

Summary
Glucose transporters are gatekeepers of cellular glucose metabolism. Understanding how their activity is regulated can provide insight into mechanisms of glucose homeostasis and diseases arising from dysregulation of glucose transport. Glucose stimulates endocytosis of the human glucose transporter GLUT1, but several important questions remain surrounding the intracellular trafficking itinerary of GLUT1. Here, we report that increased glucose availability triggers lysosomal trafficking of GLUT1 in HeLa cells, with a subpopulation of GLUT1 routed through ESCRT-associated late endosomes. This itinerary requires the arrestin-like protein TXNIP, which interacts with both clathrin and E3 ubiquitin ligases to promote GLUT1 lysosomal trafficking. We also find that glucose stimulates GLUT1 ubiquitylation, which promotes its lysosomal trafficking. Our results suggest that excess glucose first triggers TXNIP-mediated endocytosis of GLUT1 and, subsequently, ubiquitylation to promote lysosomal trafficking. Our findings underscore how complex coordination of multiple regulators is required for fine-tuning of GLUT1 stability at the cell surface.
Subject areas: Biological sciences, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Substrate transport triggers GLUT1 endocytosis and trafficking to lysosomes
-
•
TXNIP regulates GLUT1 trafficking via interactions with clathrin and ubiquitin ligases
-
•
Ubiquitination of GLUT1 is regulated by glucose levels
-
•
GLUT1 cytosol-facing lysines are required for glucose-stimulated lysosomal trafficking
Biological sciences; Molecular biology; Cell biology
Introduction
Glucose transporters at the plasma membrane (PM) regulate the uptake of extracellular glucose and thus can be regarded as principal gatekeepers of cellular metabolism. In mammalian cells, two transporter families are responsible for glucose uptake: the facilitative glucose transporters, or GLUTs, and the sodium-glucose co-transporters, or SGLTs. SGLTs couple glucose uptake with sodium transport in order to drive import against a concentration gradient. Whereas SGLTs primarily function in absorption and resorption of glucose in specific tissues,1 GLUTs are widely expressed in diverse cell types where they facilitate diffusion of glucose into cells down a concentration gradient.2 The human genome encodes 14 GLUTs, which are part of the major facilitator superfamily3,4 and have a conserved topology consisting of 12 transmembrane domains, short N- and C-terminal cytosolic tails, and a large cytosolic loop between TM6 and TM7 (which we refer to as the “major cytosolic loop”). Some GLUT family members exhibit tissue-specific expression (e.g., GLUT2 in the liver and intestinal epithelia) while some are broadly expressed (e.g., GLUT1).3,5 While structures and transport mechanisms have been well characterized for a few GLUTs,6,7 very little is known about their regulation in a cellular context. One exception is GLUT4, the primary insulin-responsive glucose transporter operating in peripheral tissues, which undergoes regulated trafficking and secretion in response to insulin signaling by well-characterized mechanisms.8,9 By contrast, far less is known about regulation of other GLUT family members. For example, GLUT1 is broadly expressed and responsible for basal glucose uptake in many cell types—including pancreatic β-cells10,11,12 and endothelial cells at the blood–brain barrier13—yet post-translational mechanisms regulating GLUT1 activity, trafficking, and degradation remain poorly characterized.
In erythrocytes and endothelial cells, GLUT1 has a Km for glucose transport of around 2 mM2. Since this is below normal blood glucose concentration (∼4–5 mM), the abundance of GLUT1 at the cell surface is likely a limiting factor for glucose uptake in many cells and tissues. Previous studies have identified three regulatory events that likely contribute to the PM stability of GLUT1. First, phosphorylation of Ser226 in the major cytosolic loop by PKC was reported to promote GLUT1 PM stability and activity in Xenopus oocytes.14 Although the mechanism of positive regulation remains unclear,15 this is the only reported cytosolic post-translational modification of GLUT1. Second, GLUT1 interaction with TXNIP (thioredoxin-interacting protein), a member of the arrestin domain-containing (ARRDC) family of proteins, is reported to regulate its PM stability. Specifically, knocking down TXNIP increased steady-state levels of GLUT1 in HepG2 cells, and TXNIP co-purified with GLUT1 in a manner that was negatively regulated by AMPK activity.16 TXNIP also co-purified with clathrin and subunits of the AP2 clathrin adaptor complex, and GFP-TXNIP co-localized with clathrin assemblies in the PM of HepG2 cells.16 Third, the endosomal recycling retromer complex contributes to the PM stability of GLUT1. Specifically, GLUT1 interacts with the PDZ domain of the retromer subunit SNX27,17 and loss of retromer function results in its aberrant trafficking to lysosomes.17,18,19 Upstream factors that antagonize retromer function—including the RabGAP protein TCB1D5 and the tumor suppressor and phosphatase PTEN—inhibit endosomal recycling of GLUT1.20,21 Taken together, these findings suggest that GLUT1 is regulated by a complex trafficking itinerary that involves clathrin-mediated endocytosis and endosomal recycling, both of which are potential points of regulation for controlling the abundance of GLUT1 at the PM. However, several important questions remain: (i) Is TXNIP-mediated regulation of GLUT1 restricted to clathrin-mediated endocytosis, or does it regulate other sorting or trafficking events after internalization? (ii) What signals activate endocytosis and regulate endosomal sorting and recycling of GLUT1? (iii) Can GLUT1 bypass endosomal recycling for sorting and trafficking to alternative destinations such as lysosomes? A key limitation to addressing these questions is a lack of known regulators of GLUT1 trafficking in response to altered glucose availability.
We hypothesized that cells adapt to increased glucose availability by trafficking GLUT1 to lysosomes. To test this, we analyzed GLUT1 trafficking during a glucose shift time course, and we observed a subpopulation of GLUT1 that traffics to lysosomes as cells adapt to increased glucose availability. We further hypothesized that TXNIP and ubiquitin modification play a role in the regulation of GLUT1 trafficking. However, specific ubiquitin modifications of GLUT1 have not been previously reported. Here, we present evidence that GLUT1 is ubiquitylated in response to glucose stimulation, and we determine that GLUT1 is ubiquitin modified in its major cytosolic loop. Our results suggest that both TXNIP and ubiquitylation are critical for GLUT1 lysosomal trafficking, but TXNIP is dispensable for GLUT1 ubiquitylation. Based on our data, we propose a model where glucose triggers sequential regulation of GLUT1, first by TXNIP-mediated endocytosis and subsequently by ubiquitin modification of GLUT1 to promote trafficking to lysosomes. By elucidating the regulatory mechanisms that govern GLUT1 endocytic sorting and trafficking, these studies provide new insights into how cells adapt to changing glucose availability.
Results
Extracellular glucose stimulates GLUT1 endocytic trafficking to lysosomes
To investigate GLUT1 regulation in response to changing glucose availability, we performed surface biotinylation/capture assays to measure surface levels of endogenous GLUT1 in HeLa cells during a glucose shift time course (i.e., shifting from no glucose to 25 mM glucose at t = 0). We chose these concentrations to represent physiological extremes, ranging from the glucose-replete conditions of peripheral tissues to conditions endothelial cells might encounter during hyperglycemia. This assay revealed significant decline in the levels of surface-localized GLUT1 after 24 h in high glucose media (Figures 1A, 1B and S1A-S1D). In contrast, surface levels of the Na+/K+ ATPase were unchanged during the glucose shift time course (Figures 1A, 1C, and S1A–S1C). Thus, switching from glucose depleted to glucose replete media triggers surface clearance of GLUT1 over a 24 h period of adaptation.
Figure 1.

Excess glucose availability promotes GLUT1 clearance from the plasma membrane
(A) HeLa cells were cultured for 24 h in media with no glucose, then switched to high glucose media (25 mM) for the indicated amount of time. Biotin-labeling was performed at the post-glucose shift time points. Following biotinylation, labeled cells were lysed and surface proteins were affinity purified with NeutrAvidin beads (Thermo Scientific). Analysis was performed by SDS-PAGE and immunoblot with antibodies that recognize GLUT1, Na+/K+ ATPase, and GAPDH.
(B,C) Quantification of captured GLUT1 (B) and Na+/K+ ATPase (C) for the experiment shown in (A) was performed over multiple biological replicates (n ≥ 3). GLUT1 measurements were taken of the whole lane using FIJI. Immunoblots for biological replicate experiments are provided in Figure S1.
(D) HeLa cells stably expressing mCherry-CaaX (red) were cultured in no glucose media for 24 h (top row) then shifted to high glucose (25 mM) for 24 h (bottom row), at which point the samples were fixed for immunofluorescence detection with GLUT1 antibody (green). Zoomed images provided in the far right column correspond to the yellow dashed-line inset boxes in the "MERGE" image to the left.
(E) Quantification of co-localization shown in (D) was measured by Pearson correlation on Softworx software (n = 30 cells), p = 3.75 × 10-8.
(F) HeLa cells stably expressing GLUT1-GFP (green) were cultured using the conditions indicated in (D). Prior to imaging, cells were pulse-labeled with FM4-64 (red), a lipophilic tracer dye that inserts into the outer leaflet of the cell membrane. Live cells were incubated on ice in 8 μM cold FM4-64 for ∼5 min before imaging. Zoomed images provided in the far right column correspond to the yellow dashed-line inset boxes in the "MERGE" image to the left.
(G) Quantification of the results shown in (F). Pearson correlation coefficient was measured using Softworx software (n = 30 cells), p = 1.69 × 10-24. For all experiments, p values were computed using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM.
To further examine this response to glucose, we performed immunofluorescence detection of GLUT1 in HeLa cells stably expressing mCherry-CaaX, a marker for the PM. We observed that cells grown in media lacking glucose exhibit a large population of GLUT1 that co-localizes with mCherry-CaaX at the PM and this co-localization is decreased following glucose repletion (Figures 1D and 1E). Although mCherry-CaaX localizes primarily to the PM, it also exhibits some localization to internal puncta, which limits its fidelity as a specific marker of the PM. To address this limitation, we used pulse labeling of the lipophilic tracer dye FM4-64 to specifically label the PM. Although this improves specificity of PM labeling, FM4-64 is not fixable and thus must be imaged in live cells (i.e., it is incompatible with immunofluorescence). To accommodate this limitation, we generated HeLa cells stably expressing GLUT1-GFP and analyzed its surface localization during glucose adaptation by measuring co-localization with pulsed FM4-64 in live cells. This analysis revealed a significant decrease in co-localization of GLUT1-GFP with FM4-64 as cells adapt to increased glucose availability (Figures 1F and 1G). Combined, surface biotinylation assays and analysis of GLUT1 subcellular localization reveal significant internalization of GLUT1 in response to increased glucose availability.
We hypothesized that the glucose-stimulated clearance of GLUT1 from the PM corresponds to endocytosis and trafficking of GLUT1 to lysosomes. To test this, we performed immunofluorescence imaging of GLUT1 in HeLa cells during a glucose repletion time course. This analysis revealed significant internalization of GLUT1 and increasing co-localization with LAMP1, a lysosomal marker, as cells adapt to increased glucose availability (Figure 2A). We performed similar analysis of HeLa cells stably expressing GLUT1-GFP and similarly observed internalization and co-localization with LAMP1 over the glucose repletion time course (Figure 2B). Using automated image analysis tools,22 we also measured the fraction of total GLUT1-GFP co-localizing with LAMP1. This analysis revealed that in media lacking glucose 11.2% (±7.2) of GLUT1-GFP signal co-localizes with LAMP1, while a shift to high glucose media results in 36.4% (±10.0) of GLUT1-GFP co-localizing with LAMP1 (see Statistical Reporting document for Figure S2B). Glucose repletion also increased co-localization of GLUT1-GFP with LBPA and Lysotracker, two other markers of late endosomes and lysosomes (Figures S2A and S2B). Given the potential for artifacts associated with GFP tagging, particularly at the C-terminus of GLUT1 which harbors a PDZ-binding motif,23 we performed similar analysis with an N-terminal GFP-GLUT1 variant and also observed increased LAMP1 co-localization following glucose repletion (Figures S2C and S2D). To avoid any potential artifacts associated with GFP tagging on cytosol-facing termini of GLUT1, we also analyzed the subcellular localization of a GLUT1 variant harboring a FLAG epitope inserted into the first exofacial loop of the protein. Using immunofluorescence detection of the FLAG epitope, we found that glucose repletion triggered internalization and increasing co-localization of the FLAG-GLUT1 variant with LAMP1 (Figure 2C). Glucose-stimulated GLUT1 endocytosis and trafficking to lysosomes was also observed in MDA-MB-231 breast cancer cells (Figures S2E and S2F). Taken together, these results indicate that GLUT1 undergoes glucose-stimulated endocytosis and trafficking to lysosomes.
Figure 2.

Glucose-stimulated clearance of GLUT1 results in trafficking to lysosomes
(A) Imaging of endogenous GLUT1 was performed in HeLa cells cultured in no glucose media for 24 h then switched to high glucose media (25 mM) for the indicated time and fixed. Samples were imaged via immunofluorescence and probed with GLUT1 antibody (green) and LAMP1 antibody (red). A schematic of the detection strategy is shown in the top left of the panel. Quantification of co-localization (as measured by Pearson correlation on Softworx software (n = 30 cells) is shown in the graph at the top right of the panel. ∗∗ indicates p < 1 × 10-5.
(B) Imaging of stably expressed GLUT1-GFP (green) was performed in HeLa cells cultured in no glucose media for 24 h then switched to high glucose media (25 mM) for the indicated time and fixed. Samples were imaged via immunofluorescence and probed with LAMP1 antibody (red). A schematic of the detection strategy is shown in the top left of the panel. Quantification of co-localization as measured by Pearson correlation on Softworx software (n = 30 cells) is shown in the graph at the top right of the panel. ∗∗ indicates p < 1 × 10-6.
(C) Imaging of stably expressed GLUT1-FLAG, which harbors a FLAG tag on its first exofacial loop, was performed in HeLa cells cultured in no glucose media for 24 h then switched to high glucose media (25 mM) for the indicated time and fixed. Samples were imaged via immunofluorescence and probed with FLAG antibody (green) and LAMP1 antibody (red). A schematic of the detection strategy is shown in the top left of the panel. Quantification of co-localization as measured by Pearson correlation on Softworx software (n = 30 cells) is shown in the graph at the top right of the panel. ∗∗ indicates p < 0.004. For all experiments, p values were computed using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM.
A subpopulation of GLUT1 traffics through ESCRT-associated endosomes
To better understand the glucose-stimulated GLUT1 trafficking itinerary, we analyzed GLUT1 subcellular localization using a panel of endosomal markers with the three GLUT1 detection strategies described in Figure 2 (immunodetection of endogenous GLUT1, stable expression of GLUT1-GFP, and stable expression of a GLUT1 variant harboring a FLAG epitope in the first exofacial loop (FLAG-GLUT)). Previously, GLUT1 was found to co-localize with Vps35,17 a retromer subunit and a marker of endosomal recycling. We observed glucose-stimulated co-localization of GLUT1 with Vsp35 (Figures 3A and 3B), as well the early endosomal markers EEA1 (Figure S3) and transferrin receptor (TfR) (Figure S4) and the late endosomal markers CD63 (Figures 3C and 3D) and VPS4A (Figure S5). In most cases, co-localization was observed when endogenous GLUT1 was detected by immunofluorescence, while co-localization with exogenous GLUT1-GFP or FLAG-GLUT1 was unchanged or slightly changed in response to glucose stimulation. One exception was the late endosomal marker CD63, which exhibited increased co-localization with both endogenous GLUT1 and GLUT1-GFP, and to a lesser extent with FLAG-GLUT1. We organized co-localization data into heat maps that depict subcellular localization of endogenous GLUT1, GLUT1-GFP, and FLAG-GLUT1 over the glucose stimulation time course (Figure 3E). Although we cannot exclude the possibility that trafficking of tagged GLUT1 variants (e.g., GLUT1-GFP or FLAG-GLUT1) is altered due to tag-associated artifacts, these variants recapitulate the lysosomal trafficking of endogenous GLUT1 in response to glucose (Figure 3E). Overall, the data support an itinerary where a subpopulation of GLUT1 is routed to late endosomal and lysosomal compartments as cells adapt to increased glucose availability.
Figure 3.

Characterization of the GLUT1 trafficking itinerary stimulated by excess glucose availability
HeLa cells expressing the three versions of GLUT1 described in Figure 2 were cultured in media lacking glucose for 24 h then either fixed or switched to high glucose media and fixed at the indicated time points. Cells were then probed by immunofluorescence for the endosomal proteins VPS35 (A-B) or CD63 (C-D) (red). In each case, co-localization was analyzed for endogenous GLUT1 (left, green), GLUT1-GFP (middle, green), or GLUT1-FLAG (right, green). Co-localization of GLUT1 signal with VPS35 (B) and CD63 (D) was quantified over the glucose stimulation time course. Co-localization measurements were made in Softworx using Pearson correlation coefficient (n = 30 cells). ∗∗ indicates p < 0.002.
(E) Summarized profile of the GLUT1 trafficking itinerary stimulated by excess glucose availability. Heat maps showing co-localization of endogenous GLUT1 (top), GLUT1-GFP (middle), and exofacial GLUT1-FLAG (bottom) with different markers along the endocytic/endosomal trafficking route. For each time point and each marker, at least 21 measurements were made of the Pearson coefficient of correlation using Softworx software. The color in each box is weighted based on the average Pearson coefficient (n ≥ 21) at the indicated time point.
(F) HeLa cells harboring a doxycycline-inducible dominant-negative VPS4 variant (VPS4E228Q-HA) were cultured in no glucose media + 1 μg/ml doxycycline for 24 h then fixed or switched to high glucose media + doxycycline and fixed at the indicated time point. Cells were then imaged for immunofluorescence detection of HA (red) and GLUT1 (green). VPS4E228Q is a dominant-negative mutant that accumulates on late-endosomal compartments responsible for sorting cargo into intraluminal vesicles. GLUT1 puncta that co-localize with, and are surrounded by, VPS4E228Q-HA are marked with white arrows.
(G) Quantification of the experiments represented in (F) by measuring the Pearson coefficient of correlation (n = 30 cells) using Softworx software. ∗∗ indicates p < 0.001.
(H) HeLa cells stably expressing both GLUT1-GFP (green) and doxycycline-inducible VPS4E228Q-HA were cultured as described in (F) and then imaged for immunofluorescence detection of HA (red). GLUT1-GFP puncta that co-localize with, and are surrounded by, VPS4E228Q-HA are marked with white arrows.
(I) Quantification of the experiments represented in (H) by measuring the Pearson coefficient of correlation (n = 30 cells) using Softworx software. ∗∗ indicates p < 0.02. For all experiments, p values were computed using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM.
Based on these observations, we hypothesized that glucose stimulation increases GLUT1 trafficking to the lysosome via the ESCRT (endosomal sorting complex required for transport) sorting pathway. However, our analysis of GLUT1 trafficking did not reveal significant co-localization with VPS4A, a marker of late endosomes involved in ESCRT-mediated trafficking to lysosomes. VPS4A dynamically associates with the membrane of late endosomal compartments to regulate ESCRT-III disassembly, which is consistent with the diffuse, cytosolic staining pattern we observed by immunofluorescence imaging (Figure S5A). To test if GLUT1 traffics to lysosomes via the ESCRT pathway, we generated a HeLa cell line with a stably integrated vector for inducible expression of a dominant-negative Vps4A variant (Vps4AE228Q-HA) which localizes to ESCRT sorting compartments.24 Doxycycline induction of this cell line resulted in expression of the Vps4A-HA fusion protein (Figure S6A) which was detected as subcellular puncta by immunofluorescence (Figure S6B). Using this cell line, we found that glucose stimulation increased co-localization of both endogenous GLUT1 (Figures 3F and 3G) and GLUT1-GFP (Figures 3H and 3I) with Vps4AE228Q-HA over a glucose stimulation time course. In addition to co-localized GLUT1 and Vps4AE228Q-HA puncta, we observed GLUT1/GLUT1-GFP puncta that appear to be encapsulated by a shell of Vps4AE228Q-HA (Figures 3F and 3H, white arrows), which may represent ESCRT sorting compartments frustrated by the accumulation of the dominant-negative Vps4A variant. Taken together, our data reveal that glucose stimulation triggers internalization of GLUT1 from the PM and increases localization to ESCRT sorting compartments and lysosomes.
Endocytic trafficking of GLUT1 is stimulated by substrate transport
Since cells encounter a wide range of glucose concentrations, and since complete absence of glucose and 25 mM glucose are conditions seldom experienced in physiological contexts, we wanted to examine GLUT1 trafficking in response to a range of glucose levels. After starving HeLa cells of glucose for 24 h, shifting to media containing 5, 15, or 25 mM glucose resulted in similar kinetics of GLUT1 trafficking to lysosomes (Figures S7A and S7B). These concentrations represent a normal physiological range for glucose in human blood, but glucose concentrations encountered in peripheral tissues may be much lower. To simulate a range cells might experience in peripheral tissues, we analyzed GLUT1 trafficking in HeLa cells shifted from 1.5 to 15 mM glucose. In 1.5 mM glucose, GLUT1 localized primarily to the PM, while shifting to 15 mM triggered endocytic trafficking to lysosomes (Figures S7C–S7F). Notably, 1.5 mM is below the reported Km for GLUT1 (around 2 mM2) suggesting that endocytic trafficking of GLUT1 is triggered at concentrations of glucose above the Km.
Based on these findings, we hypothesized that substrate transport by GLUT1 is a trigger for its endocytic trafficking. Alternatively, GLUT1 trafficking could result from a negative feedback response to increased glucose metabolism. To distinguish between these possibilities, we compared GLUT1 trafficking stimulated by either glucose or 3-O-methyl-d-glucose (3-OMG), a glucose analog transported by GLUT1 but not metabolized by the cell. We observed that 3-OMG triggered GLUT1 endocytic clearance and lysosomal trafficking that mimicked the response to glucose (Figures S8A–S8D). These results indicate that substrate transport is sufficient to trigger GLUT1 endocytic trafficking and delivery to lysosomes.
GLUT1 trafficking requires both clathrin- and E3-binding domains of TXNIP
A previous study reported that TXNIP promotes GLUT1 PM clearance via clathrin-mediated endocytosis.16 To further explore the role of TXNIP in GLUT1 downregulation, we generated a stable HeLa cell line expressing both GLUT1-GFP (from a constitutive CMV promoter) and TXNIP (from a doxycycline-inducible (Tet-On) promoter). Induced TXNIP expression in this cell line (Figure S9A) led to glucose-independent trafficking of GLUT1-GFP to lysosomes (Figures S9B–S9C). Similarly, induced TXNIP expression in HeLa cells led to glucose-independent trafficking of endogenous GLUT1 to lysosomes (Figures 4A and 4B). Thus, induced TXNIP expression mimics the physiological response to glucose.
Figure 4.

The clathrin-binding motif and PY motifs of TXNIP are required for glucose-mediated GLUT1 trafficking to lysosomes
(A) HeLa cells stably expressing a doxycycline-inducible expression vector were cultured using the conditions described in Figure 1D and, for the induced samples, additionally treated with 1 μg/ml doxycycline for the last 24 h before fixation. Cells were fixed and imaged for immunofluorescence detection of GLUT1 (green) and LAMP1 (red), a marker of lysosomal compartments. Zoomed images provided in the bottom row correspond to the blue dashed-line inset boxes of the top row.
(B) Quantification of the experiments represented in panel (A) was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition shown.
(C) HeLa cells and txnip knockout equivalents (clone 2) stably expressing GLUT1-GFP (green) were cultured as indicated in (A) then fixed for immunofluorescence detection of LAMP1 (red), a marker of lysosomal compartments.
(D) Quantification of the experiments represented in panel (C) was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition shown.
(E) Schematic representation of TXNIP illustrating the predicted arrestin fold domain (yellow), the clathrin-binding motif (orange), and the two PY motifs (green).
(F) Complementation analysis of HeLa cells stably expressing GLUT1-GFP (green) with the txnip gene knocked out via CRISPR/Cas9. The knockout cells were stably transfected with either an empty vector or a vector expressing wild-type TXNIP, a clathrin-binding mutant (cb), or a py motif mutant (py) expressed from a doxycycline-inducible promoter. Cells were cultured as indicated in Figure 1D with doxycycline added the last 24 h to induce expression of the indicated protein. Cells were fixed and imaged by immunofluorescence for detection of LAMP1 (red), a marker of lysosomal compartments.
(G) Quantification of the results shown in panel (F) was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) for each condition indicated. The dashed line and area shaded in red indicate the average Pearson’s coefficient and standard deviation (respectively) for the condition in which there is no glucose and TXNIP expression is not induced. All p values were measured using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM. All measurements of Pearson coefficient of correlation were performed using Softworx software.
To test if TXNIP is required for glucose-stimulated GLUT1 endocytic trafficking, we generated HeLa cells lacking TXNIP using CRISPR/Cas9 with TXNIP-targeting gRNAs (Figure S9D). We isolated and characterized two distinct txnip knockout HeLa cell lines (Figures S9E–S9F) (which also expressed GLUT1-GFP), both of which exhibited defects in glucose-stimulated lysosomal trafficking of GLUT1 (Figures 4C, 4D, and S9G–S9H). We used txnip knockout clone 1 for additional experiments described in the remainder of this study.
To test if TXNIP expression restores glucose-stimulated GLUT1 trafficking in txnip knockout cells, we stably transfected an inducible TXNIP expression vector into txnip knockout cells. Induction of TXNIP expression in these cells resulted in TXNIP protein levels greater than that detected for endogenous TXNIP in parental HeLa cells (Figure S9I) and complemented the GLUT1 trafficking defect in txnip knockout cells, as evidenced by increased GLUT1-GFP co-localization with LAMP1 (Figures 4E–4G) and decreased co-localization with PM-localized FM4-64 (Figures S9J–S9K) upon induction of TXNIP expression. We decided to use this complementation system to characterize the structural features of TXNIP involved in regulating GLUT1 endocytic trafficking.
TXNIP contains an N-terminal arrestin fold domain and C-terminal motifs that bind to clathrin and NEDD4 family E3 ubiquitin ligases (Figure 4E). Specifically, TXNIP binds to clathrin via a di-leucine motif near its C-terminus (PLLDDM355) and mutation of this motif (L351A, L352A) decreased TXNIP localization to sites of clathrin-mediated endocytosis and decreased the rate of GLUT1 endocytosis.16 PY motifs at the C-terminus of TXNIP (PPCY334 and PPTY378) interact with WW domains of NEDD4 family E3 ubiquitin ligases, including ITCH25,26 and WWP127,28 although their contribution to GLUT1 trafficking remains unknown. To determine how these C-terminal features of TXNIP contribute to GLUT1 trafficking, we induced expression of TXNIP variants harboring mutations that disrupt clathrin binding (TXNIPcb: L351A, L352A) or E3 binding (TXNIPpy: PPCY334 → PACA334 and PPTY378 → PATA378) in txnip knockout cells. Compared to the wild-type variant, both TXNIPcb and TXNIPpy exhibited increased stability following induced expression (Figure S9I). Interestingly, TXNIPpy stabilization was observed for a lower mobility form of the protein, which was previously reported and attributed to phosphorylated TXNIP.16 In contrast, TXNIPcb stabilization was observed without apparent alterations to mobility by SDS-PAGE as detected by immunoblot (Figure S9I). We next tested if induced expression of these TXNIP variants restored GLUT1-GFP trafficking in txnip knockout cells. Both TXNIP mutations conferred partial loss of function, with GLUT1-GFP exhibiting significantly lower co-localization with the lysosomal marker LAMP1 (Figures 4F and 4G) and increased co-localization with PM-localized FM4-64 (Figures S9J–S9K) compared to wild-type TXNIP. Taken together, these results indicate that both di-leucine and PY motifs contribute to TXNIP-mediated trafficking of GLUT1.
TXNIP interacts with WWP1 via its C-terminal PY motifs
Since GLUT1 co-purifies with TXNIP16,29 and TXNIP interacts with ITCH25 and WWP1,28 both E3 ubiquitin ligases of the NEDD4 family, we hypothesized that TXNIP functions as an E3 ubiquitin ligase adaptor. Previous studies reported that the second PY motif of TXNIP (PY2, PPTY378) binds to the WW domains of ITCH,25 but the basis for TXNIP interaction with WWP1 has not been determined. To test if the PY motifs of TXNIP are critical for interaction with WWP1, we incubated recombinant FLAG-WWP1 with lysates from txnip knockout HeLa cells harboring inducible TXNIP expression constructs (WT, cb, and py variants). Affinity purification of FLAG-WWP1 from these lysates revealed co-purification with WT TXNIP and the cb variant which does not bind clathrin (Figure 5A). In contrast, mutations disrupting the C-terminal PY motifs of TXNIP resulted in loss of co-purification with FLAG-WWP1 (Figure 5A). Similar experiments using FLAG-tagged TXNIP as bait also revealed co-purification with WWP1 that was lost in a TXNIPpy mutant (Figure 5B). This analysis reveals that TXNIP interacts with WWP1 via its C-terminal PY motifs. It also reveals that GLUT1 interacts with both TXNIP and WWP1 independently and in a manner that does not require the TXNIP-WWP1 interaction (Figures 5A and 5B).
Figure 5.

TXNIP is dispensable for GLUT1 ubiquitin modification
(A) HeLa cells stably expressing GLUT1-GFP were stably transfected with either empty vector (pINDUCER20) or vector expressing wild type, clathrin-binding mutant (cb), or py motif mutant (py) TXNIP under the control of Tet-on gene expression system. 1 μg/ml doxycycline was added to induce expression of TXNIP for 24 h prior to collection of cell lysate. Cell lysates were incubated with recombinant WWP1-FLAG at 4°C overnight then WWP1-FLAG was pulled down using αFLAG magnetic beads. Elution was performed using FLAG peptide. Lysates and eluates were resolved by SDS-PAGE and analyzed by immunoblot. Immunoblotting of GAPDH was performed as a loading control.
(B) HeLa cells stably expressing GLUT1-GFP were transiently transfected with either a wild type or py mutant TXNIP-FLAG expression plasmid. When cells reached 100% confluence, they were collected in lysis buffer and incubated with αFLAG magnetic beads for 1 h at 4°C with rotation. TXNIP-FLAG was eluted with FLAG peptide and samples were resolved by SDS-PAGE then analyzed by immunoblot. GAPDH was used as a loading control.
(C) HeLa cells stably expressing GLUT1-GFP and a dox-inducible clathrin-binding mutant TXNIP were transiently transfected with either wild-type WWP1-FLAG or a mutant WWP1-FLAG with all 4 ww domains mutated. TXNIPCB was induced with 1 μg/ml doxycycline 24 h before collecting lysates. Cells were then collected in lysis buffer and lysates were incubated with αFLAG magnetic beads for 1 h at 4°C with rotation. WWP1 was eluted using FLAG peptide and samples were resolved by SDS-PAGE and analyzed by immunoblot. GAPDH was used as a loading control.
(D) HEK293T cells stably expressing FLAG-Ub were split into either 1) regular 25 mM glucose DMEM media, 2) DMEM media with no glucose, or 3) no glucose DMEM media and switched to 25 mM glucose media 2 h before collection. All cells were transiently transfected with a GLUT1-GFP expression plasmid. When cells reached 100% confluency, sample 3 cells were switched to high glucose (25 mM) DMEM media and lysates were collected 2 h later then incubated with magnetic FLAG affinity beads for 1 h at 4°C with rotation. FLAG-Ub was eluted using FLAG peptide; samples were resolved by SDS-PAGE, and analyzed by immunoblot. α-Tubulin was used as a loading control.
(E) Quantification of the eluate GLUT1 signal for three biological replicates (n = 3) of the experiment shown in (D). ∗∗ indicates a significant difference (p < 0.05) compared to the no glucose condition (lane 2).
(F) txnip knockout HeLa cells stably expressing constitutive GLUT1-FLAG and a dox-inducible TXNIP expression plasmid were transiently transfected with HA-Ub. 24 hours before collecting lysates, cells were either mock-treated (sample 2) or treated with 1 µg/ml doxycycline (sample 3) to induce TXNIP expression. As a control, HeLa cells with a stably integrated empty vector (i.e., endogenous TXNIP but no GLUT1-FLAG expression) were also analyzed (sample 1). Lysates were incubated with magnetic αFLAG affinity beads for 1 h at 4°C with rotation and eluted using FLAG peptide. Samples were resolved by SDS-PAGE and analyzed by immunoblot. GAPDH was used as a loading control.
(G) Quantification of the eluate HA-Ub signal in four biological replicates (n = 4) of the experiment shown in (F).
(H) HeLa cells stably expressing constitutive GLUT1-FLAG and dox-inducible TXNIP vectors were transiently transfected with either empty vector, HA-Ub, and/or WWP1 as indicated in the figure. 24 hours after inducing TXNIP with 1 μg/ml doxycyxline, cells were collected and lysed. Lysates were incubated with magnetic αFLAG affinity beads for 1 h at 4°C with rotation. GLUT1-FLAG was eluted with FLAG peptide and samples were resolved by SDS-PAGE then analyzed by immunoblot. GAPDH was used as a loading control.
(I) Quantification of HA-Ub signal for at least three biological replicates (n ≥ 3) of the experiments shown in (H). Double asterisk (∗∗) indicates a significant difference (p < 0.05) compared to the empty vector control. All p-values were measured using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM.
To determine if TXNIP interacts with the WW domains of WWP1, we transiently transfected vectors expressing full-length FLAG-WWP1 (wild type or a 4ww mutant (W377F, P380A, W409F, P412A, W484F, P487A, F524A, and P527A) which disrupts PY motif binding at all four WW domains)28 and analyzed co-purification with TXNIP. We observed co-purification of TXNIP with wild-type FLAG-WWP1, but not the 4ww mutant (Figure 5C), indicating that TXNIP binds to the WW domains of WWP1. By comparison, GLUT1 co-purified with both wild-type WWP1 and the 4ww variant, indicating the GLUT1-WWP1 interaction does not require intact WW domains. Together, these data indicate that GLUT1 interactions with TXNIP and WWP1 are mutually exclusive, which is inconsistent with the hypothesis that TXNIP functions as an adaptor that recruits WWP1 to modify GLUT1.
GLUT1 ubiquitin modification is regulated by glucose availability
Based on our results, we hypothesized that ubiquitin modification of GLUT1 promotes its trafficking to the lysosome for degradation. However, GLUT1 ubiquitylation has not previously been reported. Our analysis of GLUT1 in cell lysates by SDS-PAGE and immunoblot detected GLUT1 across a wide range of mobilities (Figure 5), suggesting many modified forms despite the fact that few post-translational modifications of GLUT1 have been reported. One documented modification of GLUT1 is N-linked glycosylation at Asn45 in the first extracellular loop.30,31 Indeed, we found that treatment of affinity-purified GLUT1 with PNGase F (a deglycosylating enzyme specific for cleavage of N-glycans) altered its mobility by SDS-PAGE (Figure S10A), leading to accumulation of a lower molecular weight (MW) form (around 40 kDa) which could represent unmodified GLUT1 (although the predicted MW of unmodified GLUT1 is 54 kDa). Interestingly, several high MW species persisted after PNGase F treatment (Figure S10A), suggesting the existence of high MW forms of GLUT1 that cannot be attributed to N-linked glycosylation.
To test the possibility that GLUT1 may be regulated by ubiquitin modification, we transiently transfected a vector expressing HA-tagged ubiquitin into HeLa cells expressing GLUT1-FLAG and then affinity purified GLUT1-FLAG from cell lysates. SDS-PAGE and immunoblot analysis revealed ubiquitin co-purifying with GLUT1-FLAG (Figure S10B). Similarly, using HEK293 cells stably expressing FLAG-Ubiquitin, we observed GLUT1 co-purifying with FLAG-Ub—and this co-purification was elevated when GLUT1-GFP was expressed exogenously in these cells (Figure S10C). Additionally, our analysis of affinity-purified GLUT1-FLAG revealed high molecular weight species that were decreased following treatment with the deubiquitylating enzyme USP2 (Figure S10D). In order to determine if GLUT1 ubiquitylation is regulated by glucose availability, we generated HEK293 cells stably expressing FLAG-Ubiquitin and measured GLUT1 co-purification with FLAG-Ubiquitin in the context of changing glucose availability. We observed a significant decrease (∼50%) in the amount of GLUT1 co-purifying with FLAG-Ubiquitin following a switch from high (25 mM) glucose media to media without glucose (Figures 5D and 5E). Additionally, glucose repletion of starved cells triggered a significant increase in ubiquitin modification of GLUT1 (Figures 5D and 5E), indicating that ubiquitin modification of GLUT1 is increased in conditions that promote its endocytic trafficking.
Since GLUT1 ubiquitylation correlated with TXNIP expression (Figure 5D), we hypothesized that TXNIP may promote GLUT1 ubiquitylation in the presence of glucose. Indeed, high glucose levels are reported to increase TXNIP expression.16,32 To test this hypothesis, we transiently expressed HA-tagged ubiquitin in txnip knockout HeLa cells which stably harbor (i) a doxycycline-inducible TXNIP expression vector, and (ii) a vector for constitutive expression of FLAG-tagged GLUT1. In these cell lysates, HA-ubiquitin co-purified with FLAG-GLUT1, but the amount of ubiquitin co-purification did not significantly change when TXNIP expression was induced (Figures 5F and 5G). To test if NEDD4 family E3 ubiquitin ligases regulate the ubiquitin modification of GLUT1, we performed the same experiment except we included a plasmid expressing either NEDD4L (a NEDD4 family member not known to interact with GLUT1 or TXNIP) or WWP1. We found that expression of either WWP1 or NEDD4L increased GLUT1 ubiquitin modification, but this increase was only statistically significant when TXNIP expression was induced (Figures 5H, 5I, and S10E). Taken together, these results indicate that GLUT1 ubiquitylation can be induced by co-expression of WWP1 or NEDD4L with TXNIP, but given the relatively modest effect, these experiments do not strongly support an E3 adaptor function for TXNIP.
GLUT1 is ubiquitin modified on its major cytosolic loop
While our findings are consistent with ubiquitin modification of GLUT1, they do not exclude the possibility that GLUT1 may co-purify with interacting proteins that are ubiquitin modified. However, in an unbiased analysis of the ubiquitin-modified proteome of MDA-MB-231 cells, we resolved a ubiquitin remnant (diGly) peptide from GLUT1 corresponding to ubiquitylation at Lys245 (Figure S11A). This position occurs within the large cytosolic loop of GLUT1, which contains multiple other lysine residues (Figure S11B). Due to the distribution of Lys and Arg residues, much of the GLUT1 major cytosolic loop is a tryptic “blind spot” for proteomic detection (Figure S11B). Thus, while our results indicate that GLUT1 can be modified at Lys245, we cannot exclude the possibility that other lysine residues in the major cytosolic loop are also ubiquitin modified, or that additional ubiquitin modification occurs at other cytosol-facing Lys residues on the N- and C-terminal cytosolic tails.
Cytosol-facing lysines in GLUT1 regulate lysosomal trafficking in response to glucose
Given the role that ubiquitylation plays in regulating the trafficking of signaling receptors like GPCRs and EGFR,33,34 we hypothesized that ubiquitylation of GLUT1 contributes to the regulation of its endocytosis and lysosomal trafficking in response to glucose stimulation. To test this, we generated HeLa cell lines stably expressing either wild-type GLUT1-GFP or a variant with Lys245 mutated to arginine (K245R) and we analyzed lysosomal trafficking in response to glucose stimulation. The K245R GLUT1 variant robustly trafficked to lysosomes following glucose stimulation, albeit with slightly lower LAMP1 co-localization compared to wild-type GLUT1 (Figures S12A and S12B). This result suggests that K245 is not required for glucose-stimulated lysosomal trafficking of GLUT1, although there may be multiple redundant ubiquitylation sites on GLUT1. To examine this possibility, we characterized the glucose-stimulated trafficking of a GLUT1 variant with all cytosolic lysine residues substituted for arginine (11Kcyto→R; see Table S1 for specific description of mutations). This variant exhibited a defect in glucose-stimulated lysosomal trafficking (Figures 6A and 6B) and was instead partially retained at the PM with some 11Kcyto→R GLUT1-GFP internalized (Figures S12C–S12D). We hypothesized this internalized pool may localize to early endosomal compartments, given the low level of co-localization with LAMP1 (Figures 6A and 6B). Indeed, this GLUT1 variant exhibited increased co-localization with EEA1 following glucose stimulation (Figures S12E–S12F), consistent with the interpretation that a portion of the 11Kcyto→R GLUT1-GFP is retained at early endosomal compartments.
Figure 6.

Mapping of cytosolic lysines required for lysosomal trafficking of GLUT1
(A) HeLa cells stably expressing either wild-type GLUT1-GFP or GLUT1-GFP with all cytosolic lysines mutated to arginine (11Kcyto→R) were cultured in media lacking glucose for 24 h then cultured for another 24 h in fresh media lacking glucose (“no glucose”) or shifted to fresh media with high glucose (25 mM) for 24 h (“high glucose”) prior to fixation and imaging for immunofluorescence detection of LAMP1 (red). Zoomed images in the bottom row correspond to the blue dashed line inset boxes of the image above.
(B) Quantification of the results shown in (A) was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition indicated. ∗∗ indicates p < 1x10-5.
(C) Schematic of GLUT1 illustrating the primary amino acid sequence of N-terminal and C-terminal cytosolic tails. Lysine residues in the N-terminal and C-terminal cytosolic tails are highlighted in red. A similar schematic illustrating the lysine residues in the major cytosolic loop is shown in Figure S11B.
(D) HeLa cells stably expressing either wild-type GLUT1-GFP, GLUT1-GFP with all cytosolic lysines mutated to arginine (11Kcyto→R), GLUT1-GFP with the 6 lysines on the major cytosolic loop mutated to arginine (6Kloop→R ), or GLUT1-GFP with the 5 cytosolic lysines outside of the major loop mutated to arginine (5Ktails→R) were cultured as in (A) prior to fixation and imaging for immunofluorescence detection of LAMP1 (red). Zoomed images in the bottom row correspond to the blue dashed line inset boxes of the image above.
(E) Quantification of the results shown in panel D was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition indicated. The dashed line and area shaded in red indicate the average Pearson’s coefficient and standard deviation for the WT GLUT1-GFP co-localization with LAMP1 under glucose-starved conditions. All measurements of Pearson coefficient of correlation were performed using Softworx software. All p values were measured using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM.
To test if K245 in the major cytosolic loop is sufficient to restore lysosomal trafficking of the 11Kcyto→R GLUT1 variant, we analyzed glucose-stimulated trafficking of a GLUT1 variant harboring K245 as the only cytosol-facing Lys residue. This GLUT1 variant (designated 1K245) exhibited a defect in glucose-stimulated lysosomal trafficking compared to wild-type GLUT1 (Figures S13A–S13B), although this trafficking response was slightly greater compared to the 11cytoK→R variant (Figures 6A and 6B). Similarly, the 1K245 variant exhibited some retention at the plasma membrane compared to wild-type GLUT1 (Figures S13C–S13D), although the defect was not as severe as that observed for the 11cytoK→R variant (Figures S12C–S12D). These findings reveal that a single Lys residue in the 245 position of the cytosolic loop can facilitate some endocytic trafficking, although it is not sufficient to restore normal lysosomal trafficking in response to glucose stimulation.
In addition to the six Lys residues present in its major cytosolic loop (Figure S11B), GLUT1 also harbors two Lys residues at its N-terminal cytosolic tail and three Lys residues at its C-terminal cytosolic tail (Figure 6C). No other cytosol-facing Lys residues are present in the canonical GLUT1 isoform sequence used in this study, although we cannot exclude the possibility that other cytosol-facing Lys residues may exist in other isoforms or natural variants in human populations. To better understand how Lys residue position contributes to glucose-stimulated trafficking of GLUT1, we generated GLUT1 variants where all Lys residues in the major cytosolic loop or all N- and C-terminal tail Lys residues were substituted with Arg (6Kloop→R or 5Ktails→R, respectively) (Table S1). For the GLUT1 variant lacking Lys residues in the major cytosolic loop (6Kloop→R), we observed defects in glucose-stimulated lysosomal trafficking (Figures 6D and 6E) despite observing a significant decrease in PM localization (Figures S14A and S14B). These findings suggest retention of the GLUT1 6Kloop→R variant in endosomal compartments, which is consistent with an increase in observed co-localization with EEA1 (Figures S14C–S14D). In contrast, the GLUT1 variant lacking Lys residues at N- and C-terminal tails (5Ktails→R) exhibited slight PM retention but no defect in lysosomal trafficking (Figures 6D, 6E, and S14A–S14B). The 5Ktails→R variant did not exhibit early endosomal retention, based on co-localization with EEA1 (Figures S14C–S14D). Taken together, our results indicate that Lys residues in the major cytosolic loop appear to regulate endosomal sorting of GLUT1, while Lys residues at the N- and C-terminal tails may contribute to the regulation of GLUT1 endocytosis. However, loss of all cytoplasmic-facing Lys residues results in a much more dramatic trafficking defect, indicating that Lys residues at each of these locations contributes to GLUT1 trafficking following glucose stimulation.
Cytosol-facing lysines in GLUT1 are required for its TXNIP-mediated endocytic trafficking
Our results indicate that glucose-stimulated trafficking of GLUT1 requires both TXNIP and ubiquitin modification, but the relationship between these two factors remains unclear—particularly given the lack of evidence supporting an E3 adaptor function for TXNIP. To better understand the relationship between TXNIP and ubiquitination in the regulation of GLUT1 trafficking, we tested if lysosomal trafficking of GLUT1 induced by TXNIP overexpression (as shown in Figures 4A, 4B, and S9B–S9C) requires ubiquitin modification, taking advantage of the 11Kcyto→R GLUT1 variant which lacks cytosol-facing Lys residues. We found that the 11Kcyto→R GLUT1 variant was unresponsive to induced TXNIP overexpression, failing to traffic to lysosomes (Figures 7A and 7B) and instead exhibiting increased PM retention (Figures 7C and 7D). Thus, TXNIP-mediated GLUT1 endocytosis and lysosomal trafficking requires cytosol-facing Lys residues of GLUT1. These results are consistent with a role for ubiquitin modification in the regulation of GLUT1 downstream of TXNIP; however, we cannot exclude the possibility that other lysine modifications (e.g., acetylation) may also play a role in the regulation of GLUT1 trafficking.
Figure 7.

TXNIP-mediated trafficking of GLUT1 requires its cytosolic lysine residues
(A) HeLa cells stably expressing either wild-type GLUT1-GFP or GLUT1-GFP with all cytosolic lysines mutated to arginine (11Kcyto→R) were stably transfected with a doxycycline-inducible vector harboring wild-type TXNIP. Cells were cultured as indicated in Figure 1D prior to fixation and imaging for immunofluorescence detection of LAMP1 (red), a marker of lysosomal compartments. Zoomed images in the bottom row correspond to the blue dashed line inset boxes of the image above.
(B) Quantification of the results shown in panel A was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition indicated.
(C) HeLa cells stably expressing either wild-type GLUT1-GFP or GLUT1-GFP with all cytosolic lysines mutated to arginine (11Kcyto→R GLUT1) were stably transfected with a doxycyclin-inducible vector harboring wild-type TXNIP. Cells were cultured as indicated in Figure 1D. Prior to imaging, cells were placed on ice and switched to cold (4°C) buffer containing the lipophilic tracer dye FM4-64 (8 μM) (red) in order to label the plasma membrane. Live cells were imaged in cold buffer immediately to ensure retention of FM4-64 at the plasma membrane. Zoomed images in the bottom row correspond to the blue dashed line inset boxes of the image above.
(D) Quantification of the results shown in panel C was performed by measuring the Pearson coefficient of correlation for 30 cells (n = 30) with each condition indicated. All p values were measured using a two sample Student’s t-Test in Microsoft Excel. A P value < 0.05 was considered statistically significant and is indicated by ∗∗. Data are represented as mean +/- SEM. All measurements of Pearson coefficient of correlation were performed using Softworx software.
Discussion
By characterizing the trafficking itinerary of GLUT1 during cellular adaptation to increased glucose availability, we have identified factors that regulate GLUT1 trafficking to lysosomes. We found that glucose concentrations above the Km trigger GLUT1 endocytosis and trafficking to lysosomes, and that substrate transport is sufficient to trigger this response. Furthermore, we report that (i) TXNIP variants defective for binding to NEDD4 E3 ubiquitin ligases are impaired for glucose-stimulated lysosomal trafficking of GLUT1, (ii) ubiquitin modifications associated with GLUT1 are regulated by glucose availability and are promoted by NEDD4L and WWP1, (iii) GLUT1 is ubiquitin modified on its major cytosolic loop, and multiple cytosol-facing Lys residues are involved in the regulation of GLUT1 trafficking, and (iv) these cytosol-facing Lys residues are required for TXNIP-mediated endocytic trafficking of GLUT1. Our results are consistent with a model of serial regulation of GLUT1 by TXNIP and ubiquitin modification, with a critical role for ubiquitin that occurs downstream of TXNIP-mediated regulation.
Regulation of GLUT1 trafficking by TXNIP
TXNIP is reported to regulate GLUT1 endocytosis via its interaction with clathrin,16 and our analysis of a TXNIP variant defective for clathrin binding (TXNIPcb) is consistent with this previous analysis (Figure 4). However, we also found that a TXNIP variant defective for binding to NEDD4 family E3 ubiquitin ligases (TXNIPpy) exhibited less glucose-stimulated GLUT1 lysosomal trafficking than both TXNIPWT and TXNIPcb variants (Figure 4)—despite the fact that both mutant variants are stabilized compared to wild-type TXNIP (Figure S9I). Expression of either TXNIP variant resulted in increased PM retention of GLUT1 compared to wild type (Figures S9J–S9K), although GLUT1 PM retention was slightly greater in the case of TXNIPcb expression. Indeed, when the TXNIPpy variant is expressed, GLUT1 appears to exhibit increased accumulation inside the cell (Figures 4F and S9J) suggesting it may be retained on early endosomal compartments. These findings indicate that TXNIP plays multiple roles at distinct steps in the GLUT1 trafficking itinerary, with clathrin binding important for endocytic clearance from the PM and PY motifs important for regulating endosomal sorting and the decision to recycle or commit to lysosomal trafficking. Since TXNIP has also been implicated in the insulin-stimulated trafficking of GLUT4,35 it will be interesting for future studies to determine if clathrin-binding and E3-binding motifs of TXNIP also contribute to the regulation of GLUT4.
Based on studies of arrestin-related proteins in yeast,33,36 we hypothesized that TXNIP may function as an adaptor for human NEDD4 family members. As expected, mutation of the C-terminal PY motifs in TXNIP abolished binding to WWP1 (Figures 5A and 5B) and ITCH,25,26 and this mutant variant of TXNIP was impaired for glucose-stimulated GLUT1 trafficking (Figure 4). Unexpectedly, we found that ubiquitin modification of GLUT1 and its association with WWP1 occurs independently of TXNIP. These findings do not support an adaptor function for TXNIP, despite revealing a critical role for TXNIP PY motifs in glucose-stimulated clearance of GLUT1. One possible explanation is that TXNIP, although dispensable for recruitment of NEDD4 family E3s to GLUT1, might promote activation of E3 activity,37,38 perhaps by relieving an inhibitory state in response to glucose. Similarly, TXNIP may not contribute to recruitment of NEDD4 family E3 ligases to GLUT1 but may instead confer glucose-sensitive regulation of E3 activity toward GLUT1. However, since the PY motifs of TXNIP may not interact exclusively with NEDD4 family E3 ubiquitin ligases, we cannot exclude the possibility that they regulate GLUT1 trafficking via other regulatory interactions which have not yet been characterized. It will be important to determine how the PY motifs of TXNIP contribute to the regulation of GLUT1, since this mechanism appears to be distinct and independent of clathrin binding.
Regulation of GLUT1 trafficking by ubiquitylation
Despite its broad expression and importance for basal glucose uptake in many cells and tissues, not much is known about post-translational mechanisms of GLUT1 regulation, in part because few GLUT1 post-translational modifications (PTMs) have been reported and characterized. To our knowledge, the only PTMs previously reported for GLUT1 are N45 glycosylation30 and S226 phosphorylation.14 In this study, we present evidence that GLUT1 is subject to additional regulation by ubiquitylation, with specific ubiquitin modification detected on Lys245 within the major cytosolic loop. Since cytosol-facing Lys residues are required for glucose-stimulated trafficking of GLUT1, we propose that ubiquitin conjugation to GLUT1 at multiple Lys residues is critical for this response. In particular, Lys residues of the major cytosolic loop are critical for glucose-stimulated GLUT1 trafficking (Figure 6)—although Lys245 by itself is dispensable for GLUT1 trafficking to the lysosome in HeLa cells (Figures S12A–S12B). Our analysis of GLUT1 mutant variants targeting different combinations of cytosol-facing Lys residues reveals the potential for complex regulation of GLUT1 by ubiquitin conjugation at multiple sites.
GLUT1 mutations are associated with GLUT1 deficiency syndrome (GDS),39,40,41 and we examined patient mutations to determine if any mapped to Lys residues in the major cytosolic loop. Interestingly, one patient with GDS was reported to harbor the K256V mutation40 in the major cytosolic loop of GLUT1. Some have speculated this mutation could affect GLUT1 interactions with negatively charged lipid head groups.39 However, the K256V mutation could also prevent ubiquitin modification—although further experimental analysis will be required to distinguish between these possibilities.
Our data also reveal a role for NEDD4 family E3 ubiquitin ligases in the regulation of GLUT1 and its associated ubiquitin modifications (Figures 5H and 5I). Specifically, both NEDD4L and WWP1 promoted ubiquitylation associated with GLUT1, and this activity was slightly enhanced in the presence of TXNIP. We cannot exclude the possibility that other ARRDC family proteins may function redundantly in the targeting of GLUT1 for ubiquitylation. Additional experiments will be needed to address the role of WWP1, NEDD4L, and other NEDD4 family E3 ubiquitin ligases in the regulatory trafficking of GLUT1.
An emerging model for GLUT1 trafficking in response to glucose stimulation
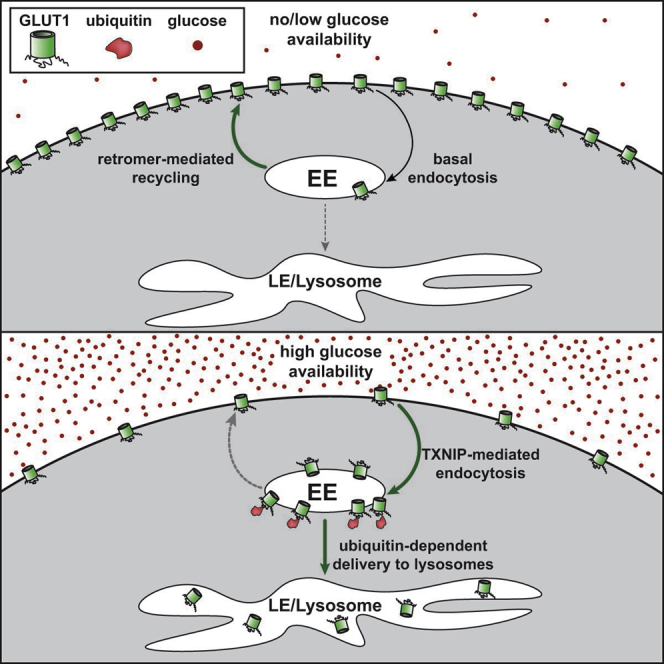
Despite established roles for GLUT1 regulation by TXNIP16 and retromer,17,20,21 many gaps remain in our understanding of GLUT1 trafficking. In this study, we address some of those gaps by identifying a role for ubiquitin modification as a regulator of GLUT1 lysosomal trafficking. Our data suggest that TXNIP has multiple functions that regulate GLUT1 trafficking—including clathrin binding and binding to E3 ubiquitin ligases. Likewise, mutational analysis of GLUT1 suggests that ubiquitin modifications are involved in the regulation of endosomal sorting and lysosomal trafficking. Based on our results, we propose the following model for glucose-regulated GLUT1 trafficking. In conditions of glucose starvation, TXNIP is degraded and ubiquitin association with GLUT1 is low, leading to low levels of endocytic clearance from the PM and a higher efficiency of retromer-mediated recycling on endosomes. In this state, GLUT1 remains relatively stable at the PM. In conditions of high extracellular glucose, TXNIP is stabilized and ubiquitylation of GLUT1 is increased, leading to an increased rate of endocytosis and decreased efficiency of retromer-mediated recycling on endosomes. It is possible that ubiquitylation of GLUT1 on its cytosolic loop interferes with recognition by retromer while also facilitating capture by ubiquitin-binding elements of the ESCRT pathway, sorting it into the MVB pathway for delivery to lysosomes. Together, TXNIP association and ubiquitin modification both contribute to the steady-state localization of GLUT1, toggling between a state of PM stability and one of targeting for delivery to lysosomes. Lysosomal delivery is typically associated with degradation; however, we cannot exclude the possibility that GLUT1 may transport glucose across lysosomal membranes. But many important questions remain surrounding the mechanisms that regulate GLUT1 ubiquitin modification in response to changing glucose levels. Ultimately, deeper understanding of how glucose availability and sensing pathways toggle between these states will facilitate new insights into mechanisms of disease and possibly identify new targets for therapeutic intervention.
Limitations of the study
The interpretation of results in this study is based primarily on experiments performed in HeLa cells. Although some results were validated in MDA-MB-231 breast cancer cells, results reported in this study may be cell line specific and future studies should address applicability of these results to other cell lines of interest. Additionally, experiments in this study were conducted using concentrations of glucose that represent physiological extremes. When interpreting the results presented in this paper, it is important to consider that the concentrations of glucose experienced by cells in a physiological context may be quite different than those examined in this study. Finally, analysis of GLUT1 subcellular localization in this study was accomplished using a combination of C-terminal, N-terminal, and exofacial tagging approaches as well as immunofluorescence detection using antibodies. Each of these approaches carries limitations. Tagging approaches provide confidence that the signal detected is specific for GLUT1, but may confer artifacts from exogenous expression or by disruption of protein interactions that normally occur in proximity to the tag. We attempted to mitigate this limitation by using tags at the N-terminus, C-terminus, and in an exofacial loop of GLUT1. However, we still cannot exclude the possibility that tagging of GLUT1 introduces artifacts that alter its subcellular localization. We further mitigated this limitation by validating key results using commercial GLUT1 antibodies to characterize endogenous GLUT1 subcellular localization by immunofluorescence microscopy. However, in these experiments, we cannot exclude the possibility that some of the signal detected by this antibody is non-specific—especially since GLUT family transporters are highly conserved.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD63 antibody [MEM-259] | abcam | Abcam Cat# ab8219, RRID:AB_306364 |
| VPS35 antibody | abcam | Abcam Cat# ab10099, RRID:AB_296841 |
| Glucose Transporter GLUT1 antibody [EPR3915] | abcam | Abcam Cat# ab115730, RRID:AB_10903230 |
| Rabbit Anti-Sodium Potassium ATPase Monoclonal Antibody, Unconjugated, Clone EP1845Y | abcam | Abcam Cat# ab76020, RRID:AB_1310695 |
| EEA1 | BD Biosciences | BD Biosciences Cat# 610457, RRID:AB_397830 |
| VPS4A Antibody (A-11) | Santa Cruz Biotechnology | Santa Cruz Biotechnology Cat# sc-393428, RRID:AB_2773025 |
| WWP1 monoclonal antibody (M01A), clone 1A7 | Abnova | Abnova Cat# H00011059-M01A, RRID:AB_1717151 |
| GLUT1 antibody | Proteintech | Proteintech Cat# 21829-1-AP, RRID:AB_10837075 |
| Rabbit Anti-NEDD4L Polyclonal Antibody, Unconjugated | Cell Signaling Technology | Cell Signaling Technology Cat# 4013, RRID:AB_1904063 |
| EEA1 (C45B10) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 3288, RRID:AB_2096811 |
| Purified Mouse Anti-LBPA (BMP) | Echelon Biosciences | Echelon Biosciences Cat# Z-PLBPA, RRID:AB_11129226 |
| Rabbit Anti-GAPDH Monoclonal Antibody, Unconjugated, Clone 14C10 | Cell Signaling Technology | Cell Signaling Technology Cat# 2118, RRID:AB_561053 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Millipore | Cat #: F1804; RRID:AB_262044 |
| Mouse anti-Tubulin | Vanderbilt Antibody and Protein Resource Core | N/A |
| Mouse anti-HA | Vanderbilt Antibody and Protein Resource Core | N/A |
| DYKDDDDK Tag Polyclonal Antibody | ThermoScientific | Cat #: PA1-984B RRID: AB_347227 |
| Chemicals, peptides, and recombinant proteins | ||
| MG-132 | APExBIO | Cat# A2585 |
| 3-O-methyl-d-glucopyranose | Millipore Sigma | Cat# M4879 |
| EZ-Link NHS-SS-Biotin | Thermo Fisher | Cat# 21331 |
| Phenanthroline | Sigma-Aldrich | Cat# P9375 |
| Iodoacetamide | Sigma-Aldrich | I1149 |
| WWP1 Active human recombinant, expressed in baculovirus infected insect cells | Sigma-Aldrich | Cat# SRP0229 |
| Recombinant Human Usp2 Catalytic Domain | Boston Biochem | Cat# E-504 |
| PNGase F | Promega | V483A |
| recombinant Shrimp Alkaline Phosphatase | New England BioLabs | Cat# M0371S |
| Experimental models: Cell lines | ||
| Human cells: MDA-MB-231 cells | ATCC | Cat #: MDA-MB-231 (ATCC® HTB-26); RRID:CVCL_0062 |
| Human cells: HEK293T cells | ATCC | Cat# CRL-3216 RRID: CVCL_0063 |
| Human cells: HeLa cells | ATCC | Cat# CCL-2 RRID: CVCL_0030 |
| Human cells: MDA-MB-231 cells stably expressing GLUT1-GFP | This study | N/A |
| Human cells: HEK293T cells stably expressing FLAG-Ub | This study | N/A |
| Human cells: HeLa cells stably expressing GLUT1-GFP (WT, 1K245-- > R,11Kcyto-- > R, 6Kloop-- > R, 5Ktails-- > R, 1K245) | This study | N/A |
| Human cells: HeLa cells stably expressing GLUT1-FLAG | This study | N/A |
| Human cells: HeLa cells stably expressing FLAGexofacial-GLUT1 | This study | N/A |
| Human cells: HeLa cells stably expressing GFP-GLUT1 | This study | N/A |
| Human cells: HeLa cells stably expressing GLUT1-GFP + TXNIP (WT, cb, py) | This study | N/A |
| Human cells: HeLa cells stably expressing GLUT1-FLAG + TXNIP (WT, cb, py) | This study | N/A |
| Oligonucleotides | ||
| ojam5373- txnip KO diagnostic primer (F), GGAGGGTGAAAGCTGATTAG | This study | N/A |
| ojam5374- txnip KO diagnostic primer (R), CACATGCTCACTGCACATTG | This study | N/A |
| Recombinant DNA | ||
| pQCXIN | ClonTech | Cat# 631514 |
| pENTR1A | addgene | Cat# 17398 RRID: Addgene_17398 |
| pInducer20 | addgene | Cat# 44012 RRID: Addgene_4401 |
| pRK5-HA-Ubiquitin-WT | addgene | Cat# 17608 RRID: Addgene_17608 |
| WT FLAG-WWP1 | Nielsen, C.P. et al.28 | N/A |
| 4ww FLAG-WWP1 (W377F, P380A, W409F, P412A, W484F, P487A, F524A, P527A) | Nielsen, C.P. et al.28 | N/A |
| WT WWP1 | this study | N/A |
| WT Nedd4L | this study | N/A |
| WT TXNIP | this study | N/A |
| TXNIPcb | this study | N/A |
| TXNIPpy | this study | N/A |
| WT TXNIP-FLAG | this study | N/A |
| TXNIPpy-FLAG | this study | N/A |
| WT GLUT1-GFP | this study | N/A |
| WT GLUT1-FLAG | this study | N/A |
| WT FLAGexofacial-GLUT1 | this study | N/A |
| HA-VPS4A-E228Q | this study | N/A |
| 1K245-- > R GLUT1-GFP | this study | N/A |
| 11Kcyto-- > R GLUT1-GFP | this study | N/A |
| 1K245 GLUT1-GFP | this study | N/A |
| 6Kloop-- > R GLUT1-GFP | this study | N/A |
| 5Ktails-- > R GLUT1-GFP | this study | N/A |
| VDUP1 CRISPR/Cas9 KO plasmid (h) | Santa Cruz Biotechnology | Cat# sc-400664 |
| Software and algorithms | ||
| Softworx | GE | RRID:SCR_019157 |
| MaxQuant | Max Planck Institute of Biochemistry | RRID:SCR_014485 |
| FIJI | NIH | RRID:SCR_002285 |
| Image Studio Lite software | LI-COR | RRID:SCR_013715 |
Resource availability
Lead contact and materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jason MacGurn (jason.a.macgurn@vanderbilt.edu).
Experimental model and subject details
Cell lines
HeLa cells (Female), MDA-MB-231 cells (Female), and HEK293T (Female) cells were purchased from American Type Culture Collection (ATCC). HeLa and HEK293T cell lines were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. MDA-MB-231 cells were cultured in RPMI with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. Cells stably expressing GLUT1-GFP (WT, 1K245-- > R,11Kcyto-- > R, 6Kloop-- > R, 5Ktails-- > R, 1K245), GLUT1-FLAG, FLAG-Ub, FLAGEXOFACIAL-GLUT1, and GFP-GLUT1 were generated using the Retro-X vectors pQCXIP and pQCXIN (Clontech) retroviral vector system. Cells stably expressing TXNIP (WT, cb, py) and DN VPS4-HA were generated using lentiviral packaging plasmids pMD2 and Pax2 along with the pInducer20 vector. The HeLa txnip knockout cells were generated using the CRISPR/Cas-9 editing system.
Method details
Assays for visualizing GLUT1
GLUT1 trafficking colocalization imaging assays
Cells were split onto 25mm glass coverslips (or glass bottom dishes for live imaging) in regular high glucose media. Cells were switched to no glucose media for 24 h and then switched to the appropriate glucose concentration. At the indicated time point, cells were fixed in 4% PFA for 10 min or washed with PBS and prepared for live imaging.
Cell surface protein biotinylation
Biotinylation protocol was adapted from Singh et al..42 HeLa cells were washed twice with cold DPBS+ then incubated with gentle rocking at 4°C with 0.5 mg/ml EZ-Link NHS-SS-Biotin (Thermo Scientific) twice for 20 min. Biotin was quenched with five 5-minute washes with cold 100mM glycine, 0.2% BSA in PBS. Cells were washed twice with cold DPBS+ and then lysed with lysis buffer (50mM Tris/HCl pH7.4, 150mM NaCl, 1mM EDTA, 1% Triton X-100, protease inhibitor tablet). Lysates were rotated at 4°C for an hour and then centrifuged for 15 min at 4°C, 16,000 x g. 1mg of protein lysate was incubated overnight with NeutrAvidin UltraLink beads (Thermo Scientific) at 4°C. Beads were washed once with cold lysis buffer, three times with cold lysis buffer without Triton X-100 and once with cold 0.1% Triton X-100, 350mM NaCl, 5mM EDTA. Biotinylated proteins were eluted from the beads by heating for 10 min at 70°C in Laemmli sample buffer (Bio-Rad). Input samples and eluates were analyzed by immunoblot.
Transfections
Plasmid transfections were performed using Lipojet (SignaGen, SL100468) and Lipofectamine LTX with PLUS reagent (Thermo Fisher, 15338100) according to manufacturer’s protocol.
Cloning
All GLUT1 plasmids were subcloned into modified pQCXIP vectors from glut1 cDNA (Transomic). Mutations were introduced via geneblocks (IDT) or primer amplification. The inducible TXNIP vector was generated using Gateway cloning with the pENTR1A donor vector and the pInducer20 destination vector. The txnip cDNA was purchased from Transomic. The E228Q VPS4 was constructed with geneblocks (IDT) cloned into the pINDUCER20, using the same Gateway method as described for TXNIP vectors.
CRISPR/Cas9 gene deletion of txnip
CRISPR/Cas9 txnip knockout plasmids were purchased from Santa Cruz Biotechnologies. HeLa cells were electroporated with the KO plasmids and a Cas9-NLS plasmid twice then single cells were sorted into 96 well plates. KO clones were screened by Western blot and PCR. The diagnostic primers used were: 5’GGAGGGTGAAAGCTGATTAG and 5’CACATGCTCACTGCACATTG.
Fluorescence microscopy
All microscopy images were acquired using the DeltaVison Elite System (GE Healthcare) and processed using SoftWoRx software. Pearson Correlation Coefficient was measured using SoftWorx or the JACoP (BIOP version) FIJI plugin. Alternate colocalization for data in Figure S2B was determined using a FIJI macro (https://github.com/dsrichardson/fiji_macros/blob/master/2D_object_colocalization). The macro identifies the center of mass of each vesicle (6 pixels in size) in the GFP channel then determines the nearest neighbor in the AF-594 channel. Centers of mass less that the average diameter were considered colocalized.
Live cell imaging
When doing live cell imaging, cells were plated onto glass bottom dishes and switched to media without phenol red for imaging. For FM4-64 staining of the plasma membrane, cells were washed with ice cold PBS and kept on ice. 8mM FM4-64 was diluted 1:1000 in ice cold HBSS and added to cells 5 min before imaging. For LysoTracker imaging, LysoTracker Deep Red (Thermo Fisher) was diluted to 50nM in culture media and cells were incubated with the dye for 2 h at 37°C. Cells were then washed with PBS and switched to phenol red-free DMEM media for imaging.
Fixed cell immunofluorescence
Cells were seeded on glass coverslips and fixed when they reached ∼50% confluency with 4% paraformaldehyde for 10 min at room temperature and washed with PBS three times. Cells were permeabilized and blocked with 10% Normal Donkey Serum (Jackson Immunoresearch), 0.1% saponin in PBS then incubated with primary antibody for 1-2 h at room temperature in 1% NDS, 0.05% Tween-20, PBS. Cells were then washed 3 times with 1% NDS, 0.05% Tween-20 in PBS and incubated with Alexa Fluor- conjugated secondary antibody at room temperature for 1 h. After washing, coverslips were mounted with ProLong Diamond mountant with DAPI (ThermoFisher).
Immunoblots and co-immunoprecipitation
Cells were collected in lysis buffer (50mM Tris-HCl, 150mM NaCl, 5mM EDTA, 1% NP-40, 20mM MG132, 1mM PMSF, 10mM Iodoacetamide, 1mM 1,10-Phenanthroline monohydrate, Roche Complete protease Inhibitor tablets, Roche Phos-Stop phosphatase inhibitor tablets) and centrifuged to isolate protein. Protein concentration was measured using Bradford Assay and a 1 mg/ml protein solution was made. For Ub co-immunoprecipitation experiments, cells were transiently transfected with HA-Ub plasmid 24 hours before lysing. 1ml of 1 mg/ml solution was added to α-FLAG magnetic beads (Sigma). For whole cell lysates, Laemmli buffer was added and samples were put at 65°C for 10 min. Co-IPs were incubated at 4°C for 1 h with rotation, washed 3 times with wash buffer (25mM Tris pH 7.5, 2.5% glycerol, 150mM NaCl), and eluted by incubation with FLAG peptide for 30 min at 4°C twice. Protein samples were run on 12% polyacrylamide gels, transferred onto PVDF membrane, and imaged after antibody incubation on the LiCor Odyssey CLx Infrared imager.
Quantification and statistical analysis
Quantification
Pearson’s coefficient of correlation measurements
When determining the colocalization, at least 20 cells from 3 different images were measured for each condition using either Softworx software or the JACoP (BIOP version) plugin for FIJI. The mean Pearson’s coefficient and standard deviation are represented in the quantifications shown.
Immunoblot quantification of GLUT1 biotinylation
Details of the quantification and normalization can be found in the statistical reporting document for Figure S1D. For each time point, FIJI was used to quantifiy the whole lane for both the input and IP samples. Next, the IP measurement was divided by the input measurement. To normalize, the [IP/input] values for each time point were divided by the T = 0 [IP/input] value. This was done for at least 3 biological replicates and presented in Figures 1B and S1D with means and standard deviations.
Quantification of FLAG co-immunoprecipitations (GLUT1-FLAG and FLAG-Ub)
Details of the quantification and normalization can be found in the statistical reporting document for Figures 5E–5I. The entirety of each input and eluate/IP lane was measured for all α-FLAG, α-Ubiquitun, and α-GLUT1 immunoblots using FIJI. The measurement for the protein of interest (POI) (GLUT1 or HA-Ub) was then divided by the corresponding FLAG measurement. To normalize, each “Eluate POI/FLAG” was divided by the “POI/FLAG” of the control condition (e.g.in Figure 5E, all eluate conditions are divided by the no glucose “GLUT1/FLAG-Ub” measurement). This was done for at least 3 biological replicates and presented in Figures 5E, 5G, 5I with means and standard deviations.
Statistical analysis
Detailed statistical analysis can be found in the accompanying statistical reporting document (Data S1). For each statistical analysis, a Student’s t test was used to test for a statistically significant difference between the means of the two variables of interest. The alpha value for each experiment was set at 0.05 and a p value was calculated using the Student’s t test function in Microsoft Excel to determine statistical significance. All quantified western blot and colocalization data are the mean of the indicated number of independent experiments. Each coIP and biotinylation figure with statistical analysis represents n ≥ 3 where n represents biological replicates. For quantification of all microscopy-based experiments, n ≥ 20 where n represents single cells. For all figures with statistics, error bars represent standard deviation from the mean. Details of statistical analysis for specific experiments can be found in the figure legends. ∗∗ indicated statistical significance.
Acknowledgments
We are grateful to T. Graham and N. Hepowit for critical reading of the manuscript. We are also grateful to K. Rose, W. Tansey, S. Lorey, O. McDonald, Q. Zhang, R. Coffey, J. Franklin, J. Goldenring, L. Lapierre, and Y. Mao for technical advice and insightful discussions. This study was funded by NIH grants R35GM144112 (to JAM) and a Vanderbilt-Ingram Cancer Center GI-SPORE pilot award (P50CA236733).
Author contributions
S.J.Q.-H. and J.A.M. conceived of the study, designed the experiments, and wrote the manuscript. S.J.Q.-H. and C.P.N. performed experiments and collected and analyzed data presented in this study. All authors discussed experimental results and contributed to manuscript revisions and editing.
Declaration of interests
The authors declare no competing interests.
Published: February 6, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106150.
Supplemental information
Data and code availability
-
•
Microscopy data reported in this paper can be found in the statistical reporting document. Original western blot and microscopy images reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gyimesi G., Pujol-Giménez J., Kanai Y., Hediger M.A. Sodium-coupled glucose transport, the SLC5 family, and therapeutically relevant inhibitors: from molecular discovery to clinical application. Pflugers Arch. 2020;472:1177–1206. doi: 10.1007/s00424-020-02433-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holman G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflugers Arch. 2020;472:1155–1175. doi: 10.1007/s00424-020-02411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueckler M., Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013;34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pao S.S., Paulsen I.T., Saier M.H. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander C.M., Martin J.A., Oxman E., Kasza I., Senn K.A., Dvinge H. Alternative splicing and cleavage of GLUT8. Mol. Cell Biol. 2020;41 doi: 10.1128/MCB.00480-20. e00480-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng D., Sun P., Yan C., Ke M., Jiang X., Xiong L., Ren W., Hirata K., Yamamoto M., Fan S., Yan N. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526:391–396. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 7.Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 8.Brumfield A., Chaudhary N., Molle D., Wen J., Graumann J., McGraw T.E. Insulin-promoted mobilization of GLUT4 from a perinuclear storage site requires RAB10. Mol. Biol. Cell. 2021;32:57–73. doi: 10.1091/mbc.E20-06-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klip A., McGraw T.E., James D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019;294:11369–11381. doi: 10.1074/jbc.REV119.008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Vos A., Heimberg H., Quartier E., Huypens P., Bouwens L., Pipeleers D., Schuit F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 1995;96:2489–2495. doi: 10.1172/JCI118308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCulloch L.J., van de Bunt M., Braun M., Frayn K.N., Clark A., Gloyn A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011;104:648–653. doi: 10.1016/j.ymgme.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 12.Coppieters K.T., Wiberg A., Amirian N., Kay T.W., von Herrath M.G. Persistent glucose transporter expression on pancreatic beta cells from longstanding type 1 diabetic individuals. Diabetes. Metab. Res. Rev. 2011;27:746–754. doi: 10.1002/dmrr.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maher F., Vannucci S.J., Simpson I.A. Glucose transporter proteins in brain. FASEB J. 1994;8:1003–1011. doi: 10.1096/fasebj.8.13.7926364. [DOI] [PubMed] [Google Scholar]
- 14.Lee E.E., Ma J., Sacharidou A., Mi W., Salato V.K., Nguyen N., Jiang Y., Pascual J.M., North P.E., Shaul P.W., et al. A Protein Kinase C Phosphorylation Motif in GLUT1 Affects Glucose Transport and is Mutated in GLUT1 Deficiency Syndrome. Mol. Cell. 2015;58:845–853. doi: 10.1016/j.molcel.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siska P.J., Rathmell J.C. PKCs Sweeten Cell Metabolism by Phosphorylation of Glut1. Mol. Cell. 2015;58:711–712. doi: 10.1016/j.molcel.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu N., Zheng B., Shaywitz A., Dagon Y., Tower C., Bellinger G., Shen C.H., Wen J., Asara J., McGraw T.E., et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg F., Gallon M., Winfield M., Thomas E.C., Bell A.J., Heesom K.J., Tavaré J.M., Cullen P.J. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat. Cell Biol. 2013;15:461–471. doi: 10.1038/ncb2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans A.J., Daly J.L., Anuar A.N.K., Simonetti B., Cullen P.J. Acute inactivation of retromer and ESCPE-1 leads to time-resolved defects in endosomal cargo sorting. J. Cell Sci. 2020;133:jcs246033. doi: 10.1242/jcs.246033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kvainickas A., Orgaz A.J., Nägele H., Diedrich B., Heesom K.J., Dengjel J., Cullen P.J., Steinberg F. Retromer- and WASH-dependent sorting of nutrient transporters requires a multivalent interaction network with ANKRD50. J. Cell Sci. 2017;130:382–395. doi: 10.1242/jcs.196758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy S., Leidal A.M., Ye J., Ronen S.M., Debnath J. Autophagy-Dependent Shuttling of TBC1D5 Controls Plasma Membrane Translocation of GLUT1 and Glucose Uptake. Mol. Cell. 2017;67:84–95.e5. doi: 10.1016/j.molcel.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinde S.R., Maddika S. PTEN Regulates Glucose Transporter Recycling by Impairing SNX27 Retromer Assembly. Cell Rep. 2017;21:1655–1666. doi: 10.1016/j.celrep.2017.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson D.S., Gregor C., Winter F.R., Urban N.T., Sahl S.J., Willig K.I., Hell S.W. SRpHi ratiometric pH biosensors for super-resolution microscopy. Nat. Commun. 2017;8:577. doi: 10.1038/s41467-017-00606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieman H.L., Horn S.R., Jacobs S.R., Altman B.J., Kornbluth S., Rathmell J.C. An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem. J. 2009;418:345–367. doi: 10.1042/BJ20081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Votteler J., Ogohara C., Yi S., Hsia Y., Nattermann U., Belnap D.M., King N.P., Sundquist W.I. Designed proteins induce the formation of nanocage-containing extracellular vesicles. Nature. 2016;540:292–295. doi: 10.1038/nature20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P., Wang C., Gao K., Wang D., Mao J., An J., Xu C., Wu D., Yu H., Liu J.O., Yu L. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J. Biol. Chem. 2010;285:8869–8879. doi: 10.1074/jbc.M109.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y., Lau J., Li W., Tempel W., Li L., Dong A., Narula A., Qin S., Min J. Structural basis for the regulatory role of the PPxY motifs in the thioredoxin-interacting protein TXNIP. Biochem. J. 2016;473:179–187. doi: 10.1042/BJ20150830. [DOI] [PubMed] [Google Scholar]
- 27.Rauch S., Martin-Serrano J. Multiple interactions between the ESCRT machinery and arrestin-related proteins: implications for PPXY-dependent budding. J. Virol. 2011;85:3546–3556. doi: 10.1128/JVI.02045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen C.P., Jernigan K.K., Diggins N.L., Webb D.J., MacGurn J.A. USP9X Deubiquitylates DVL2 to Regulate WNT Pathway Specification. Cell Rep. 2019;28:1074–1089.e5. doi: 10.1016/j.celrep.2019.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dykstra H., LaRose C., Fisk C., Waldhart A., Meng X., Zhao G., Wu N. TXNIP interaction with GLUT1 depends on PI(4, 5)P. Biochim. Biophys. Acta. Biomembr. 2021;1863:183757. doi: 10.1016/j.bbamem.2021.183757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueckler M., Caruso C., Baldwin S.A., Panico M., Blench I., Morris H.R., Allard W.J., Lienhard G.E., Lodish H.F. Sequence and structure of a human glucose transporter. Science. 1985;229:941–945. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- 31.Wollscheid B., Bausch-Fluck D., Henderson C., O'Brien R., Bibel M., Schiess R., Aebersold R., Watts J.D. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devi T.S., Yumnamcha T., Yao F., Somayajulu M., Kowluru R.A., Singh L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open. 2019;8:bio038521. doi: 10.1242/bio.038521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacGurn J.A., Hsu P.C., Emr S.D. Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 34.Lauwers E., Erpapazoglou Z., Haguenauer-Tsapis R., André B. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 2010;20:196–204. doi: 10.1016/j.tcb.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Waldhart A.N., Dykstra H., Peck A.S., Boguslawski E.A., Madaj Z.B., Wen J., Veldkamp K., Hollowell M., Zheng B., Cantley L.C., et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017;19:2005–2013. doi: 10.1016/j.celrep.2017.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikko E., Pelham H.R.B. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic. 2009;10:1856–1867. doi: 10.1111/j.1600-0854.2009.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagaki K., Yamamura H., Shimada S., Saito T., Hisanaga S.i., Taoka M., Isobe T., Ichimura T. 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry. 2006;45:6733–6740. doi: 10.1021/bi052640q. [DOI] [PubMed] [Google Scholar]
- 38.Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., Isobe T. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 2005;280:13187–13194. doi: 10.1074/jbc.M412884200. [DOI] [PubMed] [Google Scholar]
- 39.Raja M., Kinne R.K.H. Mechanistic Insights into Protein Stability and Self-aggregation in GLUT1 Genetic Variants Causing GLUT1-Deficiency Syndrome. J. Membr. Biol. 2020;253:87–99. doi: 10.1007/s00232-020-00108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D., Kranz-Eble P., De Vivo D.C. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum. Mutat. 2000;16:224–231. doi: 10.1002/1098-1004(200009)16:3<224::AID-HUMU5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 41.Koch H., Weber Y.G. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019;91:90–93. doi: 10.1016/j.yebeh.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Singh B., Bogatcheva G., Starchenko A., Sinnaeve J., Lapierre L.A., Williams J.A., Goldenring J.R., Coffey R.J. Induction of lateral lumens through disruption of a monoleucine-based basolateral-sorting motif in betacellulin. J. Cell Sci. 2015;128:3444–3455. doi: 10.1242/jcs.170852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Microscopy data reported in this paper can be found in the statistical reporting document. Original western blot and microscopy images reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.