

Abstract
Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the LIPA gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation.
We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the LIPA gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved.
When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the LIPA gene.
Keywords: LIPA, lysosomal acid lipase deficiency, variant of uncertain significance
Introduction
Lysosomal acid lipase (LAL) is a 399-amino acid protein encoded by the LIPA gene (1). Pathogenic variants in LIPA cause a rare autosomal-recessive disease belonging to the family of lysosomal storage disorders (LSDs) (2). Two types of lysosomal acid lipase deficiency (LAL-D) were recognized: early onset form (Wolman’s disease) and late-onset type (cholesteryl ester storage disease - CESD) (3). This classification is mostly replaced by the understanding that LAL-D includes a spectrum ranging from severe manifestations in infancy to more chronic course and later onset (4). The severity of clinical features of LAL-D is not entirely dependent on the level of enzyme activity and specific causative genetic variants (5,6). Severe infantile phenotype includes hepatosplenomegaly, malabsorption, adrenal calcifications and psychomotor regression, while patients with residual enzyme activity are usually recognized due to chronic hepatosplenomegaly with associated laboratory abnormalities: elevated serum low density lipoprotein (LDL) cholesterol and triglycerides (TG), low serum high density lipoprotein (HDL) cholesterol and moderately elevated serum transaminases (5, 6, 7, 8, 9). Liver histopathology typically reveals microvesicular steatosis (cytoplasmic lipid vesicles stained by Oil-red, Sudan black, Sudan IV), lysosomal accumulation of cholesteryl esters, and TG, along with Maltese cross-type birefringent needle-shaped cholesteryl ester crystals in frozen sections. Immunohistochemical use of lysosomal markers in fixed paraffin-embedded samples facilitates the diagnosis of CESD (10,11). Additional biomarker findings that are useful for the diagnosis of LAL-D are high plasma concentration of chitotriosidase as well as elevated oxysterols (12,13). Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation and stem cell transplantation (14,15).
Approximately 120 disease-causing variants in LIPA have been reported to date, mostly located in exon 8 (16). The most frequently encountered pathogenic variant is NM_000235.4:c.894G>A, responsible for the skipping of exon 8 and the CESD phenotype (5). To date, clinical implications for heterozygous carriers of LIPA variants have been scarcely studied (16). There have been reports of reevaluation of variants detected in the LIPA gene by next generation sequencing in the context of the clinical phenotype (17).
We present four Serbian patients from two families with clinical manifestations which are highly suggestive of LAL-D, harboring a novel LIPA variant with preserved enzyme activity. Our study was approved by the Institutional Ethics Committee and informed consent was obtained by the involved patients.
Case series presentation
Family 1
Patient 1 presented with hepatosplenomegaly at the age of three years. Initial laboratory evaluation showed elevated serum transaminases with preserved synthetic liver function, mild leukopenia and thrombocytopenia. He is the second child of non-consanguineous parents, from an uneventful pregnancy and perinatal period and normal psychomotor development. His plasma lipid profile revealed very low high density lipoprotein (HDL) cholesterol, elevated TG and normal total cholesterol. Significant liver and spleen enlargement were verified via an abdominal ultrasound exam. A biomarker study was indicative of LSD with high chitotriosidase, ACE and acid phosphatase levels in the blood. On account of hepatosplenomegaly, lipid profile abnormalities and biomarker findings, LAL-D was suspected. Enzyme diagnostics for LAL-D and other LSDs (Gaucher disease, Niemann-Pick disease type A/B and C, Farber disease) were performed at the Metabolic Laboratory of the University of Mainz and results returned negative. Over the following years, hepatosplenomegaly progressed with a further decrease in platelet count. Repeated plasma measurements of chitotriosidase showed elevated values, reaching the highest level at 8083 nmol/ml/hr. With the advent of next generation sequencing, patient 1 was tested by clinical exome sequencing (Quantitative Genomic Medicine Laboratories, SL., Barcelona), and compound heterozygosity for variants in the LIPA gene was found. This consisted of a pathogenic variant NM_000235.4(LIPA):c.419G>A (p.Trp140Ter) and a novel variant of uncertain significance NM_000235.4(LIPA):c.851C>T (p.Ser284Phe). Since enzyme activity remained normal in the leukocytes over repetitive analyses, whole genome sequencing was used to discern changes in the LIPA gene, and findings from the Centogene lab confirmed the previous result from Barcelona, thus categorizing Ser284Phe as a VUS. The enzyme activity of lysosomal acid lipase in a dried blood spot was measured at the Biochemistry Department of NHS Greater Glasgow and Clyde at Glasgow, UK and was reported as normal (0.14 nmol/punch/hour).
Patient 2 (the younger sibling) presented with hepatosplenomegaly upon clinical examination at the age of one and a half. He is the third child from uneventful pregnancy, with a normal perinatal period and normal psychomotor development. Elevated liver enzymes with preserved liver function and thrombocytopenia were observed during the initial laboratory evaluation. A lipidogram revealed reduced HDL cholesterol, along with hypertriglyceridemia, and normal total cholesterol (Table 1). An echographic abdominal evaluation confirmed hepatosplenomegaly, observed in clinical examination. Taking clinical findings and similarity with patient 1 into consideration, enzyme testing on a dry blood spot for LAL-D was performed and reported as normal. In the following years, hepatosplenomegaly showed progression, with persistent thrombocytopenia and consistent finding of reduced HDL cholesterol and elevated TG. Clinical exome sequencing was carried out (Quantitative Genomic Medicine Laboratories, SL., Barcelona) and patient 2 was found to be a compound heterozygote for the same variants in the LIPA gene as the older brother. Whole genome sequencing confirmed results previously obtained from Barcelona, categorizing p.Ser284Phe as a variant of uncertain significance, nevertheless vouching for the genetic diagnosis of CESD should be considered.
Table 1.
Genetic, hematologic and biochemical findings in four Serbian patients with chronic hepatosplenomegaly and variants detected in the LIPA gene
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| LIPA gene variant #1 | c.419G>A (p.Trp140Ter) | c.419G>A (p.Trp140Ter) | c.851C>T (p.Ser284Phe) | c.851C>T (p.Ser284Phe) |
| LIPA gene variant #2 | c.851C>T (p.Ser284Phe) | c.851C>T (p.Ser284Phe) | c.851C>T (p.Ser284Phe) | c.851C>T (p.Ser284Phe) |
| Enzyme activity | 0.14 nmol/punch/hr (normal >0.1) | Not available | 0.32 nmol/punch/h (normal >0.1) | Not performed |
| Total cholesterol (mmol/L) | 3.1 - 4.4 | 4.13 - 4.3 | 2.4 - 5.44 | 3.56 - 5.12 |
| LDL cholesterol (mmol/L) | 2.04 - 2.6 | 2.66 - 2.9 | 1.35 - 4.47 | 2. 44 - 3.9 |
| HDL cholesterol (mmol/L) | 0.14 - 0.42 | 0.13 - 0.14 | 0.23 - 0.73 | 0.07 - 0.51 |
| TG (mmol/L) | 1.34 - 6.6 | 2.94 - 3.18 | 0.61 - 1.63 | 1.7 - 3.22 |
| AST (IU/L) | 61 - 153 | 109 - 128 | 55 - 200 | 60 - 154 |
| ALT (IU/L) | 49 - 98 | 50 - 76 | 40 - 105 | 38 - 102 |
| Hemoglobin (g/L) | 107-126 | 92-115 | 80-118 | 75-127 |
| WBC count | 3.65 - 5.7 | 5.4 -5.8 | 2.66 - 14.4 | 2.6 - 8.54 |
| Platelet count | 57.7 -203 | 14 - 204 | 73 - 399 | 66 - 406 |
| ACE (IU/L) | 120 - 223 | ND | ND | ND |
| Chitotriosidase (nmol/ml/hr) | 2106 - 8083 | ND | 10805 | ND |
| Oxysterols (mcmol/L) | 0.597 (normal < 0.05) | ND | ND | ND |
LDL – low density lipoprotein, HDL – high density lipoprotein, TG – triglycerides, WBC – white blood cells, ACE – angiotensin converting enzyme
Family 2
Patient 3 presented with hepatosplenomegaly at two months of age. Initial laboratory evaluation showed only slightly elevated serum transaminases. He is the third child of non-consanguineous parents with an uneventful perinatal period and normal psychomotor development. A lipid profile revealed normal total cholesterol, with HDL cholesterol below normal and elevated TG. An abdominal ultrasound confirmed hepatic enlargement with structural dyshomogeneity and hyperechogenicity. Chitotriosidase was extremely elevated (Table 1). The enzyme activity of LAL was tested in a dry blood spot sample in the University Medical Center Hamburg-Eppendorf lab and revealed sufficient activity. On the basis of a clinical suspicion of a diagnosis of LSD, a liver biopsy was done at the age of five. The specimen appeared bright yellow-orange in color. Standard HE and special staining were performed on paraffin and frozen sections. Microvesicular steatosis with pronounced liver fibrosis was established, and characteristic groups of uniform, foamy macrophages and Kupffer cells with PAS-positive, granular cytoplasm were found in portal spaces and focally in fibrous septa (figure 1A). The analysis of the Sudan IV stained frozen sections under polarized light revealed numerous, diffuse infiltrate of birefringent, anisotropic, needle-like crystals (figure 1B, 1C). The crystals were lost after heating the tissue sections in a thermostat to 60 degrees Celsius, which demonstrated the thermal sensitivity of the crystals. Hepatosplenomegaly showed progression over the course of time, levels of transaminases increased with development of splenomegaly, thrombocytopenia and a constant finding of low levels of HDL cholesterol (Table 1). Clinical exome sequencing (Illumina TruSight One, Illumina MiSeq) revealed that the patient 3 is homozygous for a missense variant of uncertain significance, NM_000235.4(LIPA):c.851C>T (p.Ser284Phe) in the LIPA gene.
Figure 1.
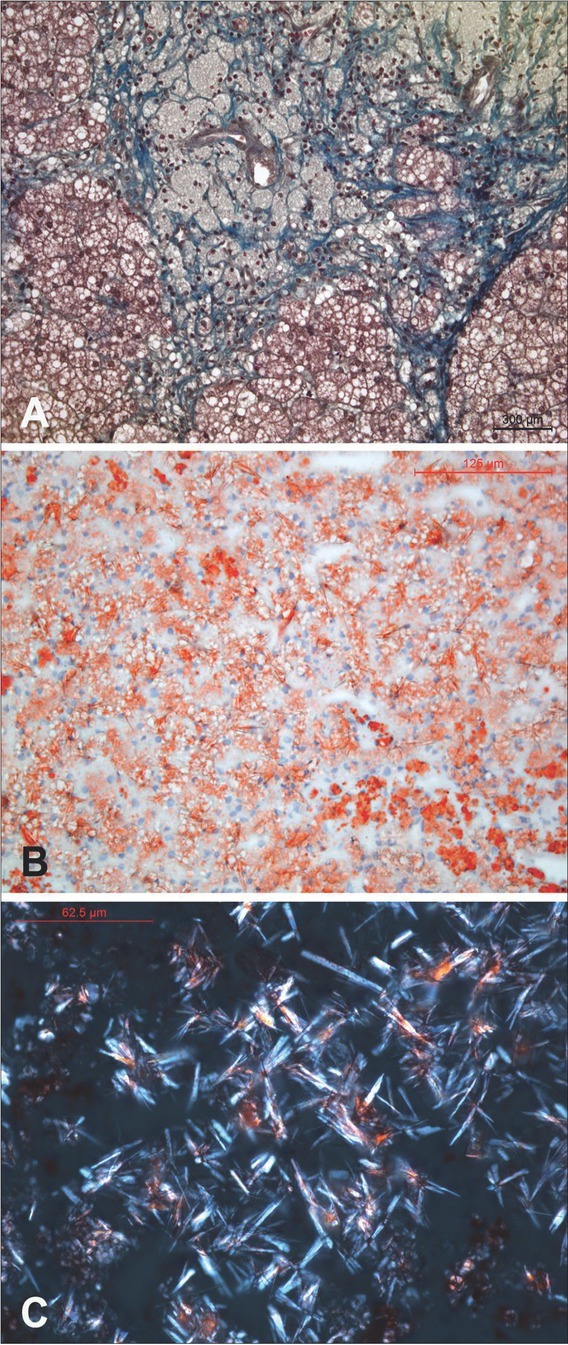
Liver histology of patient 3. 1A. Foamy macrophages with pyknotic nuclei loaded in the portal space and uniform microvesicular steatosis with accompanying marked fibrosis. (Paraffin section stained with Masson’s trichrome, x200). 1B. Histological picture of liver steatosis with evenly distributed, Sudan IV positive microvesicles in hepatocytes and crystalloid clefts of cholesteryl ester that are only hinted at due to light birefringence. (Frozen section stained with Sudan IV, x25). 1C. Birefringent, anisotropic crystals are seen under polarized light on frozen sections stained by the Sudan IV method. Maltese cross-type birefringence is visible here and there. (Frozen section stained with Sudan IV, polarized light, x40).
Patient 4 is the older sister of patient 3 and she also presented with hepatosplenomegaly at the age of 2.5 years. Laboratory evaluation showed elevated transaminases, dyslipidemia (elevated total cholesterol and LDL cholesterol, low HDL cholesterol and elevated TG) alongside anemia. A liver biopsy, performed at 3 years of age, revealed microvesicular steatosis with pronounced portal fibrosis and the presence of thermal-sensitive birefringent crystals in frozen tissue sections under polarized light. After the biopsy findings, a diagnosis of CESD was established.
In the years that followed, hepatosplenomegaly showed regression, with persistent findings of elevated transaminases, TG, and low HDL cholesterol. Leukopenia and thrombocytopenia developed as well. Upon receiving the results of patient’s brother’s exome sequencing, genetic testing confirmed that patient 4 was homozygous for the same variant as her brother.
Discussion
It has been observed that the clinical spectrum of LAL deficiency varies from the classic Wolman phenotype of early infantile cholestasis to a non-specific presentation of chronic liver disease with much later onset (2). However, a pattern of clinical (hepatosplenomegaly) and biochemical (dyslipidemia, elevated liver enzymes) abnormalities is present in most of the patients (2,7,8). The disease which affected the two pairs of siblings that we present herein fit well into the pattern of LAL deficiency, especially after the exclusion of LSDs with a more common occurrence among the Serbian population. It has been reported that approximately two thirds of CESD patients have a disease onset before the age of 5 years. This corresponds to the age range of our case series (2 months – 3 years) (18). Clinically, all patients developed progressive hepatosplenomegaly, complicated by hypersplenism. Esophageal varices were present in one pair of siblings. Furthermore, three of our patients manifested with growth faltering, which has been previously described as a relatively common finding in late onset LAL-D (19). A disturbed serum lipid profile is one of the biochemical hallmarks of LAL deficiency (8). All of the patients in our series have persistently low serum HDL cholesterol concentration (Table 1), corresponding to the proposed diagnosis. Patients 3 and 4 also had elevated serum concentrations of total cholesterol and LDL fraction in most of the measurements. Normal serum total and LDL cholesterol in family 1 does not exclude the possibility of LAL-D, especially in the presence of typically low HDL cholesterol (20). Liver enzyme elevation in the serum is present in the whole series we presented herein and these findings correspond to previous clinical reports of LAL-D patients (21). In two of our patients (one from each couple of siblings), chitotriosidase activity in plasma was obtained, and in both cases it showed starkly elevated values. Chitotriosidase is a well-known and widely used marker of LSDs (12,22). Although not entirely specific to metabolic disorders, high chitotriosidase in pediatric patients with familial occurrence of hepatosplenomegaly should direct further testing toward LSDs. Due to the absence of neurologic manifestations in our patients, we suspected the possibility of Gaucher disease type 1 and LAL-D. After negative results of specific enzyme assays for LSDs, whole exome sequencing revealed the aforementioned variants in the LIPA gene, hereby excluding other LSDs. Plasma oxysterol concentration was available only for patient 1, and its elevation was in line with the previously described biochemical abnormalities in certain LSDs, including LAL-D (13).
Uniform microvesicular steatosis on light microscopy, liquid crystals of cholesteryl esters with Maltese cross-type birefringence in frozen section and staining with Oil Red O or Sudan Black are the features of liver pathology that strongly suggest CESD (10). Foamy macrophages rich in lipid and ceroid content are one of the hallmarks of histopathology in younger patients (2). Immunohistochemistry reveals lysosomal markers, such as cathepsin D, LAMP1, LAMP2 and LIMP2 (10).
Histology changes in the liver biopsies of patients 3 and 4 were entirely in accordance with the CESD diagnosis. Progression to fibrosis and micronodular cirrhosis is probable in untreated patients (2), and despite the early age at the time of biopsy of patients 3 and 4 (3 and 5 years, respectively), portal fibrosis was pronounced. The case of patient 4 has already been reported in an article describing the usefulness of histopathology in diagnosis of this particular lysosomal storage disorder (23).
We have referred samples of one patient from each pair of siblings (patients 1 and 3) to referent metabolic laboratories (Mainz, Glasgow, Hamburg) and received results stating that LAL activity in plasma was below the normal range in both cases. However, LAL activity was above the cut-off value defined as diagnostic for CESD. Different methods of activity measurement have been employed as diagnostic tools for LAL deficiency worldwide, making it difficult to compare the findings of individual patients. A residual enzyme activity of 2-11% is considered as diagnostic for CESD (24). However, there has been a report of severe LAL-D phenotype associated with substantially higher LAL-D of 16% (25). Therefore, the predictive value of enzymatic activity in regard to the clinical course is not firmly established. Certain authors suggest that precise cutoffs for low LAL activity are still dubious for late-onset CESD, and that clinicians dealing with LSDs should take into account other biomarkers (chitotriosidase, serum transaminases, lipid profile) during the diagnostic process (26, 27, 28, ). Over the years, assays of enzyme activity employed in the diagnostics of different lysosomal storage disorders have been subjected to critical evaluation, pointing out the reduced possibility of false negativity with a more recent methodology (29). Therefore, a borderline enzyme result in the case of LSD suspicion, such as in cases from our report, is recommended for further evaluation by molecular genetic testing (30).
In patients 1 and 2 from our report, two variants were detected in the LIPA gene in a compound heterozygous state by clinical exome sequencing: NM_000235.4:c.419G>A (p.Trp140Ter) (designated as pathogenic) and NM_000235.4:c.851C>T (p.Ser284Phe) (designated as VUS), while segregation testing confirmed the carrier status of the parents. Both brothers share the clinical pattern resembling LAL and have the same genotype. The same VUS was detected in a homozygous state in siblings from family 2 (patients 3 and 4) presented in this study. However, the final diagnosis could not be made, despite clinical and biochemical findings, in absence of an indisputable enzyme deficiency and in the context of uncertain nature of the c.851C>T (p.Ser284Phe) variant in the LIPA gene. In a very recent study of infants treated for severe LAL deficiency, a diagnosis based on a clinical phenotype and supported by the variant of uncertain significance (VUS) was reported in at least one case (31). More authors address the possibility of the impact of VUS in patients with probable lysosomal storage disease (32). There are estimates that approximately a quarter of variants designated as VUS contribute significantly to enzyme deficiency in Mucopolysaccharidosis type III (33).
The pathogenic c.419G>A (p.Trp140Ter) variant in the LIPA gene, present in heterozygous state in one pair of siblings from our study, is considered as a severe null variant (24). As recently stated by Reiner et al., the clinical importance of heterozygosity (carriership) for variants in the LIPA gene has yet to be elucidated in the future (34). On the other hand, pathogenicity of LIPA gene variants is a subject of critical reevaluation, primarily by means of in vitro functional studies (17). There have been reports that even polymorphisms in the LIPA gene can contribute significantly to the occurrence of metabolic complications in patients with obesity (35). The interpretation of genetic findings in the context of the clinical condition is further complicated in CESD due to the lack of genotypephenotype correlation in this specific disorder (9).
The possibility of incongruence between genetic findings and enzyme activity, as observed in this series of patients, prompts further investigations. Nonsense, frameshift, and splice mutations typically cause severe (Wolman) forms of the disease (36) while the effect of missense mutations (typically causing the milder CESD) has recently been shown to be related with the structural domain compromised (37). In the case of the missense mutation described herein, we could speculate that it could be related to the compromised ability of the mutated enzyme to reach the optimal subcellular location, while the in vitro biological activity remains normal, since the catalytic domain is not compromised. A similar pathophysiologic scenario has been described in several different rare genetic diseases (38,39).
When confronted with a challenge of confirming the diagnosis of a treatable inherited metabolic disorder, a clinician should consider a multitude of aspects: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. We hope that this report calls cases with considerable discrepancy between those aspects to attention.
Acknowledgement
No sources or grants contributed to the completion of this research. A. Sarajlija contributed to the study concept, data accumulation, thorough medical documentation and literature review, and article writing. L. Dulcet, A. Maver, and B. Peterlin contributed to article writing, article critical revision, and especially analysis and interpretation of genotype to phenotype discrepancies. D. Prokic, I. Kitic, and M. Cehic contributed to data accumulation, literature review and critical revision. S. Djuricic contributed to article writing and critical revision.
Footnotes
Conflict of interest
Conflict of interest statement: All authors declare that they have no conflict of interest.
Data availability statement: The data that support the findings of our study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Ameis D, Merkel M, Eckerskorn C, Greten H. Purification, characterization and molecular cloning of human hepatic lysosomal acid lipase. Eur J Biochem. 1994;219(3):905–14. doi: 10.1111/j.1432-1033.1994.tb18572.x. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230–43. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33(1):85–93. doi: 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- 4.Wilson DP, Patni N. Feingold KR, Anawalt B, Boyce A. https://www.ncbi.nlm.nih.gov/books/NBK395569/ Endotext [Internet]; South Dartmouth (MA): Lysosomal Acid Lipase Deficiency.MDText.com Updated 2020 Jan 18. et al., editors. Inc.; 2000-. Available from. [Google Scholar]
- 5.Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958–65. doi: 10.1002/hep.26327. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cappuccio G, Donti TR, Hubert L, Sun Q, Elsea SH. Opening a window on lysosomal acid lipase deficiency: Biochemical, molecular, and epidemiological insights. J Inherit Metab Dis. 2019;42(3):509–18. doi: 10.1002/jimd.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pisciotta L, Tozzi G, Travaglini L, Taurisano R, Lucchi T, Indolfi G. Molecular and clinical characterization of a series of patients with childhood-onset lysosomal acid lipase deficiency. Retrospective investigations, follow-up and detection of two novel LIPA pathogenic variants. Atherosclerosis. 2017;265:124–32. doi: 10.1016/j.atherosclerosis.2017.08.021. et al. [DOI] [PubMed] [Google Scholar]
- 8.Zimetti F, Favari E, Cagliero P, Adorni MP, Ronda N, Bonardi R. Cholesterol trafficking-related serum lipoprotein functions in children with cholesteryl ester storage disease. Atherosclerosis [Internet] 2015;242(2):443–9. doi: 10.1016/j.atherosclerosis.2015.08.007. et al. [DOI] [PubMed] [Google Scholar]
- 9.Pericleous M, Kelly C, Wang T, Livingstone C, Ala A. Wolman’s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol. 2017;2(9):670–9. doi: 10.1016/S2468-1253(17)30052-3. [DOI] [PubMed] [Google Scholar]
- 10.Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107–13. doi: 10.1111/j.1365-2559.2011.04164.x. [DOI] [PubMed] [Google Scholar]
- 11.Thompson RJ, Portmann BC, Roberts EA. Burt AD, Portmann BC, Ferrell LD. MacSween’s pathology of the liver (6th edition) Edinburgh: Churchil Livingstone Elsevier; 2012. Genetic and metabolic liver disease; pp. 157–259. [Google Scholar]
- 12.vom Dahl S, Harzer K, Rolfs A, Albrecht B, Niederau C, Vogt C, van Weely S, Aerts J, Müller G, Häussinger D. Hepatosplenomegalic lipidosis: what unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol. 1999;31(4):741–6. doi: 10.1016/s0168-8278(99)80356-0. [DOI] [PubMed] [Google Scholar]
- 13.Boenzi S, Deodato F, Taurisano R, Goffredo BM, Rizzo C, Dionisi-Vici C. Evaluation of plasma cholestane-3β,5α,6β-triol and 7-ketocholesterol in inherited disorders related to cholesterol metabolism. J Lipid Res. 2016;57(3):361–7. doi: 10.1194/jlr.M061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet. 2016;9:157–67. doi: 10.2147/TACG.S86760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastores GM, Hughes DA. Lysosomal Acid Lipase Deficiency: Therapeutic Options. Drug Des Devel Ther. 2020;14:591–601. doi: 10.2147/DDDT.S149264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter A, Brackley SM, Gao J, Mann JP. The global prevalence and genetic spectrum of lysosomal acid lipase deficiency: A rare condition that mimics NAFLD. J Hepatol. 2019;70(1):142–50. doi: 10.1016/j.jhep.2018.09.028. [DOI] [PubMed] [Google Scholar]
- 17.Del Angel G, Hutchinson AT, Jain NK, Forbes CD, Reynders J. Large-scale functional LIPA variant characterization to improve birth prevalence estimates of lysosomal acid lipase deficiency. Hum Mutat. 2019;40(11):2007–20. doi: 10.1002/humu.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Porto AF. Cholesteryl ester storage disease: protean presentations of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2013;56(6):682–5. doi: 10.1097/MPG.0b013e31828b36ac. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Andrés C, Sellés E, Arias A, Gort L. Spanish LAL Deficiency Working Group. Lysosomal Acid Lipase Deficiency in 23 Spanish Patients: High Frequency of the Novel c.966+2T>G Mutation in Wolman Disease. JIMD Rep. 2017;37:7–12. doi: 10.1007/8904_2017_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman EP, Barr ML, Giovanni MA, Murray MF. Adam MP, Ardinger HH, Pagon RA. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993-2021. Lysosomal Acid Lipase Deficiency. 2015 Jul 30 [Updated 2016 Sep 1]https://www.ncbi.nlm.nih.gov/books/NBK305870/ et al., editors. Available from. [PubMed] [Google Scholar]
- 21.Pullinger CR, Stock EO, Movsesyan I, Malloy MJ, Frost PH, Tripuraneni R. Identification and metabolic profiling of patients with lysosomal acid lipase deficiency. J Clin Lipidol. 2015;9(5):716–26. doi: 10.1016/j.jacl.2015.07.008. et al. e1. [DOI] [PubMed] [Google Scholar]
- 22.Aerts JM, Hollak CE, van Breemen M, Maas M, Groener JE, Boot RG. Identification and use of biomarkers in Gaucher disease and other lysosomal storage diseases. Acta Paediatr Suppl. 2005;94(447):43–6. doi: 10.1111/j.1651-2227.2005.tb02110.x. discussion 37-8. [DOI] [PubMed] [Google Scholar]
- 23.Zlatkovic M, Stankovic I, Prokic D, Plamenac P. Patohistoloska dijagnostika bolesti nagomilavanja estra holesterola [Histopathologic diagnosis of cholesterol ester storage disease] Srp Arh Celok Lek. 2001;129(7-8):207–10. [PubMed] [Google Scholar]
- 24.Fasano T, Pisciotta L, Bocchi L, Guardamagna O, Assandro P, Rabacchi C. Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab. 2012;105(3):450–6. doi: 10.1016/j.ymgme.2011.12.008. et al. [DOI] [PubMed] [Google Scholar]
- 25.Arterburn JN, Lee WM, Wood RP, Shaw BW, Markin RS. Orthotopic liver transplantation for cholesteryl ester storage disease. J Clin Gastroenterol. 1991;13(4):482–5. doi: 10.1097/00004836-199108000-00028. [DOI] [PubMed] [Google Scholar]
- 26.Andrade J, Waters PJ, Singh RS, Levin A, Toh BC, Vallance HD. Screening for Fabry disease in patients with chronic kidney disease: limitations of plasma alpha-galactosidase assay as a screening test. Clin J Am Soc Nephrol. 2008;3(1):139–45. doi: 10.2215/CJN.02490607. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma J, Thomas DC, Kasper DC, Sharma S, Puri RD, Bijarnia-Mahay S. Inherited Metabolic Disorders: Efficacy of Enzyme Assays on Dried Blood Spots for the Diagnosis of Lysosomal Storage Disorders. JIMD Rep. 2017;31:15–27. doi: 10.1007/8904_2016_548. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masi S, Chennamaneni N, Turecek F, Scott CR, Gelb MH. Specific Substrate for the Assay of Lysosomal Acid Lipase. Clin Chem. 2018;64(4):690–6. doi: 10.1373/clinchem.2017.282251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukacs Z, Barr M, Hamilton J. Best practice in the measurement and interpretation of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2017;471:201–5. doi: 10.1016/j.cca.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 30.Monserrat L, Gimeno-Blanes JR, Marín F, Hermida-Prieto M, García-Honrubia A, Pérez I. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50(25):2399–403. doi: 10.1016/j.jacc.2007.06.062. et al. [DOI] [PubMed] [Google Scholar]
- 31.Vijay S, Brassier A, Ghosh A, Fecarotta S, Abel F, Marulkar S. Long-term survival with sebelipase alfa enzyme replacement therapy in infants with rapidly progressive lysosomal acid lipase deficiency: final results from 2 open-label studies. Orphanet J Rare Dis. 2021;16(1):13. doi: 10.1186/s13023-020-01577-4. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mashima R, Okuyama T, Ohira M. Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry. Int J Mol Sci. 2020;21(8):2704. doi: 10.3390/ijms21082704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark WT, Yu GK, Aoyagi-Scharber M, LeBowitz JH. Utilizing ExAC to assess the hidden contribution of variants of unknown significance to Sanfilippo Type B incidence. PLoS One. 2018;13(7):e0200008. doi: 10.1371/journal.pone.0200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S. Lysosomal acid lipase deficiency-an under-recognized cause of dyslipidemia and liver dysfunction. Atherosclerosis. 2014;235(1):21–30. doi: 10.1016/j.atherosclerosis.2014.04.003. et al. [DOI] [PubMed] [Google Scholar]
- 35.Guénard F, Houde A, Bouchard L, Tchernof A, Deshaies Y, Biron S. Association of LIPA gene polymorphisms with obesity-related metabolic complications among severely obese patients. Obesity (Silver Spring) 2012;20(10):2075–82. doi: 10.1038/oby.2012.52. et al. [DOI] [PubMed] [Google Scholar]
- 36.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–77. doi: 10.1007/s00439-017-1779-6. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinje T, Laerdahl JK, Bjune K, Leren TP, Strøm TB. Characterization of the mechanisms by which missense mutations in the lysosomal acid lipase gene disrupt enzymatic activity. Hum Mol Genet. 2019;28(18):3043–52. doi: 10.1093/hmg/ddz114. [DOI] [PubMed] [Google Scholar]
- 38.Oetjen S, Kuhl D, Hermey G. Revisiting the neuronal localization and trafficking of CLN3 in juvenile neuronal ceroid lipofuscinosis. J Neurochem. 2016;139(3):456–70. doi: 10.1111/jnc.13744. [DOI] [PubMed] [Google Scholar]
- 39.Björses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J, Ellonen P. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J Hum Genet. 2000;66(2):378–92. doi: 10.1086/302765. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]