

Abstract
Second primary cancer (SPC) is one of the most life‐threatening late effects of childhood cancers. We investigated the incidence and survival outcomes of SPC in childhood cancer patients in Japan. Data were obtained from the population‐based Osaka Cancer Registry. Individuals diagnosed with cancer at age 0–14 years during 1975–2014 and survived 2 months or longer were followed through December 2015. The risk of developing SPC was assessed with standardized incidence ratio (SIR), excess absolute risk (EAR, per 100,000 person‐years), and cumulative incidence. Multivariable Poisson regression analysis was carried out to assess relative risks of SPC by treatment method. Survival analysis was undertaken using the Kaplan–Meier method. Of 7229 childhood cancer survivors, 101 (1.4%) developed SPC after a median of 11.6 years. Overall SIR was 5.0, which corresponded with 84.3 EAR. The cumulative incidence was 0.9%, 2.1%, and 3.4% at 10, 20, and 30 years, respectively. Among all SPCs, the type that contributed most to the overall burden was cancers in the central nervous system (EAR = 28.0) followed by digestive system (EAR = 15.1), thyroid (EAR = 8.3), and bones and joints (EAR = 7.8); median latency ranged from 2.0 years (lymphomas) to 26.6 years (skin cancers). Patients treated with radiotherapy alone were at a 2.58‐fold increased risk of developing SPC compared to those who received neither chemotherapy nor radiotherapy. Among patients who developed SPCs, 5‐year and 10‐year survival probabilities after SPC diagnosis were 61.7% and 52.0%, respectively. Risk‐based long‐term follow‐up planning is essential to inform survivorship care and help reduce the burden of SPCs in childhood cancer survivors.
Keywords: cancer registry, childhood cancer, population‐based analysis, second primary cancer
We investigated the incidence and survival outcomes of SPC in childhood cancer patients in Japan using the population‐based Osaka Cancer Registry. During 1975‐2015, childhood cancer patients who survived ≥2 months were at a five‐fold increased risk of developing a new cancer relative to the general population of Osaka with a median of 11.6 years of latency. Risk‐based long‐term follow‐up planning is essential to inform survivorship care and help reduce the burden of SPCs in childhood cancer survivors.
Abbreviations
- AYA
adolescent and young adult
- CI
confidence interval
- CNS
central nervous system
- DCO
death certificate only
- EAR
excess absolute risk
- FAP
familial adenomatous polyposis
- FPC
first primary cancer
- ICCC‐3
International Classification of Childhood Cancers, third edition
- IQR
interquartile range
- OCR
Osaka Cancer Registry
- PYR
person‐years at risk
- RR
relative risk
- SIR
standardized incidence ratio
- SPC
second primary cancer
1. INTRODUCTION
Survival for childhood cancer has seen a substantial improvement over the past several decades, largely due to the advances in treatments and health‐care infrastructure. 1 , 2 , 3 , 4 , 5 However, the improved survival has resulted in an increasing number of childhood cancer survivors suffering from lifelong adverse health effects that are attributable to either their cancer or its treatment. 6
Development of subsequent cancers is one of the most life‐threatening consequences. In the reports from the Childhood Cancer Survivor Study, second primary cancers were found to be the second most common cause of late mortality next to recurrence/progressive disease, accounting for 18.5% of all deaths among 5‐year survivors of childhood cancer. 7 , 8 , 9 While a number of previous studies have reported that childhood cancer survivors are at a higher risk of developing a new cancer, at 3–20 times than the general population, 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 the existing evidence mostly comes from North American or European countries. Only a handful assessments have been carried out in Asian populations; some of them were hospital‐based studies for which external validity was unconfirmed, 16 , 17 or population‐based analyses with a relatively short duration of follow‐up. 18
The development of a long‐term follow‐up system for childhood cancer survivors is listed as one of the major tasks in the third term Basic Plan to Promote Cancer Control Programs in Japan, effective since 2018. 19 Given that childhood cancer survivors often have a longer life expectancy than adult cancer survivors, understanding long‐term risk patterns for subsequent cancers is essential to inform better survivorship care, particularly with regard to screening, surveillance, treatment, and prevention. In light of the above, the purpose of this study was to assess the incidence, relative risks, and survival outcomes of second primary cancers in patients diagnosed with cancer during childhood, using data from a Japanese population. In Japan, the national cancer registry has only been in operation since 2016 and does not have sufficient data accumulation to assess the long‐term risk of subsequent cancers. Thus, we utilized a long‐term, large‐scale subnational cancer registry to yield population‐based estimates.
2. MATERIALS AND METHODS
2.1. Data and study cohort
For this retrospective cohort study, data were extracted from the population‐based OCR. The OCR has a 60‐year history since 1962 and is one of the few cancer databases in Japan that meet international qualifications for comparability, validity, timeliness, and completeness. 20 Covering the entire population of Osaka, the third most populated prefecture (8.8 million as of 2015) 21 in Japan, the OCR collects patients’ information including sex, age, date of diagnosis and death, cancer site and histology, stage at diagnosis, and type of initial treatment. The vital status of registered patients is routinely followed using death certificates every year and official resident registries up until 10 years from diagnosis. The study cohort for the present study included individuals diagnosed with cancer before the age of 15 years during 1975–2014 and who survived at least 2 months from the date of initial cancer diagnosis.
2.2. Statistical analysis
First primary cancer was defined as any, except in‐situ, malignancies and nonmalignant CNS tumors defined in the International Classification of Disease for Oncology, third edition (ICD‐O‐3) 22 that were diagnosed first for each patient. Second primary cancer was defined as a subsequent primary cancer that occurred 2 months or more after the date of initial cancer diagnosis. Recurrence of the first cancer and third or any higher‐order cancers were not included in the analysis. To estimate the risk of developing SPC, PYR were calculated as the time from FPC diagnosis until the earliest date of the following: (i) December 31, 2015, (ii) diagnosis of SPC, (iii) death, or (iv) 30 years after FPC diagnosis. The expected number of SPC was estimated by multiplying the PYR and the cancer incidence rate of the general population of Osaka for each of the population subgroups stratified by sex, age (5‐year group), and calendar year. Standardized incidence ratio was computed as the ratio of the observed to expected numbers of SPC. The 95% CI was estimated assuming a Poisson distribution. 23 To measure the overall burden of SPC, EAR was calculated as the difference between the observed and expected numbers of SPC per 100,000 PYR. To allow comparison with the results from previous studies, we also estimated the incidence of SPC in individuals who survived at least 5 years after FPC diagnosis. The results were stratified by sex (man/woman), age at initial cancer diagnosis (0–4/5–9/10–14 years), and cancer type; FPCs were classified into 12 diagnostic groups according to ICCC‐3, 24 and SPCs were categorized into 14 sites in accordance with the Surveillance, Epidemiology, and End Results Program. 25
Cumulative incidence of SPC at 5, 10, 20, and 30 years after the initial cancer diagnosis was assessed as a function of time after the first cancer diagnosis considering death as a competing risk. The results were stratified by calendar year (1975–1984/1985–1999/2000–2014) in addition to the characteristics listed in the previous paragraph. We also assessed cumulative incidence by attained age (age at diagnosis of SPC for childhood cancer survivors or any first or later cancers for the general population of Osaka) to allow comparison with the age‐specific cumulative incidence in the general population of Osaka. Among childhood cancer survivors, cumulative incidence for attained ages 0–4 years and 5–9 years was calculated by restricting the analysis to individuals first diagnosed with cancer before age 5 years (n = 3477) and 10 years (n = 5320), respectively. The analysis for attained age 10 years or older included all survivors (n = 7229). Cumulative incidence in the general population of Osaka was computed by dividing the number of cancer cases by the total number of individuals at risk during the period 1975–2015 combined. For childhood cancer patients who developed SPC, Kaplan–Meier survival analysis was carried out to estimate survival probabilities at 5 and 10 years after SPC diagnosis.
To investigate the risk of SPC by treatment method, the analysis was restricted to patients for whom treatment information was available. Patients were categorized as follows: neither chemotherapy nor radiotherapy, chemotherapy alone, radiotherapy alone, and both chemotherapy and radiotherapy. We calculated the RR of SPC using a Poisson regression model with the “neither chemotherapy nor radiotherapy” group as the referent. The model was adjusted for sex, age at initial cancer diagnosis, and ICCC‐3 diagnostic group of FPC. All statistical tests were two‐tailed, and statistical significance was set for p values <0.05. All analyses were undertaken using R version 4.0.3.
2.3. Ethics approval
This study was approved by the Institutional Review Board of the Osaka International Cancer Institute (approval number: 19143). We obtained the dataset with no personally identifiable information from the OCR and independently processed it under the Act on Promotion of Cancer Registries. The OCR data is not publicly accessible and is available only on request due to privacy and ethical restrictions.
3. RESULTS
Through December 2015, SPCs were diagnosed in 101 (1.4%) of 7229 childhood cancer patients who survived at least 2 months after the initial cancer diagnosis (Table 1). The overall SIR was 5.0 (95% CI, 4.0–6.0) which corresponded with 84.3 EAR per 100,000 PYR. The median latency between FPC and SPC was 11.6 years (IQR, 5.0–19.5). Compared to women, men had a higher SIR (6.8 [95% CI, 5.2–8.7] vs. 3.5 [95% CI, 2.5–4.7], respectively) and EAR (101.8 vs. 63.5, respectively). The SIR was highest in patients diagnosed at age 0–4 years (SIR = 6.7 [95% CI, 5.0–8.8]) and decreased with increasing age (SIR = 4.7 [95% CI, 2.9–7.1] at age 5–9 years; SIR = 3.3 [95% CI, 2.2–4.9] at age 10–14 years). By diagnostic group of FPC, elevated risks of developing SPC were observed in all groups except “malignant bone tumors” (group VIII) and “germ cell tumors, trophoblastic tumors, and neoplasms of gonads” (group X). The highest SIR was seen in “other and unspecified malignant neoplasms” (group XII) (SIR = 17.0 [95% CI, 6.4–36.9]), followed by “intracranial and intraspinal embryonal tumors” (group III.c) (SIR = 14.9 [95% CI, 4.1–38.1]) and “hepatic tumors” (group VII) (SIR = 13.6 [95% CI, 4.4–31.8]). Among ≥5‐year survivors of childhood cancer (n = 4557), 76 (1.7%) developed SPC after a median of 16.2 (IQR, 10.1–21.4) years (Table S1). The overall SIR in this population was 3.8 (95% CI, 3.0–4.8), and similar risk patterns were seen to those of the ≥2‐month survivors.
TABLE 1.
Profile of childhood cancer patients who survived ≥2 months after initial cancer diagnosis, Osaka Cancer Registry, 1975–2015
| Characteristic | First primary cancer | Second primary cancer | Standardized incidence ratio | Excess absolute risk | Median latency |
|---|---|---|---|---|---|
| n | n (%) | SIR (95% CI) | per 100,000 PYR | Year (IQR) | |
| Overall | 7229 | 101 (1.4) | 5.0 (4.0–6.0) | 84.3 | 11.6 (5.0–19.5) |
| Sex | |||||
| Man | 3977 | 62 (1.6) | 6.8 (5.2–8.7) | 101.8 | 12.4 (5.0–19.0) |
| Woman | 3252 | 39 (1.2) | 3.5 (2.5–4.7) | 63.5 | 10.1 (5.1–20.0) |
| Age at initial cancer diagnosis (years) | |||||
| 0–4 | 3477 | 53 (1.5) | 6.7 (5.0–8.8) | 90.7 | 7.4 (4.7–18.7) |
| 5–9 | 1843 | 22 (1.2) | 4.7 (2.9–7.1) | 76.2 | 15.6 (10.2–19.6) |
| 10–14 | 1909 | 26 (1.4) | 3.3 (2.2–4.9) | 78.5 | 11.4 (3.8–24.6) |
| Diagnostic group of first primary cancers a | |||||
| I. Leukemias, myeloproliferative diseases, and myelodysplastic diseases | 2301 | 24 (1.0) | 4.8 (3.1–7.2) | 75.2 | 14.8 (7.1–19.1) |
| I.a. Lymphoid leukemias | 1509 | 18 (1.2) | 5.1 (3.0–8.1) | 80.2 | 13.0 (6.5–19.7) |
| I.b. Acute myeloid leukemias | 537 | 4 (0.7) | 4.6 (1.2–11.7) | 70.2 | 15.4 (14.8–16.9) |
| II. Lymphomas and reticuloendothelial neoplasms | 693 | 8 (1.2) | 3.7 (1.5–7.4) | 61.2 | 16.7 (13.4–19.1) |
| II.a. Hodgkin's lymphomas | 49 | 0 (0.0) | – | – | – |
| II.b. Non‐Hodgkin's lymphomas | 344 | 2 (0.6) | – | – | – |
| III. CNS and miscellaneous intracranial and intraspinal neoplasms | 1437 | 20 (1.4) | 4.7 (2.8–7.2) | 84.7 | 13.7 (5.0–24.1) |
| III.a. Ependymomas and choroid plexus tumors | 131 | 1 (0.8) | – | – | – |
| III.b. Astrocytomas | 312 | 7 (2.2) | 9.0 (3.6–18.5) | 184.4 | 6.3 (3.4–9.6) |
| III.c. Intracranial and intraspinal embryonal tumors | 204 | 4 (2.0) | 14.9 (4.1–38.1) | 261.3 | 18.7 (13.1–22.3) |
| IV. Neuroblastoma and other peripheral nervous cell tumors | 693 | 7 (1.0) | 4.4 (1.8–9.1) | 54.4 | 6.8 (4.0–14.9) |
| V. Retinoblastoma | 254 | 4 (1.6) | 5.1 (1.4–13.0) | 66.6 | 12.1 (5.1–21.7) |
| VI. Renal tumors | 223 | 5 (2.2) | 7.4 (2.4–17.4) | 110.0 | 10.4 (4.6–16.7) |
| VII. Hepatic tumors | 172 | 5 (2.9) | 13.6 (4.4–31.8) | 237.3 | 10.1 (2.7–23.4) |
| VIII. Malignant bone tumors | 295 | 3 (1.0) | 2.8 (0.6–8.1) | 53.1 | 10.1 (9.3–18.1) |
| VIII.a. Osteosarcomas | 179 | 2 (1.1) | – | – | – |
| VIII.c. Ewing tumors and related bone sarcomas | 79 | 0 (0.0) | – | – | – |
| IX. Soft tissue and other extraosseous sarcomas | 414 | 8 (1.9) | 6.4 (2.7–12.5) | 120.7 | 6.4 (5.4–19.7) |
| IX.a. Rhabdomyosarcomas | 197 | 3 (1.5) | 6.9 (1.4–20.1) | 111.7 | 5.5 (5.3–12.6) |
| X. Germ cell tumors, trophoblastic tumors, and neoplasms of gonads | 525 | 5 (1.0) | 2.2 (0.7–5.1) | 31.0 | 9.3 (2.3–13.8) |
| XI. Other malignant epithelial neoplasms and malignant melanomas b | 131 | 6 (4.6) | 9.9 (3.6–21.4) | 271.0 | 6.9 (1.5–14.8) |
| XII. Other and unspecified malignant neoplasms c | 91 | 6 (6.3) | 17.0 (6.4–36.9) | 375.1 | 1.6 (0.8–9.2) |
Note: The analysis included individuals diagnosed with cancer before the age of 15 years during 1975–2014. Person‐years at risk (PYR) were calculated as the time from first primary cancer diagnosis until the earliest date of the following: (i) December 31, 2015, (ii) diagnosis of second primary cancer, (iii) death, or (iv) 30 years after the initial cancer diagnosis. First primary cancer was defined as any, except in‐situ, cancer that was diagnosed first for each patient. Second primary cancer was defined as a subsequent primary cancer that occurred ≥2 months after the date of initial cancer diagnosis. Estimates not reported for second primary cancers with <3 cases (−).
Abbreviations: CI, confidence interval; CNS, central nervous system; IQR, interquartile range; SIR, standardized incidence ratio.
Classification in accordance with the International Classification of Childhood Cancers, third edition.
Breakdown: a, Adrenocortical carcinomas (n = 5); b, thyroid carcinomas (n = 31); c, nasopharyngeal carcinomas (n = 11); d, malignant melanomas (n = 20); e, skin carcinomas (n = 10); f, other and unspecified carcinomas (n = 54).
Breakdown: a.2. Pancreatoblastoma (n = 12); a.3. pulmonary blastoma and pleuropulmonary blastoma (n = 5); b. other unspecified malignant tumors (n = 74).
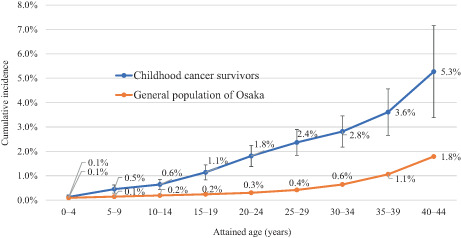
Among all ≥2‐month survivors, the cumulative incidence of SPC was 0.5% at 5 years, 0.9% at 10 years, 2.1% at 20 years, and 3.4% at 30 years of follow‐up (Table 2). The 30‐year cumulative risk ranged from 2.9% in women to 3.8% in men, 3.2% among those diagnosed at age 0–4 years to 4.0% among those diagnosed at age 10–14 years, and 1.6% for “germ cell tumors, trophoblastic tumors, and neoplasms of gonads” (group X) to 8.7% for “hepatic tumors” (group VII). By diagnostic period, the cumulative incidence was higher in the more recent period at any point of follow‐up; for example, the 10‐year cumulative risk increased from 0.7% during 1975–1984 to 1.2% during 2000–2014. Stratified by attained age, childhood cancer survivors were at a higher risk of developing SPC compared with the general population of Osaka (Figure 1). The proportion of childhood cancer patients who had developed SPC by age 24 years was 1.8%, whereas it took until age 44 years for 1.8% of the general population to develop a new cancer.
TABLE 2.
Cumulative incidence of second primary cancers in childhood cancer survivors at 5, 10, 20, and 30 years after the initial cancer diagnosis, Osaka Cancer Registry, 1975–2015
| Characteristic | Time from initial cancer diagnosis | |||
|---|---|---|---|---|
| 5 years (%) | 10 years (%) | 20 years (%) | 30 years (%) | |
| Overall | 0.5 | 0.9 | 2.1 | 3.4 |
| Sex | ||||
| Man | 0.5 | 0.9 | 2.4 | 3.8 |
| Woman | 0.4 | 0.9 | 1.6 | 2.9 |
| Age at initial cancer diagnosis (years) | ||||
| 0–4 | 0.6 | 1.1 | 2.1 | 3.2 |
| 5–9 | 0.2 | 0.4 | 2.2 | 3.4 |
| 10–14 | 0.5 | 0.8 | 1.9 | 4.0 |
| Calendar year at initial cancer diagnosis | ||||
| 1975–1984 | 0.3 | 0.7 | 1.4 | 2.4 |
| 1985–1999 | 0.5 | 0.8 | 1.8 | 4.0 |
| 2000–2014 | 0.6 | 1.2 | – | – |
| Diagnostic group of first primary cancers | ||||
| I. Leukemias, myeloproliferative diseases, and myelodysplastic diseases | 0.2 | 0.6 | 2.0 | 3.6 |
| II. Lymphomas and reticuloendothelial neoplasms | 0.0 | 0.0 | 2.4 | 3.0 |
| III. CNS and miscellaneous intracranial and intraspinal neoplasms | 0.5 | 1.0 | 1.7 | 3.6 |
| IV. Neuroblastoma and other peripheral nervous cell tumors | 0.4 | 0.8 | 1.4 | 2.1 |
| V. Retinoblastoma | 0.4 | 0.9 | 1.6 | 2.7 |
| VI. Renal tumors | 1.1 | 1.1 | 2.5 | 3.4 |
| VII. Hepatic tumors | 1.8 | 1.8 | 3.1 | 8.7 |
| VIII. Malignant bone tumors | 0.0 | 0.7 | 1.4 | 2.9 |
| IX. Soft tissue and other extraosseous sarcomas | 0.4 | 2.0 | 3.3 | 4.5 |
| X. Germ cell tumors, trophoblastic tumors, and neoplasms of gonads | 0.4 | 0.7 | 1.1 | 1.6 |
| XI. Other malignant epithelial neoplasms and malignant melanomas | 2.6 | 2.6 | 5.3 | 7.8 |
| XII. Other and unspecified malignant neoplasms | 4.7 | 4.7 | 8.6 | 8.6 |
Abbreviation: CNS, central nervous system.
Note: The analysis included individuals diagnosed with cancer before the age of 15 years during 1975–2014. First primary cancer was defined as any, except in‐situ, cancer that was diagnosed first for each patient (classification in accordance with the International Classification of Childhood Cancers, third edition). Second primary cancer was defined as a subsequent primary cancer that occurred ≥2 months after the date of initial cancer diagnosis.
FIGURE 1.
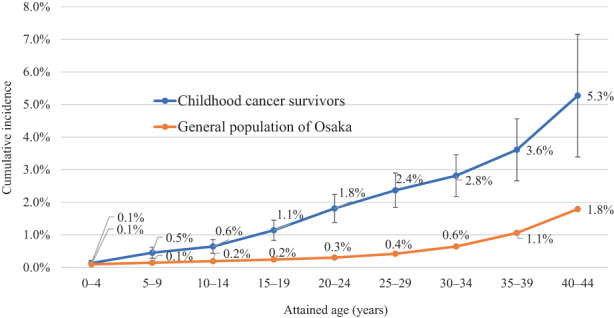
Cumulative incidence of second primary cancers by attained age, Osaka Cancer Registry, 1975–2015. Among childhood cancer survivors, cumulative incidence for attained ages 0–4 and 5–9 was calculated by restricting the analysis to individuals first diagnosed with cancer before age 5 years (n = 3477) and 10 years (n = 5320), respectively. The analysis for attained age 10 or older included all survivors (n = 7229). Person‐years at risk were calculated as the time from first primary cancer diagnosis until the earliest date of the following: (i) December 31, 2015; (ii) diagnosis of second primary cancer; (iii) death; or (iv) 30 years after the initial cancer diagnosis. First primary cancer was defined as any except in‐situ cancer that was diagnosed first for each patient. Second primary cancer was defined as a subsequent primary cancer that occurred ≥2 months after the date of initial cancer diagnosis
Table 3 presents the profile of SPCs. Among the observed 101 SPCs, the most frequent cancer site was the CNS (n = 29 [28.7%]), followed by the digestive system (n = 17 [16.8%]) and thyroid (n = 9 [8.9%]). Of the 84.3 EAR (per 100,000 PYR) for all SPCs combined, CNS cancers accounted for the largest portion (EAR = 28.0 [33.2%]), followed by cancers in the digestive system (EAR = 15.1 [17.9%]) and thyroid (EAR = 8.3 [9.9%]). The SIR was highest for cancers in the bones and joints (13.6 [95% CI, 5.9–29.6]), followed by CNS (12.9 [95% CI, 8.7–18.6]), skin (11.6 [95% CI, 3.2–29.6]), and urinary system (11.5 [95% CI, 3.7–26.9]). The median latency ranged from 2.0 years (IQR, 2.0–2.4) for subsequent lymphomas to 26.6 years (IQR, 26.4–27.5) for subsequent skin cancers.
TABLE 3.
Profile of second primary cancers in childhood cancer survivors, Osaka Cancer Registry, 1975–2015
| Diagnostic group of first primary cancers a | Second primary cancer site b | |||||||
|---|---|---|---|---|---|---|---|---|
| Oral cavity and pharynx | Digestive system | Respiratory system | Bones and joints | Skin | Soft tissue | Breast | Female genital system | |
| I. Leukemias, myeloproliferative diseases, and myelodysplastic diseases (n = 2301) | 2 | 1 | 1 | 4 | ||||
| II. Lymphomas and reticuloendothelial neoplasms (n = 693) | 3 | 2 | ||||||
| III. CNS and miscellaneous intracranial and intraspinal neoplasms (n = 1437) | 2 | 1 | 3 | 1 | ||||
| IV. Neuroblastoma and other peripheral nervous cell tumors (n = 693) | ||||||||
| V. Retinoblastoma (n = 254) | 1 | 3 | ||||||
| VI. Renal tumors (n = 223) | 2 | |||||||
| VII. Hepatic tumors (n = 172) | 3 | |||||||
| VIII. Malignant bone tumors (n = 295) | 3 | |||||||
| IX. Soft tissue and other extraosseous sarcomas (n = 414) | 1 | 1 | 1 | 1 | ||||
| X. Germ cell tumors, trophoblastic tumors, and neoplasms of gonads (n = 525) | 2 | 1 | ||||||
| XI. Other malignant epithelial neoplasms and malignant melanomas (n = 131) | 2 | 1 | ||||||
| XII. Other and unspecified malignant neoplasms (n = 91) | 2 | 1 | ||||||
| Total (n) | 5 | 17 | 4 | 8 | 4 | 1 | 4 | 2 |
| Standardized incidence ratio (95% CI) | 8.9 (2.9–20.8) | 6.6 (3.9–10.6) | 5.0 (1.4–12.8) | 13.6 (5.9–26.9) | 11.6 (3.2–29.6) | – | 1.7 (0.5–4.5) | – |
| Excess absolute risk, per 100,000 PYR | 4.6 | 15.1 | 3.3 | 7.8 | 3.8 | – | 1.8 | – |
| Median latency, year (IQR) | 15.0 (13.6–16.3) | 16.7 (11.1–20.8) | 7.5 (4.6–9.3) | 5.0 (3.1–5.9) | 26.6 (26.4–27.5) | – | 18.1 (9.7–26.7) | – |
| Diagnostic group of first primary cancers a | Second primary cancer site b | |||||||
|---|---|---|---|---|---|---|---|---|
| Urinary system | CNS | Thyroid | Other endocrine | Lymphoma | Leukemia | All sites | ||
| I. Leukemias, myeloproliferative diseases, and myelodysplastic diseases (n = 2301) | 1 | 11 | 3 | 1 | 24 | |||
| II. Lymphomas and reticuloendothelial neoplasms (n = 693) | 3 | 8 | ||||||
| III. CNS and miscellaneous intracranial and intraspinal neoplasms (n = 1437) | 9 | 2 | 2 | 20 | ||||
| IV. Neuroblastoma and other peripheral nervous cell tumors (n = 693) | 2 | 2 | 1 | 2 | 7 | |||
| V. Retinoblastoma (n = 254) | 4 | |||||||
| VI. Renal tumors (n = 223) | 2 | 1 | 5 | |||||
| VII. Hepatic tumors (n = 172) | 1 | 1 | 5 | |||||
| VIII. Malignant bone tumors (n = 295) | 3 | |||||||
| IX. Soft tissue and other extraosseous sarcomas (n = 414) | 4 | 8 | ||||||
| X. Germ cell tumors, trophoblastic tumors, and neoplasms of gonads (n = 525) | 1 | 1 | 5 | |||||
| XI. Other malignant epithelial neoplasms and malignant melanomas (n = 131) | 2 | 1 | 6 | |||||
| XII. Other and unspecified malignant neoplasms (n = 91) | 1 | 1 | 1 | 6 | ||||
| Total (n) | 5 | 29 | 9 | 2 | 3 | 8 | 101 | |
| Standardized incidence ratio (95% CI) | 11.5 (3.7–26.9) | 12.9 (8.7–18.6) | 8.6 (3.9–16.4) | – | 2.8 (0.6–8.0) | 2.6 (1.1–5.2) | 5.0 (4.0–6.0) | |
| Excess absolute risk (per 100,000 PYR) | 4.8 | 28.0 | 8.3 | – | 2.0 | 5.2 | 84.3 | |
| Median latency, years (IQR) | 7.4 (4.6–10.4) | 12.8 (5.1–19.7) | 16.1 (12.1–18.8) | – | 2.0 (2.0–2.4) | 2.7 (2.2–9.1) | 11.6 (5.0–19.5) | |
Note: The analysis included individuals diagnosed with cancer before the age of 15 years during 1975–2014. Person‐years at risk (PYR) were calculated as the time from first primary cancer diagnosis until the earliest date of the following: (i) December 31, 2015, (ii) diagnosis of second primary cancer, (iii) death, or (iv) 30 years after the initial cancer diagnosis. First primary cancer was defined as any, except in‐situ, cancer that was diagnosed first for each patient. Second primary cancer was defined as a subsequent primary cancer that occurred ≥2 months after the date of initial cancer diagnosis. Estimates not reported for second primary cancers with <3 cases (−).
Abbreviations: CI, confidence interval; CNS, central nervous system; IQR, interquartile range; PYR, person‐year at risk.
Classification in accordance with the International Classification of Childhood Cancers, third edition.
Classification in accordance with the Surveillance, Epidemiology, and End Results Program.
When we restricted the analysis to patients whose treatment information was available (n = 4882), those who did not undergo chemotherapy or radiotherapy had the lowest SIR (2.8 [95% CI, 1.1–6.2]), EAR (50.2/100,000 PYR), and the shortest latency to the diagnosis of SPC (6.3 [IQR, 4.5–12.9] years) (Table 4). Using this group as the referent, patients who were initially treated with radiotherapy alone had a 2.58 (95% CI, 1.09–6.89) times increased risk of developing SPC in the multivariable analysis. The other treatment groups also had, although not statistically significant, higher estimated risks of SPC relative to patients who received neither treatment (RR = 1.45 [95% CI, 0.76–3.86] for those with chemotherapy alone; RR = 1.34 [95% CI, 0.60–3.01] for those with both chemotherapy and radiotherapy). The cumulative incidence was similar across all groups up to 10 years of follow‐up; however, patients who received either chemotherapy or radiotherapy or both had nearly double the cumulative incidence of the neither‐treatment group at 30 years (2.1% among those with neither treatment vs. 4.0%, 3.8%, and 3.6% among those with radiotherapy alone, chemotherapy alone, and both, respectively).
TABLE 4.
Standardized incidence ratios and relative risks of subsequent primary cancers by treatment method, Osaka Cancer Registry, 1975–2015
| Treatment | First primary cancer | Subsequent primary cancer | Standardized incidence ratio | Excess absolute risk | Relative risk | Median latency, | Cumulative incidence | |||
|---|---|---|---|---|---|---|---|---|---|---|
| n | n (%) | (95% CI) | per 100,000 PYR | (95% CI) | years (IQR) | 5 years (%) | 10 years (%) | 20 years (%) | 30 years (%) | |
| Neither chemotherapy nor radiotherapy | 692 | 6 (0.9) | 2.8 (1.1–6.2) | 50.2 | Ref. | 6.3 (4.5–12.9) | 0.3 | 0.8 | 1.4 | 2.1 |
| Chemotherapy alone | 2457 | 31 (1.3) | 5.8 (4.0–8.2) | 89.8 | 1.45 (0.76–3.86) | 14.6 (3.2–20.4) | 0.5 | 0.6 | 1.8 | 3.8 |
| Radiotherapy alone | 470 | 10 (2.1) | 6.8 (3.4–12.3) | 95.3 | 2.58 (1.09–6.89) | 19.5 (16.1–25.0) | 0.0 | 0.7 | 2.0 | 4.0 |
| Both chemotherapy and radiotherapy | 1263 | 15 (1.2) | 5.6 (3.1–9.4) | 82.5 | 1.34 (0.60–3.01) | 16.7 (5.1–26.3) | 0.5 | 0.8 | 2.0 | 3.6 |
Note: The analysis included individuals diagnosed with cancer before the age of 15 years during 1975–2014. Person‐years at risk (PYR) were calculated as the time from first primary cancer diagnosis until the earliest date of the following: (i) December 31, 2015, (ii) diagnosis of second primary cancer, (iii) death, or (iv) 30 years after the initial cancer diagnosis. First primary cancer was defined as any, except in‐situ, cancer that was diagnosed first for each patient. Second primary cancer was defined as a subsequent primary cancer that occurred ≥2 months after the date of initial cancer diagnosis.
Abbreviations: CI, confidence interval; IQR, interquartile range; Ref., reference.
Figure 2 presents survival probabilities for childhood cancer patients who developed SPC. Survival probabilities after SPC diagnosis were 61.7% (95% CI, 52.8–72.1) at 5 years and 52.0% (95% CI, 41.8–64.7) at 10 years.
FIGURE 2.
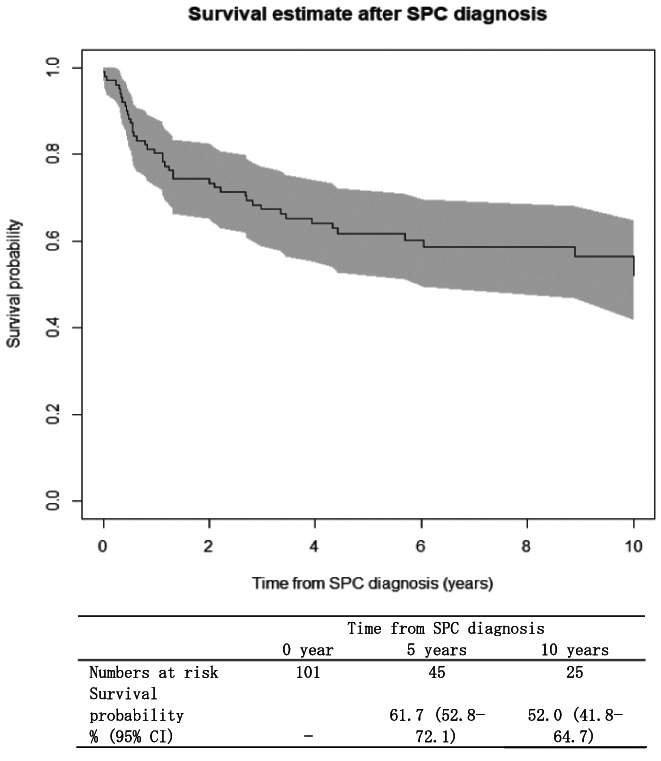
Kaplan–Meier survival curves for overall survival in patients who developed second primary cancers (SPC), Osaka Cancer Registry, 1975–2015. The analysis included individuals who were first diagnosed with cancer before age 15 years, survived ≥2 months, and developed SPC during 1975–2015 (n = 101). Gray shading represents 95% confidence interval (CI)
4. DISCUSSION
Through 41 years of observation, 101 (1.4%) of the 7229 childhood cancer survivors developed SPC after a median of 11.6 years. Patients who survived ≥2 months were at a five‐fold increased risk, and those who survived ≥5 years were at a 3.8‐fold increased risk of developing a new cancer relative to the general population of Osaka. These estimates were within the SIR range of 3–20 reported by previous studies. 10 , 11 , 12 , 13 , 14 , 16 , 17 , 18 The overall cumulative incidence of SPC was 0.9% at 10 years, 2.1% at 20 years, and 3.4% at 30 years after the initial cancer diagnosis. These estimates were similar to those reported in a previous Japanese hospital‐based study. 16 The cumulative risk of SPC showed an upward trend during the study period, which could be attributed to the substantial improvement in both childhood cancer survival 1 , 2 , 3 and the completeness of the population‐based cancer registry (i.e., fewer cases notified by DCO) since the 1970 s. During the study period, survival probabilities after SPC diagnosis were 61.7% and 52.0% at 5 and 10 years, respectively. These findings highlight the need for long‐term screening, surveillance, and prevention planning.
This study yielded a few notable results. First, of the 101 SPCs observed in this study, the largest portion was represented by cancers of the CNS (n = 29), which may be partly due to the late effect of treatment for FPC. An increased risk of subsequent CNS cancers such as gliomas, meningiomas, and sarcomatous lesions has been reported as a consequence of irradiation for CNS cancers and leukemia. 26 , 27 , 28 , 29 In line with this, the most common subsequent CNS cancer in the present study was glioma (n = 16), followed by meningioma (n = 6). Recognition of patients at risk for developing radiation‐induced neoplasms could facilitate early diagnosis through careful clinical observation and follow‐up screening. Second, we found that survivors of hepatic tumors (group VII) had a 13.6‐fold elevated risk of developing a new cancer compared to the general population and the highest cumulative incidence (8.7%) among the 12 diagnostic groups. Although we observed only five SPCs in this population, it would be noteworthy that two of them were rectal cancers preceded by hepatoblastoma, which might suggest the presence of FAP. Clinicians should be aware that hepatoblastoma in early childhood could be the first manifestation of FAP; previous studies recommend children with hepatoblastoma undergo APC mutation screening and colorectal surveillance regardless of family history in order to detect this rare but serious hereditary cancer predisposition syndrome. 30 , 31 Third, we did not observe any SPC among survivors of Hodgkin's lymphoma, likely due to their small population size (n = 49). The global patterns of Hodgkin's lymphoma show a lower incidence in Asian populations. 32 While previous studies in other countries have reported an increased risk of SPC, particularly breast cancer, thyroid cancer, and sarcoma after treatment for Hodgkin's lymphoma, 8 , 12 evidence is limited in Asian populations. There could be racial differences in the incidence of SPCs as well as in the profile of FPCs.
We observed a greater burden (measured as SIR and EAR) of SPC among patients who underwent either or both chemotherapy and radiotherapy than those who received neither treatment. They also had a longer latency to SPC; the cumulative risk diverged from that of the neither‐treatment group over time. Radiation has been documented as a strong risk factor for subsequent cancers of the breast, thyroid, CNS, salivary gland, bone, and stomach. 29 , 33 , 34 , 35 , 36 , 37 , 38 Chemotherapeutic agents such as alkylators, anthracyclines, and epipodophyllotoxins are also known risk factors for SPC, 39 , 40 , 41 , 42 although our multivariable analysis did not show a statistically significantly increased risk for patients who received chemotherapy. This might be due to the relatively small number of patients with treatment information and the lack of detailed treatment information, which did not allow for an adjusted analysis that took into account the types and cumulative dose of anticancer drugs used. Other risk factors include hematopoietic stem cell transplantation, 43 , 44 , 45 however, information for this is not collected in the OCR. In Japan and worldwide, efforts have been made to minimize the short‐term and long‐term treatment‐related adverse health effects, including subsequent neoplasms, by balancing toxicity and efficacy. It will be of importance to investigate whether the adoption of more contemporary treatment protocols is accompanied by a reduction in the incidence of treatment‐related SPCs. Detailed information on individual treatment methods is desired for future studies.
In this study, secondary hematological malignancies occurred earlier than other types of SPC with a median latency of 2.0 years for lymphomas and 2.7 years for leukemias, consistent with reports from other countries. 13 , 15 , 16 , 17 , 18 , 33 , 46 Median latency for other SPCs ranged from 5.0 years (cancers in the bones and joints) to 26.6 years (skin cancers), underscoring the need for long‐term screening planning for childhood cancer survivors that takes into account the time since initial cancer diagnosis. For example, a routine blood test is warranted for the first several years to detect subsequent hematological neoplasms, then the focus may shift to subsequent cancers with longer latency and heavier burden, such as cancers in the CNS and digestive system. In Japan, national guidelines recommend annual or biennial screening for five common cancers for people above certain age: ≥50 years for stomach cancer, ≥20 years for cervical cancer, and ≥40 years for lung cancer, breast cancer, and colorectal cancer. 47 In this study, the analysis of cumulative risk by attained age indicated that, even before reaching these ages, childhood cancer survivors had a comparable risk of developing a new cancer to that of adults in the general population who were old enough to be recommended for regular screening. Educational interventions are also important for survivors to build health literacy regarding risk perception of SPC, adherence to follow‐up screening, and practice of cancer‐preventing behaviors. 48 , 49
The development of a long‐term follow‐up system for childhood cancer survivors is listed as one of the major tasks in the third term Basic Plan to Promote Cancer Control Programs in Japan. This study found that the median latency to SPC diagnosis was 11.6 years, indicating that many childhood cancer survivors developed SPC in the AYA generation, and the 10‐year survival probability after SPC was 52.0%. In Japan, 15 hospitals have been designated as childhood cancer care hospitals by the Ministry of Health, Labour, and Welfare since 2012, promoting the centralization of childhood cancer care. However, the health‐care structure to provide optimal care to childhood cancer survivors in the AYA generation has not yet been established. Building networks to promote collaboration across pediatric and adult cancer hospitals and community health‐care providers is key to information sharing. Such networks facilitate long‐term follow‐up to ensure that survivors receive seamless care at accessible facilities in their own communities and comprehensive support to meet various clinical and psychosocial needs as they transition through their life stages. Our findings will inform efforts to plan and implement risk‐based clinical follow‐up and provide the basis to help specify the roles of pediatric and adult cancer care facilities.
This is the first population‐based assessment of subsequent primary cancers in childhood cancer patients in Japan. The OCR has a 60‐year history and has accumulated data on cancer patients of all ages, which allowed us to follow survivors into their 40s. Despite this strength, there are several limitations in this study. First, the data were extracted from the cancer registry of a single prefecture in Japan. This resulted in a small number of cases for some cancer types and may limit the generalizability of findings to the overall childhood cancer survivors in Japan. A nationwide, long‐term surveillance is needed to better understand the risk patterns of subsequent cancers. However, the population‐based nature of this study is an important advantage over most hospital‐based studies, given that the survivor cohort is less likely to be affected by referral patterns and lost to follow‐up. Second, the number of hematological SPCs in this study was likely underreported. We observed only 11 hematological SPCs (3 lymphomas and 8 leukemias), which accounted for 10.9% of all SPCs. This proportion was lower than those previously reported in other Asian studies: 67 of 340 SPCs (19.7%) in a recent Korean population‐based assessment, 18 58 of 128 SPCs (45.3%) in a Japanese hospital‐based assessment, 16 and 62 of 99 SPCs (62.6%) in a Taiwanese hospital‐based assessment. 17 This could be due in part to the difference in follow‐up duration and the reporting rule; myelodysplastic syndrome, a type of complication of cytotoxic chemotherapy, 50 , 51 , 52 , 53 was not recorded in population‐based cancer registries prior to 2001 in Japan. Third, the risk estimates in this study might be conservative due to the under‐ascertainment of new cancers among long‐term survivors who migrated outside Osaka or invalid registrations (i.e., DCO cases). The impact of this limitation is not likely to be large because our estimate of approximately a five‐fold risk of SPC in overall survivors was within the range previously reported worldwide. 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 To check for completeness of the OCR, we computed the percentage of DCO cases in our study population. While we confirmed it met the international standard (<10%), we observed a gradual improvement throughout the study period (i.e., 6.4% during 1975–1984, 5.1% during 1985–1999, and 2.1% during 2000–2015). Thus, the finding that the cumulative risk of SPC increased over time should be interpreted with caution given the fact that under‐reporting was more common in the past, which possibly underestimated the incidence of SPCs in the earlier years. Fourth, the 10‐year survival probabilities and cumulative incidence for the 2000–2014 diagnostic period might be underestimated due to end‐of‐study censoring (i.e., patients who did not complete the entire follow‐up interval by the end of 2015) that did not reflect the potential impact of recent improvement in survival or incidence of late‐onset SPCs. Fifth, treatment information was missing for one‐third of childhood cancer survivors, although this is unlikely to affect the direction of association in the adjusted multivariable analyses or the overall implication of the findings. One reason for the lack of treatment information is that reporting of radiation and chemotherapy was not mandatory in regional cancer registries in earlier years. While this issue will be resolved in the national cancer registry, which requires treatment information for all records, our results provide an overview of treatment late effects among Japanese childhood cancer survivors and may serve as a milestone for future research. Finally, we did not have complete, detailed treatment information such as irradiation dose, cumulative dose of anticancer drugs, hematopoietic stem cell transplantation, and other known or potentially carcinogenic treatments. Comprehensive data (e.g., data linked to clinical records) is desired to yield more accurate estimates of burdens and evaluate the ongoing efforts for minimizing treatment‐related SPCs.
In conclusion, survivors of childhood cancer had a five‐fold increased risk of developing a new cancer compared to the general population. The latency, burden, and relative risk of SPC varied considerably depending on the cancer type and treatment method. Risk‐based clinical follow‐up planning, in combination with implementation of educational interventions, is important to inform better survivorship care that helps reduce the burden and mortality of SPCs in childhood cancer survivors.
AUTHOR CONTRIBUTIONS
Conceptualization – KN, YH, JH, and KK. Methodology – SO and KN. Validation – all authors. Formal analysis – SO and KN. Resources – all authors. Data curation – all authors. Writing (original draft) – SO. Writing (review and editing) – all authors. Visualization – SO and KN. Supervision – KN. Project administration – all authors. Funding acquisition – SO, KN, TM, and IM.
ACKNOWLEDGMENTS
We would like to thank all the patients and their families and medical staff who cooperate with the Osaka Cancer Registry.
FUNDING INFORMATION
This work was supported by a KAKENHI grant (grant number JP22K10548 [recipients SO, TM, and IM]) and Grant‐in‐Aid for Early‐Career Scientists KAKENHI (grant number JP20K18952 [recipient KN]) from the Japan Society for the Promotion of Science, and Health, Labour and Welfare Science Research Grants (grant numbers H30‐Gantaisaku‐Ippan‐009 [recipients KN, TM, and IM] and 20EA1026 [recipients KN and IM]) from the Ministry of Health, Labour and Welfare, Japan.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: This study was approved by the Institutional Review Board of the Osaka International Cancer Institute (approval number: 19143).
Supporting information
Table S1.
Odani S, Nakata K, Inoue M, et al. Incidence of second primary cancers among survivors of childhood cancer: A population‐based study, Osaka, Japan, 1975–2015. Cancer Sci. 2023;114:1142‐1153. doi: 10.1111/cas.15640
REFERENCES
- 1. Nakata K, Ito Y, Magadi W, et al. Childhood cancer incidence and survival in Japan and England: a population‐based study (1993‐2010). Cancer Sci. 2018;109(2):422‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nakata K, Okawa S, Fuji S, et al. Trends in survival of leukemia among children, adolescents, and young adults: a population‐based study in Osaka Japan. Cancer Sci. 2021;112(3):1150‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakata K, Hiyama E, Katanoda K, et al. Cancer in adolescents and young adults in Japan: epidemiology and cancer strategy. Int J Clin Oncol. 2022;27(1):7‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saletta F, Seng MS, Lau LMS. Advances in paediatric cancer treatment. Translational Pediatrics. 2014;3(2):156‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Applebaum MA, Cohn SL. Surveillance of childhood cancer survivors: a lifelong affair. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33(31):3531‐3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morton LM, Onel K, Curtis RE, Hungate EA, Armstrong GT. The rising incidence of second cancers: patterns of occurrence and identification of risk factors for children and adults. Am Soc Clin Oncol Educ Book. 2014;34(1):e57‐e67. [DOI] [PubMed] [Google Scholar]
- 7. Armstrong GT, Liu W, Leisenring W, et al. Occurrence of multiple subsequent neoplasms in long‐term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol. 2011;29(22):3056‐3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meadows AT, Friedman DL, Neglia JP, et al. Second neoplasms in survivors of childhood cancer: findings from the childhood cancer survivor study cohort. J Clin Oncol. 2009;27(14):2356‐2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Armstrong GT, Liu Q, Yasui Y, et al. Late mortality among 5‐year survivors of childhood cancer: a summary from the childhood cancer survivor study. J Clin Oncol. 2009;27(14):2328‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedman DL, Whitton J, Leisenring W, et al. Subsequent neoplasms in 5‐year survivors of childhood cancer: the childhood cancer survivor study. J Natl Cancer Inst. 2010;102(14):1083‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. MacArthur AC, Spinelli JJ, Rogers PC, Goddard KJ, Phillips N, McBride ML. Risk of a second malignant neoplasm among 5‐year survivors of cancer in childhood and adolescence in British Columbia Canada. Pediatr Blood Cancer. 2007;48(4):453‐459. [DOI] [PubMed] [Google Scholar]
- 12. Jenkinson HC, Hawkins MM, Stiller CA, Winter DL, Marsden HB, Stevens MCG. Long‐term population‐based risks of second malignant neoplasms after childhood cancer in Britain. Br J Cancer. 2004;91(11):1905‐1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reulen RC, Frobisher C, Winter DL, et al. Long‐term risks of subsequent primary neoplasms among survivors of childhood cancer. JAMA. 2011;305(22):2311‐2319. [DOI] [PubMed] [Google Scholar]
- 14. Inskip PD, Curtis RE. New malignancies following childhood cancer in the United States, 1973–2002. Int J Cancer. 2007;121(10):2233‐2240. [DOI] [PubMed] [Google Scholar]
- 15. Youlden DR, Baade PD. The relative risk of second primary cancers in Queensland, Australia: a retrospective cohort study. BMC Cancer. 2011;11(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ishida Y, Qiu D, Maeda M, et al. Secondary cancers after a childhood cancer diagnosis: a nationwide hospital‐based retrospective cohort study in Japan. Int J Clin Oncol. 2016;21(3):506‐516. [DOI] [PubMed] [Google Scholar]
- 17. Ho W‐L, Hung G‐Y, Yen H‐J, et al. Characteristics and outcomes of second cancers in patients with childhood cancer: a report from the Taiwan pediatric oncology group. J Formos Med Assoc. 2022;121(1):350‐359. [DOI] [PubMed] [Google Scholar]
- 18. Ju HY, Moon E‐K, Lim J, et al. Second malignant neoplasms after childhood cancer: a nationwide population‐based study in Korea. PLoS One. 2018;13(11):e0207243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Japanese Ministry of Health, Labour and Welfare . Third Term Basic Plan to Promote Cancer Control Programs in Japan (in Japanese). Japanese Ministry of Health, Labour and Welfare; 2018. Accessed March 22, 2022. https://www.mhlw.go.jp/file/06‐Seisakujouhou‐10900000‐Kenkoukyoku/0000196975.pdf [Google Scholar]
- 20. Bray F, Colombet M, Mery L, et al. Cancer Incidence in Five Continents, Vol XI (electronic version). International Agency for Research on Cancer; 2017. [Google Scholar]
- 21. Osaka Prefectural Government . Census 2015 (in Japanese). Osaka Prefectural Government; 2021. Accessed March 22, 2022. https://www.pref.osaka.lg.jp/toukei/top_portal/oosakahujinnkou.html [Google Scholar]
- 22. WHO . International Classification of Diseases for Oncology (ICD‐O). 3rd ed. World Health Organization (WHO); 2000. [Google Scholar]
- 23. Breslow NE, Day NE. Statistical Methods in Cancer Research. Volume II‐the Design and Analysis of Cohort Studies. 1‐406. IARC Scientific Publications; 1987. [PubMed] [Google Scholar]
- 24. Steliarova‐Foucher E, Colombet M, Ries LAG, et al. International incidence of childhood cancer, 2001–10: a population‐based registry study. Lancet Oncol. 2017;18(6):719‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. U.S. National Cancer Institute . Site Recode ICD‐O‐3/WHO. Definition. U.S. National Cancer Institute; 2008. Accessed March 22, 2022. https://seer.cancer.gov/siterecode/icdo3_dwhoheme/ [Google Scholar]
- 26. Ueda T, Migita M, Itabashi T, et al. Therapy‐related secondary malignancies after treatment for childhood malignancy: cases from a single institution. J Nippon Med Sch. 2019;86:207‐214. [DOI] [PubMed] [Google Scholar]
- 27. Agnihotri S, Suppiah S, Tonge PD, et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat Commun. 2017;8(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettorini BL, Park Y‐S, Caldarelli M, Massimi L, Tamburrini G, di Rocco C. Radiation‐induced brain tumours after central nervous system irradiation in childhood: a review. Childs Nerv Syst. 2008;24(7):793‐805. [DOI] [PubMed] [Google Scholar]
- 29. Sigurdson AJ, Ronckers CM, Mertens AC, et al. Primary thyroid cancer after a first tumour in childhood (the childhood cancer survivor study): a nested case‐control study. The Lancet. 2005;365(9476):2014‐2023. [DOI] [PubMed] [Google Scholar]
- 30. Trobaugh‐Lotrario AD, López‐Terrada D, Li P, Feusner JH. Hepatoblastoma in patients with molecularly proven familial adenomatous polyposis: clinical characteristics and rationale for surveillance screening. Pediatr Blood Cancer. 2018;65(8):e27103. [DOI] [PubMed] [Google Scholar]
- 31. Aretz S, Koch A, Uhlhaas S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer. 2006;47(6):811‐818. [DOI] [PubMed] [Google Scholar]
- 32. Singh D, Vaccarella S, Gini A, Silva NDP, Steliarova‐Foucher E, Bray F. Global patterns of Hodgkin lymphoma incidence and mortality in and a prediction of the future burden in 2040. Int J Cancer. 2020;150(12):1941‐1947. [DOI] [PubMed] [Google Scholar]
- 33. Neglia JP, Friedman DL, Yasui Y, et al. Second malignant neoplasms in five‐year survivors of childhood cancer: childhood cancer survivor study. J Natl Cancer Inst. 2001;93(8):618‐629. [DOI] [PubMed] [Google Scholar]
- 34. de Vathaire F, Hardiman C, Shamsaldin A, et al. Thyroid carcinomas after irradiation for a first cancer during childhood. Arch Intern Med. 1999;159(22):2713‐2719. [DOI] [PubMed] [Google Scholar]
- 35. Schneider AB, Lubin J, Ron E, et al. Salivary gland tumors after childhood radiation treatment for benign conditions of the head and neck: dose‐response relationships. Radiat Res. 1998;149(6):625‐630. [PubMed] [Google Scholar]
- 36. le Vu B, de Vathaire F, Shamsaldin A, et al. Radiation dose, chemotherapy and risk of osteosarcoma after solid tumours during childhood. Int J Cancer. 1998;77(3):370‐377. [DOI] [PubMed] [Google Scholar]
- 37. Newton WA Jr, Meadows AT, Shimada H, Bunin GR, Vawter GF. Bone sarcomas as second malignant neoplasms following childhood cancer. Cancer. 1991;67(1):193‐201. [DOI] [PubMed] [Google Scholar]
- 38. Bhatia S, Robison LL, Oberlin O, et al. Breast cancer and other second neoplasms after childhood Hodgkin's disease. N Engl J Med. 1996;334(12):745‐751. [DOI] [PubMed] [Google Scholar]
- 39. Bhatia S, Krailo MD, Chen Z, et al. Therapy‐related myelodysplasia and acute myeloid leukemia after ewing sarcoma and primitive neuroectodermal tumor of bone: a report from the children's oncology group. Blood. 2007;109(1):46‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hawkins MM, Wilson LM, Stovall MA, et al. Epipodophyllotoxins, alkylating agents, and radiation and risk of secondary leukaemia after childhood cancer. Br Med J. 1992;304(6832):951‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guérin S, Hawkins M, Shamsaldin A, et al. Treatment‐adjusted predisposition to second malignant neoplasms after a solid cancer in childhood: a case‐control study. J Clin Oncol. 2007;25(19):2833‐2839. [DOI] [PubMed] [Google Scholar]
- 42. de Vathaire F, Hawkins M, Campbell S, et al. Second malignant neoplasms after a first cancer in childhood: temporal pattern of risk according to type of treatment. Br J Cancer. 1999;79(11):1884‐1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Curtis RE, Metayer C, Rizzo JD, et al. Impact of chronic GVHD therapy on the development of squamous‐cell cancers after hematopoietic stem‐cell transplantation: an international case‐control study. Blood. 2005;105(10):3802‐3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Landgren O, Gilbert ES, Rizzo JD, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009;113(20):4992‐5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rizzo JD, Curtis RE, Socié G, et al. Solid cancers after allogeneic hematopoietic cell transplantation. Blood. 2009;113(5):1175‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de Vathaire F, Francois P, Hill C, et al. Role of radiotherapy and chemotherapy in the risk of second malignant neoplasms after cancer in childhood. Br J Cancer. 1989;59(5):792‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. National Cancer Center J . About cancer screening. National Cancer Center, Japan; 2022. Accessed March 22, 2022. https://ganjoho.jp/public/pre_scr/screening/about_scr02.html [Google Scholar]
- 48. Berg CJ, Stratton E, Esiashvili N, Mertens A. Young adult cancer survivors' experience with cancer treatment and follow‐up care and perceptions of barriers to engaging in recommended care. J Cancer Educ. 2016;31(3):430‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oeffinger KC, Wallace WHB. Barriers to follow‐up care of survivors in the United States and the United Kingdom. Pediatr Blood Cancer. 2006;46(2):135‐142. [DOI] [PubMed] [Google Scholar]
- 50. Godley LA, Larson RA. Therapy‐related myeloid leukemia. Semin Oncol. 2008;35(4):418‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morton LM, Dores GM, Schonfeld SJ, et al. Association of chemotherapy for solid tumors with development of therapy‐related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5(3):318‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morton LM, Swerdlow AJ, Schaapveld M, et al. Current knowledge and future research directions in treatment‐related second primary malignancies. EJC Suppl. 2014;12(1):5‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shenolikar R, Durden E, Meyer N, Lenhart G, Moore K. Incidence of secondary myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients with ovarian or breast cancer in a real‐world setting in the United States. Gynecol Oncol. 2018;151(2):190‐195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.