesticide NPM.
Predicting the site of ozone attack
As discussed above, ozone could attack at either or both of the C═C and/or amino functional groups. Computational insight was obtained from local ionization energy (IE) calculations of the model compounds and NPM. The local IE provides an indication of the energy required to remove an electron from a position in a molecule. When plotted on a molecular surface, locations with a minimum IE can indicate labile reaction sites for electrophilic reactions such as ozonolysis (64–66). Figure 5 shows the average local IE surfaces for DPA, DMAA, DMNE, and NPM and the local minimum values of IE at the C═C, and the amine nitrogen sites for each compound are shown in fig. S18. The values of minimum IE (red) at the amine and C═C sites on DPA are similar, suggesting that ozone attacks both the C═C and amino groups, consistent with the products identified from both reaction sites. For DMAA, the IE at the C═C site is much larger than that at the amine site, indicating that the amine site is more favorable, also consistent with the identified products. For DMNE and NPM, both the products measured and the IE are consistent with the C═C site being more reactive to ozone. In summary, the combination of the electron-withdrawing/donating effects of substituents and their multiple reaction sites provides a path forward for predicting multifunctional alkene ozonolysis rate constants and reaction mechanisms and, hence, a predictive capability that is critical for assessing their environmental fates.
Fig. 5. Average local IE surfaces for DPA, DMAA, DMNE, and NPM calculated at 0.001 electrons/bohr3 density surface using Spartan′20 with B3LYP/6-311+G** in a nonpolar solvent.

DISCUSSION
The EC and model compounds in Fig. 1 can partition into aqueous layers or into organic media in the environment such as surface organic layers on water (19–21), waxy plant surfaces, organic coatings on indoor and outdoor environments and soils (22, 23), and airborne organic particles (24). Octanol-water partitioning coefficients for the model compounds and some EC are given in table S1. It is seen that the model compounds and EC will partition mainly into the nonpolar organic phase, while some EC prefer water and some will partition between the phases. The studies reported here are directly relevant to EC that partition into the organic phase.
Our results show that a nitro group in the vinyl position of an alkene markedly slows the ozonolysis by about three orders of magnitude compared to that of the corresponding alkene. However, if there is an additional amino group on the opposite side of the C═C (e.g., DMNE), then the reaction becomes even faster than that of the corresponding alkene. The amine is a strong electron-donating group and, in addition, is itself quite reactive toward ozone. This is highlighted by the DPA reaction where ozone attacks both the amino and C═C groups. Unexpectedly, in the case of DMNE with an ─NO2 group on the opposite side of the C═C from the amine, ozone seems to mainly attack the C═C group instead of the amine nitrogen, while the amino group becomes more reactive to ozone if it is not attached to the C═C double bond, for example, in DMAA.
Ozonolysis of alkenes has been reported to be several times to one order of magnitude slower in the gas phase or in CCl4 compared to that in water (36, 67). However, the trend in the kinetics is expected to qualitatively hold. Thus, the dramatic slowing of the ozonolysis due to a vinyl ─NO2 group and the increase due to an amino group is consistent with the results of recent studies of the gas phase reactions of the model compounds (53). Note that the reaction rate constant and pathway for amine compounds in water vary depending on the pH, because the reaction rate constants for protonated amines with ozone are much smaller than those for neutral amines (36, 54). Thus, ozone will likely attack the C═C instead of protonated amine nitrogen during the amino-alkene ozonolysis under acidic conditions (54).
For both the model compounds and EC, the product distributions in aqueous environmental media are expected to differ somewhat from those in CCl4, because water reacts with Criegee intermediates generated in the first steps (68, 69). However, on the basis of the mechanisms proposed here (Fig. 4 and fig. S2), the production of DMF, formic acid, N-oxides, and aldehydes is still expected. N-Nitroso compounds have also been reported as common products of the reactions of secondary and tertiary amines with HONO in acidic environments (60, 61, 70), where HONO was formed from the decomposition of nitroformaldehyde (62, 63) and •OOCH(•)NO2 Criegee intermediate (18).
Products of these reactions include a number of species of concern from the point of view of health effects, including NDMA and DMF (59). Thus, environmental assessments of the impacts of EC that are multifunctional alkenes such as heartburn medications and pesticides shown in Fig. 1A should take into account not only the toxicity of the parent compound but also its reaction products. The results presented here have even wider applicability because ozone is often used in the preparation of drugs, synthetic lubricants, and many other commercially useful organic compounds (71, 72) as well as water treatment (16). The similarity in the chemistry of the model compound DMNE and the neonicotinoid pesticide NPM supports the use of simpler model compounds for predicting the chemistry of EC. Similar studies of additional model compounds will help to further refine this approach to assessing the fates of EC.
MATERIALS AND METHODS
Preparation of ozone solutions
HPLC-grade CCl4 (≥99.9%; Sigma-Aldrich) was used as a nonparticipating solvent that avoids OH generation from ozone reactions with water (73–75) or organic solvents containing a C-H bond (47). Ozone was generated by passing oxygen (99.993%; Praxair) through an ozonizer (Polymetrics Inc.) and bubbled into CCl4 at a flow rate of ~300 ml min−1 at room temperature for 20 min. Dissolved O3 concentrations were measured using an ultraviolet-visible (UV/Vis) spectrometer (Hewlett Packard model 8452A; path length of 1 cm) or a stopped-flow spectrophotometer (SX-18MV, Applied Photophysics, Leatherhead, UK) with molar absorption coefficients from Nakagawa et al. (76). Kinetics studies were carried out under a range of O3 concentrations as shown in table S3 to verify that measured rate constants remained constant over the range. For product studies, O3 solutions (0.15 to 0.20 mM) were prepared by flowing pure O2 (100 ml min−1) through a low-pressure mercury lamp (78-2046-5, Jelight Co. Inc.) into CCl4.
Model compounds and neonicotinoid NPM
The model compounds NTP (95%; AK Scientific), MNP (96%; MuseChem), DMAA (≥99%; Sigma-Aldrich), DMNE (97%; Sigma-Aldrich), and NPM (99.5%; Sigma-Aldrich) were used as received. DPA was synthesized by condensation of anhydrous DMA with propionaldehyde following the procedure of Ellenberger et al. (77). More details regarding the synthesis are found in text S1. Synthesized DPA was determined to be the E-isomer (78) by 1H nuclear magnetic resonance (NMR), and gas chromatography–MS analysis showed a purity of ~93% (fig. S19). Each of the model compounds was dissolved in CCl4 to give concentrations in the range of 66 μM to 20 mM. Because DMNE and NPM had limited solubility in CCl4, DMNE or NPM was first added to the CCl4 and mixed for 30 min. The supernatant containing small concentrations of DMNE or NPM was withdrawn for the experiments. To quantify the DMNE or NPM concentrations in CCl4, a known amount of DMNE or NPM was first dissolved in acetonitrile (≥99.9%; Sigma-Aldrich) and then diluted by CCl4 more than 300 times to generate a calibration curve.
Stopped-flow experiments
Kinetics measurements in CCl4 were performed using a stopped-flow spectrophotometer that allowed effective data collection starting at 2 ms. The CCl4 solutions of the model compound or NPM and ozone were loaded into individual syringes. A known volume of each reactant was forced into a quartz reaction cell (20 μl of volume and 1 cm of path length) by a pneumatically controlled ram. A photodiode array detector or photomultiplier was used to monitor the reaction by measuring the decay of ozone and/or the model compound (fig. S20). The stopped-flow experiments were carried out at controlled temperatures ranging from 278 to 298 K using a circulating water bath.
Table S3 summarizes the analyte whose loss was followed for each model compound, the wavelengths used, the reaction conditions, and the range of concentrations of the reactants. The UV/Vis (HP 8452A UV/Vis spectrometer) absorption spectra of NTP, MNP and DMAA, and O3 overlap at the wavelengths used to monitor O3 (fig. S21). More details about the calculations of reaction rate constants for these five model compounds and NPM with ozone were described in text S2. While the ozone concentrations were higher in these studies than would be the case for ambient air levels, this is not expected to affect the measurements of the fundamental rate constants.
Product measurements
Solutions of O3 and the model compounds or NPM in CCl4 were mixed rapidly in a 25-ml round-bottom flask. The use of higher than ambient ozone concentrations could affect the distribution of products if, for example, ozone reacts rapidly with Criegee intermediates or initially formed products. However, the O3 concentrations in the product studies were kept at less than half the model compounds to minimize secondary oxidation reactions, and the products identified are consistent with the proposed mechanisms without having to invoke these secondary reactions. After the reaction, the CCl4 solutions were injected into a liquid IR cell with NaCl windows and a path length of 0.08 mm, and the IR spectra were recorded using FTIR (Mattson, RS Series) with a resolution of 1 cm−1 and 32 co-added scans. For some experiments, after the reaction was complete, the solutions were heated to 55° to 65°C using a water bath for half an hour to probe for thermal decomposition of the SOZ. The solutions were then cooled down to room temperature for the FTIR measurements. In some experiments, gaseous O3/O2 was flowed over the top of the model compound in CCl4 solution, allowing the gas to diffuse into the solution. Standards—including DMF (99.9%; Alfa Aesar), acetone (99.5%; Fisher Chemical), formic acid (≥95%; Sigma-Aldrich), and CO2 (99.99%; Oxygen Service Co.)—were used as is for reference and/or quantification.
Accurate mass measurements of the products were performed using a Thermo Q Exactive Plus Orbitrap high-resolution mass spectrometer (Thermo Fisher Scientific) with a mass resolving power of 140,000 at m/z 200 and a scanning range of m/z 50 to 750. The Orbitrap mass spectrometer was coupled to a Vanquish Horizon UHPLC platform (Thermo Fisher Scientific) equipped with a Luna Omega 1.6-μm Polar C18 150-mm by 2.1-mm (Phenomenex) column. All data were processed using Thermo Fisher Scientific FreeStyle 1.6.
Quantification of some products was performed on a separate LC-MS platform equipped with an Acquity ultra performance LC (UPLC; Waters) system coupled to a Xevo TQD mass spectrometer (Waters). The separation was performed using a Waters Acquity BEH C18 column (1.7 μm, 50 mm by 2.1 mm). This method was used for the quantification of DMA from DMNE ozonolysis using a commercial standard (40 weight % DMA in H2O; Sigma-Aldrich) for calibration. All data were acquired using Masslynx v4.2 and TargetLynx for quantification.
DART-MS (DART-SVP with Vapur Interface; Ionsense) (18, 79) was also used to measure DPA and its ozonolysis reaction products. Reactions with O3 were performed in CCl4. Silicon strips were dipped into the solutions before and after reaction and placed directly into the DART-MS source for sample analysis in the positive ion mode. The DART ion source was coupled to a Xevo TQS triple quadrupole mass spectrometer (Waters). The DART parameters used were as follows: helium reagent gas flow, 3.1 liter/min; helium gas temperature was varied from room temperature to 300°C; and grid electrode voltage, 350 V.
For some experiments, water was used to extract the products from the CCl4 solvent before and after the reaction, and the extracts were analyzed by IC (940 Professional IC Vario, Metrohm) equipped with an anionic column (Metrosep A Supp 5). This was used to identify nitrite ions expected from HONO as a product of DMNE and NPM ozonolysis. Because the extraction efficiency was unknown, nitrite production yield was not quantified here. More information on the various analytical platforms used can be found in text S3.
Local IE surface calculations
Structures of all compounds were optimized, and local IE surfaces were calculated on the density surface (0.001 electron/bohr3) using Spartan′20 (version 1.1.4) with DFT/B3LYP/6-311+G** (64, 66) in a nonpolar solvent using the conductor-like polarizable continuum model (C-PCM) (80). The B3LYP functional is useful for a broad range of organic species (81), and the extended Pople basis set and diffuse function are appropriate for polarizable systems (82).
Supplementary Material
Acknowledgments
We thank P. Richard Dennison for assistance with NMR experiment and analysis and R. B. Gerber and N. Karimova for helpful discussion about the local IE calculations.
Funding: We are grateful to the National Science Foundation for support (grant nos. 2002909 and 1707883 and equipment grant nos.1920242 and 1337080), the Army Research Office for an equipment grant (#W911NF2010064), and the National Institutes of Health grant (GM131920).
Author contributions: B.J.F.-P. designed research. X.W., W.W., J.G., and T.L.P. conducted stopped-flow experiments. X.W., W.W., L.M.W., and V.P. performed product measurements. X.W. and W.W. analyzed data. L.M.W. did Spartan analysis. X.W., W.W., M.J.E., and B.J.F.-P. proposed mechanism. and X.W., W.W., and B.J.F.-P. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Correction (24 July 2023): Due to a production error, an incorrect version of the supplemental PDF was included at the time of publication. This previous version included display errors in figures S3 to S5, S8, S10, and S12 that did not affect the results or conclusions of the paper. These have been corrected.
Supplementary Materials
This PDF file includes:
Texts S1 to S3
Tables S1 to S3
Figs. S1 to S25
References
REFERENCES AND NOTES
- 1.Schnoor J. L., Re-emergence of emerging contaminants. Environ. Sci. Technol. 48, 11019–11020 (2014). [DOI] [PubMed] [Google Scholar]
- 2.OW/ORD Emerging Contaminants Workgroup, Aquatic life criteria for contaminants of emerging concern: General challenges and recommendations (U.S. Environmental Protection Agency, 2008).
- 3.Barroso P. J., Santos J. L., Martín J., Aparicio I., Alonso E., Emerging contaminants in the atmosphere: Analysis, occurrence and future challenges. Crit. Rev. Environ. Sci. Technol. 49, 104–171 (2019). [Google Scholar]
- 4.Nakaike Y., Asahara H., Nishiwaki N., Construction of push-pull systems using β-formyl-β-nitroenamine. Russ. Chem. Bull. 65, 2129–2142 (2016). [Google Scholar]
- 5.Mital A., Synthetic nitroimidazoles: Biological activities and mutagenicity relationships. Sci. Pharm. 77, 497–520 (2009). [Google Scholar]
- 6.Nishiwaki N., A walk through recent nitro chemistry advances. Molecules 25, 3680–3684 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon-Delso N., Amaral-Rogers V., Belzunces L. P., Bonmatin J. M., Chagnon M., Downs C., Furlan L., Gibbons D. W., Giorio C., Girolami V., Goulson D., Kreutzweiser D. P., Krupke C. H., Liess M., Long E., Mcfield M., Mineau P., Mitchell E. A., Morrissey C. A., Noome D. A., Pisa L., Settele J., Stark J. D., Tapparo A., Van Dyck H., Van Praagh J., Van Der Sluijs J. P., Whitehorn P. R., Wiemers M., Systemic insecticides (neonicotinoids and fipronil): Trends, uses, mode of action and metabolites. Environ. Sci. Pollut. Res. 22, 5–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.B. J. Finlayson-Pitts, J. N. Pitts Jr., Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications (Academic Press, 2000). [Google Scholar]
- 9.Bailey P. S., The reactions of ozone with organic compounds. Chem. Rev. 58, 925–1010 (1958). [Google Scholar]
- 10.Williamson D. G., Cvetanovic R. J., Rates of ozone-olefin reactions in carbon tetrachloride solutions. J. Am. Chem. Soc. 90, 3668–3672 (1968). [Google Scholar]
- 11.Williamson D. G., Cvetanović R. J., Rates of reactions of ozone with chlorinated and conjugated olefins. J. Am. Chem. Soc. 90, 4248–4252 (1968). [Google Scholar]
- 12.Pryor W. A., Giamalva D., Church D. F., Kinetics of ozonation. 3. Substituent effects on the rates of reaction of alkenes. J. Am. Chem. Soc. 107, 2793–2797 (1985). [Google Scholar]
- 13.Pryor W. A., Giamalva D., Church D. F., Kinetics of ozonation. 1. Electron-deficient alkenes. J. Am. Chem. Soc. 105, 6858–6861 (1983). [Google Scholar]
- 14.Atkinson R., Baulch D. L., Cox R. A., Crowley J. N., Hampson R. F., Hynes R. G., Jenkin M. E., Rossi M. J., Troe J., Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II—Gas phase reactions of organic species. Atmos. Chem. Phys. 6, 3625–4055 (2006). [Google Scholar]
- 15.Mcgillen M. R., Carey T. J., Archibald A. T., Wenger J. C., Shallcross D. E., Percival C. J., Structure-activity relationship (SAR) for the gas-phase ozonolysis of aliphatic alkenes and dialkenes. Phys. Chem. Chem. Phys. , 1757–1768 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Lim S., McArdell C. S., von Gunten U., Reactions of aliphatic amines with ozone: Kinetics and mechanisms. Water Res. 157, 514–528 (2019). [DOI] [PubMed] [Google Scholar]
- 17.C. von Sonntag, U. von Gunten, Chemistry of Ozone in Water and Wastewater Treatment: From Basic Principles to Applications (IWA Publishing, 2015). [Google Scholar]
- 18.Wang W., Ezell M. J., Lakey P. S. J., Aregahegn K. Z., Shiraiwa M., Finlayson-Pitts B. J., Unexpected formation of oxygen-free products and nitrous acid from the ozonolysis of the neonicotinoid nitenpyram. Proc. Natl. Acad. Sci. U.S.A. 117, 11321–11327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.G. Gade, M. Hühnerfuss, H. Korenowski, Marine Surface Films (Springer, 2006). [Google Scholar]
- 20.Carpenter L. J., Nightingale P. D., Chemistry and release of gases from the surface ocean. Chem. Rev. 115, 4015–4034 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Donaldson D. J., Vaida V., The influence of organic films at the air-aqueous boundary on atmospheric processes. Chem. Rev. 106, 1445–1461 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Ma’Shum M., Tate M. E., Jones G. P., Oades J. M., Extraction and characterization of water–repellent materials from Australian soils. J. Soil Sci. 39, 99–110 (1988). [Google Scholar]
- 23.Capriel P., Beck T., Borchert H., Gronholz J., Zachmann G., Hydrophobicity of the organic matter in arable soils. Soil Biol. Biochem. 27, 1453–1458 (1995). [Google Scholar]
- 24.Jimenez J. L., Canagaratna M. R., Donahue N. M., Prevot A. S. H., Zhang Q., Kroll J. H., DeCarlo P. F., Allan J. D., Coe H., Ng N. L., Aiken A. C., Docherty K. S., Ulbrich I. M., Grieshop A. P., Robinson A. L., Duplissy J., Smith J. D., Wilson K. R., Lanz V. A., Hueglin C., Sun Y. L., Tian J., Laaksonen A., Raatikainen T., Rautiainen J., Vaattovaara P., Ehn M., Kulmala M., Tomlinson J. M., Collins D. R., Cubison M. J., Dunlea E. J., Huffman J. A., Onasch T. B., Alfarra M. R., Williams P. I., Bower K., Kondo Y., Schneider J., Drewnick F., Borrmann S., Weimer S., Demerjian K., Salcedo D., Cottrell L., Griffin R., Takami A., Miyoshi T., Hatakeyama S., Shimono A., Sun J. Y., Zhang Y. M., Dzepina K., Kimmel J. R., Sueper D., Jayne J. T., Herndon S. C., Trimborn A. M., Williams L. R., Wood E. C., Middlebrook A. M., Kolb C. E., Baltensperger U., Worsnop D. R., Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Huang Y., Mahrt F., Xu S., Shiraiwa M., Zuend A., Bertram A. K., Coexistence of three liquid phases in individual atmospheric aerosol particles. Proc. Natl. Acad. Sci. U.S.A. 118, e2102512118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid J. P., Dennis-Smither B. J., Kwamena N. O. A., Miles R. E. H., Hanford K. L., Homer C. J., The morphology of aerosol particles consisting of hydrophobic and hydrophilic phases: Hydrocarbons, alcohols and fatty acids as the hydrophobic component. Phys. Chem. Chem. Phys. 13, 15559–15572 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Freedman M. A., Phase separation in organic aerosol. Chem. Soc. Rev. 46, 7694–7705 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Biń A., Ozone solubility in liquids. Ozone Sci. Eng. 28, 67–75 (2006). [Google Scholar]
- 29.U.S. Environmental Protection Agency, CompTox Chemicals Dashboard;https://comptox.epa.gov/dashboard/).
- 30.Todey S. A., Fallon A. M., Arnold W. A., Neonicotinoid insecticide hydrolysis and photolysis: Rates and residual toxicity. Environ. Toxicol. Chem. 37, 2797–2809 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Vehabovic M., Hadzovic S., Stambolic F., Hadzic A., Vranjes E., Haracic E., Stability of ranitidine in injectable solutions. Int. J. Pharm. 256, 109–115 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Haag W. R., Spanggord R., Mill T., Podoll R. T., Chou T.-W., Tse D. S., Harper J. C., Aquatic environmental fate of nitroguanidine. Environ. Toxicol. Chem. 9, 1359–1367 (1990). [Google Scholar]
- 33.Hoigné J., Bader H., Rate constants of reactions of ozone with organic and inorganic compounds in water—II. Water Res. 17, 185–194 (1983). [Google Scholar]
- 34.J. C. Ianni, A comparison of the Bader-Deuflhard and the Cash-Karp Runge-Kutta integrators for the GRI-MECH 3.0 model based on the chemical kinetics code Kintecus, in Computational Fluid and Solid Mechanics 2003 (Elsevier, 2003), pp. 1368–1372. [Google Scholar]
- 35.Ezell M. J., Wang W., Shemesh D., Ni A., Gerber R. B., Finlayson-Pitts B. J., Experimental and theoretical studies of the environmental sensitivity of the absorption spectra and photochemistry of nitenpyram and analogs. ACS Earth Sp. Chem. 3, 2063–2075 (2019). [Google Scholar]
- 36.Pryor W. A., Giamalva D. H., Church D. F., Kinetics of ozonation. 2. Amino acids and model compounds in water and comparisons to rates in nonpolar solvents. J. Am. Chem. Soc. 106, 7094–7100 (1984). [Google Scholar]
- 37.Hoigné J., Bader H., Rate constants of reactions of ozone with organic and inorganic compounds in water-I. Non-dissociating organic compounds. Water Res. 17, 173–183 (1983). [Google Scholar]
- 38.Hammett L. P., The effect of structure upon the reactions of organic compounds. Temperature and solvent influences. J. Chem. Phys. 4, 613–617 (1936). [Google Scholar]
- 39.Taft R. W. Jr., Concerning the electron-withdrawing power and the electronegativity of groups. J. Chem. Phys. 26, 93–96 (1957). [Google Scholar]
- 40.Hansch C., Leo A., Taft R. W., A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991). [Google Scholar]
- 41.Taft R. W. Jr., The general nature of the proportionality of polar effects of substituent groups in organic chemistry. J. Am. Chem. Soc. 75, 4231–4238 (1953). [Google Scholar]
- 42.Criegee R., Mechanism of ozonolysis. Angew. Chemie Int. Ed. English. 14, 745–752 (1975). [Google Scholar]
- 43.P. S. Bailey, Ozonation in Organic Chemistry I: Olefinic Compounds (Academic Press, 1978). [Google Scholar]
- 44.Epstein S. A., Donahue N. M., The kinetics of tetramethylethene ozonolysis: Decomposition of the primary ozonide and subsequent product formation in the condensed phase. J. Phys. Chem. A 112, 13535–13541 (2008). [DOI] [PubMed] [Google Scholar]
- 45.McIntire T. M., Ryder O., Finlayson-Pitts B. J., Secondary ozonide formation from the ozone oxidation of unsaturated self-assembled monolayers on zinc selenide attenuated total reflectance crystals. J. Phys. Chem. C 113, 11060–11065 (2009). [Google Scholar]
- 46.Nøjgaard J. K., Nørgaard A. W., Wolkoff P., On-line analysis of secondary ozonides from cyclohexene and d-limonene ozonolysis using atmospheric sampling townsend discharge ionization mass spectrometry. Atmos. Environ. 41, 8345–8354 (2007). [Google Scholar]
- 47.Gomez A. L., Lewis T. L., Wilkinson S. A., Nizkorodov S. A., Stoichiometry of ozonation of environmentally relevant olefins in saturated hydrocarbon solvents. Environ. Sci. Technol. 42, 3582–3587 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Kuczkowski R. L., Formation and structure of ozonides. Acc. Chem. Res. 16, 42–47 (1983). [Google Scholar]
- 49.Choe J. I., Srinivasan M., Kuczkowski R. L., Mechanism of the ozonolysis of propene in the liquid phase. J. Am. Chem. Soc. 105, 4703–4707 (1983). [Google Scholar]
- 50.G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts (John Wiley & Sons, 2004). [Google Scholar]
- 51.Finlayson-Pitts B. J., Pham T. T. H., Lai C. C., Johnson S. N., Lucio-Gough L. L., Mestas J., Iwig D., Thermal decomposition of phospholipid secondary ozonides: Implications for the toxicity of inhaled ozone. Inhal. Toxicol. 10, 813–830 (1998). [Google Scholar]
- 52.Hull L. A., Hisatsune I. C., Heicklen J., Vapor-phase thermal decomposition of some simple ozonides. J. Phys. Chem. 76, 2659–2665 (1972). [Google Scholar]
- 53.Wang W., Wang X., Lakey P. S. J., Ezell M. J., Shiraiwa M., Finlayson-Pitts B. J., Gas phase and gas–solid interface ozonolysis of nitrogen containing alkenes: Nitroalkenes, enamines, and nitroenamines. J. Phys. Chem. A 126, 5398–5406 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knoop O., Hohrenk L. L., Lutze H. V., Schmidt T. C., Ozonation of tamoxifen and toremifene: Reaction kinetics and transformation products. Environ. Sci. Technol. 52, 12583–12591 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Zimmermann S. G., Schmukat A., Schulz M., Benner J., Von Gunten U., Ternes T. A., Kinetic and mechanistic investigations of the oxidation of tramadol by ferrate and ozone. Environ. Sci. Technol. 46, 876–884 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Tuazon E. C., Atkinson R., Aschmann S. M., Arey J., Kinetics and products of the gas-phase reactions of O3 with amines and related compounds. Res. Chem. Intermed. 20, 303–320 (1994). [Google Scholar]
- 57.W. Lijinsky, Chemistry and Biology of N-nitroso Compounds (Cambridge Univ. Press, 1992). [Google Scholar]
- 58.R. N. Loeppky, C. J. Michejda, Nitrosamines and Related N-nitroso Compounds (ACS Publications, 1994), pp. 1–18, vol. 553. [Google Scholar]
- 59.IARC, Agents classified by the IARC monographs, volumes 1–114 (2015).
- 60.J.-P. Anselme, The organic chemistry of N-nitrosamines: A brief review, in The Organic Chemistry of Sugars (ACS Publication, 1979), pp. 1–12. [Google Scholar]
- 61.Smith P. A. S., Loeppky R. N., Nitrosative cleavage of tertiary amines. J. Am. Chem. Soc. 89, 1147–1157 (1967). [Google Scholar]
- 62.Guo Y., Smith S. C., Moore C. B., Melius C. F., Kinetics and product branching ratios for the reaction HCO + NO2. J. Phys. Chem. 99, 7473–7481 (1995). [Google Scholar]
- 63.Dammeier J., Colberg M., Friedrichs G., Wide temperature range (T = 295 K and 770–1305 K) study of the kinetics of the reactions HCO + NO and HCO + NO2 using frequency modulation spectroscopy. Phys. Chem. Chem. Phys. 9, 4177 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Brown J. J., Cockroft S. L., Aromatic reactivity revealed: Beyond resonance theory and frontier orbitals. Chem. Sci. 4, 1772–1780 (2013). [Google Scholar]
- 65.Galabov B., Koleva G., Hadjieva B., Schaefer H. F., π-Hydrogen bonding probes the reactivity of aromatic compounds: Nitration of substituted benzenes. J. Phys. Chem. A 123, 1069–1076 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Lundstedt A., Webb M. J., Grennberg H., Ozonolysis of polycyclic aromatic hydrocarbons in participating solvents. RSC Adv. 7, 6152–6159 (2017). [Google Scholar]
- 67.Dowideit P., Von Sonntag C., Reaction of ozone with ethene and its methyl- and chlorine-substituted derivatives in aqueous solution. Environ. Sci. Technol. 32, 1112–1119 (1998). [Google Scholar]
- 68.Long B., Bao J. L., Truhlar D. G., Atmospheric chemistry of Criegee intermediates: Unimolecular reactions and reactions with water. J. Am. Chem. Soc. 138, 14409–14422 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Chao W., Hsieh J.-T., Chang C.-H., Lin J. J.-M., Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor. Science 347, 751–754 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Yang L., Chen Z., Shen J., Xu Z., Liang H., Tian J., Ben Y., Zhai X., Shi W., Li G., Reinvestigation of the nitrosamine-formation mechanism during ozonation. Environ. Sci. Technol. 43, 5481–5487 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Cook G. A., Industrial uses of ozone. J. Chem. Educ. 59, 392 (1982). [Google Scholar]
- 72.Van Ornum S. G., Champeau R. M., Pariza R., Ozonolysis applications in drug synthesis. Chem. Rev. 106, 2990–3001 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Yasui K., Tuziuti T., Kanematsu W., Mechanism of OH radical production from ozone bubbles in water after stopping cavitation. Ultrason. Sonochem. 58, 104707 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Takahashi M., Chiba K., Li P., Formation of hydroxyl radicals by collapsing ozone microbubbles under strongly acidic conditions. J. Phys. Chem. B 111, 11443–11446 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Takahashi M., Horibe H., Matsuura K., Tatera K., Effect of microbubbles on ozonized water for photoresist removal. J. Photopolym. Sci. Technol. 28, 293–298 (2015). [Google Scholar]
- 76.Nakagawa T. W., Andrews L. J., Keefer R. M., The kinetics of ozonization of polyalkylbenzenes. J. Am. Chem. Soc. 82, 269–276 (1960). [Google Scholar]
- 77.Ellenberger M. R., Dixon D. A., Farneth W. E., Proton affinities and the site of protonation of enamines in the gas phase. J. Am. Chem. Soc. 103, 5377–5382 (1981). [Google Scholar]
- 78.Sauer J., Prahl H., Basenkatalysierte Doppelbindungs-Isomerisierungen, II. Synthese einfacher cis-Enamine. Chem. Ber. 102, 1917–1927 (1969). [Google Scholar]
- 79.Cody R. B., Laramée J. A., Durst H. D., Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal. Chem. 77, 2297–2302 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Cossi M., Rega N., Scalmani G., Barone V., Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Shao Y., Molnar L. F., Jung Y., Kussmann J., Ochsenfeld C., Brown S. T., Gilbert A. T. B., Slipchenko L. V., Levchenko S. V., O’Neill D. P., DiStasio R. A., Lochan R. C., Wang T., Beran G. J. O., Besley N. A., Herbert J. M., Yeh Lin C., Van Voorhis T., Hung Chien S., Sodt A., Steele R. P., Rassolov V. A., Maslen P. E., Korambath P. P., Adamson R. D., Austin B., Baker J., Byrd E. F. C., Dachsel H., Doerksen R. J., Dreuw A., Dunietz B. D., Dutoi A. D., Furlani T. R., Gwaltney S. R., Heyden A., Hirata S., Hsu C. P., Kedziora G., Khalliulin R. Z., Klunzinger P., Lee A. M., Lee M. S., Liang W., Lotan I., Nair N., Peters B., Proynov E. I., Pieniazek P. A., Min Rhee Y., Ritchie J., Rosta E., David Sherrill C., Simmonett A. C., Subotnik J. E., Lee Woodcock H., Zhang W., Bell A. T., Chakraborty A. K., Chipman D. M., Keil F. J., Warshel A., Hehre W. J., Schaefer H. F., Kong J., Krylov A. I., Gill P. M. W., Head-Gordon M., Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 8, 3172–3191 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Krishnan R., Binkley J. S., Seeger R., Pople J. A., Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980). [Google Scholar]
- 83.Moller B., Rarey J., Ramjugernath D., Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions. J. Mol. Liq. 143, 52–63 (2008). [Google Scholar]
- 84.Leo A., Hansch C., Elkins D., Partition coefficients and their uses. Chem. Rev. 71, 525–616 (1971). [Google Scholar]
- 85.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.National Institute of Standards and Technology , NIST Chemistry WebBook. SRD 69, (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Texts S1 to S3
Tables S1 to S3
Figs. S1 to S25
References