

Abstract
Globally, the human respiratory syncytial virus (RSV) is one of the major causes of lower respiratory tract infections (LRTIs) in children. The scarcity of complete genome data limits our understanding of RSV spatiotemporal distribution, evolution, and viral variant emergence. Nasopharyngeal samples collected from hospitalized pediatric patients from Buenos Aires tested positive for RSV LRTI during four consecutive outbreaks (2014–2017) were randomly subsampled for RSV complete genome sequencing. Phylodynamic studies and viral population characterization of genomic variability, diversity, and migration of viruses to and from Argentina during the study period were performed. Our sequencing effort resulted in one of the largest collections of RSV genomes from a given location (141 RSV-A and 135 RSV-B) published so far. RSV-B was dominant during the 2014–2016 outbreaks (60 per cent of cases) but was abruptly replaced by RSV-A in 2017, with RSV-A accounting for 90 per cent of sequenced samples. A significant decrease in RSV genomic diversity—represented by both a reduction in genetic lineages detected and the predominance of viral variants defined by signature amino acids—was observed in Buenos Aires in 2016, the year prior to the RSV subgroup predominance replacement. Multiple introductions to Buenos Aires were detected, some with persistent detection over seasons, and also, RSV was observed to migrate from Buenos Aires to other countries. Our results suggest that the decrease in viral diversity may have allowed the dramatic predominance switch from RSV-B to RSV-A in 2017. The immune pressure generated against circulating viruses with limited diversity during a given outbreak may have created a fertile ground for an antigenically divergent RSV variant to be introduced and successfully spread in the subsequent outbreak. Overall, our RSV genomic analysis of intra- and inter-outbreak diversity provides an opportunity to better understand the epochal evolutionary dynamics of RSV.
Keywords: respiratory syncytial virus (RSV), genome, evolution, molecular epidemiology, migration, genotype, phylogeography, next-generation sequencing (NGS)
Introduction
Human respiratory syncytial virus (RSV) is the leading cause of acute lower respiratory tract infection (LRTI) in children globally (Shi et al. 2017). Recently reclassified as an orthopneumovirus in the pneumoviridae family, RSV is a non-segmented negative-strand RNA virus whose genome (∼15,000 nucleotide) contains ten genes that encode eleven proteins (Rima et al. 2017). Two antigenic subgroups (RSV-A and RSV-B) can be distinguished by polyclonal and monoclonal antibodies (Coates, Alling, and Chanock 1966; Anderson et al. 1985). Despite its significant disease burden and public health importance, there is no approved RSV vaccine although there are several RSV vaccine candidates (Mazur et al. 2018). Palivizumab, a monoclonal antibody against the fusion protein (F), one of the two surface glycoproteins, is the only prophylactic treatment available (Beeler and van Wyke Coelingh 1989; Johnson et al. 1997). The other glycoprotein is the attachment glycoprotein (G). F and G are the main targets of the anti-RSV humoral immune response. Still, the G protein has the highest degree of variability at both the amino acid and nucleotide levels (Knipe and Howley 2013). Historically, the G gene has been extensively used for RSV genotype classification (Peret et al. 1998; Venter et al. 2001; Goya et al. 2020; Muñoz-Escalante et al. 2021). Within the two major antigenic subgroups of RSV (RSV-A and RSV-B), a diverse range of genotypes can co-circulate in a given outbreak and replace each other (Schobel et al. 2016; Viegas, Goya, and Mistchenko 2016). Interestingly, over a short time, genotypes can replace the pre-existing ones and become predominant (Viegas, Goya, and Mistchenko 2016; Otieno et al. 2018; Kamau et al. 2020).
RSV acquires genomic and antigenic diversity mainly by accumulating point mutations (genetic drift) and, to a lesser extent, by small nucleotide insertions and deletions that usually do not generate frameshifts (Knipe and Howley 2013). Interestingly, a twenty-amino acid duplication in the C-terminal of the RSV-B G protein was reported in Buenos Aires in 1999 (BA strains). Later, at a similar genome location, a twenty-four-amino acid duplication in RSV-A (ON1 strains) emerged in 2010 (Trento et al. 2006; Eshaghi et al. 2012). Since their independent emergence, the BA and ON1 strains became the only ones circulating as they have displaced all other RSV strains, suggesting a selective advantage of the G protein duplication (van Niekerk and Venter 2011; Viegas, Goya, and Mistchenko 2016; Otieno et al. 2018). In vitro experiments in RSV-B have shown that the duplication in the G gene may increase viral fitness by improving viral protein attachment to the host cell (Hotard et al. 2015).
While extensive sequence data are available to study the evolutionary dynamics of the RSV G gene (and to some extent F gene), overall RSV genome sequencing efforts are far limited compared to other RNA viruses (e.g. influenza or Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)). Few studies were based on RSV complete genome sequences, and none focused on the South American region (Rebuffo-Scheer et al. 2011; Schobel et al. 2016; Otieno et al. 2018; Trovão et al. 2021). This relative paucity of sequences from a broad spatiotemporal origin, combined with a lack of associated clinical metadata, impedes our effort to understand the (1) global sequence diversity, circulation patterns, and seasonality; (2) emergence of new genotypes; (3) association with disease severity; (4) role of other genes in the viral evolution; and (5) epistatic interactions and co-evolution of viral genes of RSV (Lyons and Lauring 2018). With many vaccine candidates in various stages of clinical trials, it is now of greater importance to surveil RSV circulation and genomic diversity for rational vaccine development (Mazur et al. 2018).
The current work addresses the epidemiological and evolutionary dynamics of RSV in the metropolitan area of Buenos Aires, Argentina, the second largest metropolitan city in South America. Recent reports show significant changes in seasonality patterns of RSV after the implementation of public health measures, e.g. social distancing and masking, after the Corona Virus Disease-19 (COVID-19) pandemic (Dolores et al. 2022; Saravanos et al. 2022), and this study establishes the pre-pandemic baseline for South America RSV seasonality. Additionally, our data show intra- and inter-outbreak RSV sequence diversity driving viral subgroup switch and overall provides an opportunity to better understand the epochal evolutionary dynamics of RSV.
Materials and methods
Ethics statement
The project was reviewed and approved by the Medical Ethics and Research Committees of Ricardo Gutiérrez Children’s Hospital, Buenos Aires, Argentina (Institutional Review Board No. 17.21). As the samples were anonymized, parental informed consent was not requested.
Clinical samples and data collection
Nasopharyngeal aspirates (NPAs) were obtained from children under 15 years of age hospitalized with acute LRTI for diagnostic purposes. Clinical samples were tested by immunofluorescence assay for RSV, influenza A and B (Flu A and FluB, respectively), parainfluenza (PIV) 1, 2, and 3, and adenovirus (AdV) as part of the routine hospital diagnosis. Retrospective random subsampling for 403 samples was performed from RSV-positive NPAs collected between 2014 and 2017 (∼20 per cent of RSV-positive samples per year) for whole-genome sequencing (WGS). All samples were stored at −70˚C until RNA extraction.
Viral RNA extraction and WGS
The method for RSV WGS was performed as described previously (Schobel et al. 2016). In brief, the extraction of viral RNA from the NPA was performed using the QIAamp Viral RNA Mini Kit (Qiagen). cDNA was generated using the SuperScript III First-Strand Synthesis System (Thermo Fisher) using the four pooled forward RSV primers. Four independent PCR reactions were performed from the cDNA product to generate four overlapping ∼4 kb amplicons covering the entire genome. Amplicons were quantified using the SYBR Green double stranded DNA detection assay (SYBR Green I Nucleic Acid Gel Stain; Thermo Fisher) and pooled before library preparation using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina. Libraries were sequenced using a 2 × 250 paired-end run in an Illumina MiSeq sequencer at the VANTAGE core, Vanderbilt University Medical Center.
Assembly and annotation of viral consensus genomes
Raw data were demultiplexed by barcode. Raw FASTQ file quality was assessed with FastQC (Babraham Bioinformatics 2022). Barcodes and primer sequences were trimmed using Cutadapt (Martin 2011). Trimmomatic was used for quality filtering of FASTQ: right and left read’s ends were trimmed according to the FastQC assessment of raw data, and reads with length lower than fifty and quality lower than Q20 were removed (Bolger, Lohse, and Usadel 2014). FastQC assessments were also performed on the filtered FASTQ files. BWA-MEM was used for the mapping assembly using either subgroup RSV-A or RSV-B (GenBank accession numbers: KY883567 and KY883569, respectively) as the reference sequence, depending on the sample subgroup assignment (Li and Durbin 2009). The mapping quality was visualized using Tablet software, mainly around known G gene duplications, before extracting the final majority consensus genome sequence using samtools and bcftools (Milne et al. 2013; Danecek et al. 2021).
Sequence data availability
RSV WGS generated in this study was submitted to EpiRSV (GISAID) (ID: EPI_ISL_1074025–EPI_ISL_1074300) and GenBank (ID: ON237083–ON237358).
Phylogenetic and phylodynamic analyses
Genotyping was performed based on the G gene sequence using the reference alignments (Goya et al. 2020). After alignment using multiple sequence comparison by log-expectation (MUSCLE), the most suitable nucleotide substitution model for each dataset was selected with IQ-TREE v1.6.11 software according to the Akaike information criterion (Edgar 2004; Nguyen et al. 2015; Kalyaanamoorthy et al. 2017). Then, maximum-likelihood (ML) trees were inferred with IQ-TREE v1.6.11 software with 1,000 ultrafast bootstrap replicates plus SH-like approximate likelihood ratio test as statistical support (SH-alrt) (Hoang et al. 2018). Statistical values ≥80 per cent were considered for well-supported phylogenetic clades.
The evolutionary rate calculation and effective population size (Ne) calculation of the analyzed RSV sequences were performed with BEAST package v1.10.4 and Tracer v1 7 (Rambaut et al. 2018; Suchard et al. 2018). The Bayesian phylogeographic diffusion analysis in discrete space was performed to study the migration of the ON1 and BA Argentine RSV genomes. For the analysis, in addition to the Argentine genomes, reference genomes downloaded from GenBank were included: 272 RSV-A genomes (270 ON1 genomes and 2 non-ON1 genomes) and 375 BA genomes (Supplementary Table S1). The selection of those reference genomes was phylogenetically based: from a phylogenetic tree with all ON1 and BA genomes published up to December 2021, the sequences associated with the Argentine clades were selected by weighing the geographic representation and collection dates between 2010 and 2018 (Supplementary trees RSV-A and RSV-B). All the alignments were performed with MUSCLE and were visually inspected to detect and correct bioinformatic artifacts. The phylogeographic inference was performed with BEAST package v1.10.4, setting a strict molecular clock and Coalescent Bayesian Skyline tree prior. More than 100 million generations of Markov Chain Monte Carlo were performed with an appropriate sample frequency to obtain 10,000 sampled trees. The convergence of the analysis was assessed by estimating the effective sampling size after a 10 per cent burn-in using Tracer v1.7 and evaluating the highest posterior density (HPD) interval (95 per cent HPD) (Rambaut et al. 2018). TreeAnnotator was used to summarize the information from a sample of trees produced by BEAST onto a single tree (the maximum clade credibility tree). Phylogenetic clades were considered statistically well supported when a posterior probability was ≥0.8.
Results
Viral epidemiology during four consecutive outbreaks in Buenos Aires
A total of 7,845 NPAs from hospitalized children with LRTI at Ricardo Gutiérrez Children’s Hospital between 2014 and 2017 were analyzed by immunofluorescence assay. Of these samples, 1,818 NPAs tested positive for a virus, with an average of 454 viral-positive cases per year (383–487 cases). RSV was the most predominant virus every year, representing an average of 70.4 per cent of the positive cases per year, followed by PIV-3 (15.5 per cent), Flu A (6.6 per cent), and AdV (5.1 per cent). PIV-1, PIV-2, and Flu B reached less than 1 per cent of the total positive cases. Coinfections were detected in twelve RSV-positive cases: eight RSV and PIV-3, three RSV and Flu A, and one RSV and AdV. Figure 1A shows the seasonal pattern of RSV, showing RSV incidences and temperature during a given month. The peak of RSV cases overlapped with the winter when the minimum temperature was lower than 10°C in a given month (in June in most years).
Figure 1.
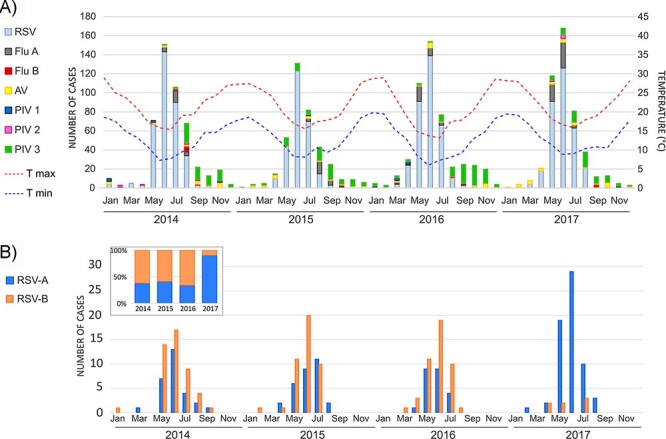
Seasonality of RSV cases in children hospitalized with lower respiratory tract infection in Buenos Aires between 2014 and 2017. (A) A cumulative bar graph of the number of positive cases of respiratory viruses detected per month per year. Dotted lines represent the minimum and maximum temperatures recorded per month per year published by the Argentine National Weather Service. (B) The seasonality of the RSV-positive cases for subgroups A and B.
A total of 403 RSV-positive NPAs were randomly selected without filtering them by viral load value (Ct) to reach 20 per cent of yearly cases. Complete or almost complete RSV genomes were obtained for a total of 286 samples (71 per cent of samples attempted to sequence) with coverage greater than 99 per cent, covering the whole open reading frames and with an average depth of coverage of 13038.5X (range: 544.9x - 13038.5X), and the sequences covered the complete open reading frames of the virus genomes (Supplementary Table S2). The final number of RSV genomes per year was seventy-four from 2014, seventy-three from 2015, sixty-eight from 2016, and seventy-one from 2017, and the total number of RSV-A and RSV-B sequenced was similar (141 RSV-A and 135 RSV-B). Nevertheless, the analysis per year showed that subgroup B predominated from 2014 to 2016 with 60 per cent of the cases, but in 2017, it was displaced by subgroup A, which increased from 40 per cent prior year to 90 per cent of the total analyzed cases in 2017 (Fig. 1B). The seasonality analysis showed the co-circulation of both RSV subgroups with nearly overlapped annual epidemiological peaks.
Genetic diversity of RSV within and between outbreaks
The RSV genotyping analysis showed that all RSV-A Argentine strains were classified as the GA2 genotype and GA2.3 sub-genotype, with multiple genetic lineages. Except for three sequences, all the RSV-A sequences had the characteristic 72-nt duplication in the G gene (ON1 strains). Most classified in the GA2.3.5 genetic lineage and the others were classified in the ON1 divergent lineages, i.e. GA2.3.6a and GA2.3.6b (Fig. 2A). The three sequences without the 72-nt duplications in the G gene were detected in 2015 and were classified as the GA2.3.4 genetic lineage. Similarly, all RSV-B study sequences had the 60-nt duplication in the C-terminal (BA strains) and were classified as the GB5 genotype, GB5.0 sub-genotype with multiple genetic lineages GB5.0.2 and GB5.0.4a, and the divergent GB5.0.5a and GB5.0.4c (Fig. 2B).
Figure 2.
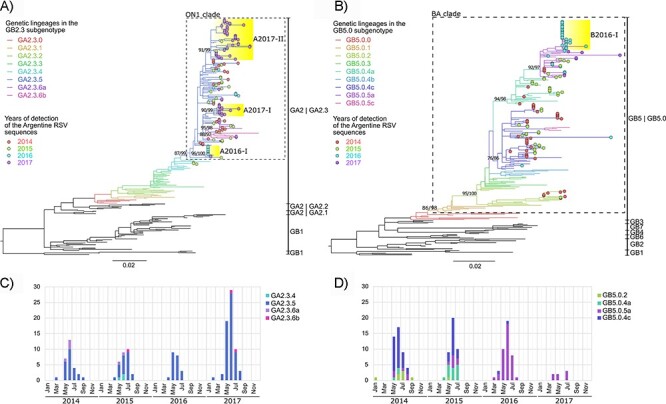
RSV-A and RSV-B genotyping. Maximum-likelihood trees for RSV genotyping based on the G gene for RSV-A (A) and RSV-B (B). Genotypes and sub-genotypes are detailed at the right of each tree. Color in branches denoted genetic lineages where Argentine sequences were associated. Colored dots at the tips indicate the Argentine sequences per year. Yellow highlight in phylogenetic clades denotes the RSV viral variants (see details in the manuscript). Statistical support (SH-alrt/ultrafast (UF)-Bootstrap) is shown in phylogenetic nodes of ON1 or BA clades, genetic lineages where Argentine sequences are associated, and the viral variants. Bar plots below the phylogenetic trees show the seasonality of the genetic lineages detected in Buenos Aires between 2014 and 2017 for RSV-A (C) and RSV-B (D). The number of cases of each genetic lineage detected per month per year is detailed.
The seasonality of the genetic lineages showed that not all of them were co-circulated (Fig. 2C and D). While the RSV-A genetic lineage GA2.3.5 predominated during the entire study period, for RSV-B, GB5.0.4c predominated during 2014 and co-circulated with GB5.0.4a in 2015, and finally, both GB5.0.4a and GB5.0.4c were subsequently replaced by GB5.0.5a in 2016. After 2017, GB5.0.5a was the only RSV-B lineage detected although the number of RSV-B cases in 2017 was minimal as that year was dominated by RSV-A, comprising 90 per cent of the cases.
Well-supported monophyletic clades of Argentine sequences were detected mainly from a specific annual outbreak. The characterization of those Argentine clades was assessed by defining their signature amino acids to better comprehend those viral variants and how they were locally replaced across the years (Harvey et al. 2021). Within the study dataset, the most frequent amino acid in each subgroup’s alignment (A or B) was used as the reference amino acid to identify the viral variant-defining amino acids in Argentina. Multiple viral variants were placed in the GA2.3.5 lineage (named A2016-I, A2017-I, and A2017-II to highlight the RSV subgroup and the year of detection) and the GB5.0.5a lineage (named B2016-I) clustering to specific annual outbreaks (Fig. 2 and Table 1).
Table 1.
Variant-specific signature amino acids in RSV.
| Lineage | Viral variant (# seq) | N | P | SH | G | M2-2 | L |
|---|---|---|---|---|---|---|---|
| GA2.3.5 | A2016-I (11 seq) |
– | – | – | Ser105Pro + Pro222Gln + Thr249Ile + Tyr273His + Asn318Thr | – | Ser141Pro + Ile234Val |
| A2017-I (14 seq) |
– | – | – | – | – | Asn143Asp + Thr179Ser + Ile1653Val + Lys1661Asn | |
| A2017-II (47 seq) |
– | – | – | K134I + I243S + E262K | – | Asp1731Gly | |
| GB5.0.5a | B2016-I (33 seq) |
Ile65Val | Glu68Asp | Asn64Asp | Thr220Pro | Ile82Met | Ile1716Val + Asn1727Thr + Phe1982Leu |
The haplotypes of amino acid substitutions that characterize the predominant viral variants described in 2016 and 2017 are shown. The genetic lineage, the number of sequences, and the variant-specific signature amino acids in each viral protein (when appropriate) are detailed for each viral variant. The ‘+’ symbol indicates that all the amino acid substitutions together are present in that viral variant.
While A2016-I and B2016-I included sequences mainly from the 2016 outbreak, A2016-I represented 52 per cent of RSV-A cases in 2016, and B2016-I represented 78 per cent of the RSV-B instances in the same year. The A2016-I variant had signature amino acids in the G and L protein sequences, while the B2016-I variant had signature amino acids in the SH, G, N, P M2-2, and L protein sequences. During the 2017 outbreak, A2017-I comprised 21 per cent, and A2017-II comprised 73 per cent of the RSV-A cases. The A2017-I variant had specific variant-defining substitutions in the L protein and A2017-II in the G and L proteins. All the variants of RSV-A (A2016-I, A2017-I, and A2017-II) had divergent origins, despite being classified in the same genetic lineage (Fig. 2A).
RSV evolutionary rate and effective population size dynamics
The evolutionary rates estimated for the RSV genomes in this study were similar for both subgroups: RSV-A: 7.6 × 10−4 subs/site/year (95 per cent HPD: 6.63 × 10−4 to 8.58 × 10−4) and RSV-B: 7.7 × 10−4 subs/site/year (95 per cent HPD: 6.66 × 10−4 to 8.77 × 10−4), similarly to previously reported global RSV evolutionary rates (Trovão et al. 2021). Similar to published findings, evolutionary rates per Open Reading Frame showed a range from 6 × 10−4 to 4 × 10−3 subs/site/year, where the highest evolutionary rate was found in the G gene with 2.86 × 10−3 subs/site/year (95 per cent HPD: 2.27 × 10−3 to 3.43 × 10−3) for RSV-A and 3.93 × 10−3 subs/site/year (95 per cent HPD: 2.84 × 10−3 to 5.06 × 10−3) for RSV-B (Supplementary Fig. S1) (Schobel et al. 2016).
Demographic reconstruction for the analyzed Argentine RSV sequences showed an annual pattern of the effective population size (Ne; interpreted as a measure of viral diversity) with a maximum by the middle of each year (during the winter) and a minimum at the end of the year (during the summer) (Fig. 2). In addition, despite the annual fluctuation, a general decrease was observed during the entire analyzed period in which the Ne at the end of 2017 was significantly lower for both subgroups (Fig. 2).
Evidence of multiple RSV introductions and local persistence of RSV in Buenos Aires
To understand if RSV introductions to Argentina coincided with the decrease in viral diversity in Buenos Aires, as found in Fig. 3, and the abrupt change in the prevalence of viral subgroups detected between 2016 and 2017, Bayesian phylogeographic inferences were performed. Our analysis showed that the Argentine sequences were associated with sequences from multiple countries, suggesting multiple viral introductions to Buenos Aires each year. In addition, some of those introduced viruses were detected for extended periods and circulated locally, as evidenced by the presence of sequences over multiple annual outbreaks. For example, a total of fourteen introductions of RSV-A were detected, of which five introductions were persistently detected (Fig. 4, Supplementary Fig. S2, Table S2). Similarly, thirteen introductions were detected for RSV-B, from which nine were detected for more than one annual epidemiological outbreak (Fig. 4, Supplementary Fig. S3, Table S3). To support our findings, we further analyzed the monophyly of the RSV introductions in Argentina by ML inference of the G gene, including all the G gene sequences available in GenBank up to December 2020 (Supplementary phylogenetic trees).
Figure 3.
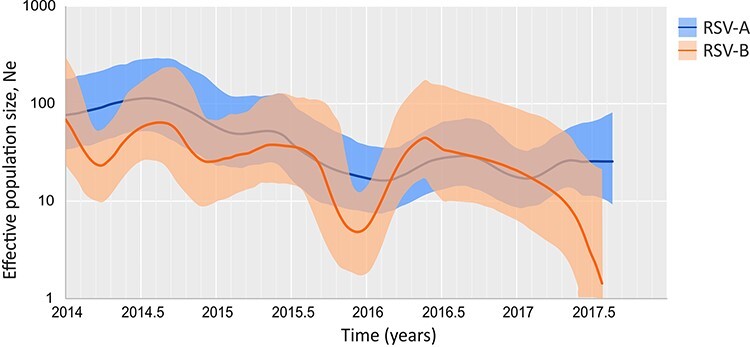
The phylodynamics of RSV in Buenos Aires between 2014 and 2017. The reconstruction of the effective population size (Ne) concerning the time for RSV circulating in Buenos Aires. The lines show the median value, and the colored areas denote the 95 per cent HPD ranges (light blue: RSV-A and orange: RSV-B).
Figure 4.
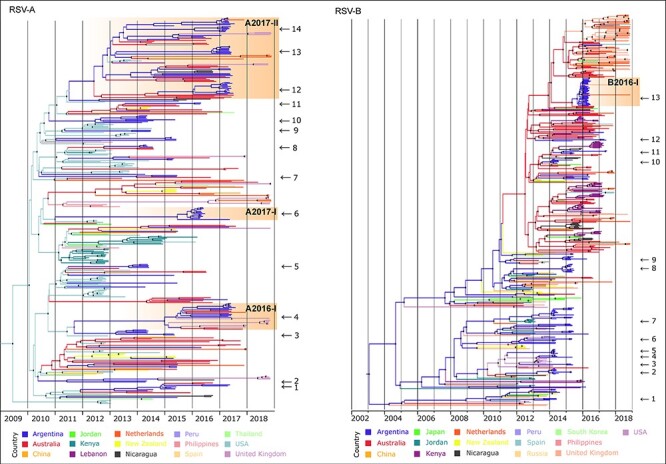
The Bayesian phylogeographic discrete analysis. Maximum clade credibility tree for RSV-A and RSV-B. The branches’ colors represent the MRCA’s location (described in the legend). Black dots in the nodes denote statistical support above 0.7 posterior probability. The time scale in years is detailed at the bottom. The viral introductions described in this study are marked with an arrow and identified with a number (details in Table 2). RSV viral variants discussed in the text are highlighted in orange, including the variant name at the top. Complete full-detailed phylogeographic trees are in the Supplementary material.
The locations of the most recent common ancestor (MRCA) for most introductions of both RSV-A and RSV-B were the USA and Australia. However, bias in the global RSV genome sharing in public databases leads to uncertainty degree in the actual location of MRCA. In some cases, the time of the MRCA (tMRCA) of the introductions occurred several years before the Argentine sequences were detected. For example, the tMRCA in introduction #1 (Table 3) was 5 years before the detection of the first RSV-B sequence in Buenos Aires, likely evidencing the limitation of the surveillance of this study that began in 2014. On the other hand, other introductions were detected months after the inferred tMRCA; for example, introduction #10 of RSV-B was detected 3 months after the tMRCA (Table 3). Interestingly, the number of introductions detected decreased over time.
Table 3.
RSV-B introductions detected in Buenos Aires.
| # | tMRCA (95% HPD) | No. of ARG sequences | Collection date of ARG | Introduced from (PP) | Genetic lineage | Viral variant |
|---|---|---|---|---|---|---|
| 1 | 2009.02 (2008.47–2009.64) | 7 | 2014 | Argentina (0.82*) | GB5.0.2 | – |
| 2 | 2013.32 (2012.97–2013.62) | 8 | 2014 and 2015 | Australia (0.47) and Nicaragua (0.29) | GB5.0.4c | – |
| 3 | 2015.15 (2012.97–2013.62) | 2 | 2015 | USA (0.67) | GB5.0.4c | – |
| 4 | 2013.46 (2013.19–2013.71) | 4 | 2014 and 2015 | USA (0.87) | GB5.0.4c | – |
| 5 | 2014.20 (2014.08–2014.30) | 7 | 2014 | USA (0.61) | GB5.0.4c | – |
| 6 | 2009.69 (2009.19–2010.18) | 6 | 2014 and 2015 | Argentina (0.80*) | GB5.0.4c | – |
| 7 | 2014.1 (2013.99–2014.19) | 11 | 2014 and 2015 | Argentina (1*) | GB5.0.4c | – |
| 8 | 2014.76 (2014.55–2014.91) | 11 | 2014 and 2015 | Argentina (1*) | GB5.0.4a | – |
| 9 | 2014.87 (2014.63–2015.07) | 5 | 2015 and 2016 | Argentina (0.87*) | GB5.0.4a | – |
| 10 | 2014.17 (2014.03–2014.27) | 6 | 2014 and 2015 | Australia (0.99) | GB5.0.5a | – |
| 11 | 2014.81 (2014.53–2015.06) | 5 | 2015 and 2017 | Australia (0.99) | GB5.0.5a | – |
| 12 | 2014.28 (2013.93–2014.68) | 3 | 2015 and 2017 | Australia (0.99) | GB5.0.5a | – |
| 13 | 2015.46 (2015.17–2015.70) | 35 | 2016 | Australia (0.72) | GB5.0.5a | B2016-I |
The characterization of each viral introduction indicated in Fig. 4 (also in Supplementary Fig. S3) is described. The table details the tMRCA with the 95 per cent HDP range, the number of Argentine (ARG) sequences, and their collection years. The location of the MRCA and the posterior probability of the ancestral state (PP) are given in the ‘Introduced from’ column. When the support was similar for two locations, both countries were reported. The symbol * indicates that the low statistical support of the ancestral node to the introduction did not allow to identify the probable location of origin. Finally, the genetic lineage associated and, when appropriate, the viral variant are also given.
A detailed analysis of the RSV viral variants in our study (Fig. 2, Table 1) showed that A2016-I and A2017-I for RSV-A (#6 and #4 from Table 2, respectively) and B2016-I for RSV-B (#13 from Table 3) arose from single different introductions to the country. A2016-I and B2016-I were introduced in mid-2015, a year before their detection. A2016-I was not detected in other countries, but B2016-I was detected in the UK in 2017 and the Netherlands in 2018. A2017-I was also detected in Australia in 2016, Spain in 2018, the UK in 2018, and the Netherlands in 2018. On the other hand, the A2017-II variant was introduced multiple times to Buenos Aires (#12, #13, and #14 in Table 2, Supplementary Fig. S2), all cases closely related to a monophyletic clade with high statistical support, suggesting that the A2017-II variant had been circulating globally. Specifically, A2017-II was introduced in Buenos Aires for the first time in mid-2013 (#14 in Table 2, Supplementary Fig. S2). Detailed analyses showed that the variants with the same specific sequence substitutions identified in this study were also identified from other countries (Supplementary Table S3).
Table 2.
RSV-A introductions detected in Buenos Aires.
| # | tMRCA (95% HPD) | No. of ARG sequences | Collection year of ARG | Introduced from (PP) | Genetic lineage | Viral variant |
|---|---|---|---|---|---|---|
| 1 | 2013.25 (2012.88–2013.65) | 7 | 2014, 2015, and 2017 | USA (0.90) | GA2.3.5 | – |
| 2 | 2011.52 (2011.19–2011.96) | 2 | 2014 | USA (0.82) | GA2.3.5 | – |
| 3 | 2013.21 (2012.84–2013.51) | 6 | 2014 and 2015 | USA (0.51) and Argentina (0.46) | GA2.3.5 | – |
| 4 | 2015.41 (2015.11–2015.69) | 13 | 2017 | Argentina (0.92*) | GA2.3.6a | A2017-I |
| 5 | 2013.78 (2013.50–2014.03) | 4 | 2014 | USA (0.86) | GA2.3.5 | – |
| 6 | 2015.65 (2015.20–2015.85) | 13 | 2016 and 2017 | USA (0.99) | GA2.3.5 | A2016-I |
| 7 | 2013.21 (2012.75–2013.68) | 2 | 2015 | USA (1) | GA2.3.5 | – |
| 8 | 2014.16 (2014.00–2014.35) | 3 | 2014 | USA (1) | GA2.3.5 | – |
| 9 | 2013.89 (2013.59–2014.17) | 2 | 2014 | USA (1) | GA2.3.5 | – |
| 10 | 2012.56 (2012.17–2012.89) | 8 | 2014 and 2015 | USA (0.9) | GA2.3.5 | – |
| 11 | 2016.18 (2015.98–2016.33) | 2 | 2016 | Australia (0.97) | GA2.3.5 | – |
| 12 | 2016.13 (2015.80–2016.44) | 13 | 2017 | Australia (0.59) | GA2.3.5 | A2017-II |
| 13 | 2016.7 (2016.46–2016.94) | 8 | 2017 | Argentina (1*) | GA2.3.5 | A2017-II |
| 14 | 2013.49 (2013.17–2013.94) | 18 | 2015, 2016, and 2017 | Argentina (0.95*) | GA2.3.5 | A2017-II |
The characterization of each viral introduction indicated in Fig. 4 (also in Supplementary Fig. S2) is described. The table details the tMRCA with the 95 per cent HDP range, the number of Argentine (ARG) sequences, and their collection years. The location of the MRCA and the posterior probability of the ancestral state (PP) are given in the ‘Introduced from’ column. When the support was similar for two locations, both countries are reported. The symbol * indicates that the low statistical support of the ancestral node to the introduction did not allow the identification of the probable origin location. Finally, the genetic lineage associated and, when appropriate, the viral variant are also given.
Discussion
Despite being associated with subsequent early-life recurrent wheezing and asthma and one of the most significant pathogens causing LRTIs in children, there are very few studies on RSV molecular epidemiology and evolutionary dynamics using complete genome sequences (Ding et al. 2020). This study utilizes one of the largest collections of complete RSV genomes reported from a given location to understand RSV seasonality and molecular evolution in one of the major metropolitan cities in the southern hemisphere, Buenos Aires, over four consecutive annual outbreaks (2014–2017). Our epidemiologic analysis showed RSV to be the most frequently identified virus in children hospitalized with LRTI, with higher incidences during the winter season of the southern hemisphere (June to September) (Tang and Loh 2014; Gentile et al. 2019). There were multiple RSV introductions from other countries, where some of the introduced viruses persisted locally over multiple annual epidemics (Viegas, Goya, and Mistchenko 2016). There was an abrupt switch in the RSV subgroup from RSV-B dominance from 2014 to 2016 to RSV-A dominance in 2017. Interestingly, prior to the subgroup switch, a significant decrease in the overall circulating RSV diversity was detected.
While RSV subgroup dominance switched between RSV-A and RSV-B, as previously reported, at any given time, both subgroups can co-circulate in the same localities (Salimi et al. 2016; Rojo et al. 2017). Reports from the USA and Germany also found RSV-B predominance up to 2016, while in other countries (Colombia, Australia, South Africa, and India), the opposite was found (Ruzin et al. 2018; Streng et al. 2019; Chadha et al. 2020; Londono-Avendano et al. 2021; Robertson et al. 2021). However, none of the published studies showed such a dramatic switch of RSV-B to RSV-A in 2017, highlighting complex local versus global epidemiological dynamics of RSV transmission.
A twenty-amino acid duplication in the RSV-B C-terminal of the G protein first emerged in 1999 in Buenos Aires, named BA strains. Since 2005, these BA strains have completely replaced non-duplicated RSV-B (Trento et al. 2006), suggesting a selective advantage, which has been confirmed using in vitro studies (Hotard et al. 2015). Similarly, RSV-A acquired a twenty-four-amino acid duplication in almost the same region of the G protein, first identified in Ontario, Canada (ON1 strain) in 2010, which now dominates RSV-A global circulation (Goya et al. 2020). In the present study of Argentine genomes, all except three RSV-A genomes had the G gene duplication (ON1 strain), and all the three non-ON1 strains were found only during the 2015 outbreak, clustering in the GA2.3.4 genetic lineage (the ancestor from which the ON1 strains diverged). Our findings concur with what others have reported, demonstrating the diminishing presence of non-ON1 strains in global circulation (Lee et al. 2021; Valley-Omar et al. 2022). This result suggests that the ON1 strains completely replaced all other non-duplicated RSV-A viruses globally around 2015–2016. However, this hypothesis should be tested using a comprehensive global RSV genomic surveillance study.
We also reported a decrease in the genetic diversity of circulating RSV in Buenos Aires throughout the analysis period. Not only did we observe a decline in the number of genetic lineages detected, but there was a viral homogeneity given by the detection and predominance of viral variants defined by signature amino acids (A2016-I, A2017-I, and A2017-II in RSV-A and B2016-I in RSV-B). Most of these variant-defining substitutions were located in either the G (attachment glycoprotein) or the L (RNA-polymerase RNA-dependent) proteins. However, none of these unique amino acid substitutions have been previously linked to viral mutations that affect viral fitness, such as transmission, antiviral escape, or disease severity (Bakker et al. 2013; Knipe and Howley 2013; Galloux et al. 2015; Gilman et al. 2019). Future experimental work is needed to confirm whether some or any of these mutations have any selective advantage, similar to what has been found for other viruses such as influenza and SARS-CoV-2 (Cotter, Jin, and Chen 2014; Zhang et al. 2020). Interestingly, none of the variant-defining substitutions were in the F protein, the target protein of the only prophylactic treatment based on monoclonal antibodies against RSV.
The effective population size (Ne) accounts for both the viability of the observed population and the environmental stochasticity, which include, for example, environmental factors such as transmission bottlenecks and the introduction or emergence of new viral variants (Pearson 2013). The Ne oscillated between annual seasons but also generally decreased over the study period, and this decrease was more pronounced in RSV-B. In the period analyzed, annual periodicity was reflected by increased genetic diversity during the winter seasons, with higher transmission rates and viral introductions from other parts of the world.
Phylogeographic analyses showed that viral variants were introduced from out of the country, some as one introduction, while other variants were introduced multiple times. The A2016-I, A2017-I, and B2016-I viral variants were introduced to the region during the 2015 outbreak but were detected in 2016. A2017-II was introduced for the first time in 2013 but was reintroduced multiple times in the following seasons. Except for a few instances, most viral variant cases expanded dramatically after the introduction in one given outbreak and were replaced by another introduction during the next annual epidemic. The dramatic expansion (>50 per cent of total cases) of the RSV-B variant B2016-I in 2016 reduced the overall local viral genetic diversity and potentially created a fertile ground for the introduction/emergence of the genetically divergent variants A2017-I and A2017-II during the next outbreak (2017).
Based on our findings, we hypothesize that the annual viral variant replacement or even the RSV subgroup switch may be associated with a decrease in the overall viral diversity during the prior outbreak. There could be multiple factors simultaneously or independently driving it. For example, (1) the locally persisted virus over an outbreak was introduced early in the RSV outbreak and had an increased advantage in transmission locally, a phenomenon defined as the founder effect (Díez-Fuertes et al. 2021). (2) The genetic drift and accumulation of mutations eventually improve the viral fitness and transmission, displacing the other viral variants, a process called clonal interference or the ‘selective sweeps’ described in the case of influenza A virus (Strelkowa and Lässig 2012). In addition, some substitutions might not be directly beneficial but rather as an epistatic interaction or merely a genetic hitchhiking (Kaplan, Hudson, and Langley 1989). (3) Herd immunity (immune pressure from the community) can act as a bottleneck for the virus circulating in a specific annual outbreak (Agoti et al. 2012). For example, the circulation of a single variant for a prolonged period in a community could induce herd immunity against that variant. The introduction of an antigenically different variant or subtype (with selective advantage) can force the prior circulating variant to extinction and replacement. Although the immunity generated against RSV after infection is not long lasting and reinfections occur throughout life (Falsey, Singh, and Walsh 2006), it is most likely long enough to mount selection pressure on the RSV during the subsequent outbreak.
To support part of our hypothesis, with the COVID-19 pandemic, new factors, such as the mitigation measures implemented against the pandemic, served as a bottleneck. Consequently, a significant decrease in RSV cases was reported in 2020, even reaching non-detection in several countries, such as Argentina (Dolores et al. 2022). In 2021, RSV reemerged in Argentina with a delayed seasonal activity, a lower number of cases than usual, and a low viral diversity led by the viral introduction from other countries (Dolores et al. 2022).However, it is worth mentioning that the results from this study need to be confirmed with a comprehensive global genomic epidemiologic study in the future. RSV surveillance in outpatients and asymptomatic cases should take greater importance, considering the possibility of persistent circulation. To explain how a viral introduction would remain in a given community from one annual outbreak to the next, in the context of decreasing case detection during the inter-outbreak period, it is critical to identify which age groups may be potential reservoirs of the virus. Additionally, immunosuppressed patients may also serve as reservoirs of RSV with prolonged infection and intra-host viral evolution, as has been reported (Grad et al. 2014).
While our findings cannot be generalized, as it was only focused on pediatric patients hospitalized in Buenos Aires, future studies in this region and elsewhere will allow us to identify if the described patterns are repeated or location specific. Additionally, our findings are contingent on improving the spatiotemporal representation of RSV genomes in public databases (Supplementary Fig. S4). Here, we reported the study of the largest dataset of RSV genomes in South America, describing the evolutionary dynamics of RSV in Buenos Aires and improving our understanding of RSV diversity from one annual outbreak to the next. This study shows how genomic surveillance and detailed evolutionary analysis are essential to understanding future viral outbreaks and how circulating viral diversity may determine the RSV molecular epidemiology profile from one annual outbreak to the next.
Supplementary Material
Acknowledgements
We would like to especially thank the health-care workers who perform the routine diagnosis at the Virology Laboratory of Ricardo Gutiérrez Children’s Hospital.
Contributor Information
Stephanie Goya, Virology Laboratory, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina; National Scientific and Technical Research Council, Godoy Cruz 2290, Buenos Aires 1425, Argentina; Department of Medicine, Vanderbilt University Medical Center, 1161 21st Ave S, Nashville, TN 37232, USA.
Maria Florencia Lucion, Department of Epidemiology, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina.
Meghan H Shilts, Department of Medicine, Vanderbilt University Medical Center, 1161 21st Ave S, Nashville, TN 37232, USA.
María del Valle Juárez, Department of Epidemiology, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina.
Angela Gentile, Department of Epidemiology, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina.
Alicia S Mistchenko, Virology Laboratory, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina.
Mariana Viegas, Virology Laboratory, Ricardo Gutiérrez Children’s Hospital, Gallo 1330, Buenos Aires 1425, Argentina; National Scientific and Technical Research Council, Godoy Cruz 2290, Buenos Aires 1425, Argentina.
Suman R Das, Department of Medicine, Vanderbilt University Medical Center, 1161 21st Ave S, Nashville, TN 37232, USA; Department Otolaryngology—Head and Neck Surgery, Vanderbilt University Medical Center, 1215 21st Ave S, Nashville, TN 37232, USA.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
The sample collection for this work was supported by ANPCyT, Ministerio de Ciencia, Tecnología e Innovación de la Nación Argentina (grant number PICT03443/2014). The sequencing work was supported NIAID, NIH with grant numbers R21AI154016, R21AI149262, and R21AI142321 and by the Centers for Disease Control and Prevention (grant number 75D3012110094). The support for the Vanderbilt Technologies for Advanced Genomics Core was received from the National Institutes of Health under award numbers UL1RR024975, P30CA68485, P30EY08126, and G20RR030956. The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest:
The authors declare no conflict of interest.
References
- Agoti C. N. et al. (2012) ‘Genetic Relatedness of Infecting and Reinfecting Respiratory Syncytial Virus Strains Identified in a Birth Cohort from Rural Kenya’, The Journal of Infectious Diseases, 206: 1532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. J. et al. (1985) ‘Antigenic Characterization of Respiratory Syncytial Virus Strains with Monoclonal Antibodies’, Journal of Infectious Diseases, 151: 626–33. [DOI] [PubMed] [Google Scholar]
- Babraham Bioinformatics . FastQC A Quality Control Tool for High Throughput Sequence Data (cited 18 Feb 2022) <https://www.bioinformatics.babraham.ac.uk/projects/fastqc/> accessed 24 Sep 2022.
- Bakker S. E. et al. (2013) ‘The Respiratory Syncytial Virus Nucleoprotein-RNA Complex Forms a Left-Handed Helical Nucleocapsid’, Journal of General Virology, 94: 1734–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler J. A., and van Wyke Coelingh K. (1989) ‘Neutralization Epitopes of the F Glycoprotein of Respiratory Syncytial Virus: Effect of Mutation upon Fusion Function’, Journal of Virology, 63: 2941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics (Oxford, England), 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha M.et al. , WHO RSV Surveillance Group . (2020) ‘Human Respiratory Syncytial Virus and Influenza Seasonality Patterns-Early Findings from the WHO Global Respiratory Syncytial Virus Surveillance’, Influenza and Other Respiratory Viruses, 14: 638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates H. V., Alling D. W., and Chanock R. M. (1966) ‘An Antigenic Analysis of Respiratory Syncytial Virus Isolates by a Plaque Reduction Neutralization Test’, American Journal of Epidemiology, 83: 299–313. [DOI] [PubMed] [Google Scholar]
- Cotter C. R., Jin H., and Chen Z. (2014) ‘A Single Amino Acid in the Stalk Region of the H1N1pdm Influenza Virus HA Protein Affects Viral Fusion, Stability and Infectivity’, PLoS Pathogens, 10: e1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P. et al. (2021) ‘Twelve Years of SAMtools and BCFtools’, GigaScience, 10: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Fuertes F. et al. (2021) ‘A Founder Effect Led Early SARS-CoV-2 Transmission in Spain’, Journal of Virology, 95: e01583–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q. et al. (2020) ‘Comparison of Clinical Features of Acute Lower Respiratory Tract Infections in Infants with RSV/HRV Infection, and Incidences of Subsequent Wheezing or Asthma in Childhood’, BMC Infectious Diseases, 20: 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolores A. et al. (2022) ‘RSV Reemergence in Argentina since the SARS-CoV-2 Pandemic’, Journal of Clinical Virology, 149: 105126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004) ‘MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity’, BMC Bioinformatics, 5: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi A. et al. (2012) ‘Genetic Variability of Human Respiratory Syncytial Virus A Strains Circulating in Ontario: A Novel Genotype with A 72 Nucleotide G Gene Duplication’, PLoS One, 7: e32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey A. R., Singh H. K., and Walsh E. E. (2006) ‘Serum Antibody Decay in Adults Following Natural Respiratory Syncytial Virus Infection’, Journal of Medical Virology, 78: 1493–7. [DOI] [PubMed] [Google Scholar]
- Galloux M. et al. (2015) ‘Identification and Characterization of the Binding Site of the Respiratory Syncytial Virus Phosphoprotein to RNA-Free Nucleoprotein’, Journal of Virology, 89: 3484–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile A. et al. (2019) ‘Burden of Respiratory Syncytial Virus Disease and Mortality Risk Factors in Argentina: 18 Years of Active Surveillance in a Children’s Hospital’, Pediatric Infectious Disease Journal, 38: 589–94. [DOI] [PubMed] [Google Scholar]
- Gilman M. S. A. et al. (2019) ‘Structure of the Respiratory Syncytial Virus Polymerase Complex’, Cell, 179: 193–204.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goya S. et al. (2020) ‘Toward Unified Molecular Surveillance of RSV: A Proposal for Genotype Definition’, Influenza and Other Respiratory Viruses, 14: 274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad Y. H. et al. (2014) ‘Within-Host Whole-Genome Deep Sequencing and Diversity Analysis of Human Respiratory Syncytial Virus Infection Reveals Dynamics of Genomic Diversity in the Absence and Presence of Immune Pressure’, Journal of Virology, 88: 7286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. T. et al. (2021) ‘SARS-CoV-2 Variants, Spike Mutations and Immune Escape’, Nature Reviews. Microbiology, 19: 409–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotard A. L. et al. (2015) ‘Functional Analysis of the 60-Nucleotide Duplication in the Respiratory Syncytial Virus Buenos Aires Strain Attachment Glycoprotein’, Journal of Virology, 89: 8258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. et al. (1997) ‘Development of a Humanized Monoclonal Antibody (MEDI-493) with Potent In Vitro and In Vivo Activity against Respiratory Syncytial Virus’, The Journal of Infectious Diseases, 176: 1215–24. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamau E. et al. (2020) ‘Evolution of Respiratory Syncytial Virus Genotype BA in Kilifi, Kenya, 15 Years On’, Scientific Reports, 10: 21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N. L., Hudson R. R., and Langley C. H. (1989) ‘The “Hitchhiking Effect’’ Revisited’, Genetics, 123: 887–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe D. M., and Howley P. (2013) Fields Virology. Lippincott Williams & Wilkins. [Google Scholar]
- Lee C.-Y. et al. (2021) ‘Genetic Diversity and Molecular Epidemiology of Circulating Respiratory Syncytial Virus in Central Taiwan, 2008–2017’, Viruses, 14: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., and Durbin R. (2009) ‘Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform’, Bioinformatics (Oxford, England), 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londono-Avendano M. A. et al. (2021) ‘Transmission of Respiratory Syncytial Virus Genotypes in Cali, Colombia’, Influenza and Other Respiratory Viruses, 15: 521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons D. M., and Lauring A. S. (2018) ‘Mutation and Epistasis in Influenza Virus Evolution’, Viruses, 10: E407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011) ‘Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads’, EMBnet Journal, 17: 10–2. [Google Scholar]
- Mazur N. I.et al. , Respiratory Syncytial Virus Network (Resvinet) Foundation . (2018) ‘The Respiratory Syncytial Virus Vaccine Landscape: Lessons from the Graveyard and Promising Candidates’, The Lancet Infectious Diseases, 18: e295–e311. [DOI] [PubMed] [Google Scholar]
- Milne I. et al. (2013) ‘Using Tablet for Visual Exploration of Second-Generation Sequencing Data’, Briefings in Bioinformatics, 14: 193–202. [DOI] [PubMed] [Google Scholar]
- Muñoz-Escalante J. C. et al. (2021) ‘Respiratory Syncytial Virus B Sequence Analysis Reveals a Novel Early Genotype’, Scientific Reports, 11: 3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otieno J. R. et al. (2018) ‘Whole Genome Analysis of Local Kenyan and Global Sequences Unravels the Epidemiological and Molecular Evolutionary Dynamics of RSV Genotype ON1 Strains’, Virus Evolution, 4: vey027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson S. M. (2013) ‘Landscape Ecology and Population Dynamics’, in S. A. Levin (ed.) Encyclopedia of Biodiversity, pp. 488–502. Waltham: Academic Press. (cited 18 Feb 2022) <https://www.sciencedirect.com/science/article/pii/B9780123847195004172> accessed 24 Sep 2022. [Google Scholar]
- Peret T. C. et al. (1998) ‘Circulation Patterns of Genetically Distinct Group A and B Strains of Human Respiratory Syncytial Virus in a Community’, Journal of General Virology, 79: 2221–9. [DOI] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebuffo-Scheer C. et al. (2011) ‘Whole Genome Sequencing and Evolutionary Analysis of Human Respiratory Syncytial Virus A and B from Milwaukee, WI 1998–2010’, PLoS One, 6: e25468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rima B.et al. , ICTV Report Consortium . (2017) ‘ICTV Virus Taxonomy Profile: Pneumoviridae’, Journal of General Virology, 98: 2912–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M. et al. (2021) ‘The Spatial-Temporal Dynamics of Respiratory Syncytial Virus Infections across the East-West Coasts of Australia during 2016–17’, Virus Evolution, 7: veab068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo G. L. et al. (2017) ‘Unravelling Respiratory Syncytial Virus Outbreaks in Buenos Aires, Argentina: Molecular Basis of the Spatio-Temporal Transmission’, Virology, 508: 118–26. [DOI] [PubMed] [Google Scholar]
- Ruzin A. et al. (2018) ‘Characterization of Circulating RSV Strains among Subjects in the OUTSMART-RSV Surveillance Program during the 2016–17 Winter Viral Season in the United States’, PLoS One, 13: e0200319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi V. et al. (2016) ‘Prevalence of Human Respiratory Syncytial Virus Circulating in Iran’, Journal of Infection and Public Health, 9: 125–35. [DOI] [PubMed] [Google Scholar]
- Saravanos G. L. et al. (2022) ‘RSV Epidemiology in Australia before and during COVID-19’, Pediatrics, 149: e2021053537. [DOI] [PubMed] [Google Scholar]
- Schobel S. A. et al. (2016) ‘Respiratory Syncytial Virus Whole-Genome Sequencing Identifies Convergent Evolution of Sequence Duplication in the C-Terminus of the G Gene’, Scientific Reports, 6: 26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.et al. , RSV Global Epidemiology Network . (2017) ‘Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Young Children in 2015: A Systematic Review and Modelling Study’, Lancet (London, England), 390: 946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkowa N., and Lässig M. (2012) ‘Clonal Interference in the Evolution of Influenza’, Genetics, 192: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streng A. et al. (2019) ‘Spread and Clinical Severity of Respiratory Syncytial Virus A Genotype ON1 in Germany, 2011–2017’, BMC Infectious Diseases, 19: 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. W., and Loh T. P. (2014) ‘Correlations between Climate Factors and Incidence—A Contributor to RSV Seasonality’, Reviews in Medical Virology, 24: 15–34. [DOI] [PubMed] [Google Scholar]
- Trento A. et al. (2006) ‘Natural History of Human Respiratory Syncytial Virus Inferred from Phylogenetic Analysis of the Attachment (G) Glycoprotein with a 60-Nucleotide Duplication’, Journal of Virology, 80: 975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovão N. S. et al. (2021) ‘Molecular Characterization of Respiratory Syncytial Viruses Circulating in a Paediatric Cohort in Amman, Jordan’, Microbial Genomics, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valley-Omar Z. et al. (2022) ‘Human Respiratory Syncytial Virus Diversity and Epidemiology among Patients Hospitalized with Severe Respiratory Illness in South Africa, 2012–2015’, Influenza and Other Respiratory Viruses, 16: 222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niekerk S., and Venter M. (2011) ‘Replacement of Previously Circulating Respiratory Syncytial Virus Subtype B Strains with the BA Genotype in South Africa’, Journal of Virology, 85: 8789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter M. et al. (2001) ‘Genetic Diversity and Molecular Epidemiology of Respiratory Syncytial Virus over Four Consecutive Seasons in South Africa: Identification of New Subgroup A and B Genotypes’, Journal of General Virology, 82: 2117–24. [DOI] [PubMed] [Google Scholar]
- Viegas M., Goya S., and Mistchenko A. S. (2016) ‘Sixteen Years of Evolution of Human Respiratory Syncytial Virus Subgroup A in Buenos Aires, Argentina: GA2 the Prevalent Genotype through the Years’, Infection, Genetics and Evolution, 43: 213–21. [DOI] [PubMed] [Google Scholar]
- Zhang L. et al. (2020) ‘SARS-CoV-2 Spike-protein D614G Mutation Increases Virion Spike Density and Infectivity’, Nature Communications, 11: 6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.