

Abstract
Increased tetrahydrobiopterin (BH4) generated in injured sensory neurons contributes to increased pain sensitivity and its persistence. GTP cyclohydrolase 1 (GCH1) is the rate-limiting enzyme in the de novo BH4 synthetic pathway, and human single-nucleotide polymorphism studies, together with mouse genetic modeling, have demonstrated that decreased GCH1 leads to both reduced BH4 and pain. However, little is known about the regulation of Gch1 expression upon nerve injury and whether this could be modulated as an analgesic therapeutic intervention. We performed a phenotypic screen using about 1000 bioactive compounds, many of which are target-annotated FDA-approved drugs, for their effect on regulating Gch1 expression in rodent injured dorsal root ganglion neurons. From this approach, we uncovered relevant pathways that regulate Gch1 expression in sensory neurons. We report that EGFR/KRAS signaling triggers increased Gch1 expression and contributes to neuropathic pain; conversely, inhibiting EGFR suppressed GCH1 and BH4 and exerted analgesic effects, suggesting a molecular link between EGFR/KRAS and pain perception. We also show that GCH1/BH4 acts downstream of KRAS to drive lung cancer, identifying a potentially druggable pathway. Our screen shows that pharmacologic modulation of GCH1 expression and BH4 could be used to develop pharmacological treatments to alleviate pain and identified a critical role for EGFR-regulated GCH1/BH4 expression in neuropathic pain and cancer in rodents.
A shared target between cancer and pain
Neuropathic pain is a chronic condition for which effective safe treatments are lacking. Polymorphisms in the GTP cyclohydrolase 1 (GCH1) gene have been associated with chronic pain severity; and modulation of the GCH1/BH4 pathway has been shown to affect neuropathic pain. Here, Cronin et al. screened over a thousand annotated and FDA-approved compounds and showed that the antipsychotic fluphenazine hydrochloride reduced GCH1 expression and had analgesic effects in a neuropathic pain model in rodents. Among the hits, EGFR/KRAS pathway modulators also affected GCH1 expression, and the authors identified a common signaling, downstream KRAS, involving GCH1, that affects chronic pain and lung cancer development in mice. The results suggest that targeting this signaling could alleviate neuropathic pain.
INTRODUCTION
Treatment of chronic pain has been notoriously challenging. The current strategy is mostly one of “trial and error” of the various currently available analgesics, because many patients do not respond to a particular treatment. Analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors, and anti-convulsant drugs such as gabapentin have shown limited efficacy in the treatment of various forms of chronic pain and carry with them considerable side effects (1–4). For example, long-term use of NSAIDs that target cyclooxygenase-2 (COX-2) can lead to gastrointestinal bleedings and cardiovascular complications (5). This has forced many clinicians to prescribe stronger pain killers such as opioids, which in themselves host a magnitude of side effects from nausea and vomiting to constipation, sedation, and dependence. Moreover, because of opioid-induced mu receptor down-regulation, higher doses are needed to elicit the desired antinociceptive effects, and this, combined with the dangerously addictive nature of these drugs, has culminated in an “opioid epidemic” leading to substantial loss of lives as well as familial and societal crises in certain regions (6, 7). Therefore, the underlying mechanisms of chronic pain need to be thoroughly investigated and more effective and safer targets identified to find the next generation of analgesics.
Tetrahydrobiopterin, BH4, is a cofactor molecule essential for the function of several enzymes with critical physiologic and metabolic functions, including the three nitric oxide synthases (neuronal, inducible, and endothelial), alkylglycerol monooxygenase, and aromatic amino acid hydroxylases (phenylalanine, tryptophan, and tyrosine hydroxylases) (8). Through these enzymes, BH4 is required for nitric oxide production, metabolism of ether lipids, phenylalanine catabolism, and synthesis of the neurotransmitters, noradrenaline, adrenaline, serotonin, and dopamine (8). BH4 can be generated through three distinct pathways—de novo, salvage, and recycling pathways—of which de novo is the main route for synthesis. Guanosine triphosphate (GTP) cyclohydrolase 1 (GCH1) is the first and rate-limiting enzyme of de novo–dependent BH4 production. We previously identified a positive correlation between GCH1/BH4 concentrations and neuropathic pain (9). A plethora of reports have confirmed the association of single-nucleotide polymorphisms in the GCH1 locus with chronic pain severity in humans (10–17). Some reports found no link between the amount of BH4 and pain sensitivity, suggesting that the type and intensity of chronic pain as well as ethnicity may play a role in this association (18, 19). However, genetically modified mice support a role for BH4 in neuropathic and inflammatory pain models; increased BH4 results in reduced pain thresholds, whereas reduced BH4 ameliorates pain hypersensitivity (9, 20–25). Therefore, targeting BH4 synthesis represents an attractive strategy for treating certain chronic pain conditions.
Here, we screened 1000 target-annotated and U.S. Food and Drug Administration (FDA)–approved drugs for regulating expression of GCH1 in injured mouse primary sensory neurons and identified and characterized several hits that reduce or enhance GCH1. We identified epidermal growth factor receptor (EGFR) and Kirsten ras sarcoma virus (KRAS) signaling as a strong trigger of GCH1/BH4 expression. Genetic induction of constitutively active KRAS in sensory neurons, in consequence, enhanced pain. Moreover, we showed that GCH1/BH4 constitutes a downstream pathway of mutant Kras-driven lung cancer in rodents.
RESULTS
PKC activation drives Gch1 expression in injured sensory neurons
Upon peripheral nerve injury, Gch1, the rate-limiting enzyme of BH4 de novo synthesis, is induced in injured sensory neurons in the dorsal root ganglion (DRG), and this drives an increase in BH4, which in turn contributes to neuropathic pain hypersensitivity (9, 24). We therefore sought to screen for compounds that can reduce this injury-dependent up-regulation of Gch1. Nerve injury can be modeled in vitro by culturing axotomized DRG cells from naïve Gch1–green fluorescent protein (GFP) transgenic reporter mice (24, 26) in which induction of Gch1 expression can be monitored through GFP expression and intensity (Fig. 1, A and B, and fig. S1, A and B) (24), and we show here that a subset of axotomized DRG cells strongly expresses Gch1-GFP (referred to as GFPHI) (Fig. 1, A and B). We characterized these GFPHI DRG cells as predominantly NeuN+, TrpV1-expressing, and IB4-binding neurons; to a lesser extent, a small proportion were also myelinated as determined by NF200 staining and express the neuropeptide CGRP (fig. S2). We used this approach to screen primary cultured DRG neurons isolated from naïve, Gch1-GFP transgenic reporter mice (24). GFP is strongly induced upon 3 days of culturing axotomized DRG neurons, which mimics the elevation in endogenous GCH1 expression (Fig. 1, B and C). Using this Gch1-GFP reporter system, we set up a primary culture platform in which ~1000 DRG neurons were added to each well of 384-well plates (day 0). On day 1, compounds at various concentrations were added, and on day 3, propidium iodide (PI) was included to identify dead cells. Each well was subdivided into 15 sections, and confocal images of neurons were taken in each section on day 3 (Fig. 1D and fig. S3).
Fig. 1. DRG drug screen identifies PKC as an inducer of Gch1 expression.
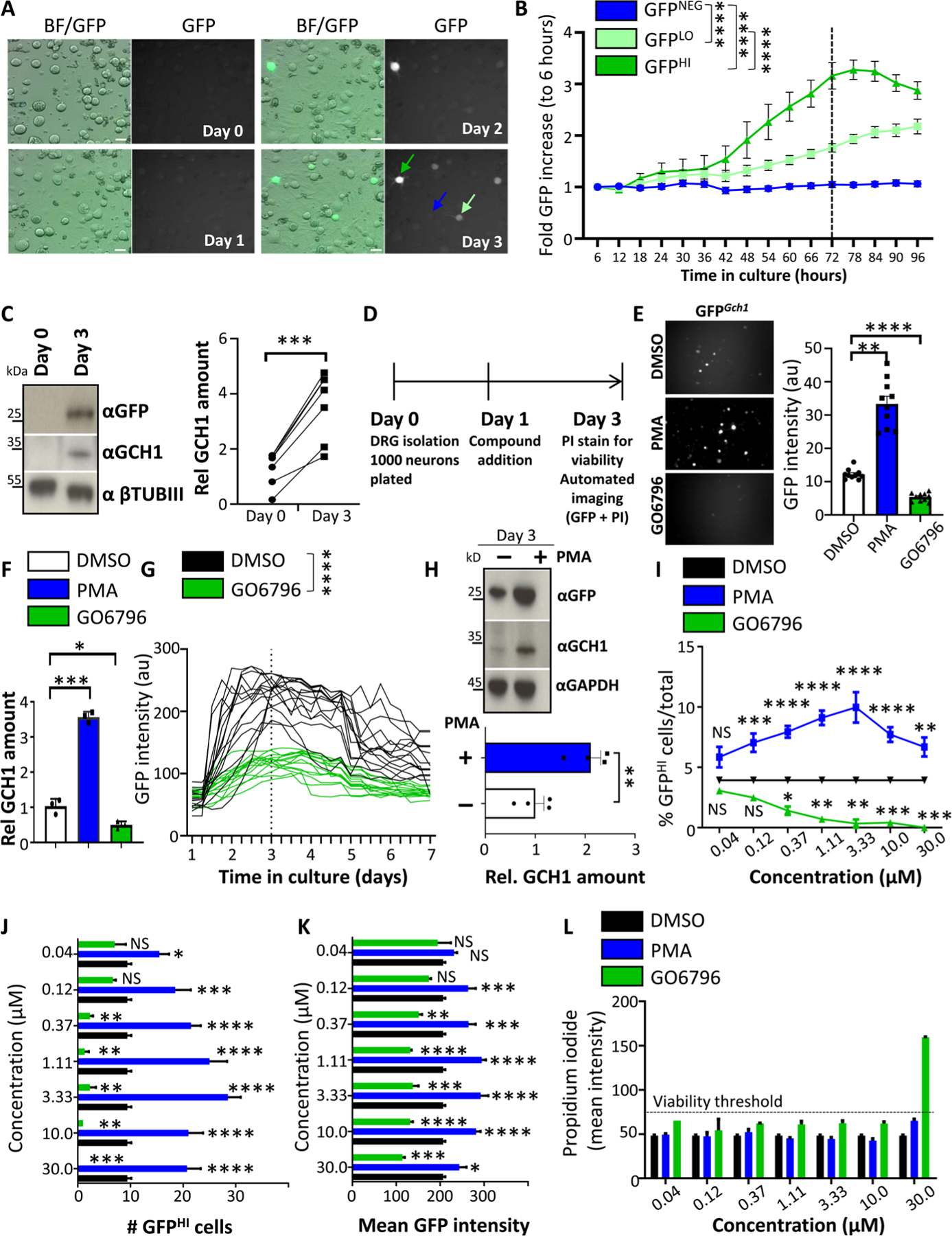
(A and B) Brightfield and fluorescent GFP images of cultured DRG neurons over time isolated from Gch1GFP reporter mice (A) and quantification of increased GFP intensity (B) from GFP high–expressing (dark green arrow), GFP low–expressing (light green arrow), and GFP-negative (blue arrow) DRG neurons. Scale bars, 30 μm. Data are shown as means ± SEM. ****P < 0.0001 (two-way repeated-measures ANOVA). (C) Representative Western blot analysis of cultured DRG neurons on days 0 and 3 blotted with specific antibodies to detect GFP (top) and GCH1 (middle blot). Neuronal-specific anti–β-tubulin III (βTubIII) was used as a loading control (bottom blot). Relative quantification of GCH1 protein on days 0 and 3 of DRG culture from wild-type animals (right). Data are shown as means ± SEM and pooled from five independent experiments. Individual samples are shown. ***P < 0.001 (paired t test). (D) Schematic time frame of the drug screen on cultured DRG neurons from Gch1GFP reporter mice. Compounds were added to the culture on day 1, and GFP as well as propidium iodide (PI) quantification and intensity were analyzed on day 3. (E) Representative GFP fluorescent images and quantification of GFP intensity from wells of cultured (day 3) DRG neurons isolated from Gch1GFP reporter mice treated with PKC modifier drugs [PMA (3 μM), which activates PKC; GO6796 (0.3 μM), which inhibits PKC] on day 1. Data are shown as means ± SEM. Individual samples are shown. **P < 0.01; ****P < 0.0001 (one-way ANOVA with Dunnett’s multiple comparison test). au, arbitrary units. (F) Validation of the effect by PKC regulation on Gch1 expression by RT-qPCR on DRG neurons from wild-type animals treated with DMSO as vehicle control, PMA (3 μM), and GO6796 (0.3 μM). Data are shown as means ± SEM. Individual samples for each treatment condition are shown. *P < 0.05; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparison test). (G) GFP intensities over time of individual GFP-expressing DRG neurons isolated from Gch1GFP reporter mice and treated with DMSO and GO6796 (0.3 μM) 24 hours after plating. Data are shown as means ± SEM. Individual intensities over time are shown. ****P < 0.0001 (two-way repeated-measures ANOVA). (H) Representative Western blot of GFP and GCH1 on DRG neurons isolated from Gch1GFP reporter mice treated with PMA (3 μM). GAPDH was used as a loading control (top). Relative quantification of GCH1 protein on day 3 of DRG culture from wild-type animals untreated and treated with PMA (3 μM) (bottom). Data are shown as means ± SEM and pooled from three independent experiments. Individual samples are shown. **P < 0.01 (paired t test). (I to K) Analysis of various parameters on day 3 of cultured Gch1GFP DRG neurons treated with the indicated doses of PMA and GO6796 as well as the DMSO vehicle control (note that DMSO concentration was kept constant at 0.1%) including % GFPHI cells among total identified neurons (I), total numbers of GFPHI neurons per well (J), and mean GFP intensity per well (K). Data are shown as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant (one-way ANOVA with Dunnett’s multiple comparison test to DMSO treatment). (L) Mean intensity of PI staining per well as a measure of toxicity. PI intensity greater than 75 was arbitrarily taken to be considered toxic (dotted black line). Note toxicity of GO6796 at a concentration of 30 μM. Data are shown as means ± SEM.
We first identified positive and negative control compounds. Very little is known about upstream factors regulating Gch1 in injured DRG neurons; however, we recently uncovered a role of protein kinase C (PKC) activation in the induction of Gch1 expression upon T cell activation (26). Pharmacological activation of PKC using phorbol 12-myristate 13-acetate (PMA) increased the number of GFPHI DRG neurons, whereas inhibition of PKC using the specific PKC inhibitor GO6796 greatly reduced the number of GFPHI neurons (Fig. 1E). These data were corroborated using reverse transcription quantitative polymerase chain reaction (RT-qPCR), in which the effect of these compounds on Gch1 mRNA was assessed (Fig. 1F). Next, we investigated the effects of control dimethyl sulfoxide (DMSO) and GO6796 on Gch1-GFP expression kinetics of individual neurons over 7 days (Fig. 1G). GO6796 strongly reduced the kinetics of Gch1-GFP expression by day 3, and by day 7, the intensity of GFP was comparable to that of baseline (Fig. 1G). Conversely, PMA increased GFP protein, as well as endogenous GCH1 expression in injured DRG neurons in vivo from Gch1-GFP reporter animals (Fig. 1H). Using these positive (PMA) and negative (GO6796) compounds, as well as vehicle (DMSO)–treated controls, we established screening parameters and readouts. Our principal readout was the percentage of GFP high-expressing (GFPHI) DRG neurons among the total number of identified neurons in each well (% GFPHI cells/total neurons per well) (Fig. 1I). In addition, we assessed total numbers of GFPHI neurons, as well as the mean GFP intensity, per well (Fig. 1, J and K). Each well was stained with PI to identify dead neurons to limit false-positive hits, in which a given compound might have induced cytotoxicity (Fig. 1L). Using this setup, we screened 1116 annotated compounds, primarily targeting kinases and signal transduction molecules, as well as ion channels, G protein–coupled receptors (GPCRs), and epigenetic modifiers (Fig. 2A and table S1). For the primary screen, two doses were chosen for each library compound within each experimental 384-well plate setup (fig. S4 and table S2). Vehicle (DMSO), positive (PMA), and negative (GO6796) controls were included to ensure quality control and limit plate-to-plate variations (Fig. 2B and fig. S4). Each well was also stained with PI before image acquisitions to identify and exclude compound toxicity. We therefore successfully set up a controlled screening platform to identify compounds that induce or reduce expression of Gch1 in DRG cultures from Gch1-GFP reporter mice.
Fig. 2. Screen of annotated drugs for regulation of GCH1 in DRG sensory neurons.
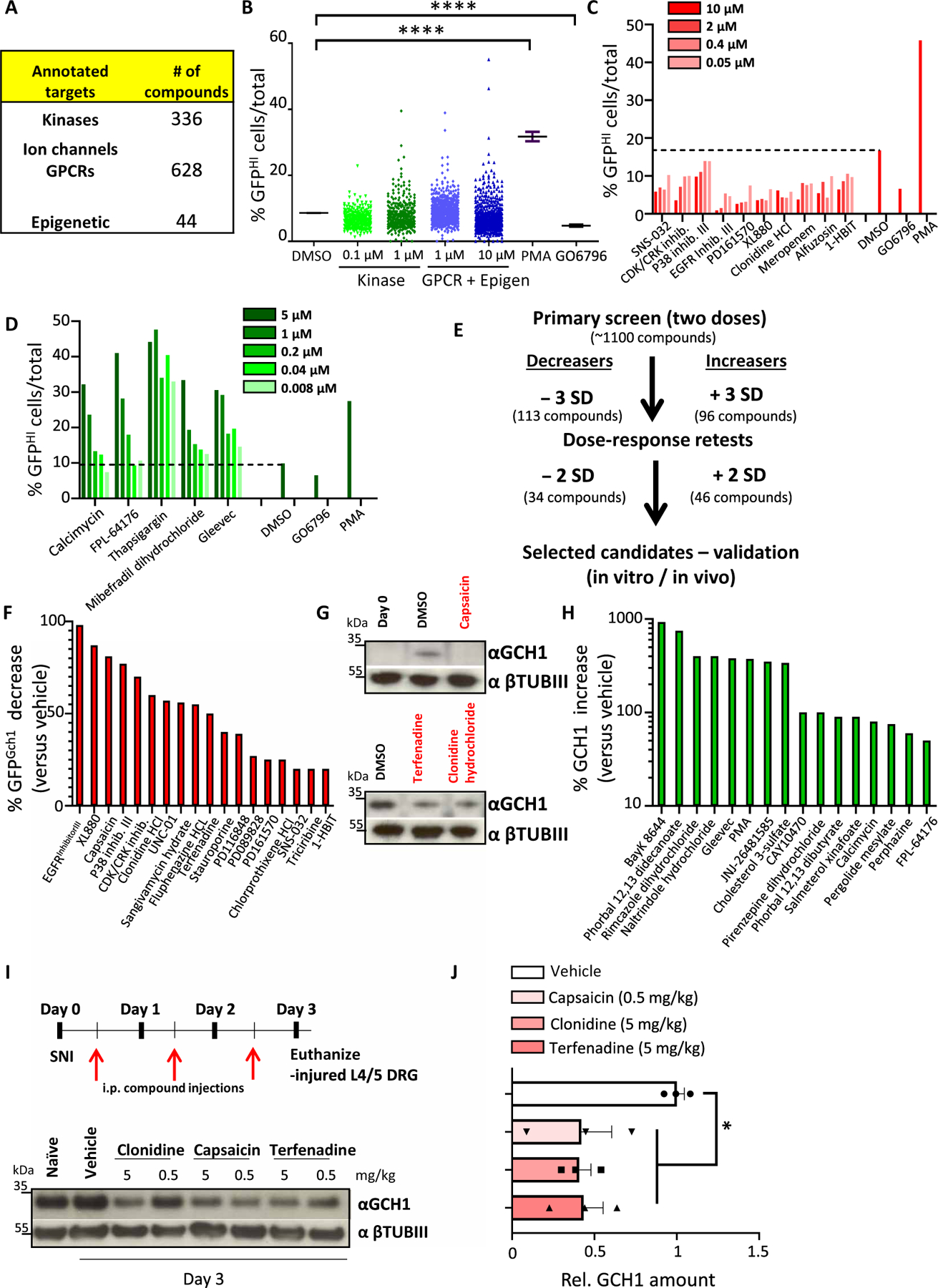
(A) Table detailing the type and number of compounds used in the screen. (B) Total screen data for each compound shown as % GFPHI cells among total identified neurons. Two concentrations were used for each compound, 0.1 and 1 μM for those in the kinase set, and 1 and 10 μM for drugs in the GPCR/Epigenetic set. DMSO as well as the control compounds PMA (3 μM) and GO6796 (0.3 μM) are also shown. ****P < 0.0001 (one-way ANOVA with Dunnett’s multiple comparison test). (C and D) Examples of dose-response retests of primary candidate hits covering both decreasers (C) and increasers (D) of % GFPHI cells in day 3 cultured DRG neurons from Gch1GFP mice. DMSO as well as PMA (3 μM) and GO6796 (0.3 μM) are also shown. (E) Schematic depicting the workflow, compound hit rates, and validation protocols for the primary screen and retests. (F and G) Western blot quantification of compounds that decrease GFP protein after 3 days of culture (F) and corresponding validation of endogenous GCH1 protein decrease compared to vehicle (DMSO) treatment of capsaicin, clonidine, and terfenadine (G). βTubIII was used as a loading control. (H) Quantification of those compounds that increase GCH1 protein compared to vehicle treatment after 3 days of culture. βTubIII was used as a loading control. Cross symbol (†) indicates potential toxicity of the compound on the neurons as determined by visual microscopic inspection and/or low βTubIII. (I and J) Schematic depicting the validation protocol for compound testing in vivo using the spared nerve injury (SNI) neuropathic pain model in mice and representative Western blot of GCH1 protein in injured L3-L5 DRG after SNI in which wild-type animals were treated with the indicated compounds and compared to vehicle treatment as well as DRG tissue isolated from naïve animals. βTubIII was used as a loading control (I). Relative quantification of GCH1 protein in injured sciatic nerve 3 days after SNI from wild-type animals treated with vehicle, capsaicin (0.5 mg/kg), clonidine (5 mg/kg), and terfenadine (5 mg/kg). Data are shown as means ± SEM and pooled from two independent experiments. Individual samples are shown. *P < 0.01 (one-way ANOVA with Dunnett’s multiple comparison test). i.p., intraperitoneal.
Repurposed drug screen to identify regulators of GCH1 expression
Compounds that exhibited a decrease or increase (see Materials and Methods for statistical parameters), at either dose, in the percentage of GFP-high neurons compared to DMSO-treated cells were selected for retesting (tables S3 and S4). Dose responses were performed for each retested candidate, and those compounds that again exhibited substantial differences from DMSO treatment were selected for further analysis (Fig. 2, C to E, and tables S5 and S6). Next, the identified candidates were carried forward for protein validation on 3-day cultured Gch1-GFP reporter DRG neurons where GFP expression was quantified and ranked in terms of maximum effect (Fig. 2F and fig. S5A). Candidates that produced considerate (>20%) GFP protein reduction were next tested on 3-day cultured DRG cells isolated from wild-type mice and blotted for endogenous GCH1 protein (Fig. 2G, fig. S5B, and table S7). Candidates that increased the percentage of GFP+ neurons were also tested for GCH1 regulation in wild-type DRG neurons (Fig. 2H, fig. S6, and table S7).
Because our primary goal was to find blockers of BH4 production through a reduction in GCH1, we focused our efforts on those compounds that decreased the amount of GFP/GCH1. Some of the hits identified have previously been characterized as compounds that reduce pain hypersensitivity such as capsaicin, clonidine, and terfenadine (Fig. 2G) (27–33). Using the spared nerve injury (SNI) model of neuropathic pain in mice, we were able to show that these compounds also reduced GCH1 expression in injured DRG tissue in vivo (Fig. 2, I and J), indicating that their analgesic effects may in part be related to lowering GCH1 protein.
The antipsychotic fluphenazine decreases GCH1/BH4 and pain hypersensitivity
Next, we performed a computational STITCH (34) analysis of the validated hits (table S7) to map protein and pathway targets (figs. S7 and S8). We first focused on those compounds that reduced Gch1-GFP expression as well as GCH1 protein as representing the most promising leads for analgesics. We detected nodes for cyclin-dependent kinases, PKC, and alpha-2A adrenergic receptors, all of which have been associated with pain (35–37). One compound that substantially decreased GCH1 was the FDA-approved drug fluphenazine hydrochloride (Figs. 2F and 3A). Fluphenazine is an antipsychotic used to treat schizophrenia (38). Its mechanism of action is not well elucidated but is proposed to reduce hallucinations and delusions by targeting the dopamine D2 receptor on postsynaptic neurons in the basal ganglia; however, it also regulates alpha-2A adrenergic signaling (39). Our data show that fluphenazine reduced GCH1 in the sciatic nerve after SNI in vivo, validating the screen (Fig. 3, B to E). Furthermore, fluphenazine treatment substantially reduced BH4 in injured sciatic nerve after SNI compared to vehicle-treated animals (Fig. 3F).
Fig. 3. The antipsychotic fluphenazine reduces GCH1 and BH4 in sensory neurons as well as neuropathic pain in mice.
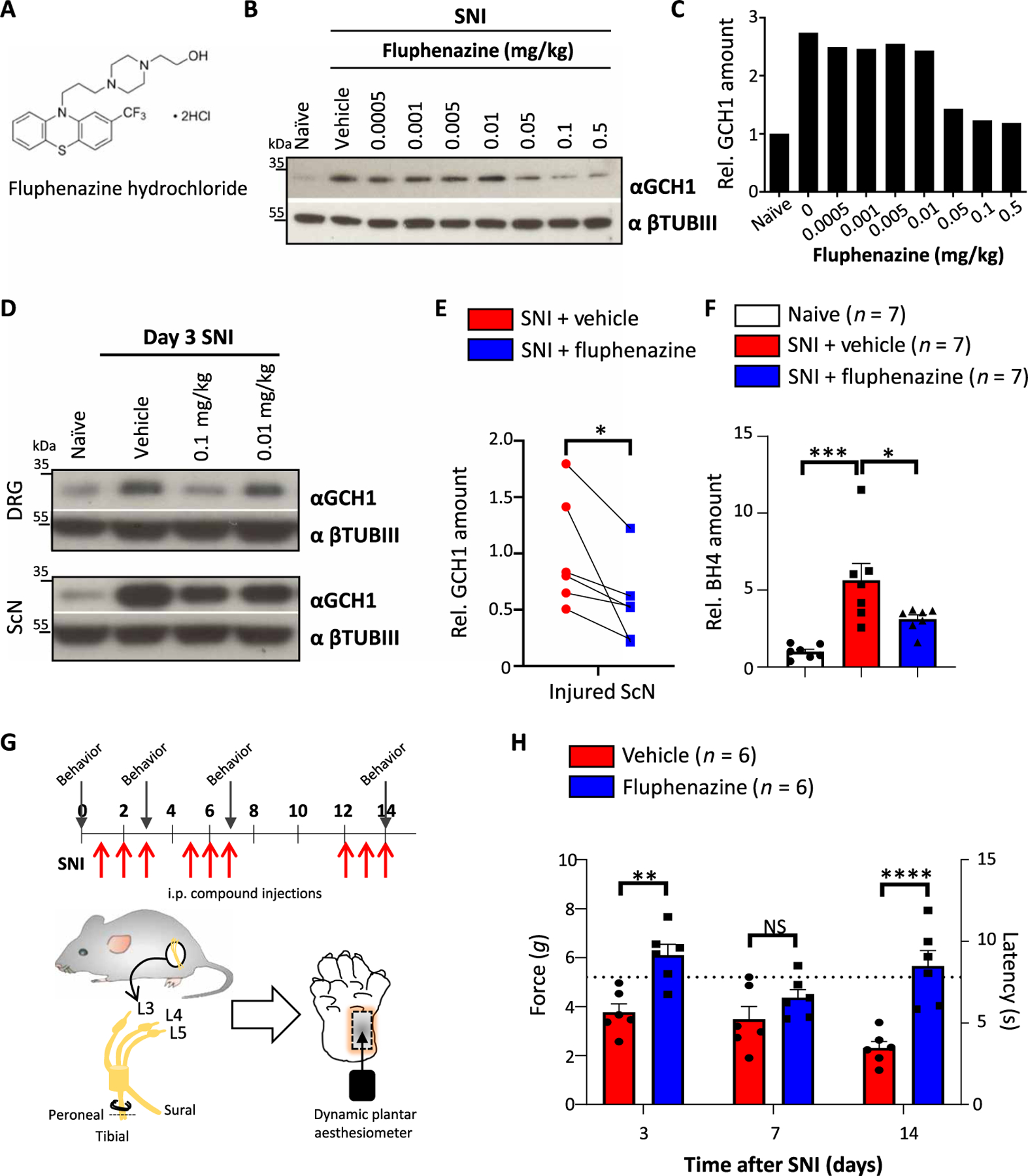
(A) Chemical structure of fluphenazine hydrochloride. (B and C) Western blot of GCH1 in injured sciatic nerve tissue 3 days after SNI in wild-type animals treated with varying doses of fluphenazine on days 0.5, 1.5, and 2.5 intraperitoneally (B) and relative quantification of GCH1 (C). βTubIII was used as a loading control. Naïve and vehicle (saline)–treated animals were included. The amounts are normalized to those in naïve tissue. (D) Western blot of GCH1 in injured L3-L5 DRG and sciatic nerve (ScN) tissue 3 days after SNI in wild-type animals treated with the indicated doses of fluphenazine on days 0.5, 1.5, and 2.5 intraperitoneally. βTubIII was used as a loading control. Naïve and vehicle (saline)–treated animals were included. (E) Relative quantification of GCH1 protein in injured sciatic nerve tissue 3 days after SNI in vehicle (saline)– and fluphenazine (0.1 mg/kg)–treated animals. Individual mice for each condition are shown. Data are shown as means ± SEM and pooled from four independent experiments. *P < 0.05 (paired t test). (F) Amount of BH4 measured in injured sciatic nerve tissue after SNI in vehicle (saline)– and fluphenazine (0.1 mg/kg)–treated animals. The amounts are normalized to those in naïve, noninjured tissue. Individual mice for each condition are shown. Data are shown as means ± SEM. *P < 0.05; ***P < 0.001 (one-way ANOVA with Dunnett’s multiple comparison test). (G) Schematic injection protocol of saline and fluphenazine (0.1 mg/kg) and of SNI mechanical pain testing of wild-type animals. Arrows indicate time points of mechanical pain testing. (H) Mechanical thresholds of SNI wild-type animals treated with saline and fluphenazine (0.1 mg/kg) as depicted in (G). Data are shown as means ± SEM. Individual mice for each genotype are shown. **P < 0.01; ****P < 0.0001 (two-way ANOVA with Sidak’s multiple comparison test). Dotted brown line indicates the force and latency of naïve wild-type animals before SNI.
We did not detect any effect of the drug on body weight or any other gross abnormalities in the observation time frames (fig. S9A). We tested other drugs related in structure to fluphenazine but failed to detect any apparent effects of these compounds on GCH1 protein regulation in cultured DRG neurons (fig. S9B). Fluphenazine-treated wild-type animals showed a slight analgesic effect to acute nociceptive noxious thermal insults, shown as increased latency at exposure to 50°C. No effect was found at 52°C (fig. S9C). In addition, there was no effect in the response to capsaicin between vehicle- and fluphenazine-treated animals (fig. S9D). This is partially in line with sensory-specific genetic BH4-deficient animals, which display responses comparable to control animals to the same acute nociceptive tests (24). However, fluphenazine treatment in vivo ameliorated the mechanical pain hypersensitivity induced by the SNI model (Fig. 3, G and H). Together, these data show that fluphenazine reduced pathologically elevated BH4 after nerve injury and alleviated neuropathic pain in mice.
Increased EGFR/KRAS signaling enhances GCH1/BH4 and pain sensitivity in sensory neurons in mice
Compounds targeting the EGFR pathway were among the most effective reducers of GCH1, and the most potent of these was EGFR inhibitor III (hereafter referred to as EGFRinh-III), followed by XL880 (also known as foretinib), which was developed as a c-MET/VEGFR2 (vascular endothelial growth factor receptor 2) blocker but which also inhibits the phosphorylation and activation of EGFR (Fig. 4A and fig. S5, A and B) (40). PD-161570 and PD-089828 also target EGFR activation (41, 42), and all four EGFR blockers reduced GCH1 protein in injured sciatic nerves after neuropathic pain induction (fig. S10A). The fact that these four compounds all target the same pathway led us to investigate EGFR signaling in sensory neurons upon injury. EGFRinh-III was the top GCH1 reducer in our screen, and we further validated its strong effects by investigating the kinetics of Gch1-GFP expression over an extended time (Fig. 4B). EGFR was strongly up-regulated in DRG cultures after 3 days (Fig. 4C), and this increase was validated in vivo in the SNI model, with a higher up-regulation of EGFR and GCH1 in the injured sciatic nerve compared to the injured DRG tissue (Fig. 4, D and E, and fig. S10, B and C). Moreover, in vivo administration of EGFRinh-III not only resulted in a reduction of GCH1 protein in the injured nerve but also reduced mechanical allodynia after SNI (Fig. 4, F and G, and fig. S10D). Thus, our screen has identified a potential role for EGFR in injured DRG cells in contributing to GCH1 modulation and neuropathic pain in mice.
Fig. 4. EGFR signaling regulates the GCH1/BH4 pathway in sensory neurons.
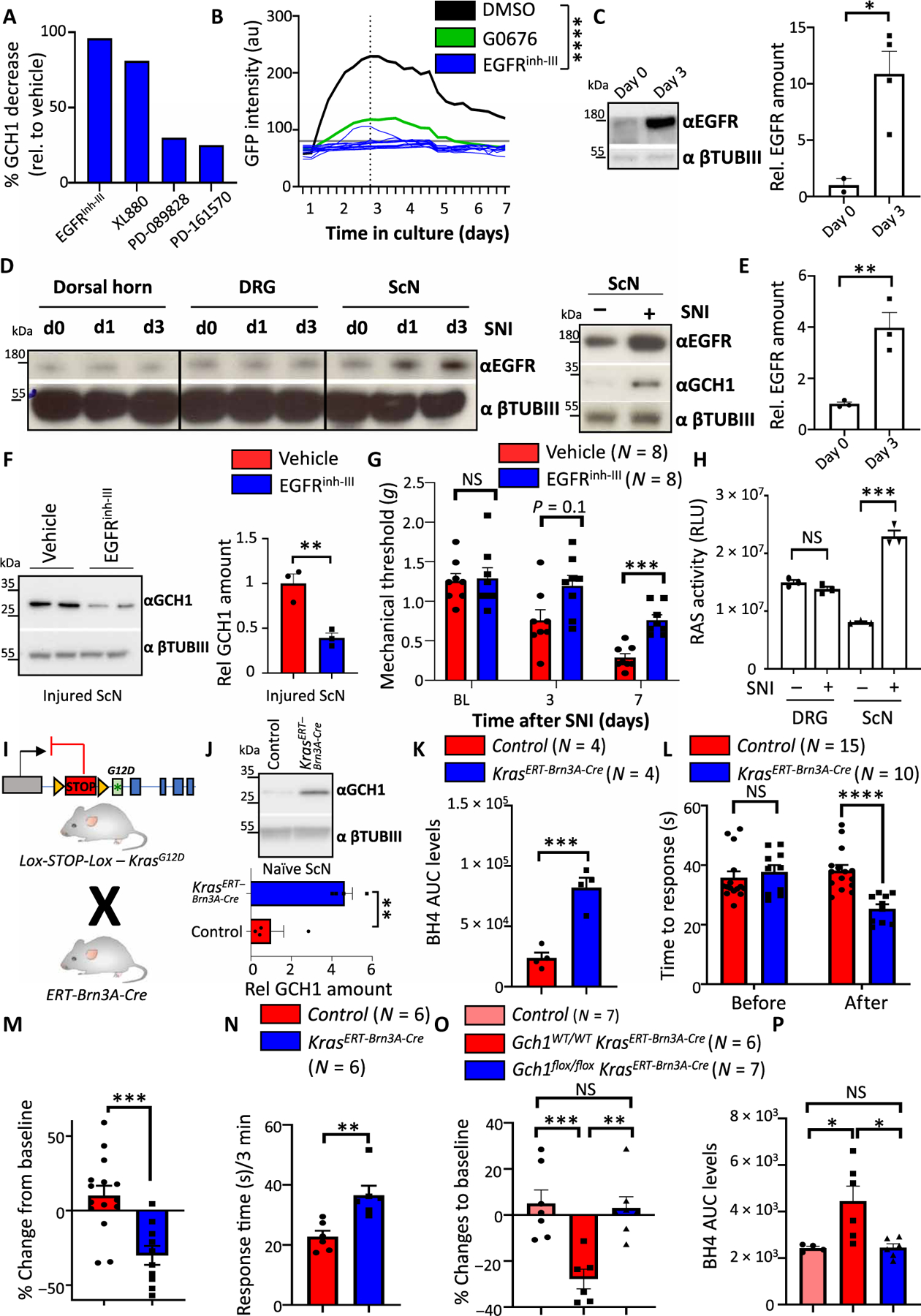
(A) Quantification of GCH1 protein reduction on DRG cultures from wild-type mice by several compounds that target EGFR signaling; see fig. S5B for Western blot. (B) GFP intensities over time of individual GFP-expressing DRG neurons treated with EGFR inhibitor III (EGFRinh-III) (10 μM) isolated from Gch1GFP reporter mice. DMSO and GO6796 (0.3 μM) treatments from Fig. 1F are also shown. Individual intensities over time are shown for EGFRinh-III. ****P < 0.0001 (two-way repeated-measures ANOVA between DMSO and EGFRinh-III). (C) Rep resentative Western blot and quantification of EGFR protein in cultured DRG cells on days 0 and 3. βTubIII was used as a loading control. Data are shown as means ± SEM and pooled from two independent experiments. Individual mice for each condition are shown. *P < 0.05 (Student’s t test). (D) Western blot of EGFR in the injured ipsilateral dorsal horn, DRGs, and sciatic nerve assayed at days 0 and 1 and 3 days after SNI (left). Western blot of EGFR and GCH1 in the DRG and sciatic nerve of naïve and 3-day SNI-treated wild-type animals (right). βTubIII was used as a loading control. (E) Relative quantification of EGFR in sciatic nerve tissue in naïve and SNI-treated animals from fig. S10C. Data are shown as means ± SEM. Individual mice for each condition are shown. **P < 0.01 (Student’s t test). (F) Representative Western blot of GCH1 in the sciatic nerve of 3-day SNI-operated wild-type animals treated with vehicle or EGFRinh-III (10 mg/kg). βTubIII was used as a loading control. Data are shown as means ± SEM and pooled from two independent experiments. Individual mice for each condition are shown. **P < 0.01 (Student’s t test). (G) Mechanical thresholds of SNI wild-type animals treated with vehicle and EGFRinh-III (10 mg/kg). Treatment was administered intraperitoneally for 2 days before each behavioral testing. Data are shown as means ± SEM. Individual mice for each genotype are shown. ***P < 0.001 (two-way ANOVA with Sidak’s multiple comparison test). (H) Ras activity in naïve and 3-day SNI-treated animals from DRG and sciatic nerve tissues. Data are shown as means ± SEM. Individual mice for each genotype are shown. ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparison test). (I) Breeding scheme for inducible expression of constitutively active KrasG12D in sensory neurons. (J) Western blot of GCH1 in naïve DRG tissue from tamoxifen-treated control and Kras; ERT-Brn3A-Cre mice. Tissue was extracted 8 weeks after tamoxifen treatment (five times daily, 2 mg per mouse). βTubIII is shown as a loading control. (K) BH4 measurements in the sciatic nerve of naïve control and Kras; ERT-Brn3A-Cre mice 8 weeks after tamoxifen administration. Data are shown as means ± SEM. Individual mice for each genotype are shown. ***P < 0.001 (Student’s t test). (L and M) Hot-plate behavioral testing of control and Kras; ERT-Brn3A-Cre mice at baseline (L) and percentage change in response (M) as determined 8 weeks after tamoxifen administration. Data are shown as means ± SEM. Individual mice for each genotype are shown. ***P < 0.001; ****P < 0.0001 [one-way ANOVA with Tukey’s multiple comparison test (L) and Student’s t test (M)]. (N) Response time to capsaicin (1 μg) intradermal paw injection of control and Kras; ERT-Brn3A-Cre mice 8 weeks after tamoxifen administration. Data are shown as means ± SEM. Individual mice for each genotype are shown. **P < 0.01 (Student’s t test). (O) Percent change in hot-plate (50°C) latencies of control, Kras; ERT-Brn3A-Cre, and Gch1flox/flox; Kras; ERT-Brn3A-Cre mice at baseline and 8 weeks after tamoxifen administration. Data are shown as means ± SEM. Individual mice for each genotype are shown. **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparison test). (P) BH4 measurements in the sciatic nerve of naïve control, Kras; ERT-Brn3A-Cre, and Gch1flox/flox; Kras; ERT-Brn3A-Cre mice 8 weeks after tamoxifen administration. Data are shown as means ± SEM. Individual mice for each genotype are shown. *P < 0.05 (one-way ANOVA with Tukey’s multiple comparison test).
Ras is a guanosine triphosphatase (GTPase) downstream of various growth factor receptors, including EGFR, whose activation enables extracellular signals to be transmitted from the receptor to the nucleus (43). Activation of RAS (for example, GTP-bound RAS) is therefore an indicator of EGFR activation (43). RAS activity was enhanced in injured sciatic nerve after nerve injury (Fig. 4H). RAS is composed of a family of three proteins, HRAS, NRAS, and KRAS. KRAS is most often associated with EGFR signaling in cancer pathogenesis, and mutated KRAS leading to constitutive activation is one of the most mutated genes in human cancers (44, 45). To investigate a role of KRAS activation in EGFR signaling in sensory neurons, we selectively expressed the constitutively active KRAS mutant, KrasG12D, in DRG neurons using a tamoxifen-inducible Cre line, ERT-Brn3A-Cre (Fig. 4I) (46). In naïve animals, KrasG12D overexpression led to increased GCH1 (Fig. 4J). BH4 was also enhanced (Fig. 4K), supporting the notion that active KRAS leads to increased GCH1 expression and BH4 synthesis. In addition, constitutive KRAS activation led to increased pain sensitivity in vivo in response to noxious heat (Fig. 4, L and M) and an increased sensitivity to capsaicin (Fig. 4N). To confirm that this enhanced thermal hypersensitivity was due to increased BH4 in the nerve, we genetically deleted Gch1 in the sensory neurons while at the same time constitutively activating KrasG12D; in this genetic rescue experiment, the thermal hypersensitivity and increased BH4 returned to baseline (Fig. 4, O and P). Together, these data indicate that upon nerve injury in mice, EGFR is up-regulated in injured sensory nerves, and this leads to KRAS activation, which in turn increases BH4 production and pain hypersensitivity. These data also suggest that targeting either EGFR stimulation or KRAS activation may lower pathological amounts of BH4 in sensory neurons, and thereby reduce neuropathic pain.
GCH1 promotes tumorigenesis in KRAS-driven lung cancer
Because KRAS activation increased GCH1 and BH4 in sensory neurons, we next wanted to assess whether this link also holds true for other KRAS-dependent systems. The three Ras genes (Kras, Nras, and Hras) are among the most mutated genes associated with cancer (mutated in 90% of pancreatic, 35% of lung, and 45% of colon cancers), and, in particular, Kras is the isoform most prevalently mutated in lung cancers (47, 48). We therefore used the KrasG12D-dependent lung cancer model, in which adenovirus-mediated Cre expression leads to constitutively active KrasG12D overexpression in lung epithelial cells (hereafter referred to as KrasADENO-CRE), which then drives cancer development (49). We excised lung tumors from KrasADENO-CRE mice treated with Cre-expressing adenovirus for 20 weeks and compared these to normal lung tissue. BH4 was considerably increased in the KrasADENO-CRE–driven lung cancer tissue compared to lung tissue from control treated (controlADENO-CRE) mice (Fig. 5A). BH4 can also be produced by immune cells such as macrophages and T cells, as well as CD31+ endothelial cells (50). To exclude the possibility that the source of the BH4 arises from these cells and not the cancer cells, we purified the lung cancerous tissue and depleted immune and endothelial cells (Fig. 5B) and still detected increased BH4 in the purified tumor cells (Fig. 5C).
Fig. 5. Active KrasG12D enhances the GCH1/BH4 pathway to drive lung cancer progression.
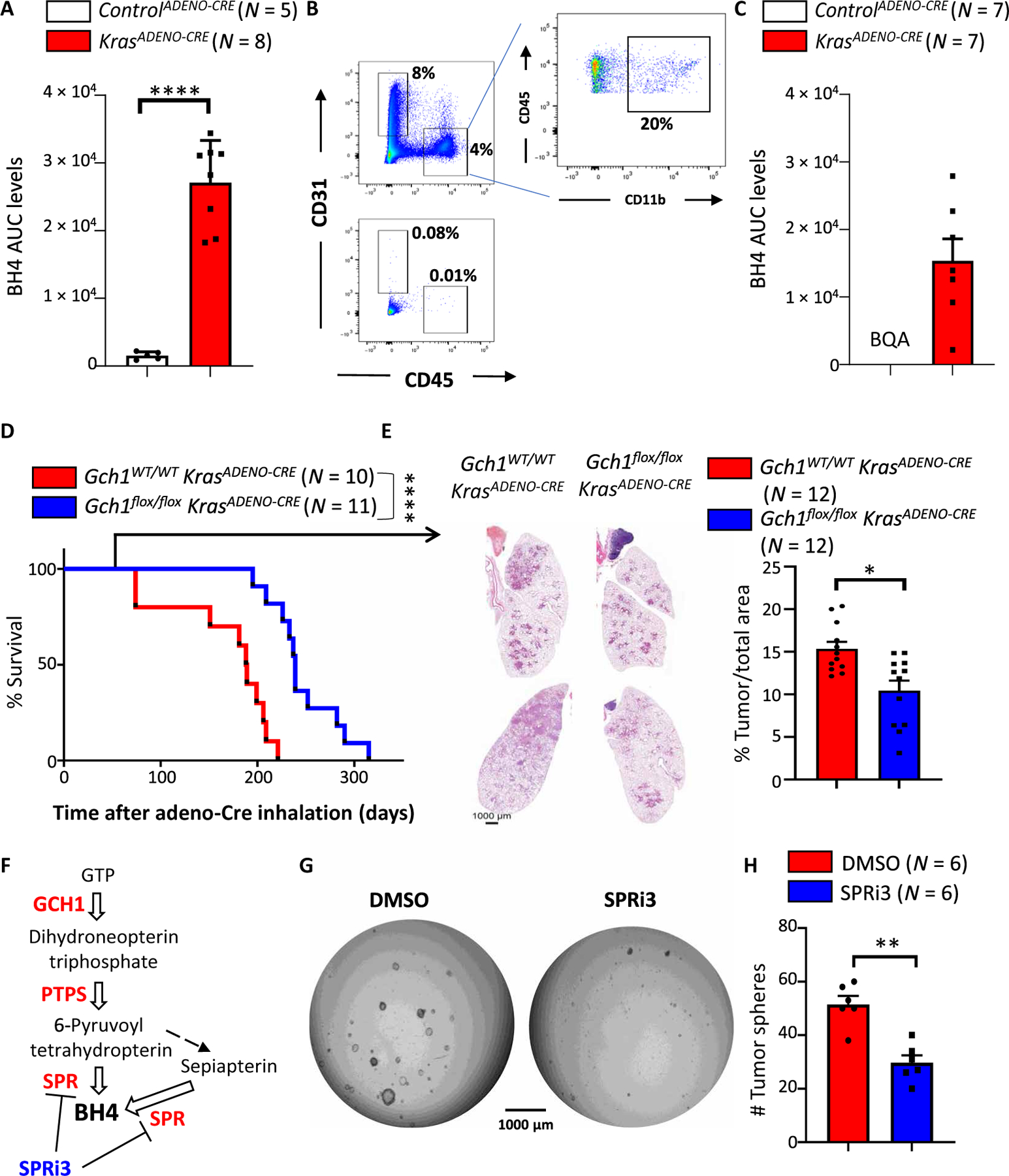
(A) BH4 measurements in lung tissue of controlADENO-CRE and KrasADENO-CRE mice 5 months after adenovirus (Ad5-CMV-Cre) inhalation. Data are shown as means ± SEM. Individual mice for each genotype are shown. ****P < 0.0001 (Student’s t test). (B and C) FACS analysis of lung tumors before (top) and after (bottom) depletion of immune cells (CD45+CD11b+) and endothelial cells (CD31+) from lung tissue of controlADENO-CRE and KrasADENO-CRE mice 5 months after adenovirus (Ad5-CMV-Cre) inhalation (B). Remaining tumor-enriched cells were subjected to BH4 measurements (C). Data are shown as means ± SEM. Individual mice for each genotype are shown. BQA, below quantifiable amount. (D) Survival curve for Gch1WT/WT KrasADENO-CRE (n = 11) versus Gch1flox/flox KrasADENO-CRE (n = 10) littermates treated with adenovirus (Ad5-CMV-Cre) inhalation. Data are pooled from two independent experiments. ****P < 0.0001 [Log-rank (Mantel-Cox) test]. (E) Representative lung tumor sections (H&E staining) in Gch1WT/WT KrasADENO-CRE and Gch1flox/flox KrasADENO-CRE littermates 12 weeks after Ad5-CMV-Cre inhalation (left) and quantification of overall tumor burden (right). Total tumor areas comprising hyperplasia and adenomas were scored automatically by a Definiens software algorithm and confirmed by a trained pathologist. Data are shown as means ± SEM. Individual lung section samples are shown pooled from three animals from each genotype. *P < 0.05 (Student’s t test). (F) Schematic pathway for BH4 biosynthesis. Enzymes are shown in red, white arrows indicate enzymatic reactions, and black arrow indicates nonenzymatic reaction. The sepiapterin reductase (SPR) inhibitor SPRi3 is shown in blue. (G) Representative images of tumor spheroids derived from purified KrasADENO-CRE primary lung tumor cells treated with vehicle control (DMSO) or the SPR inhibitor SPRi3 (50 μM). Images were acquired 4 days after cells were seeded in Matrigel (5000 primary tumor cells per well). The experiment was repeated with six different mice for each group. (H) Quantification of tumor spheroid numbers described in (G). Data are shown as means ± SEM and pooled from three independent experiments. Individual samples are shown. **P < 0.01 (Student’s t test).
Last, we ablated Gch1 in KrasG12D lung cancer cells by crossing Gch1flox/flox animals with a KrasG12D transgenic line and infecting the mice with Cre-expressing adenovirus intranasally (Gch1flox/flox; KrasADENO-CRE). The genetic inactivation of Gch1 led to increased survival of the Gch1flox/flox KrasADENO-CRE mice compared to similarly treated Gch1WT/WT; KrasG12D control animals (Gch1WT/WT KrasADENO-CRE) (Fig. 5D). Moreover, histological analysis of the lungs 8 weeks after adeno-infection showed reduced tumor burden under the Gch1-deficient conditions (Fig. 5E). An assay has been developed to determine lung cancer stem cell activity using a spheroid formation as a readout (51). Pharmacological blockage of the GCH1/BH4 pathway using SPRi3, a specific inhibitor of sepiapterin reductase (SPR) (24), an enzyme critical for BH4 biosynthesis (Fig. 5F), also resulted in reduced KrasG12D-driven tumor spheroid formation compared to vehicle treatment (Fig. 5, G and H). These data demonstrate that a KrasG12D-mediated increase in GCH1 and BH4 production not only is confined to sensory neurons after injury but also might play a role in KrasG12D-driven lung cancer tumorigenesis.
DISCUSSION
Pain is an essential physiological response that protects organisms from dangerous stimuli, allowing injury the necessary time to heal and thereby prevent additional damage to injured areas. However, in certain individuals, usually after damage to the nervous system, pain persists long after the noxious stimuli have ceased and the injured areas have healed, constituting chronic pain (52). In a 26-year study from 1990 to 2016 assessing data of ~200 countries, lower back pain, neck pain, and migraines topped the list for leading causes of disability (53). It is estimated that 20% of the adult population in Europe and the United States experience moderate to severe pain exceeding 6 months (54, 55). This substantially impairs the health and well-being of the affected individuals, one-third of which take no medication, and of those taking medication, almost 50% report an inadequacy in pain management (54, 55).
The BH4 pathway represents a pathway independent of the opioid system to combat pain hypersensitivity (9). Our group recently developed small-molecule inhibitors of SPR, the terminal enzyme in the BH4 biosynthetic pathway, which reduce pain after nerve injury and inflammation in rodents (20, 24, 26). Here, we report a phenotypic screening platform using annotated bioactive compounds to identify drugs that up-regulate or decrease GCH1 expression upon nerve injury in preclinical models. Although screening primary DRG neurons results in lower throughput than heterologous target expression assays, such an approach might offer greater translatability. Using this approach, the annotated targets of hits provided insights into the biology of GCH1 expression and BH4 synthesis in sensory neurons; in addition, any hits with FDA-approved compounds have existing safety and pharmacokinetic profiles and could therefore be readily repurposed.
To confirm the utility of our approach, we tested the top candidates in vitro and in vivo in a neuropathic nerve injury model. In our hit list, we found several known drugs that reduce neuropathic pain, such as clonidine (29, 31, 37), offering an additional explanation for their analgesic effects. A repurposing opportunity identified by the screen was fluphenazine hydrochloride. Fluphenazine is an antipsychotic used to treat schizophrenia by blocking postsynaptic D2 dopamine receptors and alpha-1 adrenergic receptors. We demonstrate here that fluphenazine reduces GCH1 and BH4 in DRG cultures and in sensory neurons after sciatic nerve injury in mice. Moreover, we show that fluphenazine treatment markedly alleviated pain hypersensitivity after nerve injury in mice, using a dose that is comparable to that used in patients (0.01 mg/kg daily intraperitoneally in our study in mice versus the range 0.006 to 0.125 mg/kg orally used in patients) (56). Side effects of fluphenazine treatment include movement disorders and depression (57–59). Because BH4 is required by tyrosine hydroxylase and tryptophan hydroxylase to produce dopamine and serotonin, respectively, these effects may point to GCH1 reduction in certain brain regions. A peripherally restricted form of fluphenazine that abrogates pathologically elevated GCH1 and BH4 in peripheral neurons after injury or inflammation could therefore be used to lower pain sensitivity, potentially reducing the risk of central nervous system side effects.
We also identified a role of EGFR and KRAS signaling in GCH1/BH4 regulation upon nerve injury. We show that EGFR is up-regulated specifically in injured nerves, and this is accompanied by increased Ras activity. Overexpressing mutant constitutively active Kras (KrasG12D) specifically in sensory neurons increased pain sensitivity in mice. Mechanistically, constitutively active KRAS resulted in increased GCH1 protein and elevated BH4 in the sensory nerves. Recently, attention has focused on the role of EGFR activation in neuropathic pain because up-regulation of EGFR in the DRG is associated with chronic pain development in rodents (60, 61). Moreover, in a case study of a patient with rectal cancer and neuropathic pain, it was observed that EGFR inhibition using the drug cetuximab resulted in pain relief despite little effect on tumor progression (62). This study was confirmed in a series of case reports in which different inhibitors of EGFR were found to offer pain relief to patients suffering from debilitating neuropathic pain (63, 64). Our data now uncovered a potential molecular explanation for the pain-reducing effects of EGFR inhibition via GCH1/BH4 regulation.
We were able to expand the link between EGFR and BH4 activation in sensory nerves to lung cancer, in which mutated, constitutively active KrasG12D is a driver of tumorigenesis. In KrasG12D-driven lung cancer, BH4 was substantially enhanced in the tumor cells, and genetic inactivation of Gch1 reduced tumor burden and increased survival in mice. Blocking EGFR activation and inhibiting active KRAS signals is a major goal for cancer therapy, and many therapeutic approaches are currently being designed to target EGFR/KRAS activation (65). Our results indicate that it will be important to determine whether these inhibitors are also analgesics, and further investigate the mechanistic role of GCH1/BH4 in KrasG12D-driven cancer. Recently, two reports have demonstrated that SPR activity and BH4 promote cancer cell proliferation in a manner similar to that we recently uncovered for T cell proliferation via regulation of mitochondrial adenosine triphosphate (ATP) synthesis (66, 67). The cell lines used in the BH4 cancer study harbor Kras mutations that lead to hyperactivity of the protein (66).
Overall, we demonstrated the value of a phenotypic screen with an annotated drug library to both identify existing drugs that may be repurposed for treating pain and uncover a biological cross-talk between cancer and pain via the GCH1/BH4 pathway. Our data also provide potential mechanistic explanations for the analgesic activities of several known pain drugs and might contribute to the development of better analgesics for chronic pain. Our results also unlock a molecular link for EGFR/KRAS-regulated pain perception and lung cancer via the GCH1/BH4 metabolic pathway, opening multiple diverse therapeutic opportunities.
Limitations of the study
Using primary axotomized mouse DRG neurons restricted our platform to low-throughput screen of only about 1100 annotated bioactive compounds and was not amenable to screen a larger diversity library. GCH1/BH4 has been linked to both neuropathic and inflammatory pain conditions in humans; in addition to using the SNI model here, going forward would also be interesting to investigate the effect of fluphenazine and EGFR inhibition on other relevant mouse models of inflammatory pain (rheumatoid or osteoarthritis) and neuropathic pain (models of diabetic neuropathy, chemotherapy-induced neuropathy, compression neuropathy, and peripheral neuritis). Last, the generation of induced pluripotent stem cell (iPSC)–derived human nociceptors has been recently described; testing our compound candidates for GCH1/BH4 regulation on human pain neurons would be a promising translational step from our mouse study to a human platform.
MATERIALS AND METHODS
Study design
The rationale for this study was that our laboratories, and others, have identified GCH1/BH4 pathway as a major contributor to neuropathic pain. We have previously shown that genetic ablation of Gch1 in sensory neurons ameliorates neuropathic pain in mice. Therefore, our idea was to screen annotated and FDA-approved drugs for regulation of Gch1 expression to find new analgesics and biology of BH4 regulation after nerve injury. In inhibitor-treatment behavioral experiments in vivo, wild-type mice were randomly allocated into each experimental group.
For in vivo behavioral experiments, the investigator was blinded to the treatment for experiments shown in Fig. 3H and unblinded for experiments shown in Fig. 4G. For nontreatment behavioral experiments, the experimenter was blinded to the genotypes used. For lung cancer in vivo experiments, the investigator was blinded to the mouse genotypes. For measurements of BH4, the experimenter was unaware of the treatment the samples received before analysis.
Replication is indicated in the figures and/or figure legends. On the graphs, individual dots represent individual samples/mice used. For each experiment, all attempts at replication were successful and our findings showed comparable results.
Mice
Mice expressing enhanced green fluorescent protein (eGFP) under the Gch1 promoter (Gch1GFP) were used to label neurons that up-regulate Gch1 after DRG axotomy (24). For loss-of-function experiments, we used Gch1 floxed (Gch1floxed) mice, which have previously been described (68). Tamoxifen-inducible ERT-Brn3A-Cre mice (46) were crossed to LSL-KrasG12D (49) and Gch1flox (68) animals. Wild-type are C57Bl6/J strain bred in-house. Tamoxifen (Sigma-Aldrich, T5648, 2 mg per mouse for four consecutive days) was used to induce KrasG12D activation and Gch1 deletion. All animal experiments were performed in accordance with institutional policies and federal guidelines. The Austrian Federal Ministry of Education, Science and Research approved the corresponding proposals GZ BMBWF-66.015/0033-V/3b/2019 and their amendments.
DRG cultures
DRG cultures were prepared as follows from naïve animals, either Gch1GFP or wild-type mice. Briefly, DRGs were extracted, dissociated using Liberase (Roche) and 0.25% trypsin, triturated, and cultured from eight 10- to 12-week-old Gch1GFP mice for each experimental day of the screen. The neurons were cultured on 384-well poly-d-lysine–coated plates (BD BioCoat, Corning 354461), which were also coated with laminin [10 μg/ml in phosphate-buffered saline (PBS), 1 hour at room temperature; Sigma-Aldrich, L2020]. Identification and counting of neurons were performed with a standard hemocytometer using trypan blue to exclude dead cells. DRG cultures were plated at a density of ~1000 neurons per well. The neurons were cultured in Neurobasal (Full NB) medium supplemented with B27 (Invitrogen) and 200 mm l-glutamine. For Western validations, a similar procedure was used for Gch1GFP and wild-type C57Bl6/J mice, where six-well plates were used. The neurons were cultured for 3 days before protein extraction, and compounds were added 24 hours after seeding.
Small-molecule libraries
The small molecules used in these screens were from the following collection: National Institutes of Health (NIH), LOPAC1280 (Sigma-Aldrich), Spectrum (Microsource Discovery Systems), and Prestwick Chemical Library. One thousand compounds were selected from these libraries to create custom compound sets targeting GPCR, ion channels, kinases, and epigenetic modifiers (Fig. 2A and table S1). Fluphenazine (Cayman Chemical, 23555, and Sigma-Aldrich, BP167) was dissolved in saline to the concentrations indicated in figure legends. EGFRinh-III (Merck, 324833) was dissolved in Tween 80/carboxymethylcellulose to the concentrations indicated in figure legends. Capsaicin (Sigma-Aldrich, M2028) was dissolved in DMSO or ethanol and subsequently in saline to the desired concentrations indicated in the figures.
Image analyses for quantification of GFP and PI intensities
Microtiter 384-well plates containing the cells were examined in a PerkinElmer/Evotec Opera QEHS model 2.0 laser spinning disk confocal fluorescence microscope. The GFP was visualized with a 488-nm laser, and the PI viability stain was visualized with a 561-nm laser. The resulting images were analyzed with scripts written in the PerkinElmer Acapella language that is packaged with the automated microscope. Subroutines for nuclei detection used the GFP channel images. The first-round candidate nuclei that were too small, large, wide, or narrow were filtered out. The remaining accepted nuclei were divided into high intensity, low intensity, or GFP negative based on the average intensities in the GFP channel. The nuclei were further subgrouped into “alive” or “dead” based on their PI fluorescence intensity. Positive and negative control chemicals (PMA and GO6796) as well as buffer with appropriately diluted DMSO unperturbed cells were included as controls on each plate to validate the algorithm thresholds and data consistency on each plate. For primary screening, three different parameters were calculated for each compound—the number of high-expressing GFP neurons (#GFPHi), the mean GFP intensity per well (mean GFP intensity), and the percent of high-expressing GFP neurons of total neurons identified per well (% GFPHI/total nuclei). Only compounds that were nontoxic were analyzed. For each parameter, three SDs above and below (±3 SD) that of the negative (DMSO) control samples were scored. For each threshold passed, the following scoring was assigned: #GFPHI = 1; mean GFP intensity = 3; % GFPHI/total nuclei = 5 (see also tables S3 and S4). The total rank sum was assigned to each compound. For “decreasers,” compounds that acquired a total score of >1 was considered a hit and proceeded to dose-response retesting; for “increasers,” we used the stricter total score of >5 to proceed to dose-response retesting. For time course experiments, Cell Discoverer CD7 (Zeiss) was used and images were acquired every 6 hours for up to 1 week. Analysis was performed using Fiji. For Western blot validation, a cross symbol (†) indicates potential toxicity of the compound on the neurons as determined by visual microscopic inspection and/or relatively (to vehicle treatment) low βTUBIII protein.
Behavioral assays
Mice were housed with their littermates (two to five mice per cage based on the litters) with food and water ad libitum. All animals were maintained under the same conditions (22° ± 1°C, 50% relative humidity, 12-hour light/dark cycle). For behavioral experiments involving transgenic mice, randomization was achieved through the breeding: At the time of weaning, mice were separated on the basis of their sex and placed in their new home cage. Only cages with a mixed representation of transgenic mice and their littermates were used for behavioral experiments. All experiments used at least two independent litters and were duplicated.
Determination of pain responses/thresholds
Contact heat pain (hot-plate test)
Mice were placed on a metallic plate heated to a set temperature (30°, 49°, 52°, or 55°C) within an acrylic container (Bioseb), and the latency for flinching, licking one of the hind paws or jumping was measured. Mice were sequentially tested for 30°, 52°, 55°, and 49°C. One temperature was tested per day.
Capsaicin
Twenty microliters of capsaicin (1 μg) (Sigma-Aldrich, M2028) diluted in 1% DMSO and saline was injected into the plantar surface of the hind paw, the mouse was placed onto a room temperature surface within a container, and the time spent licking, biting, or lifting the paw was measured.
SNI model
SNI surgery was performed under 3% induction/2% maintenance with isoflurane on adult mice (8 to 12 weeks old). The tibial and common peroneal branches of the sciatic nerve were tightly ligated with a 5.0 silk suture and transected distally, whereas the sural nerve was left intact (69). After injury, incision was sutured, and mice were allowed to recover on heated pads before being returned to their home cage. The surgeon who performed the SNI was blinded to the genotype.
Dynamic plantar test—von Frey element
The Dynamic Plantar Aesthesiometer has been designed to automate the assessment of “touch sensitivity” on the plantar surface of mice. Animals were habituated for 2 hours in the testing chamber on the gridded platform. Each mouse is given three trials on the injured hind paw, which is then averaged. It is best to alternate between mice each time and wait at least 20 s on one and the same individual. The gram is the force applied when they remove the paw. The latency is the time (in seconds) from the onset of initial paw contact until the paw is removed. The lateral region of the left paw innervated by the spared sural nerve was tested (see Fig. 3G).
KrasG12D-driven lung cancer model
Gch1floxed mice were then crossed to LSL-KrasG12D (49) mice to generate Gch1fl/flLSL-KrasG12D and Gch1+/+ LSL-KrasG12D littermate mice. Inhalation of 6- to 8-week-old mice with Ad5-CMV-Cre (VVC-U of Iowa-5) virus was performed as previously reported (49). All experimental animals were anesthetized with 10% ketasol/xylasol and placed on a heated pad. An AdCre-CaCl2 precipitate was produced by mixing 60 μl of minimum essential medium, 2.5 μl of adeno-Cre (1010 plaque-forming units/ml; University of Iowa, Gene Transfer Vector Core Iowa, USA), and 0.6 μl of CaCl2 (1 M) for each mouse and incubated for 20 min at room temperature. Survival times after inhalation were noted.
Lung tumor histology
All lung tumors were analyzed histologically as previously reported (70). Briefly, lungs were cut into 2-μm sections from at least three different planes and stained with hematoxylin and eosin. Lung sections were scanned using a Mirax slide scanner, and lung/tumor areas were scored by an algorithm programmed and executed using the Definiens software suite and visually controlled in a blinded way. Positive cells were counted on 15 randomly chosen tumor areas at ×100 magnifications in a double-blinded fashion. Quantitative analysis was performed using HistoQuest software (TissueGnostics GmbH).
Tumor spheroid cultures
A flat round drop of Matrigel (Corning) was seeded in cell culture plates followed by incubation at 37°C for 5 min. Primary lung tumor cells were mixed with the Matrigel and kept on ice until they were seeded onto the droplet of Matrigel in the plate in “a droplet on a droplet” fashion. The Matrigel plug was incubated at 37°C for 30 min and then covered with cell culture medium. SPRi3 and vehicle (DMSO) were added 24 hours after seeding. Images were acquired and analyzed 5 days later.
BH4 measurements
Metabolites were extracted from tissue or cell pellets using a methanol (MeOH):acetonitrile (ACN):0.1% dithiothreitol (DTT) in water (2:2:1, v/v) ice-cold solvent mixture by adding 500 μl of the solvent to 50 mg of tissue in an Eppendorf tube vortexed for 30 s, incubated in liquid nitrogen for 1 min, followed by vigorous vortex shaking during 5 min. Then, the cells were disrupted using a pellet mixer (2 min) at low temperature, followed by centrifugation at 4000g for 10 min at 4°C. The supernatant was collected and transferred to another tube. One hundred microliters of MeOH:ACN:0.1% DTT in water (2:2:1, v/v) was added to the pellet, which was homogenized again using a pellet mixer for 2 min with 100 μl, and then, it was centrifuged at the same conditions described above. After that, both supernatants were combined (about 600 μl) followed by another centrifugation at 4000g for 10 min at 4°C and transferred to a new tube. Reversed-phase liquid chromatography tandem mass spec-trometry (LC-MS/MS) has been used for the quantification of BH4. Briefly, 1 μl of the extract has been injected on RSLC Ultimate 3000 (Thermo Fisher Scientific) directly coupled to a TSQ Vantage mass spectrometer (Thermo Fisher Scientific) via electrospray ionization. A Kinetex C18 column was used (100 Å, 150 mm × 2.1 mm) at a flow rate of 80 μl/min. LC-MS/MS was performed by using the selected reaction monitoring mode of the instrument using the transitions (quantifiers) 242.1 mass/charge ratio (m/z) → 166.1 m/z (BH4) in the positive ion mode. A 7-min-long linear gradient was used, starting from 100% A (1% ACN and 0.1% formic acid in water) to 80% B (0.1% formic acid in ACN). Freshly prepared DTT (1 mg/ml) was used for stabilizing BH4. Authentic metabolite standards (Merck) were used for determining the optimal collision energies for LC-MS/MS and for validating experimental retention times.
Flow cytometry
Flow cytometry of lung tumors was performed 5 months after adeno-Cre inhalation on control LSL-KrasG12D–expressing mice by dissociating tumors with collagenase IV (2 mg/ml; LS004186, Worthington) and deoxyribonuclease (DNase) I (0.2 mg/ml; LS002138, Worthington) in RPMI 1640 medium for 45 min at 37°C. The collagenase/DNase solution was replaced with 10 ml of cold fluorescence-activated cell sorting (FACS) buffer (PBS, 2% fetal bovine serum), and the dissociated cells were passed through a 70-μm cell strainer and then washed with 10 ml of cold FACS buffer. The cells were stained with allophycocyanin (APC)–conjugated anti-mouse CD31 antibody (1:100; 17–0311, eBioscience), phycoerythrin (PE)–Cy7–conjugated anti-mouse CD45 (1:400; 103114, BioLegend), and fluorescein isothiocyanate (FITC)–conjugated anti-mouse CD11b (1:400; 01714D, BD), as well as an anti-mouse CD16/CD32 Fc block (1:100; 553142, BD Biosciences) and 4′,6-diamidino-2-phenylindole (DAPI) (1:500 from a 5 mg/ml stock; D1306, Thermo Fisher Scientific) all diluted in FACS buffer and incubated for 20 min at 4°C. Cells were acquired on BD LSR Fortessa. All data were analyzed with FlowJo v10.0.8r1. A tumor isolation kit (Tumor Isolation Kit, mouse, 130-110-187, Miltenyi Biotec) was used on the samples to deplete endothelial cells (CD31+) and immune cells (CD45+) so that the samples were enriched for only living tumor cells.
Protein blotting
Protein blotting was carried out using standard protocols. Blots were blocked for 1 hour with 5% milk in PBST (1× PBS and 0.1% Tween 20) and then incubated overnight at 4°C with primary antibodies diluted in 5% milk in PBST (1:1000 dilution). Blots were washed three times in PBST for 15 min and then incubated with horseradish peroxidase–conjugated secondary antibody (1:2500 dilution; GE Healthcare, NA9340V) for 45 min at room temperature, washed three times in PBST for 15 min, and visualized using enhanced chemiluminescence (ECL Plus, Pierce, 1896327). Antibodies used in the present study include GCH1 (sc-271482, Santa Cruz Biotechnology; NBP1-84949, Novus Biologicals), β-tubulin III (ab18207, Abcam; T2200, Sigma-Aldrich), GFP (ab13970, Abcam; 632375, Takara), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 2118, Cell Signaling Technology), and EGFR (06-847, Sigma-Aldrich; 4267, Cell Signaling Technology). Quantification of Western blot bands was performed using Fiji software.
Reverse transcription quantitative real-time PCR
Isolated DRG neurons treated with vehicle, PMA, and GO6796 as indicated were cultured for 3 days, and RNA was extracted by acid phenol extraction (TRIzol reagent, Invitrogen). First-strand cDNA synthesis (1 μg of total RNA per reaction) was performed with SuperScript III Reverse Transcriptase per the manufacturer’s instructions (Invitrogen). RT-qPCR was performed using the SYBR green detection system with primer sets designed on Primer Express. Specific PCR product amplification was confirmed using dissociation protocol. Transcript regulation was determined using the relative standard curve method per the manufacturer’s instructions (Applied Biosystems). Relative loading was determined before RT with RNA spectrophotometry followed by gel electrophoresis and after RT by amplification of GAPDH. Primers used for this study were as follows: GCH1, ACAAGCAAGTCCTTGGTCTCA (forward) and GTGAGGCGCTCTTGAACTTG (reverse).
Statistical analyses
All values are expressed as means ± SEM. Details of the statistical tests used are stated in the figure legends. Briefly, Student’s t test was used to compare between two groups. One-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s post hoc tests for multiple comparisons was used for analysis between multiple groups. Two-way ANOVA was used to compare two groups over time. In all tests, P < 0.05 was considered significant. GraphPad Prism v7 was used for data entry, graph construction, and data analysis.
Supplementary Material
Acknowledgments:
We acknowledge the Harvard Stem Cell Institute (HSCI) for their expertise and infrastructure and chemical libraries in running the screen. We acknowledge the Vienna BioCenter Core Facilities (VBCF) for their phenotyping and histopathology work. Metabolomics was performed by the VBCF Metabolomics Facility, which is funded by the city of Vienna through the Vienna Business Agency. The behavioral assays were performed by the Preclinical Phenotyping Facility at VBCF, a member of the Vienna BioCenter (VBC), Austria, and funded by the Austrian Federal Ministry of Education, Science and Research and the city of Vienna.
Funding:
C.J.W. is supported by NIH R35NS105076 and an HMS Blavatnik Award. J.M.P. is supported by the Austrian Federal Ministry of Education, Science and Research, the Austrian Academy of Sciences, and the city of Vienna and grants from the Austrian Science Fund (FWF) Wittgenstein award (Z 271-B19), the T. von Zastrow foundation, a Canada 150 Research Chairs Program (F18-01336), and CIHR (FRN 168899). L.L.R. is supported by an HSCI Therapeutic Screening Center (TSC) grant (no. CF-0009-17-03). A. Latremoliere is supported by NIH grant R01NS112266. A. Latini is a CNPq fellow. S.R. is supported by the National Key R&D Program of China, 2021YFE0193400.
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: The authors declare that they have no competing interests.
View the article online
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Latremoliere A, Woolf CJ, Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cherubino P, Sarzi-Puttini P, Zuccaro SM, Labianca R, The management of chronic pain in important patient subgroups. Clin. Drug Investig 32, 35–44 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Backonja MM, Anticonvulsants (antineuropathics) for neuropathic pain syndromes. Clin. J. Pain 16, S67–S72 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Patetsos E, Horjales-Araujo E, Treating chronic pain with SSRIs: What do we know? Pain Res. Manag 2016, 2020915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drazen JM, COX-2 inhibitors - A lesson in unexpected problems. N. Engl. J. Med 352, 1131–1132 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Blendon RJ, Benson JM, The public and the opioid-abuse epidemic. N. Engl. J. Med 378, 407–411 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Shipton EA, Shipton EE, Shipton AJ, A review of the opioid epidemic: What do we do about it? Pain Ther 7, 23–36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Werner ER, Blau N, Thöny B, Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J 438, 397–414 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, Wu T, Allchorne A, Diatchenko L, Binshtok AM, Goldman D, Adolph J, Sama S, Atlas SJ, Carlezon WA, Parsegian A, Lötsch J, Fillingim RB, Maixner W, Geisslinger G, Max MB, Woolf CJ, GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat. Med 12, 1269–1277 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Lötsch J, Klepstad P, Doehring A, Dale O, A GTP cyclohydrolase 1 genetic variant delays cancer pain. Pain 148, 103–106 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, Dai F, Belfer I, Banco RJ, Martha JF, Tighiouart H, Tromanhauser SG, Jenis LG, Hunter DJ, Schwartz CE, Polymorphic variation of the guanosine triphosphate cyclohydrolase 1 gene predicts outcome in patients undergoing surgical treatment for lumbar degenerative disc disease. Spine 35, 1909–1914 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Zheng N-N, Zhang R-C, Yang X-X, Zhong L-S, Association of rs3783641 single-nucleotide polymorphism in GTP cyclohydrolase 1 gene with post-herpetic neuralgia. J. Dermatol 46, 993–997 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Belfer I, Dai F, Kehlet H, Finelli P, Qin L, Bittner R, Aasvang EK, Association of functional variations in COMT and GCH1 genes with postherniotomy pain and related impairment. Pain 156, 273–279 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Nasser A, Møller LB, GCH1 variants, tetrahydrobiopterin and their effects on pain sensitivity. Scand. J. Pain 5, 121–128 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Heddini U, Bohm-Starke N, Grönbladh A, Nyberg F, Nilsson KW, Johannesson U, GCH1-polymorphism and pain sensitivity among women with provoked vestibulodynia. Mol. Pain 8, 68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadhu N, Jhun EH, Yao Y, He Y, Molokie RE, Wilkie DJ, Wang ZJ, Genetic variants of GCH1 associate with chronic and acute crisis pain in African Americans with sickle cell disease. Exp. Hematol 66, 42–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell CM, Edwards RR, Carmona C, Uhart M, Wand G, Carteret A, Kim YK, Frost J, Campbell JN, Polymorphisms in the GTP cyclohydrolase gene (GCH1) are associated with ratings of capsaicin pain. Pain 141, 114–118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasser A, Møller AT, Hellmund V, Thorborg SS, Jespersgaard C, Bjerrum OJ, Dupontd E, Nachman G, Lykkesfeldt J, Jensen TS, Møller LB, Heterozygous mutations in GTP-cyclohydrolase-1 reduce BH4 biosynthesis but not pain sensitivity. Pain 159, 1012–1024 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Kim H, Dionne RA, Lack of influence of GTP cyclohydrolase gene (GCH1) variations on pain sensitivity in humans. Mol. Pain 3, 6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita M, da Luz Scheffer D, Lenfers Turnes B, Cronin SJF, Latrémolière A, Costigan M, Woolf CJ, Latini A, Andrews NA, Sepiapterin reductase inhibition selectively reduces inflammatory joint pain and increases urinary sepiapterin. Arthritis Rheumatol. 72, 57–66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arai H, Takahashi R, Sakamoto Y, Kitano T, Mashita O, Hara S, Yoshikawa S, Kawasaki K, Ichinose H, Peripheral tetrahydrobiopterin is involved in the pathogenesis of mechanical hypersensitivity in a rodent post-surgical pain model. Pain 161, 2520–2531 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Bulmer DC, Botha CA, Wheeldon A, Grey K, Mein CA, Lee K, Knowles CH, Winchester WJ, Aziz Q, Evidence of a role for GTP cyclohydrolase-1 in visceral pain. Neurogastroenterol. Motil 27, 656–662 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Nasser A, Ali S, Wilsbech S, Bjerrum OJ, Møller LB, Intraplantar injection of tetrahydrobiopterin induces nociception in mice. Neurosci. Lett 584, 247–252 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Latremoliere A, Latini A, Andrews N, Cronin SJ, Fujita M, Gorska K, Hovius R, Romero C, Chuaiphichai S, Painter M, Miracca G, Babaniyi O, Remor AP, Duong K, Riva P, Barrett LB, Ferreirós N, Naylor A, Penninger JM, Tegeder I, Zhong J, Blagg J, Channon KM, Johnsson K, Costigan M, Woolf CJ, Reduction of neuropathic and inflammatory pain through inhibition of the tetrahydrobiopterin pathway. Neuron 86, 1393–1406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latremoliere A, Costigan M, GCH1, BH4 and pain. Curr. Pharm. Biotechnol 12, 1728–1741 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronin SJF, Seehus C, Weidinger A, Talbot S, Reissig S, Seifert M, Pierson Y, McNeill E, Longhi MS, Turnes BL, Kreslavsky T, Kogler M, Hoffmann D, Ticevic M, da Luz Scheffer D, Tortola L, Cikes D, Jais A, Rangachari M, Rao S, Paolino M, Novatchkova M, Aichinger M, Barrett L, Latremoliere A, Wirnsberger G, Lametschwandtner G, Busslinger M, Zicha S, Latini A, Robson SC, Waisman A, Andrews N, Costigan M, Channon KM, Weiss G, Kozlov AV, Tebbe M, Johnsson K, Woolf CJ, Penninger JM, The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 563, 564–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anand P, Bley K, Topical capsaicin for pain management: Therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8 patch. Br. J. Anaesth 107, 490–502 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peppin JF, Pappagallo M, Capsaicinoids in the treatment of neuropathic pain: A review. Ther. Adv. Neurol. Disord 7, 22–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar A, Maitra S, Khanna P, Baidya DK, Clonidine for management of chronic pain: A brief review of the current evidences. Saudi J. Anaesth 8, 92–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Han HJ, Beitz AJ, Lee JH, Intrathecal clonidine suppresses phosphorylation of the n-methyl-D-aspartate receptor NR1 subunit in spinal dorsal horn neurons of rats with neuropathic pain. Anesth. Analg 107, 693–700 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Maruta T, Nemoto T, Satoh S, Kanai T, Yanagita T, Wada A, Tsuneyoshi I, Dexmedetomidine and clonidine inhibit the function of NaV1.7 independent of α2-adrenoceptor in adrenal chromaffin cells. J. Anesth 25, 549–557 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Sisignano M, Angioni C, Park CK, Dos Santos SM, Jordan H, Kuzikov M, Liu D, Zinn S, Hohman SW, Schreiber Y, Zimmer B, Schmidt M, Lu R, Suo J, Zhang DD, Schäfer SMG, Hofmann M, Yekkirala AS, De Bruin N, Parnham MJ, Woolf CJ, Ji RR, Scholich K, Geisslinger G, Targeting CYP2J to reduce paclitaxel-induced peripheral neuropathic pain. Proc. Natl. Acad. Sci. U.S.A 113, 12544–12549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anoush M, Khani MRM, Evaluating the anti-nociceptive and anti-inflammatory effects of ketotifen and fexofenadine in rats. Adv. Pharm. Bull 5, 217–222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szklarczyk D, Santos A, Von Mering C, Jensen LJ, Bork P, Kuhn M, STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 44, D380–D384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pareek TK, Zipp L, Letterio JJ, Cdk5: An emerging kinase in pain signaling. Brain Disord. Ther 2013, (Suppl. 1), 003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velázquez KT, Mohammad H, Sweitzer SM, Protein kinase C in pain: Involvement of multiple isoforms. Pharmacol. Res 55, 578–589 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mannelli L, Micheli L, Crocetti L, Giovannoni M, Vergelli C, Ghelardini C, α2 adrenoceptor: A target for neuropathic pain treatment. Mini Rev. Med. Chem 17, 95–107 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Matar HE, Almerie MQ, Sampson SJ, Fluphenazine (oral) versus placebo for schizophrenia. Cochrane Database Syst. Rev 6, CD006352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curry S, Whelpton R, de Schepper P, Vranckx S, Schiff A, Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man. Br. J. Clin. Pharmacol 7, 325–331 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kataoka Y, Mukohara T, Tomioka H, Funakoshi Y, Kiyota N, Fujiwara Y, Yashiro M, Hirakawa K, Hirai M, Minami H, Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs, inhibits growth of gastric cancer cell lines by blocking inter-receptor tyrosine kinase networks. Invest. New Drugs 30, 1352–1360 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Batley BL, Doherty AM, Hamby JM, Lu GH, Keller P, Dahring TK, Hwang O, Crickard K, Panek RL, Inhibition of FGF-1 receptor tyrosine kinase activity by PD 161570, a new protein-tyrosine kinase inhibitor. Life Sci. 62, 143–150 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Dahring TK, Lu GH, Hamby JM, Batley BL, Kraker AJ, Panek RL, Inhibition of growth factor-mediated tyrosine phosphorylation in vascular smooth muscle by PD 089828, a new synthetic protein tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther 281, 1446–1456 (1997). [PubMed] [Google Scholar]
- 43.Downward J, Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Prior IA, Hood FE, Hartley JL, The frequency of ras mutations in cancer. Cancer Res. 80, 2969–2974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hajdúch M, Jančík S, Drábek J, Radzioch D, Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol 2010, 150960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shacham-Silverberg V, Sar Shalom H, Goldner R, Golan-Vaishenker Y, Gurwicz N, Gokhman I, Yaron A, Phosphatidylserine is a marker for axonal debris engulfment but its exposure can be decoupled from degeneration. Cell Death Dis. 9, 1116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prior IA, Lewis PD, Mattos C, A comprehensive survey of ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simanshu DK, Nissley DV, McCormick F, RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA, Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chuaiphichai S, Rashbrook VS, Hale AB, Trelfa L, Patel J, McNeill E, Lygate CA, Channon KM, Douglas G, Endothelial cell tetrahydrobiopterin modulates sensitivity to ang (angiotensin) II-induced vascular remodeling, blood pressure, and abdominal aortic aneurysm. Hypertension 72, 128–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tammela T, Sanchez-Rivera FJ, Cetinbas NM, Wu K, Joshi NS, Helenius K, Park Y, Azimi R, Kerper NR, Wesselhoeft RA, Gu X, Schmidt L, Cornwall-Brady M, Yilmaz ÖH, Xue W, Katajisto P, Bhutkar A, Jacks T, A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 545, 355–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.IASP Task Force on Taxonomy, Classification of Chronic Pain (International Association for the Study of Pain, 1994). [Google Scholar]
- 53.Vos T, Abajobir AA, Abbafati C, Abbas KM, Abate KH, Abd-Allah F, Abdulle AM, Abebo TA, Abera SF, Aboyans V, Abu-Raddad LJ, Ackerman IN, Adamu AA, Adetokunboh O, Afarideh M, Afshin A, Agarwal SK, Aggarwal R, Agrawal A, Agrawal S, Kiadaliri AA, Ahmadieh H, Ahmed MB, Aichour AN, Aichour I, Aichour MTE, Aiyar S, Akinyemi RO, Akseer N, Al Lami FH, Alahdab F, Al-Aly Z, Alam K, Alam N, Alam T, Alasfoor D, Alene KA, Ali R, Alizadeh-Navaei R, Alkerwi A, Alla F, Allebeck P, Allen C, Al-Maskari F, Al-Raddadi R, Alsharif U, Alsowaidi S, Altirkawi KA, Amare AT, Amini E, Ammar W, Amoako YA, Andersen HH, Antonio CAT, Anwari P, Ärnlöv J, Artaman A, Aryal KK, Asayesh H, Asgedom SW, Assadi R, Atey TM, Atnafu NT, Atre SR, Avila-Burgos L, Avokpaho EFGA, Awasthi A, Quintanilla BPA, Saleem HOB, Bacha U, Badawi A, Balakrishnan K, Banerjee A, Bannick MS, Barac A, Barber RM, Barker-Collo SL, Bärnighausen T, Barquera S, Barregard L, Barrero LH, Basu S, Battista B, Battle KE, Baune BT, Bazargan-Hejazi S, Beardsley J, Bedi N, Beghi E, Béjot Y, Bekele BB, Bell ML, Bennett DA, Bensenor IM, Benson J, Berhane A, Berhe DF, Bernabé E, Betsu BD, Beuran M, Beyene AS, Bhala N, Bhansali A, Bhatt S, Bhutta ZA, Biadgilign S, Bienhoff K, Bikbov B, Birungi C, Biryukov S, Bisanzio D, Bizuayehu HM, Boneya DJ, Boufous S, Bourne RRA, Brazinova A, Brugha TS, Buchbinder R, Bulto LNB, Bumgarner BR, Butt ZA, Cahuana-Hurtado L, Cameron E, Car M, Carabin H, Carapetis JR, Cárdenas R, Carpenter DO, Carrero JJ, Carter A, Carvalho F, Casey DC, Caso V, Castañeda-Orjuela CA, Castle CD, Catalá-López F, Chang HY, Chang JC, Charlson FJ, Chen H, Chibalabala M, Chibueze CE, Chisumpa VH, Chitheer AA, Christopher DJ, Ciobanu LG, Cirillo M, Colombara D, Cooper C, Cortesi PA, Criqui MH, Crump JA, Dadi AF, Dalal K, Dandona L, Dandona R, Neves JD, Davitoiu DV, De Courten B, De Leo D, Degenhardt L, Deiparine S, Dellavalle RP, Deribe K, Jarlais DCD, Dey S, Dharmaratne SD, Dhillon PK, Dicker D, Ding EL, Djalalinia S, Do HP, Dorsey ER, Santos KPBD, Douwes-Schultz D, Doyle KE, Driscoll TR, Dubey M, Duncan BB, El-Khatib ZZ, Ellerstrand J, Enayati A, Endries AY, Ermakov SP, Erskine HE, Eshrati B, Eskandarieh S, Esteghamati A, Estep K, Fanuel FBB, Farinha CSES, Faro A, Farzadfar F, Fazeli MS, Feigin VL, Fereshtehnejad SM, Fernandes JC, Ferrari AJ, Feyissa TR, Filip I, Fischer F, Fitzmaurice C, Flaxman AD, Flor LS, Foigt N, Foreman KJ, Franklin RC, Fullman N, Fürst T, Furtado JM, Futran ND, Gakidou E, Ganji M, Garcia-Basteiro AL, Gebre T, Gebrehiwot TT, Geleto A, Gemechu BL, Gesesew HA, Gething PW, Ghajar A, Gibney KB, Gill PS, Gillum RF, Ginawi IAM, Giref AZ, Gishu MD, Giussani G, Godwin WW, Gold AL, Goldberg EM, Gona PN, Goodridge A, Gopalani SV, Goto A, Goulart AC, Griswold M, Gugnani HC, Gupta R, Gupta R, Gupta T, Gupta V, Hafezi-Nejad N, Hailu AD, Hailu GB, Hamadeh RR, Hamidi S, Handal AJ, Hankey GJ, Hao Y, Harb HL, Hareri HA, Haro JM, Harvey J, Hassanvand MS, Havmoeller R, Hawley C, Hay RJ, Hay SI, Henry NJ, Heredia-Pi IB, Heydarpour P, Hoek HW, Hoffman HJ, Horita N, Hosgood HD, Hostiuc S, Hotez PJ, Hoy DG, Htet AS, Hu G, Huang H, Huynh C, Iburg KM, Igumbor EU, Ikeda C, Irvine CMS, Jacobsen KH, Jahanmehr N, Jakovljevic MB, Jassal SK, Javanbakht M, Jayaraman SP, Jeemon P, Jensen PN, Jha V, Jiang G, John D, Johnson CO, Johnson SC, Jonas JB, Jürisson M, Kabir Z, Kadel R, Kahsay A, Kamal R, Kan H, Karam NE, Karch A, Karema CK, Kasaeian A, Kassa GM, Kassaw NA, Kassebaum NJ, Kastor A, Katikireddi SV, Kaul A, Kawakami N, Keiyoro PN, Kengne AP, Keren A, Khader YS, Khalil IA, Khan EA, Khang YH, Khosravi A, Khubchandani J, Kieling C, Kim D, Kim P, Kim YJ, Kimokoti RW, Kinfu Y, Kisa A, Kissimova-Skarbek KA, Kivimaki M, Knudsen AK, Kokubo Y, Kolte D, Kopec JA, Kosen S, Koul PA, Koyanagi A, Kravchenko M, Krishnaswami S, Krohn KJ, Defo BK, Bicer BK, Kumar GA, Kumar P, Kumar S, Kyu HH, Lal DK, Lalloo R, Lambert N, Lan Q, Larsson A, Lavados PM, Leasher JL, Lee JT, Lee PH, Leigh J, Leshargie CT, Leung J, Leung R, Levi M, Li Y, Li Y, Kappe DL, Liang X, Liben ML, Lim SS, Linn S, Liu A, Liu PY, Liu S, Liu Y, Lodha R, Logroscino G, London SJ, Looker KJ, Lopez AD, Lorkowski S, Lotufo PA, Low N, Lozano R, Lucas TCD, Macarayan ERK, El Razek HMA, El Razek MMA, Mahdavi M, Majdan M, Majdzadeh R, Majeed A, Malekzadeh R, Malhotra R, Malta DC, Mamun AA, Manguerra H, Manhertz T, Mantilla A, Mantovani LG, Mapoma CC, Marczak LB, Martinez-Raga J, Martins-Melo FR, Martopullo I, März W, Mathur MR, Mazidi M, McAlinden C, McGaughey M, McGrath JJ, McKee M, McNellan C, Mehata S, Mehndiratta MM, Mekonnen TC, Memiah P, Memish ZA, Mendoza W, Mengistie MA, Mengistu DT, Mensah GA, Meretoja A, Meretoja TJ, Mezgebe HB, Micha R, Millear A, Miller TR, Mills EJ, Mirarefin M, Mirrakhimov EM, Misganaw A, Mishra SR, Mitchell PB, Mohammad KA, Mohammadi A, Mohammed KE, Mohammed S, Mohanty SK, Mokdad AH, Mollenkopf SK, Monasta L, Hernandez JM, Montico M, Moradi-Lakeh M, Moraga P, Mori R, Morozoff C, Morrison SD, Moses M, Mountjoy-Venning C, Mruts KB, Mueller UO, Muller K, Murdoch ME, Murthy GVS, Musa KI, Nachega JB, Nagel G, Naghavi M, Naheed A, Naidoo KS, Naldi L, Nangia V, Natarajan G, Negasa DE, Negoi I, Negoi RI, Newton CR, Ngunjiri JW, Nguyen CT, Nguyen G, Nguyen M, Le Nguyen Q, Nguyen TH, Nichols E, Ningrum DNA, Nolte S, Nong VM, Norrving B, Noubiap JJN, O’Donnell MJ, Ogbo FA, Oh IH, Okoro A, Oladimeji O, Olagunju AT, Olagunju TO, Olsen HE, Olusanya BO, Olusanya JO, Ong K, Opio JN, Oren E, Ortiz A, Osgood-Zimmerman A, Osman M, Owolabi MO, Pa M, Pacella RE, Pana A, Panda BK, Papachristou C, Park EK, Parry CD, Parsaeian M, Patten SB, Patton GC, Paulson K, Pearce N, Pereira DM, Perico N, Pesudovs K, Peterson CB, Petzold M, Phillips MR, Pigott DM, Pillay JD, Pinho C, Plass D, Pletcher MA, Popova S, Poulton RG, Pourmalek F, Prabhakaran D, Prasad N, Prasad NM, Purcell C, Qorbani M, Quansah R, Rabiee RHS, Radfar A, Rafay A, Rahimi K, Rahimi-Movaghar A, Rahimi-Movaghar V, Rahman M, Rahman MHU, Rai RK, Rajsic S, Ram U, Ranabhat CL, Rankin Z, Rao PV, Rao PC, Rawaf S, Ray SE, Reiner RC, Reinig N, Reitsma MB, Remuzzi G, Renzaho AMN, Resnikoff S, Rezaei S, Ribeiro AL, Ronfani L, Roshandel G, Roth GA, Roy A, Rubagotti E, Ruhago GM, Saadat S, Sadat N, Safdarian M, Safi S, Safiri S, Sagar R, Sahathevan R, Salama J, Salomon JA, Salvi SS, Samy AM, Sanabria JR, Santomauro D, Santos IS, Santos JV, Milicevic MMS, Sartorius B, Satpathy M, Sawhney M, Saxena S, Schmidt MI, Schneider IJC, Schöttker B, Schwebel DC, Schwendicke F, Seedat S, Sepanlou SG, Servan-Mori EE, Setegn T, Shackelford KA, Shaheen A, Shaikh MA, Shamsipour M, Islam SMS, Sharma J, Sharma R, She J, Shi P, Shields C, Shigematsu M, Shinohara Y, Shiri R, Shirkoohi R, Shirude S, Shishani K, Shrime MG, Sibai AM, Sigfusdottir ID, Silva DAS, Silva JP, Silveira DGA, Singh JA, Singh NP, Sinha DN, Skiadaresi E, Skirbekk V, Slepak EL, Sligar A, Smith DL, Smith M, Sobaih BHA, Sobngwi E, Sorensen RJD, Sousa TCM, Sposato LA, Sreeramareddy CT, Srinivasan V, Stanaway JD, Stathopoulou V, Steel N, Stein DJ, Stein MB, Steiner C, Steiner TJ, Steinke S, Stokes MA, Stovner LJ, Strub B, Subart M, Sufiyan MB, Abdulkader RS, Sunguya BF, Sur PJ, Swaminathan S, Sykes BL, Sylte DO, Tabarés-Seisdedos R, Taffere GR, Takala JS, Tandon N, Tavakkoli M, Taveira N, Taylor HR, Tehrani-Banihashemi A, Tekelab T, Shifa GT, Terkawi AS, Tesfaye DJ, Tesssema B, Thamsuwan O, Thomas KE, Thrift AG, Tiruye TY, Tobe-Gai R, Tollanes MC, Tonelli M, Topor-Madry R, Tortajada M, Touvier M, Tran BX, Tripathi S, Troeger C, Truelsen T, Tsoi D, Tuem KB, Tuzcu EM, Tyrovolas S, Ukwaja KN, Undurraga EA, Uneke CJ, Updike R, Uthman OA, Uzochukwu BSC, Van Boven JFM, Varughese S, Vasankari T, Venkatesh S, Venketasubramanian N, Vidavalur R, Violante FS, Vladimirov SK, Vlassov VV, Vollset SE, Wadilo F, Wakayo T, Wang YP, Weaver M, Weichenthal S, Weiderpass E, Weintraub RG, Werdecker A, Westerman R, Whiteford HA, Wijeratne T, Wiysonge CS, Wolfe CDA, Woodbrook R, Woolf AD, Workicho A, Hanson SW, Xavier D, Xu G, Yadgir S, Yaghoubi M, Yakob B, Yan LL, Yano Y, Ye P, Yimam HH, Yip P, Yonemoto N, Yoon SJ, Yotebieng M, Younis MZ, Zaidi Z, Zaki MES, Zegeye EA, Zenebe ZM, Zhang X, Zhou M, Zipkin B, Zodpey S, Zuhlke LJ, Murray CJL, Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D, Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur. J. Pain 10, 287–333 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Dahlhamer JM, Lucas J, Zelaya C, Nahin R, Mackey S, Debar L, Kerns R, Von Korff M, Porter L, Helmick C, Prevalence of chronic pain and high-impact chronic pain among adults—United States, 2016. Morb. Mortal. Wkly. Rep 67, 1001–1006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turk T, Zuhri Yafi R, Namous L, Alkhaledi A, Salahia S, Haider AY, Essali A, Fluphenazine (dose) for people with schizophrenia. Cochrane Database Syst. Rev 2017, CD012848 (2017). [Google Scholar]
- 57.Dreyfuss J, Beer B, Devine DD, Roberts BF, Schreiber EC, Fluphenazine-induced parkinsonism in the baboon: Pharmacological and metabolic studies. Neuropharmacology 11, 223–230 (1972). [DOI] [PubMed] [Google Scholar]
- 58.Jeong S, Cho H, Kim YJ, Il Ma H, Jang S, Drug-induced Parkinsonism: A strong predictor of idiopathic Parkinson’s disease. PLOS ONE 16, e0247354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Capstick N, Pudney H, A comparative trial of orphenadrine and tofenacin in the control of depression and extrapyramidal side-effects associated with fluphenazine decanoate therapy. J. Int. Med. Res 4, 435–440 (1976). [DOI] [PubMed] [Google Scholar]
- 60.Wang S, Liu S, Xu L, Zhu X, Liu W, Tian L, Chen Y, Wang Y, Nagendra BVP, Jia S, Liang L, Huo FQ, The upregulation of EGFR in the dorsal root ganglion contributes to chronic compression of dorsal root ganglions-induced neuropathic pain in rats. Mol. Pain 15, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitchell R, Mikolajczak M, Kersten C, Fleetwood-Walker S, ErbB1-dependent signalling and vesicular trafficking in primary afferent nociceptors associated with hypersensitivity in neuropathic pain. Neurobiol. Dis 142, 104961 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Kersten C, Cameron MG, Cetuximab alleviates neuropathic pain despite tumour progression. BMJ Case Rep. 2012, bcr1220115374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breivik H, Stubhaug A, A new treatment principle for neuropathic pain? Approved oncologic drugs: Epidermal growth factor receptor (EGFR) inhibitors dramatically relieve severe neuropathic pain in a case series. Scand. J. Pain 4, 1–2 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Kersten C, Cameron MG, Bailey AG, Fallon MT, Laird BJ, Paterson V, Mitchell R, Fleetwood-Walker SM, Daly F, Mjåland S, Relief of neuropathic pain through epidermal growth factor receptor inhibition: A randomized proof-of-concept trial. Pain Med. 20, 2495–2505 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Aran V, Omerovic J, Current approaches in NSCLC targeting K-RAS and EGFR. Int. J. Mol. Sci 20, 5701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X, Chen Y, Wang K, Tang J, Chen Y, Jin G, Liu X, The knockdown of the sepiapterin reductase gene suppresses the proliferation of breast cancer by inducing ROS-mediated apoptosis. Int. J. Clin. Exp. Pathol 13, 2228–2239 (2020). [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Du H, Zhan M, Wang H, Chen P, Du D, Liu X, Huang X, Ma P, Peng D, Sun L, Yuan S, Ding J, Lu L, Jiang J, Sepiapterin reductase promotes hepatocellular carcinoma progression via FoxO3a/Bim signaling in a nonenzymatic manner. Cell Death Dis. 11, 248 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chuaiphichai S, McNeill E, Douglas G, Crabtree MJ, Bendall JK, Hale AB, Alp NJ, Channon KM, Cell-autonomous role of endothelial GTP cyclohydrolase 1and tetrahydrobiopterin in blood pressure regulation. Hypertension 64, 530–540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Decosterd I, Woolf CJ, Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000). [DOI] [PubMed] [Google Scholar]
- 70.Rao S, Cronin SJF, Sigl V, Penninger JM, RANKL and RANK: From mammalian physiology to cancer treatment. Trends Cell Biol. 28, 213–223 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.