

Abstract
Medulloblastoma is the most common pediatric malignant brain tumor composed of four molecular subgroups. Recent intensive genomics has greatly contributed to our understanding of medulloblastoma pathogenesis. Sequencing studies identified novel mutations involved in the cyclic AMP‐dependent pathway or RNA processing in the Sonic Hedgehog (SHH) subgroup, and core‐binding factor subunit alpha (CBFA) complex in the group 4 subgroup. Likewise, single‐cell sequencing provided detailed insights into the cell of origin associated with brain development. In this review, we will summarize recent findings by sequencing analyses for medulloblastoma.
Keywords: CBFA complex, medulloblastoma, rhombic lip, RNA processing, SHH signaling pathway
Recent intensive genomics has greatly contributed to our understanding of medulloblastoma pathogenesis. Sequencing studies identified novel mutations involved in the cyclic AMP‐dependent pathway or RNA processing in the Sonic Hedgehog (SHH) subgroup, and core‐binding factor subunit alpha (CBFA) complex in the group 4 subgroup. Likewise, single‐cell sequencing provided detailed insights into the cell of origin associated with brain development.
1. INTRODUCTION
Brain tumors are the leading cause of death among all childhood cancers. 1 Medulloblastoma is the most common malignant pediatric brain tumor with a poor prognosis and is molecularly divided into four consensus subgroups: Wingless/INT1 (WNT), Sonic Hedgehog (SHH), group 3, and group 4. 2 Despite aggressive treatments, the clinical course is often disastrous where the 5‐year survival rate is approximately 60%‐80%. 3 , 4 , 5 In addition, survivors suffer from complications and adverse events due to intensive and long‐term treatments. 3 Thus, the development of novel therapies is highly needed to improve treatment outcomes.
With the development of next‐generation sequencing (NGS), international collaborative efforts for sequencing the genomes of medulloblastomas have discovered novel driver alterations. SHH medulloblastoma had been thought to be well characterized because SHH medulloblastomas usually harbor genetic alterations in the canonical SHH signaling pathway. However, novel recurrent mutations including mutations in U1 small nuclear RNA (snRNA) and ELP1 were unexpectedly detected in SHH medulloblastomas. 6 , 7 Interestingly, both U1 snRNA and ELP1 work on RNA processing, providing a new aspect of SHH medulloblastoma where disruption in RNA processing is one of the contributing mechanisms underlying SHH medulloblastoma pathogenesis. Although the pathogenesis of group 3 and group 4 medulloblastoma (hereafter group 3/4) remained less clear, the discovery of the convergence of somatic alterations on the core‐binding factor alpha (CBFA) complex in the group 4 subgroup has suggested that failure of cell differentiation in rhombic lip (RL) underlies medulloblastoma formation. 8 , 9
The identification of cells of origin for human cancers is of great interest because the detection would contribute to the discovery of developmental vulnerability leading to novel targeted therapies. Recently, large‐scale single‐cell RNA sequencing has emerged, enabling us to evaluate genome‐wide gene profiling at the individual cell level which was not detectable with the transcriptomics of conventional bulk tumor tissue. This novel technique allows us to identify cell populations in the developing cerebellum that each subgroup most closely resembles, providing evidence for the cell of origin. 8 , 10 , 11 Here, we review recent sequencing studies for medulloblastomas and provide our current understanding of medulloblastomas.
2. MOLECULAR CLASSIFICATION OF MEDULLOBLASTOMA
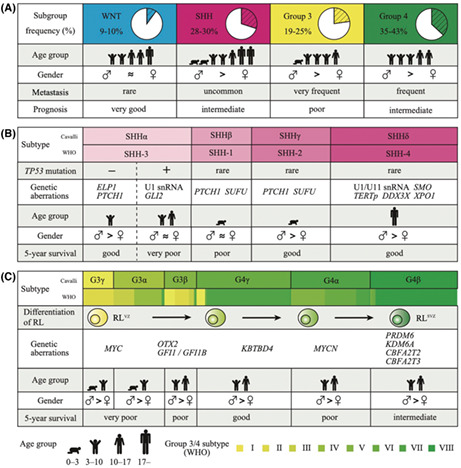
Since Cushing and Bailey initially reported medulloblastoma in 1925, 12 medulloblastoma was diagnosed based on only pathological features and was once considered a singular disease. In recent years, genome‐wide sequencing approaches have enabled the precise classification of medulloblastoma that is molecularly defined into four principal subgroups with distinct demographics, genetic aberrations, and clinical courses (Figure 1A). 2 WNT medulloblastoma accounts for 9%‐10% of cases, carries a favorable prognosis, and is characterized by activation of the WNT signaling pathway. SHH medulloblastoma represents 28%‐30% of cases with bimodal age distribution and demonstrates consecutive activation of the SHH signaling pathway. Group 3/4 medulloblastoma accounts for the remaining 19%‐25% and 35%‐43% of cases, respectively. Recently, abundant multi‐omics data produce accumulating evidence for a subsequent level of hierarchical structure defined as medulloblastoma subtype, which is biologically and clinically homogeneous compared with other subtypes within the same subgroup. 13 , 14 , 15 Cavalli et al. 13 classified four subgroups further into 12 subtypes based on DNA methylation and gene expression profiles. Northcott et al. 14 classified group 3/4 medulloblastoma into eight subtypes based on DNA methylation profiling. Although the subtype classification of medulloblastoma is still in the provisional stage, it is widely accepted that each subtype is a distinct entity supported by a subtype‐specific manner of recurrent genetic events.
FIGURE 1.
Clinical and molecular features of subgroups and subtypes of medulloblastoma. A, Schematic representation that summarizes key clinical features associated with medulloblastoma subgroups. B, C, Clinical features and genetic aberrations in Sonic Hedgehog (SHH) subtypes (B) and group 3 and group 4 subtypes (C) described by Cavalli 13 and in the 5th edition of the WHO classification. 16 The width of each subtype shows the fraction of samples per subtype in the study by Cavalli and the 5th edition of the WHO classification. Subtypes of the WHO classification are defined using MolecularNeuropathology.org (www.molecularneuropathology.org). Illustration of cells demonstrates the stages of cell differentiation in group 3 and group 4 medulloblastoma subtypes (C). RL, rhombic lip; snRNA, small nuclear RNA; TERTp, TERT promoter; SVZ, subventricular zone; VZ, ventricular zone
3. WINGLESS/INT1 MEDULLOBLASTOMA
While nearly 90% of WNT cases harbored somatic CTNNB1 mutations, the remaining cases harbored germline APC mutations. 14 The subtype classification of the WNT subgroup is still controversial: Cavalli classified WNT into two subtypes (WNTα and WNTβ), while the 5th edition of WHO classification does not adopt subtypes for WNT medulloblastoma. 13 , 16 The most common genetic mutation in the WNT subgroup except for CTNNB1 is DDX3X, which is an RNA‐binding protein of the DEAD‐box family. DDX3X acts as a tumor suppressor in medulloblastoma that regulates hindbrain development. 17 Although WNT medulloblastoma was considered a cerebellar tumor, a study using single‐cell sequencing revealed that WNT medulloblastoma is likely to arise from mossy fiber neurons of the embryonic dorsal brainstem. 11
4. SONIC HEDGEHOG MEDULLOBLASTOMA
Sonic Hedgehog medulloblastoma is classified into four subtypes: SHHα (children and adolescents; corresponds to SHH‐3 in the 5th edition of the WHO classification), SHHβ (infant, SHH‐1), SHHγ (infant, SHH‐2), and SHHδ (adult, SHH‐4) (Figure 1B). 16 In addition to clinical and transcriptional differences, each SHH subtype has distinct patterns of gene mutations, where SHHα and δ have a higher mutation burden than SHHβ and γ. 18 Some of the driver mutations are restricted to a certain subtype. For example, while mutations in TP53 and ELP1 are generally restricted to the SHHα subtype, TERT promoter mutation is exclusive to the SHHδ subtype. 6 , 7 , 13 , 14 The SHHα subtype with TP53 mutation usually harbors U1 snRNA mutation, 6 whereas ELP1 mutation is enriched in the remaining SHHα cases without TP53 mutation. 7
4.1. Mutations in the SHH signaling pathway
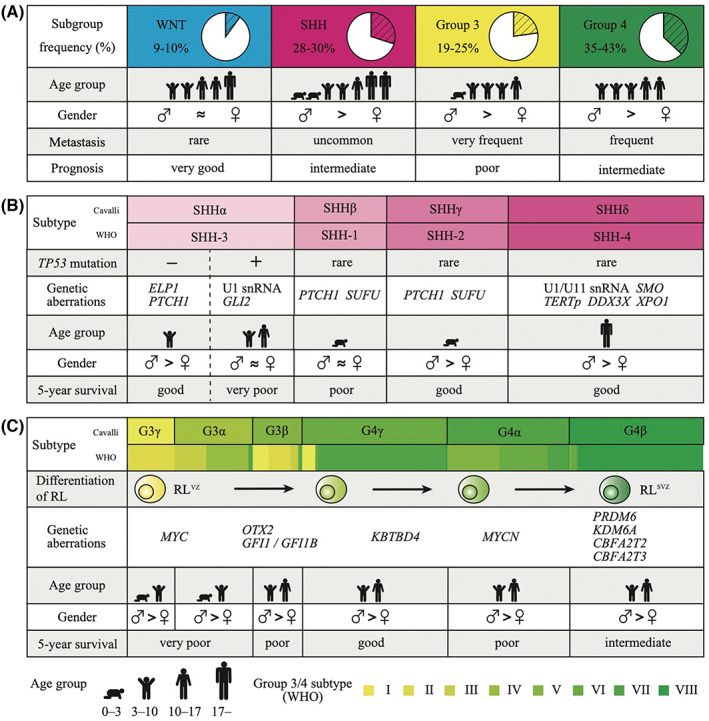
Sonic Hedgehog medulloblastoma exhibits activation of the SHH signaling pathway. SHH is one of the secreted proteins belonging to the hedgehog family that is well conserved during evolution and plays a key role as cell differentiation induction signals in the development process of the central nervous system. 19 The most frequently altered genes within the SHH signaling pathway are PTCH1 (44%‐45%), SMO (11%‐14%), SUFU (8%‐11%), and GLI2 (8%‐11%), leading to the consecutive activation of GLI2, which is the downstream target of the SHH signal (Figure 2). 14 , 18 , 20 In the canonical SHH signaling pathway, the glycoprotein SHH binds and inactivates the receptor Patched 1 (PTCH1), which inhibits the G protein–coupled transmembrane protein Smoothened (SMO). The inhibition of PTCH1 allows SMO to initiate an intracellular signaling cascade that leads to the translocation of GLI2 into the nucleus, resulting in the transcriptional activation of target genes. 21 , 22 Suppressor of fused (SUFU) is a negative intracellular regulator, which represses GLI activity by controlling the production, transport, and function of GLI proteins. 23 , 24 Thus, PTCH1 and SUFU mutations are detected as loss‐of‐function mutations. Contrarily, SMO and GLI2 alterations are identified as gain‐of‐function mutation or focal amplification, respectively. The mutation prevalence for genes in the canonical SHH signaling is different among age groups. PTCH1 mutations are detected at almost equal frequency in all generations and SHH subtypes. SMO mutations are highly enriched in adults (SHHδ), whereas SUFU mutations are almost exclusive in infants (SHHβ and SHHγ). 13 , 20 SMO and SUFU mutations are extremely rare in children and adolescents (SHHα). Involvement of altered genes in the SHH signaling pathway in the development of medulloblastoma has been scientifically demonstrated in mouse models where mice with heterozygous deletion of Ptch1 or active form of Smo develop medulloblastoma, demonstrating that mutations in the SHH signaling pathway are responsible for medulloblastoma tumorigenesis. 25 , 26
FIGURE 2.
Overview of the Hedgehog and cyclic AMP‐dependent pathways. Genetic aberrations involved in the canonical Sonic Hedgehog (SHH) signaling pathway and the cyclic AMP‐dependent pathways in SHH medulloblastoma are depicted. Red and blue boxes indicate activating and inactivating alterations, respectively. The color density of the boxes indicates each mutation rate described in the study by Skowron et al. 18 cAMP, cyclic AMP; GPCR, G protein‐coupled receptor; Gαs, G protein‐coupled transmembrane protein α subunit; PKA, protein kinase A
4.2. Mutations in the cAMP‐dependent pathway
Recently, a mutational analysis with a large cohort with SHH medulloblastoma discovered recurrent mutations in GNAS and PRKAR1A, which are involved in the cAMP‐dependent pathway (Figure 2). 18 Mutations in GNAS and PRKAR1A were found in 4.4% and 2.0% of SHH medulloblastomas, respectively; they converge on GLI2 activity, which is the key mediator of the SHH signaling pathway for medulloblastoma pathogenesis. GNAS encodes the heterotrimeric Gs protein α subunit (Gαs) and controls cell growth, survival, and motility. 27 GNAS is known to be mutated in a wide variety of tumors including growth hormone–producing pituitary tumors, corticotropin‐independent Cushing syndrome, and thyroid adenomas. 28 , 29 , 30 , 31 While GNAS mutations in the tumors of the endocrine glands are activating hotspot mutations clustering around R201 and Q227, GNAS mutations in SHH medulloblastoma are inactivating mutations clustering in the GTP/GDP binding site of Gαs, leading to inhibition of GTP binding and increase of GDP release. 32 Inhibition of GTPase activity in Gαs reduces cAMP concentration, resulting in the inactivation of protein kinase A (PKA), which is a negative regulator of the SHH signaling pathway. 33 , 34 Gnas knockout mice developed SHH medulloblastoma with 100% penetrance, supporting the role of GNAS as a driver in SHH medulloblastoma tumorigenesis. 35 PRKAR1A encodes the regulatory subunit type I‐alpha of PKA. Mutations in PRKAR1A are located within the binding pocket of the cAMP‐binding domain and reduce cAMP sensitivity, resulting in impairment of the activation of PKA. 36 Mutations in GNAS and PRKAR1A are found in a mutually exclusive manner. Furthermore, patients with alterations in GNAS or PRKAR1A barely harbor any alterations in the canonical SHH signaling pathway such as PTCH1, SUFU, SMO, and GLI2, further suggesting their imperative role in SHH medulloblastoma tumorigenesis. 18 Taken together, inactivation of the cAMP‐dependent pathway in SHH medulloblastoma is the alternative mechanism impairing the control of the SHH signaling pathway.
4.3. Mutation in RNA‐processing machinery
In recent years, successful efforts using massive parallel sequencing technology have identified highly recurrent mutations in U1 snRNA and ELP1 genes, both of which are involved in RNA processing. 6 , 7 , 37 U1 snRNA has diverse functions including splice‐site recognition. ELP1 encodes the largest subunit of the elongator complex which is required for tRNA modifications. In addition to U1 snRNA and ELP1, genes related to the RNA‐processing machinery including U11 snRNA and XPO1 are recurrently affected in SHH medulloblastoma, suggesting that aberrant RNA processing is one of the key components of SHH medulloblastoma pathogenesis. 6 , 14 , 18
4.3.1. U1 and U11 snRNA mutations
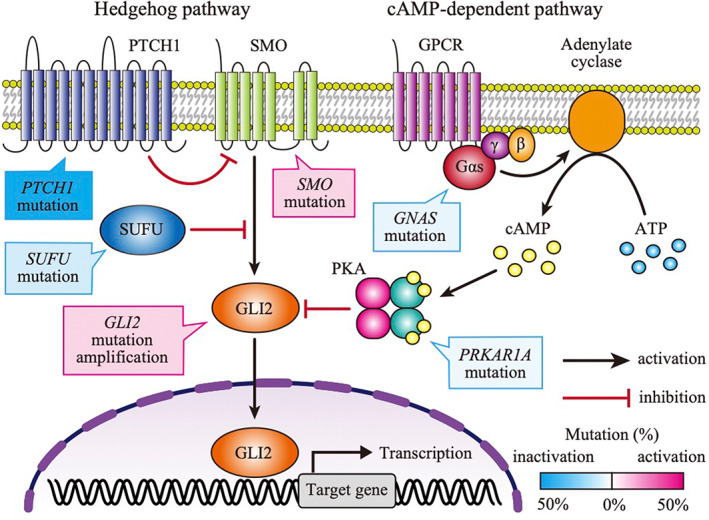
U1 snRNA is an essential component of the spliceosome and works on gene splicing. U1 snRNA mutation is the most common single‐nucleotide variant in medulloblastoma and is restricted to the SHH subgroup. 6 , 37 The mutation is a hotspot mutation with A to G substitution at the third nucleotide (g.3A > G), which forms part of the 5′ splice‐site recognition sequence (Figure 3A). 38 The U1 snRNA mutations are identified in 97% of adults (SHHδ) and 25% of adolescents (SHHα) but are rarely found in SHH medulloblastoma in infants (SHHβ and γ). In SHHα cases, U1 snRNA mutation is generally accompanied by TP53 mutation, while mutations in PTCH1, SMO, and SUFU are usually absent. 6 , 14 , 20 As U1 snRNA binds to 5′ splice site by base‐paring, mutant U1 snRNA recognizes noncanonical 5′ splice sites, resulting in excess of 5′ cryptic splicing (Figure 3A). The cryptic splicing in U1 mutant SHH medulloblastoma is detected in more than 1000 genes including several oncogenes (GLI2, CCND2) and tumor suppressor genes (PTCH1, PAX5), supporting that cryptic alternative splicing induced by U1 snRNA mutation functions as a driver in SHH medulloblastoma. Recent studies identified additional novel functions of U1 snRNA other than splice‐site recognition. It has been shown that U1 snRNA suppresses premature cleavage and polyadenylation by base pairing to pre‐mRNA. 39 Furthermore, it is reported that U1 snRNA determines the localization of RNAs to chromatin. 40 As little is yet known about how those functions are aberrant in U1 mutant SHH medulloblastoma, proof of the comprehensive roles in RNA processing of mutant U1 snRNA awaits further investigation.
FIGURE 3.
Mutations associated with RNA processing in Sonic Hedgehog (SHH) medulloblastoma. A, Schematic representation of mutations in U1 snRNA and U11 snRNA. The bases highlighted with red in the secondary structures indicate the mutational hotspots. The blue rectangle indicates the 5′ splice‐site recognition site. The upper logos are 5′ splice‐site sequences of cryptic splice sites (n = 4389) in U1 mutant SHH cases compared with U1 wildtype SHH medulloblastoma (left) and alternative splice sites (n = 5) in U11 mutant SHHδ cases compared with U11 wildtype SHHδ cases. Those splice‐site sequences were calculated using LeafCutter with RNA‐seq studies by Suzuki et al. 6 The lower logos are canonical 5′ splice‐site sequences of U2‐ and U12‐type introns from SpliceRack (http://katahdin.mssm.edu/splice/index.cgi?database=spliceNew). The lower illustrations indicate aberrant recognition of 5′ splice site by mutant U1 snRNA (left) and mutant U11 snRNA (right). SS, splice site. B, Schematic representation of tRNA modification by the elongator complex. The elongator complex modified the anticodon uridine at the wobble position (U34) indicated by the red circle and the arrow in the secondary structure of tRNA. The upper right panel shows the uridine modification in the elongator‐dependent tRNA modification pathway. ELP1 mutation causes failure of tRNA modification at the wobble position (U34), leading to an increase in AA‐ending codon usage and a decrease in AG‐ending codon usage. C, The upper panel shows the function of the nuclear export protein XPO1. The lower panel shows the mutational patterns of the XPO1 gene of SHH medulloblastoma (upper) and hematological cancer (lower) according to COSMIC v96 (https://cancer.sanger.ac.uk/cosmic). The number in the circle represents the number of cases
U11 snRNA is also recurrently mutated in SHH medulloblastoma generally along with U1 snRNA mutation, albeit rare (3.7% in SHH medulloblastoma). 6 U11 snRNA is a functional analog of U1 snRNA, where the secondary structure of U11 snRNA mimics that of U1 snRNA. 41 While U1 snRNA recognizes major (U2‐type) introns, U11 snRNA recognizes minor (U12‐type) introns which consist of 0.36% of all the splice sites in the human genome. 42 As with U1 snRNA mutation, U11 snRNA mutation is a hotspot mutation (g.5A > G) within the 5′ splice‐site recognition sequence, indicating that the U11 snRNA mutation causes mis‐splicing in U12‐type introns (Figure 3A).
4.3.2. ELP1 mutations
Mutations in ELP1 (formerly known as IKBKAP) were found in 14% of pediatric SHH medulloblastomas as the most common germline mutation. 7 The ELP1 mutations are detected as a loss‐of‐function mutations predominantly in the U1 wildtype SHHα subtype and are usually accompanied by somatic PTCH1 mutations. ELP1 mutation is mutually exclusive with mutations in TP53. Thus, cases with ELP1 mutation have a relatively good prognosis. ELP1 is a subunit of the elongator complex, which is essential for tRNA modification to uridine at the wobble position (U34), which is the first anticodon of tRNA and recognizes the third nucleotide in a codon. 43 , 44 Therefore, loss‐of‐function of ELP1 impairs elongator‐dependent tRNA modification at the wobble position. The chemical modification of uridines at the wobble position is critical for proper mRNA decoding, and its absence influences codon translation rates. 44 , 45 Therefore, mutant ELP1 tumors have a significant codon usage bias, where AA‐ending codons are inefficiently recognized but AG‐ending codons are efficiently recognized (Figure 3B). 7 , 46 The shift in codon usage gives rise to the significant upregulation of gene sets associated with RNA splicing, amino acid activation, and activation of the endoplasmic reticulum stress pathway in SHH medulloblastoma with ELP1 mutation. 7 , 46 , 47
4.3.3. XPO1 mutations
Mutations in XPO1 are observed in 8.5% of U1 mutant SHH medulloblastomas. 18 XPO1 mutations are frequently detected in primary mediastinal diffuse large B cell lymphoma and classical Hodgkin's lymphoma as a hotspot mutation (p.E571K). 48 , 49 As opposed to these hematological cancers, the majority of XPO1 mutations in SHH medulloblastoma are truncated mutations (Figure 3C). XPO1 is a nuclear export protein that carries proteins and RNAs, including snRNAs, from the nucleus to the cytoplasm, suggesting that RNA exportation and maturation are disrupted in XPO1 mutant SHH medulloblastoma. 50 Accumulating evidence demonstrates that post‐transcriptional aberration is another key component in SHH medulloblastoma.
4.4. Cell of origin of SHH medulloblastoma
Sonic Hedgehog medulloblastoma resembles the granule cell precursor (GCP) lineage transcriptionally, which is consistent with previous publications demonstrating SHH medulloblastomas arise from the GCPs at the external granular layer (Figure 4). 51 , 52 Single‐cell RNA‐seq also detected additional heterogeneity within SHH medulloblastoma, which consists of various different stages of GCP development, supporting a model in which SHH medulloblastoma evolves in a manner consistent with the GCP hierarchy. 10 Conditional Patched knockout mice using Math1‐cre/Ptc c/c which selectively generate a two‐hit inactivation of Ptc in GCPs develop medulloblastoma suggesting that GCPs can serve as the cell of origin for SHH medulloblastoma. 26 , 51
FIGURE 4.
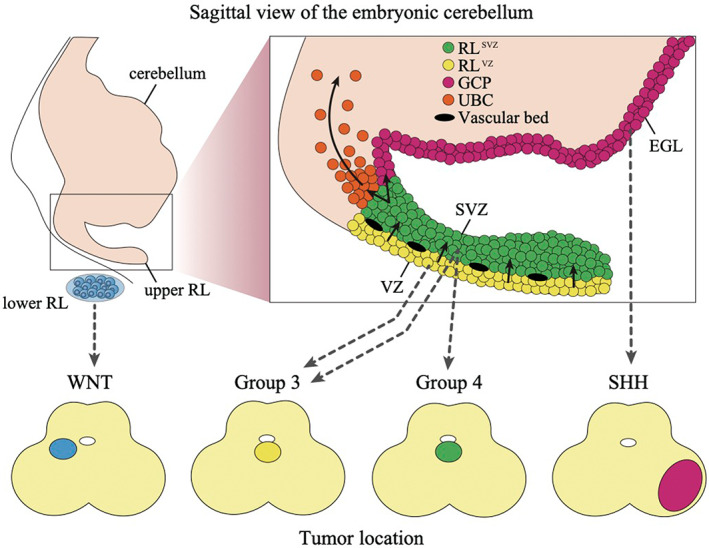
Cell of origin for each subgroup of medulloblastoma. The upper figure shows the sagittal view of the embryonic cerebellum with the stages of cell differentiation in the cerebellum. Black arrows demonstrate the differentiation process. The lower figure shows the common location of each medulloblastoma subgroup in the axial image of the cerebellum. The location of the precursors that likely give rise to each medulloblastoma subgroup is indicated by the dashed arrows. EGL, external granule layer; GCP, granule cell precursor; RL, rhombic lip; SVZ, subventricular zone; UBC, unipolar brush cell; VZ, ventricular zone
5. GROUP 3/4 MEDULLOBLASTOMA
Although several recurrent genetic events are reported in group 3/4, no convergent pathway that defines group 3/4 subgroups have been detected. 14 The subtype classification of group 3/4 by Cavalli and the 5th edition of the WHO classification have a similar tendency but are not completely concordant (Figure 1C). 13 , 16 Recent studies reported that group 3/4 medulloblastomas arise from progenitor cells of the ventricular RL (RLvz) or the subventricular RL (RLsvz), suggesting that the stage of progenitor cell differentiation could reflect subtypes of group 3/4 medulloblastoma (Figure 1C). 8 , 9 Some of the genetic events are in a subtype‐specific manner in group 3/4 medulloblastomas. MYC amplification is enriched in group 3γ, which has the poorest prognosis. 13 OTX2 amplification and activation of GFI1 or GFI1B gene expression by enhancer hijacking are common in group 3β. 9 , 13 MYCN amplification is frequently identified in group 4α. 13 Group 4β, which is considered the purest subtype consisting of only group 4 medulloblastoma frequently, has tandem duplication of SNCAIP, which causes activation of PRDM6. 13 The subtype‐specific manner of recurrent genetic events supports the idea that each subtype is a distinct entity with its own genetic features.
5.1. Genetic alterations in the CBFA complex
A recent large‐scale sequencing study identified somatic mutation of CBFA2T2 in 3.1% of group 4 medulloblastomas. 8 CBFA2T2 is a transcriptional corepressor that links transcription factors and epigenetic modifiers, and interacts with the SET and PR domain of PRDM proteins including PRDM6. 53 In addition, focal chromosome 16q24 deletions, where another CBFA family gene, CBFA2T3 is located, are enriched in cases without CBFA2T2 or PRDM6 alterations. In vitro protein interaction assays demonstrated that CBFA2T2 interacts with KDM6A, which is a known drive gene. Alterations affecting CBFA2T2, CBFA2T3, PRDM6, and KDM6A are almost mutually exclusive, providing support for their role as cancer drivers. 8
5.2. Cell of origin of group 3/4 medulloblastoma
Single‐cell analysis of the developing mouse cerebellum proposed that group 3/4 medulloblastoma arises from an earlier population of stem cells and unipolar brush cell lineage, respectively. 8 , 10 However, single‐cell analyses using the developing human and mouse cerebellum delineate the differences in developmental patterns where human RL persists longer than mouse RL, raising questions about previous insights from mouse developing cerebellum. 54 Human RL expands into the RLvz and the RLsvz by a vascular plexus at 11 post‐conception weeks. By a transcriptional comparison using the developing human cerebellum data, Hendrikse et al. revealed that group 3/4 medulloblastoma cells are most similar to the RLsvz except for a part of the group 3γ subtype which displays enrichment for the earlier RLvz. 8 CBFA2T2 and CBFA2T3 are highly expressed in the RLsvz but neither RLvz nor unipolar brush cell, suggesting that the CBFA complex determines cell fate. Another study by Smith et al using multi‐omics data delineated that group 3/4 medulloblastoma matches the molecular signatures including the gene expression pattern with progenitor cells in the RLsvz as well. 9 Those two studies by Hendrikse et al and Smith et al propose that stalled differentiation of progenitor cells in RL drives the development of group 3/4 medulloblastomas (Figure 4).
6. PERSPECTIVE
In spite of recent advances in cancer diagnostics and therapeutics, the outcome remains unfavorable for patients with medulloblastomas. 3 , 4 , 5 Because of the fundamental importance of the SHH signaling pathway in SHH medulloblastoma, targeting the SHH signaling pathway is considered an attractive strategy. SMO inhibitors targeting the SHH signaling pathway have been used clinically. 20 However, there remain some limitations including early drug resistance by the acquisition of new mutation, the ineffectiveness for cases with aberrations in downstream of the SHH signaling pathway, such as SUFU and GLI2, and premature growth plate fusion for young patients. 20 , 55 , 56 , 57 Therefore, novel targets to develop effective treatments with reduced side effects are urgently needed. Great efforts for recent genetic analyses successfully discovered novel driver alterations beyond the canonical SHH signaling pathways, opening up avenues for novel therapeutic approaches. Mutations in GNAS and PRKAR1A are identified within binding pockets which may be targetable by structure‐based drug design. Cryptic RNA processing induced by mutations in genes associated with RNA processing such as U1 snRNA and ELP1 leads to a unique form of post‐transcriptional hypermutation which should not happen in normal cells. Those tumor‐specific RNAs or neo‐epitopes are good targets by oligonucleotide or immune therapies. Single‐cell approaches enable the detection of the difference between tumor cells and their normal cells of origin, which could target transcriptional vulnerability associated with tumor development. 11 , 58 For example, targeting disrupted differentiation caused by CBFA‐complex alterations may be a good strategy considering the success of all‐trans retinoic acid for acute promyelocytic leukemia. Furthermore, screening approaches such as imaging or biomarker in the serum to detect the presence of atypical cell rests could extract a high‐risk population of medulloblastoma, having the potential for early intervention. 8 , 58 Recent sequencing studies have extended our knowledge of medulloblastoma pathogenesis with novel candidate targets, which may lead to the development of molecular‐targeted therapies for patients with medulloblastoma. In perspective, ongoing basic research efforts will further raise the hope for the improvement of patients with medulloblastoma with the transition of our knowledge into clinical applications.
DISCLOSURE
The authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: N/A.
ACKNOWLEDGEMENT
None.
Funakoshi Y, Sugihara Y, Uneda A, Nakashima T, Suzuki H. Recent advances in the molecular understanding of medulloblastoma. Cancer Sci. 2023;114:741‐749. doi: 10.1111/cas.15691
REFERENCES
- 1. Curtin SC, Minino AM, Anderson RN. Declines in cancer death rates among children and adolescents in the United States, 1999‐2014. NCHS Data Brief. 2016;257:1‐8. [PubMed] [Google Scholar]
- 2. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gajjar A, Chintagumpala M, Ashley D, et al. Risk‐adapted craniospinal radiotherapy followed by high‐dose chemotherapy and stem‐cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma‐96): long‐term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813‐820. [DOI] [PubMed] [Google Scholar]
- 4. Thompson EM, Hielscher T, Bouffet E, et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016;17:484‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Packer RJ, Zhou T, Holmes E, Vezina G, Gajjar A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children's oncology group trial A9961. Neuro Oncol. 2013;15:97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki H, Kumar SA, Shuai S, et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. 2019;574:707‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Waszak SM, Robinson GW, Gudenas BL, et al. Germline elongator mutations in sonic hedgehog medulloblastoma. Nature. 2020;580:396‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hendrikse LD, Haldipur P, Saulnier O, et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature. 2022;609:1021‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith KS, Bihannic L, Gudenas BL, et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature. 2022;609:1012‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vladoiu MC, El‐Hamamy I, Donovan LK, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jessa S, Blanchet‐Cohen A, Krug B, et al. Stalled developmental programs at the root of pediatric brain tumors. Nat Genet. 2019;51:1702‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Millard NE, De Braganca KC. Medulloblastoma. J Child Neurol. 2016;31:1341‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(737–754):e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole‐genome landscape of medulloblastoma subtypes. Nature. 2017;547:311‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18:958‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patmore DM, Jassim A, Nathan E, et al. DDX3X suppresses the susceptibility of hindbrain lineages to medulloblastoma. Dev Cell. 2020;54(455–470):e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skowron P, Farooq H, Cavalli FMG, et al. The transcriptional landscape of shh medulloblastoma. Nat Commun. 2021;12:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059‐3087. [DOI] [PubMed] [Google Scholar]
- 20. Kool M, Jones DT, Jager N, et al. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell. 2014;25:393‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pak E, Segal RA. Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. Dev Cell. 2016;38:333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino‐terminal repression domain: implication of Gli2 and Gli3 as primary mediators of shh signaling. Development. 1999;126:3915‐3924. [DOI] [PubMed] [Google Scholar]
- 23. Vaillant C, Monard D. SHH pathway and cerebellar development. Cerebellum. 2009;8:291‐301. [DOI] [PubMed] [Google Scholar]
- 24. Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319‐331. [DOI] [PubMed] [Google Scholar]
- 25. Suero‐Abreu GA, Praveen Raju G, Aristizábal O, et al. In vivo Mn‐enhanced MRI for early tumor detection and growth rate analysis in a mouse medulloblastoma model. Neoplasia. 2014;16:993‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of patched in lineage‐restricted progenitors or stem cells. Cancer Cell. 2008;14:135‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636‐1639. [DOI] [PubMed] [Google Scholar]
- 28. Sato Y, Maekawa S, Ishii R, et al. Recurrent somatic mutations underlie corticotropin‐independent Cushing's syndrome. Science. 2014;344:917‐920. [DOI] [PubMed] [Google Scholar]
- 29. O'Hayre M, Vazquez‐Prado J, Kufareva I, et al. The emerging mutational landscape of G proteins and G‐protein‐coupled receptors in cancer. Nat Rev Cancer. 2013;13:412‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH‐secreting pituitary adenomas. Nature. 1987;330:566‐568. [DOI] [PubMed] [Google Scholar]
- 31. O'Sullivan C, Barton CM, Staddon SL, Brown CL, Lemoine NR. Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol Carcinog. 1991;4:345‐349. [DOI] [PubMed] [Google Scholar]
- 32. Kan Z, Jaiswal BS, Stinson J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869‐873. [DOI] [PubMed] [Google Scholar]
- 33. Tuson M, He M, Anderson KV. Protein kinase a acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development. 2011;138:4921‐4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan Y, Wang C, Wang B. Phosphorylation of Gli2 by protein kinase a is required for Gli2 processing and degradation and the sonic hedgehog‐regulated mouse development. Dev Biol. 2009;326:177‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He X, Zhang L, Chen Y, et al. The G protein α subunit Gαs is a tumor suppressor in sonic hedgehog‐driven medulloblastoma. Nat Med. 2014;20:1035‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rhayem Y, Le Stunff C, Abdel Khalek W, et al. Functional characterization of PRKAR1A mutations reveals a unique molecular mechanism causing Acrodysostosis but multiple mechanisms causing carney complex. J Biol Chem. 2015;290:27816‐27828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shuai S, Suzuki H, Diaz‐Navarro A, et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature. 2019;574:712‐716. [DOI] [PubMed] [Google Scholar]
- 38. Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 a resolution. Nature. 2009;458:475‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaida D, Berg MG, Younis I, et al. U1 snRNP protects pre‐mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin Y, Lu JY, Zhang X, et al. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature. 2020;580:147‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol. 2003;4:960‐970. [DOI] [PubMed] [Google Scholar]
- 42. Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. Comprehensive splice‐site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955‐3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hawer H, Hammermeister A, Ravichandran KE, Glatt S, Schaffrath R, Klassen R. Roles of elongator dependent tRNA modification pathways in neurodegeneration and cancer. Genes (Basel). 2018;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johansson MJO, Xu F, Bystrom AS. Elongator‐a tRNA modifying complex that promotes efficient translational decoding. Biochim Biophys Acta Gene Regul Mech. 2018;1861:401‐408. [DOI] [PubMed] [Google Scholar]
- 45. Schack MA, Jablonski KP, Graf S, et al. Eukaryotic life without tQCUG: the role of elongator‐dependent tRNA modifications in Dictyostelium discoideum. Nucleic Acids Res. 2020;48:7899‐7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshida M, Kataoka N, Miyauchi K, et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci USA. 2015;112:2764‐2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477‐491. [DOI] [PubMed] [Google Scholar]
- 48. Puente XS, Pinyol M, Quesada V, et al. Whole‐genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Camus V, Miloudi H, Taly A, Sola B, Jardin F. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol. 2017;10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Azizian NG, Li Y. XPO1‐dependent nuclear export as a target for cancer therapy. J Hematol Oncol. 2020;13:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wallace VA. Purkinje‐cell‐derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445‐448. [DOI] [PubMed] [Google Scholar]
- 52. Wechsler‐Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron. 1999;22:103‐114. [DOI] [PubMed] [Google Scholar]
- 53. Nady N, Gupta A, Ma Z, et al. ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife. 2015;4:e10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haldipur P, Aldinger KA, Bernardo S, et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science. 2019;366:454‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee Y, Kawagoe R, Sasai K, et al. Loss of suppressor‐of‐fused function promotes tumorigenesis. Oncogene. 2007;26:6442‐6447. [DOI] [PubMed] [Google Scholar]
- 56. Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kieran MW, Chisholm J, Casanova M, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017;19:1542‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Phoenix TN. The origins of medulloblastoma tumours in humans. Nature. 2022;609:901‐903. [DOI] [PubMed] [Google Scholar]