

Abstract
Tissue availability remains an important limitation of single-cell genomic technologies for investigating cellular heterogeneity in human health and disease. BAL represents a minimally invasive approach to assessing an individual’s lung cellular environment for diagnosis and research. However, the lack of high-quality, healthy lung reference data is a major obstacle to using single-cell approaches to study a plethora of lung diseases. Here, we performed single-cell RNA sequencing on over 40,000 cells isolated from the BAL of four healthy volunteers. Of the six cell types or lineages we identified, macrophages were consistently the most numerous across individuals. Our analysis confirmed the expression of marker genes defining cell types despite background signals because of the ambient RNA found in many single-cell studies. We assessed the variability of gene expression across macrophages and defined a distinct subpopulation of cells expressing a set of genes associated with Macrophage Inflammatory Protein 1 (MIP-1). RNA in situ hybridization and reanalysis of published lung single-cell data validated the presence of this macrophage subpopulation. Thus, our study characterizes lung macrophage heterogeneity in healthy individuals and provides a valuable resource for future studies to understand the lung environment in health and disease.
Keywords: BAL, genomics, lung immunology, macrophage, heterogeneity
The lung immune milieu is comprised of cells from both the innate and adaptive immune systems. These cells play a critical role in the development and progression of many pulmonary diseases, but many are also found as tissue-resident cells in healthy lungs. Myeloid immune cells in the healthy lung primarily consist of dendritic cells, interstitial and alveolar macrophages, monocytes, and, to a lesser degree, granulocytes (neutrophils, basophils, eosinophils, and mast cells) (1, 2). Lymphoid immune cells, such as B, T, and natural killer (NK) cells, may reside in the healthy lung but are more likely to infiltrate in response to an immune threat (1, 3).
Alveolar macrophages are the main resident innate immune cells of the lung (4). Results of multiple studies in murine models under steady-state conditions have shown that alveolar macrophages can exist as a self-renewing population with minimal input from peripheral immune cells well into adulthood (5–8). However, a variety of perturbations, such as infection by respiratory viruses (e.g., severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]) or injury with bleomycin, lipopolysaccharide, inhaled particulate matter, or asbestos can deplete the tissue-resident cell population (9–13). The vacant niche enables bone marrow-derived circulating monocytes to enter the lung and move into the alveolar space, where they differentiate into macrophages (14). Monocyte-derived alveolar macrophages have been shown to contribute to the pathobiology of lung diseases in mouse models, including fibrosis and injury from viral or bacterial pneumonia (15–17). Recent single-cell RNA sequencing (scRNA-seq) data generated from human lung specimens demonstrate even greater alveolar macrophage heterogeneity under disease conditions with more variability than captured by the tissue-resident versus monocyte-derived distinction (18–21). However, less is known about the composition of the alveolar macrophage population in healthy human lungs.
BAL is a minimally invasive procedure that allows direct sampling of immune cells within the alveolar space in healthy volunteers and individuals with diseases affecting the lung. A BAL fluid cell count performed using a hemocytometer has diagnostic value for a variety of different diagnoses, including infections, inflammatory disorders such as hypersensitivity pneumonitis, and cancers (22–25). However, a BAL cell count cannot provide information about different cell subtypes or their activation states. scRNA-seq of cells obtained during BAL enables the assessment of individual leukocyte populations that occupy the human alveolar space.
In this study, we assessed the transcriptional heterogeneity of human lung cells obtained by BAL from healthy adults. We performed scRNA-seq on over 40,000 total cells obtained from four healthy volunteers and annotated distinct immune cell types, including alveolar macrophages. We also explored the impact on our analysis of ambient RNA that contaminated the transcriptional profiles of cells. Among macrophages, we defined a distinct subpopulation, which we termed MIP-1 (macrophage inflammatory protein 1) macrophage cluster, that demonstrated higher expression of a subset of genes associated with inflammation (e.g., CCL3, CCL4, and CXCL10). We then verified the presence of this subpopulation using RNA in situ hybridization (RNA-ISH) and previously published lung scRNA-seq data. Taken together, our study provides insight into the cellular milieu by sampling the alveolar spaces of healthy lungs.
Methods
Recruitment of Patients for BAL and scRNA-Seq
We recruited healthy volunteers as part of our Institutional Review Board-approved study to undergo BAL at Northwestern Memorial Hospital. BAL fluid was processed for scRNA-seq using the 10× Genomics Chromium with Single Cell 3′ v2 chemistry. Sequencing data will be made available at the time of publication.
scRNA-seq Analysis
Libraries were processed using the Cell Ranger V3.1.0 workflow and analyzed R 4.0.3 and the Seurat package V3.2.2 (26). Normalization was done using SCTransform, clustering was performed with FindClusters, and results were visualized as uniform manifold approximation and projection (27, 28). Cells annotated as macrophages were subclustered, and a gene module representing the MIP-1 macrophage cluster was created using Seurat’s AddModuleScore. GSEA software V4.1.0 is used for functional enrichment analysis (29).
Validation of MIP-1 Macrophages
A Seurat object was created using macrophage cells from the Reyfman and colleagues dataset of eight healthy donor lungs, and AddModuleScore was used to annotate the MIP-1 subset (18). Multiplex fluorescent in situ hybridization was performed using RNAScope (Advanced Cell Diagnostics), and four images of different fields from one BAL sample were captured with a Nikon A1R confocal microscope. Protein immunofluorescence was performed on the same BAL sample; images were captured using a Zeiss Axioplan 2 microscope (Carl Zeiss, Inc.).
Code Availability
All code used in the analysis and to generate figures is publicly available on GitHub: https://github.com/TheWinterLab/HealthyBAL_project.
Results
Macrophages Predominate scRNA-seq of BAL Fluid from Healthy Volunteers
Using 10× Genomics scRNA-seq technology, we assessed the transcriptional state of cells in BAL fluid from four healthy volunteers (see Methods). The volunteers were females who had never smoked tobacco, aged 24, 32, 41, and 45 years old. After filtering cells for low quality (Figure E1A in the data supplement), 41,679 cells were included in the analysis. Initial clustering produced 12 clusters with distinct expression profiles (Figure E1B and E1C and Table E1). Because seven clusters were highly similar and shared characteristics of macrophages (Figure E1D), these were combined into a single annotation (Figure 1A). Overall, we annotated six cell types plus dividing cells on the basis of the expression of lineage and cell cycle genes (Figure 1B). All cell types were represented in each individual in similar proportions, except B cells, which demonstrated increased variability (Figures 1C and E1E). Macrophages were by far the predominant cell type, accounting for 71–88% of cells. Canonical marker genes verified each cell type annotation, including alveolar macrophages (FABP4 [fatty acid binding protein 4]), T/NK cells (CCL5 [CC motif chemokine ligand 5] and LTB [lymphotoxin beta]), B cells (MS4A1 [membrane spanning 4-domains A1]), dendritic cells (CTSB [cathepsin B]), club cells (SCGB3A1 [Secretoglobin Family 3A Member 1]), ciliated cells (CAPS [Calcyphosine]), and dividing cells (TOP2A [DNA Topoisomerase II Alpha]) (Figure 1D). We noted that whereas myeloid markers, including MRC1 (mannose receptor C-type 1) and CD68 (cluster of differentiation 68), were highest in macrophages, they exhibited broad low-level expression across cell types (Figures E2A and E2B). We attribute this observation to ambient RNA, an effect we investigate further below.
Figure 1.
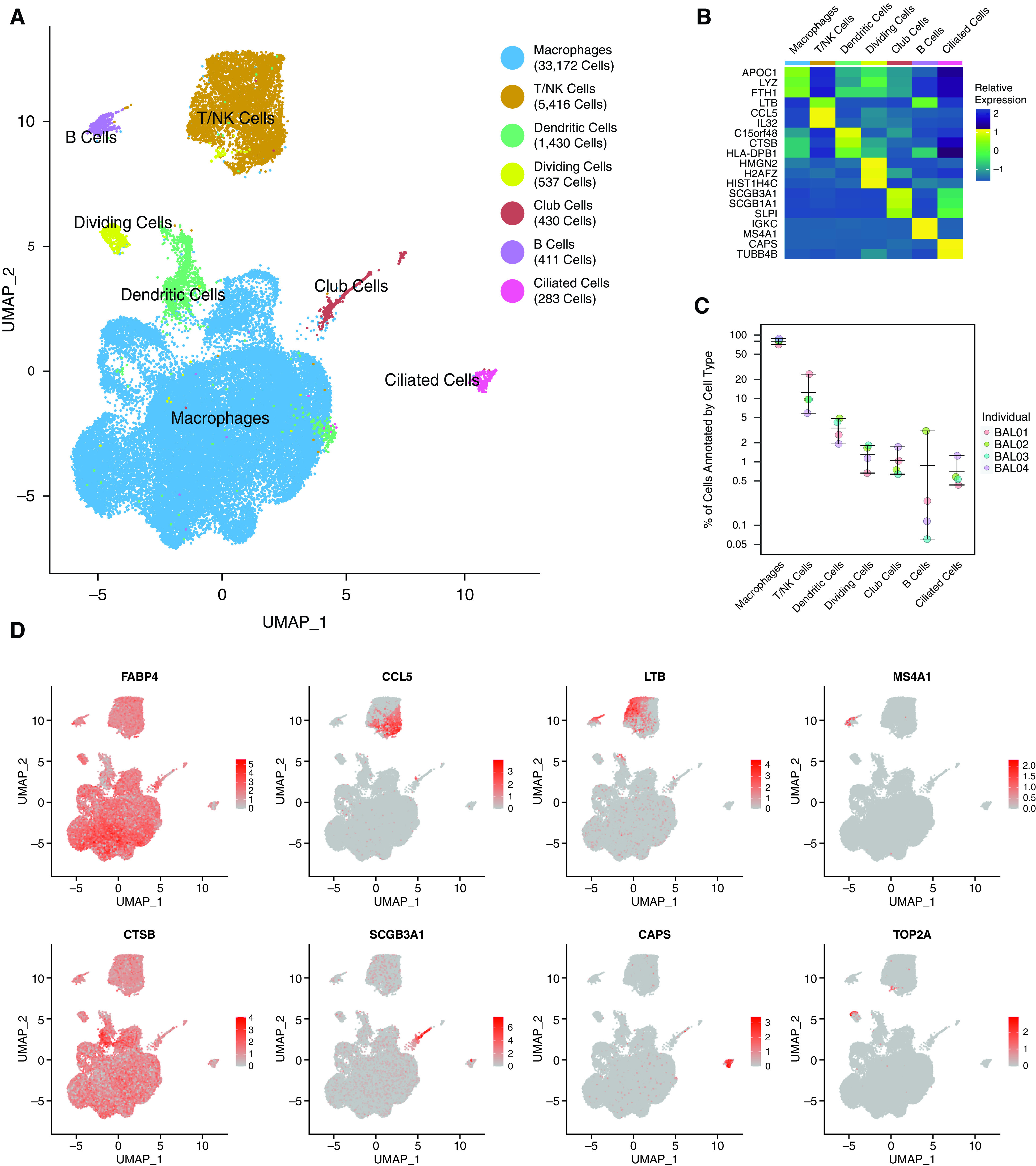
Single-cell RNA sequencing identifies diverse cell populations in BAL fluid from healthy control volunteers. (A) UMAP representation of the transcriptional profile of BAL cells manually annotated as one of six cell types or dividing cells. The total number of cells in each cell type is indicated in the legend. (B) Relative expression of the top two to three marker genes (by fold change) across cell types. (C) Percentage (in log scale) of cells in each cell type by individual sample. Whiskers represent the range of values. (D) Visualization of normalized expression for canonical cell type genes across cells. All figures represent data from BAL01-04. NK = natural killer; UMAP = uniform manifold approximation and projection.
Ubiquity of Macrophage Gene Expression Is Explained by Ambient RNA
Ambient RNA describes the pool of RNA molecules outside cells in the suspension prepared for single-cell protocols (30, 31). These RNA molecules are derived from dying or damaged cells that release their contents to create a background RNA signal. In a microfluidic technology, such as the 10× Genomics platform, these molecules are randomly included in droplets and become labeled with the barcodes of cells from which they did not originate. Thus, their presence may obfuscate the identity of individual cells and the boundary of cell-type clusters. To assess the impact of ambient RNA on our data, we retrieved the reads that were excluded from our main analysis because they originated from “cells” with too few read counts. The assumption is that these excluded low-count cells represent empty droplets filled with ambient RNA. By comparing the total counts of these excluded reads with those included in our main analysis for each cell type in each individual, we distinguished between genes associated with cell type identity and those that arise from ambient RNA. We observe that macrophage genes, such as FABP4, are among the genes most highly expressed in excluded reads; macrophage expression profiles also exhibit the highest correlation with excluded reads (Figure 2A). This may reflect that macrophages are the most dominant cell type in BAL fluid and would therefore be most likely to contribute to the ambient RNA signal. In contrast, the included counts from T/NK cells exhibit a lower correlation with excluded counts; T/NK marker genes, such as LTB and CCL5, are expressed higher than in ambient RNA (Figure 2B). Surprisingly, SCB3A1 exhibits high counts in ambient RNA despite the fact that club cells, bronchiolar exocrine cells that express high concentrations of this gene, account for less than 2% of total cells (Figure 2C). This finding may be indicative of the vulnerability of club cells to damage during the BAL procedure or sample processing.
Figure 2.
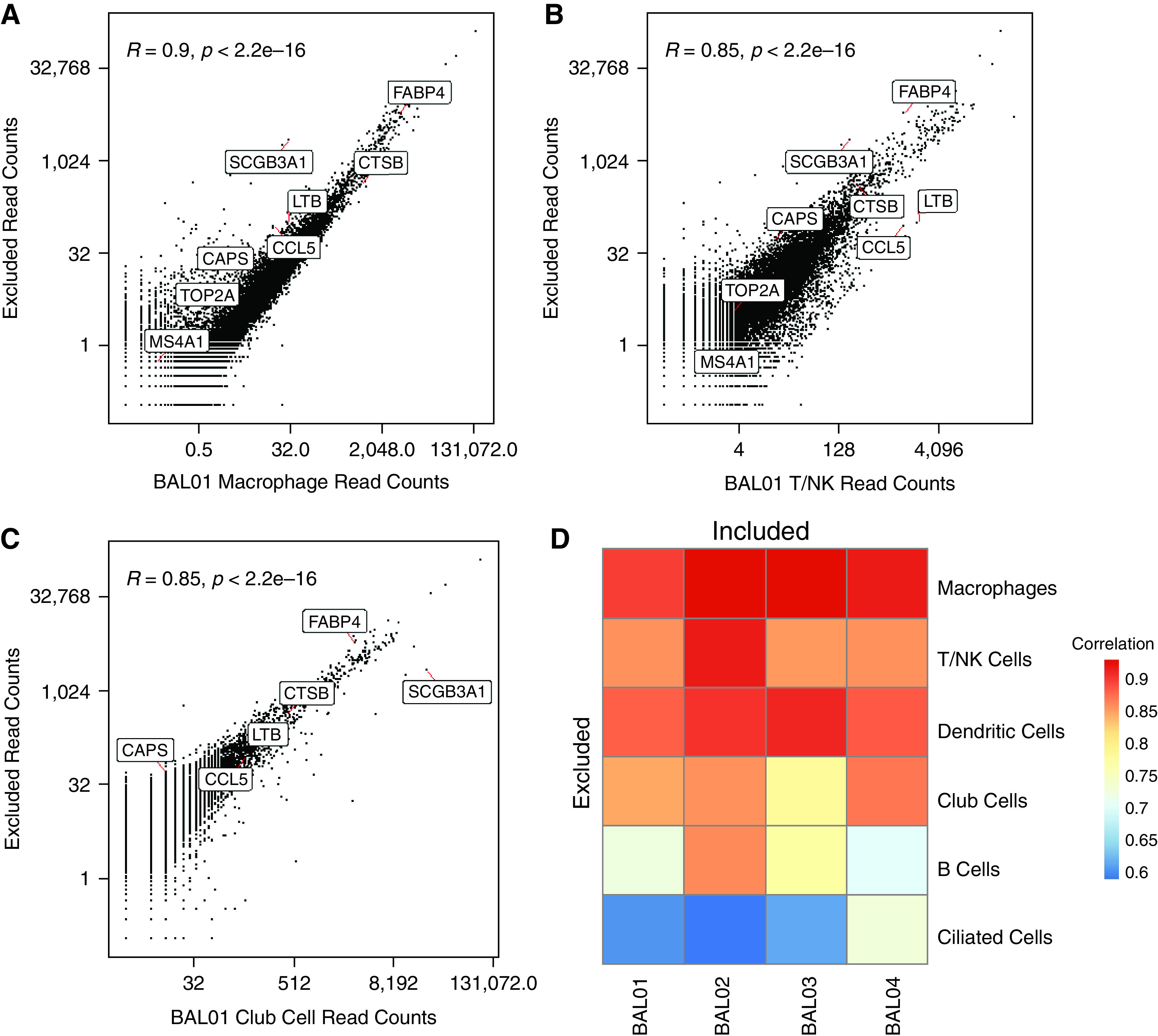
Comparison of empty droplets with cell droplets to assess ambient RNA. Scatterplot of the read counts for each gene in empty droplets that are excluded from the analysis versus cells that are annotated as (A) macrophages, (B) T/NK cells, and (C) club cells in BAL01. The Pearson correlations, R, and associated P values are indicated. A few example genes representing different cell types are annotated. (D) Pearson correlation between the read counts for genes from excluded droplets versus included and annotated as one of six cell types in individual BAL samples (BAL01–04).
Similarly, genes associated with other cell types (e.g., CAPS in ciliated cells, MS4A1 in B cells, and CTSB in dendritic cells) were found at relatively lower concentrations among the excluded reads. Similarly, with the exception of club cells mentioned above, the correlation of each cell type with ambient RNA is proportional to the number of cells annotated as that cell type in the given individual (Figure 2D). The composition of ambient RNA is specific to the sample: overall, the included reads from each individual were most highly correlated with the excluded reads from the same individual (Figure E2C). Coexpression of macrophage genes with other cell-type genes could also be indicative of doublets (32, 33), but that would affect only a subset of cells rather than leading to the widespread low expression we observe (Figure E2D). To confirm that the expression of macrophage genes in other cell types, for example, epithelial cells, is because of ambient RNA and not endogenous expression, we performed immunofluorescence on cells obtained from BAL fluid. By labeling macrophage-specific (MRC1), and shared (EPCAM1) markers, we verified that no epithelial cells simultaneously express macrophage genes (Figure E2E). Taken together, we found that ambient RNA does contribute to the transcriptional profiles in our single-cell data but does not prohibit our analysis from distinguishing specific cell types.
Macrophages Exhibit Transcriptional Heterogeneity within Individuals
Because macrophages are the dominant cell type in our BAL samples, we isolated them for further subclustering to identify potential subpopulations. To accomplish this, we first redefined the genes that were variable across the subset of macrophage cells. We found that the 50 most highly variable genes within macrophages were generally conserved across individuals and included genes related to inflammation (CXCL10, PPBP, CCL4, CCL3, and CCL20), metallothionein (MT1G, MT1X, MT1H, and MT1M), and other genes related to macrophage polarization (APOE, LYZ, and FN1) (Figures 3A and E3A–E3D). Hierarchical clustering of these genes revealed modules with similarity in expression across macrophages (Figure 3B). In particular, we defined a module comprising, among other genes, CCL3 and CCL4, which encode the peptides that form the major macrophage inflammatory protein MIP-1. Clustering of macrophages produced two clusters: a larger cluster of 31,542 cells and a smaller cluster of 1,630 cells (Figure 3C and Table E2). Because the markers of this smaller cluster included CCL3, CCL4, and other genes from the aforementioned module, we will henceforth refer to these cells as MIP-1 macrophages (Figure 3D). MIP-1 macrophages were found in each individual at an average frequency of 4.7% (Figure E3E). Gene set enrichment analysis on differentially expressed genes between the MIP-1 macrophage cluster and the other macrophages revealed significant enrichment for several gene sets related to inflammation, including TNFA signaling via NFKB (Figure 3E and Table 1). Although both clusters expressed canonical macrophage markers, such as FABP4, MRC1, and CD68, the marker genes of MIP-1 macrophages were largely specific to these cells (Figures 3F and E3F–E3I). We combined several of these genes into a module score to identify MIP-1 macrophages (Figure 3G). Our results suggest that MIP-1 macrophages represent a distinct subpopulation of macrophages in the healthy lung.
Figure 3.
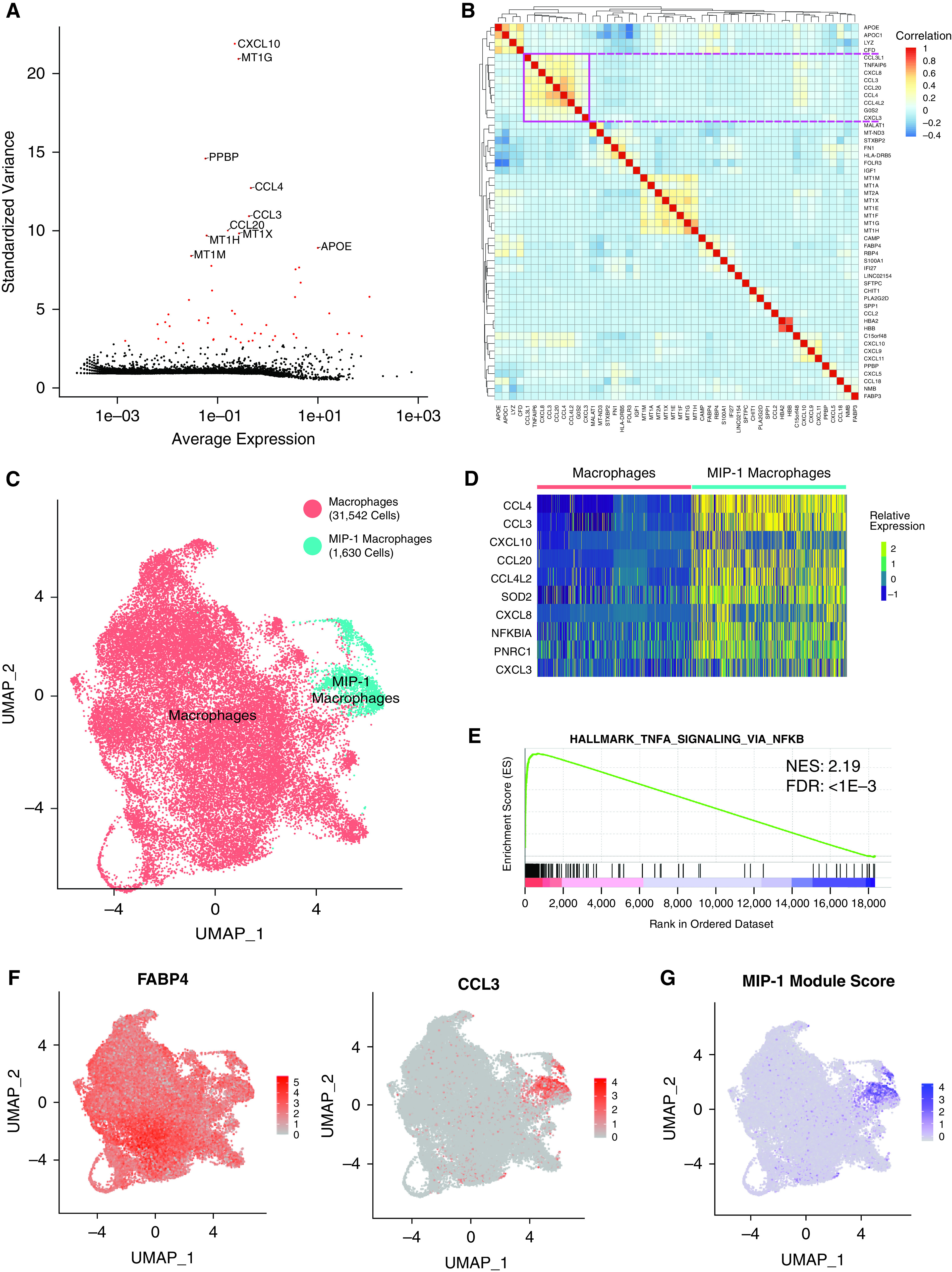
Analysis of macrophage heterogeneity reveals a MIP-1 (macrophage inflammatory protein 1) subpopulation. (A) Scatterplot showing the average normalized expression of each gene by its standardized variance across macrophage cells. The top 10 most variable genes are annotated, and the top 50 most variable genes are shown in red. (B) Pairwise Pearson correlation between the expression profiles of the top 50 most variable genes across macrophages. The pink square denotes a large clade of coregulated genes. (C) UMAP representation of the transcriptional profile of macrophage cells with a subset annotated as belonging to the MIP-1 cluster. The total number of cells in each cluster is indicated in the legend. (D) Relative expression of top 10 differentially expressed marker genes (by fold change) for the MIP-1 macrophage cluster. These largely overlap but do not exactly match those in B. (E) Representative-enriched pathway resulting from GSEA analysis of marker gene list ranked by fold-change in MIP-1 compared with other macrophages. The NES and adjusted P value for the FDR are shown. (F) Visualization of normalized expression across cells for macrophage marker, FABP4, and MIP-1 gene, CCL3. (G) Visualization of MIP-1 module score across macrophage cells. All figures represent data from BAL01–04. FDR = false discovery rate; NES = Normalized Enrichment Score.
Table 1.
GSEA Results for Macrophage Inflammatory Protein 1 Macrophages Compared with Other Macrophages
| Hallmark Name | Size | Normalized Enrichment Score | FDR |
|---|---|---|---|
| HALLMARK_TNFA_SIGNALING_VIA_NFKB | 196 | 2.20 | <0.001 |
| HALLMARK_INFLAMMATORY_RESPONSE | 187 | 2.11 | <0.001 |
| HALLMARK_IL6_JAK_STAT3_SIGNALING | 80 | 1.94 | <0.001 |
| HALLMARK_INTERFERON_GAMMA_RESPONSE | 197 | 1.90 | <0.001 |
| HALLMARK_ALLOGRAFT_REJECTION | 188 | 1.81 | 0.005 |
| HALLMARK_APOPTOSIS | 149 | 1.60 | 0.083 |
| HALLMARK_UV_RESPONSE_UP | 142 | 1.60 | 0.073 |
| HALLMARK_KRAS_SIGNALING_UP | 166 | 1.56 | 0.083 |
| HALLMARK_APICAL_SURFACE | 35 | 1.55 | 0.081 |
| HALLMARK_IL2_STAT5_SIGNALING | 190 | 1.51 | 0.100 |
| HALLMARK_INTERFERON_ALPHA_RESPONSE | 96 | 1.41 | 0.171 |
| HALLMARK_COMPLEMENT | 180 | 1.35 | 0.228 |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 157 | 1.25 | 0.345 |
| HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY | 49 | 1.21 | 0.371 |
| HALLMARK_G2M_CHECKPOINT | 193 | 1.19 | 0.368 |
| HALLMARK_ANDROGEN_RESPONSE | 96 | 1.19 | 0.351 |
| HALLMARK_HYPOXIA | 170 | 1.15 | 0.388 |
| HALLMARK_KRAS_SIGNALING_DN | 120 | 1.10 | 0.442 |
| HALLMARK_P53_PATHWAY | 192 | 1.06 | 0.480 |
| HALLMARK_ESTROGEN_RESPONSE_LATE | 176 | 1.05 | 0.476 |
| HALLMARK_TGF_BETA_SIGNALING | 51 | 1.05 | 0.458 |
| HALLMARK_HEDGEHOG_SIGNALING | 29 | 0.988 | 0.546 |
| HALLMARK_UV_RESPONSE_DN | 131 | 0.921 | 0.666 |
| HALLMARK_PEROXISOME | 94 | 0.888 | 0.712 |
| HALLMARK_BILE_ACID_METABOLISM | 93 | 0.844 | 0.786 |
| HALLMARK_PANCREAS_BETA_CELLS | 17 | 0.782 | 0.909 |
| HALLMARK_WNT_BETA_CATENIN_SIGNALING | 39 | 0.703 | 0.994 |
| HALLMARK_NOTCH_SIGNALING | 31 | 0.678 | 0.975 |
Definition of abbreviation: FDR = false discovery rate.
The MIP-1–Expressing Macrophage Subpopulation Is Present across Studies
To validate our annotation of MIP-1 macrophages, we performed RNA-ISH on cells from BAL of an additional independent, healthy female volunteer recruited from the Northwestern University research community (33 yr old, never-smoker). We found that approximately 20% of CD68+ macrophages were also CCL3+, representing the MIP-1 macrophage subset (Figures 4A, 4B, and E4A). This is consistent with the increased sensitivity of RNA-ISH compared with scRNA-seq. Very few cells expressing CCL3 did not also express CD68. Therefore, these results support our single-cell data suggesting that MIP-1 macrophages are indeed a subpopulation with a distinct transcriptional profile.
Figure 4.
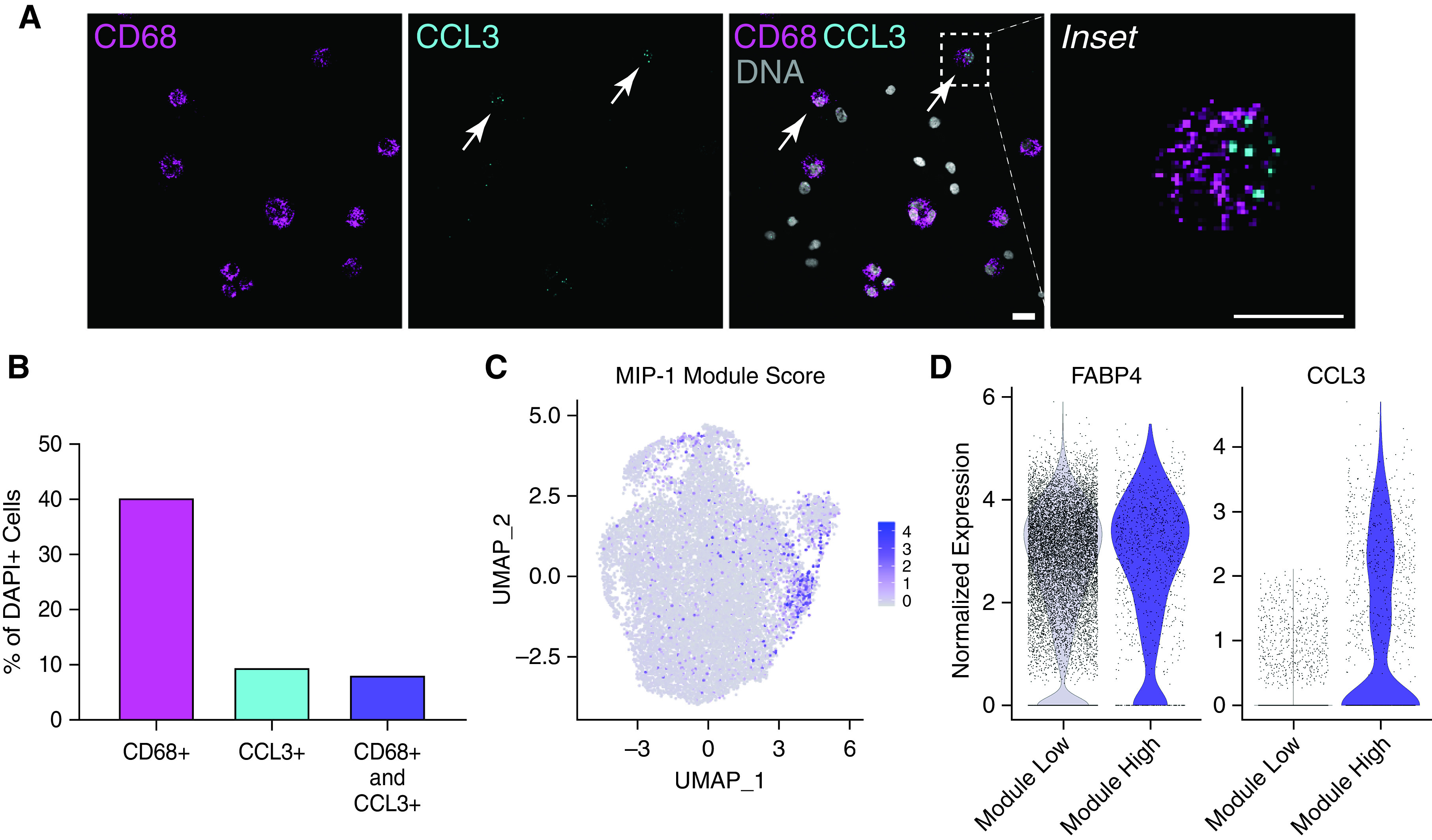
Validation of MIP-1 subpopulation in BAL fluid and another lung dataset. (A) Representative image of RNA in situ hybridization (RNA-ISH) on a BAL fluid sample from an additional individual (BAL05). Staining for macrophage marker (CD68), MIP-1 marker (CCL3), DNA (DAPI), and all channels merged (overlay). Scale bar, 40 μm. Arrows indicate CD68+ CCL3+ cells. (B) Quantification of RNA-ISH experiment above by percentage of cells from the total number of DAPI+ cells. Data represent 292 cells across four images. (C) Visualization of MIP-1 module score across cells annotated as macrophages in published data from donor transplant lung tissue (18). (D) Violin plot showing the distribution of normalized expression for FABP4 and CCL3 across macrophage cells annotated in C as either low (<0.3) or high (>0.3) module score. (C and D) Represent data from eight donor lungs.
Next, we used a previously published single-cell dataset by Reyfman and colleagues to verify that this subpopulation can be identified in whole lung tissue samples (18). Here, we restrict our analysis to the subset of macrophage cells from biopsies of eight donor lungs before transplant (Figure E4B). Using our module score from Figure 3, we annotated 1,160 cells (∼9%) with a “high module score” as MIP-1 macrophages (Figure 4C). A proportion of macrophages from each individual included in this analysis was classified as belonging to the MIP-1 macrophage subpopulation using this approach (Figure E4B). Cells with high module scores exhibited comparable expression of general macrophage genes but significantly increased expression of MIP-1 macrophage markers (Figures 4D, 4E, and E4C–E4F). In summary, we define a subpopulation of MIP-1 macrophages universally found in healthy BAL fluid and lung tissue.
Discussion
scRNA-seq of BAL fluid provides previously unavailable detail about the cellular environment in an individual’s lungs. We evaluated scRNA-seq of BAL fluid from healthy volunteers and determined that macrophages were the predominant cell type. These results provide a reference point for future scRNA-seq studies seeking to identify differences in patients across a variety of pulmonary diseases.
We enrolled volunteers who were nonsmokers, not obese, did not have current symptoms of a respiratory infection, and did not have a known history of lung disease, autoimmune disease, or other chronic condition. Obtaining cells from BAL fluid also has the advantage of requiring minimal sample processing, such as tissue digestion, which can alter the transcriptional state of cells. Although we cannot rule out undiagnosed or minimally symptomatic lung disease, our approach is more likely to yield samples that accurately reflect the steady-state human lung than other approaches, such as sampling histologically normal-appearing tissue from transplant donors (18–20), patients with lung tumors (34), or deceased individuals. However, sampling the lung using BAL is limited in that it fails to capture the vast majority of cell types composing the lung, including most epithelial, mesenchymal, vascular, and interstitial immune cells.
Our key finding is that we observed a transcriptionally distinct subpopulation of macrophages that express inflammatory genes, specifically the MIP-1–encoding genes CCL3 and CCL4. This subpopulation was present in all study participants. Moreover, the presence of MIP-1 macrophages was confirmed by RNA-ISH using a sample from an additional individual and by examining gene expression in an independent dataset that included donor lung tissue from eight individuals. Moreover, a recent publication on BAL fluid identified cells with a similar transcriptional profile among healthy lung macrophages but also defined a subpopulation associated with metallothionein genes (35). Although we observe a coregulated module of these genes, they are not assigned to a distinct cluster: this discrepancy may be because of experimental or analytical differences. The same study also reported no significant differences between males and females other than those associated with sex chromosomes. Because we only measured RNA concentrations, we cannot be certain that the genes associated with MIP-1–expressing macrophages are expressed at the protein level, and we did not assess whether this subpopulation exhibits unique function at steady-state. Given they continue to express canonical macrophage genes, MIP-1 macrophages may be primed for a response, and this could be evident at the epigenomic level (36–38). Future studies could focus on the specific isolation of these cells for further interrogation.
Existing work on macrophage heterogeneity in humans has been influenced by the dichotomy of monocyte-derived versus tissue-resident cells observed in murine disease models. However, studies in mice have suggested that, in the absence of inflammation, recruited monocyte-derived cells will eventually adapt to the local environment and become indistinguishable from tissue-resident cells (8, 9, 37, 39, 40). Because even healthy humans without diagnosed lung disease have been exposed to a lifetime of environmental and infectious challenges, the distinction between recruited and resident macrophages appears less clear: aside from the MIP-1 macrophage cluster, we do not identify a clear subpopulation of monocyte-derived cells. Moreover, single-cell studies of mouse lungs have also described multiple populations of monocytes and interstitial macrophages that coexist with alveolar macrophages (41–43). These populations may not be detected by BAL, but ongoing comparative studies (44) can identify human counterparts.
With any single-cell data set, there are inherent limitations in the conclusions that can be tested. For example, there are limits to the sensitivity of scRNA-seq, which relies on sampling from the total RNA library. Moreover, cell-type quantification is necessarily on the basis of proportion rather than absolute numbers. Because each BAL recovers a different number of cells and cell detection varies across 10× runs, it is not possible to quantify the total number of any cell type in an individual’s lungs using scRNA-seq alone. Although biases in cell recovery and sampling may alter cell composition, we find that results from our BAL samples, which were subjected to minimal processing, were consistent with those obtained from other methods, such as flow cytometry. Furthermore, gene expression in each cell is scaled to the number of unique RNA molecules detected to avoid sampling biases across cells. However, it is possible that an activated cell may express a greater number of total RNA molecules compared with a more quiescent cell. In fact, macrophages in our study tended to be associated with approximately twice as many RNA molecules as T cells, as indicated by the number of unique molecular identifiers. Other normalization techniques may better capture these differences but introduce other biases and tradeoffs. For this reason, care should be taken in interpreting comparisons of gene expression levels across cell types and samples. Similarly, ambient RNA, as introduced in our results, may impact analyses of differential gene expression when individual cells appear to coexpress genes from different cell types. Although background signal should be considered a factor in every scRNA-seq experiment, additional degrees of processing, such as isolating specific cells of interest before library construction, may serve to minimize ambient RNA. Finally, identifying variability between samples can be confounded by batch effect. It is not generally possible to determine whether differences in gene expression between individual samples result from biological differences or batch effects unless multiple samples are included across batches. Therefore, investigators should be especially vigilant in designing experiments and analyses to compare scRNA-seq samples.
Conclusions
Our results provide a valuable resource for future studies of lung biology and pathology. scRNA-seq of BAL fluid provides a powerful tool for better understanding the cellular environment of the lung and has recently been applied to study SARS-CoV-2 (12, 45). Future studies may reveal how BAL fluid changes over time as disease develops or in response to treatment. In addition, as continuing studies add more samples to the publicly available databases, we will gain a better appreciation of the variation present across individuals. In the long term, understanding the cell-type–specific changes associated with disease will aid in the development of targeted treatments.
Acknowledgments
Acknowledgment
The authors thank Hiam Abdala-Valencia and the Rheumatology/Pulmonary and Critical Care Next Generation Sequencing Facilities for assistance with sample processing, the NUSeq Core Facility for sequencing services, and the Center for Advanced Microscopy (NCI CCSG P30 CA060553, S10 RR031680, and S10OD016342) at Northwestern University. The authors also thank the staff of the NM bronchoscopy suite. Finally, the authors would like to acknowledge the participants for volunteering to provide us with access to the study samples.
Footnotes
Supported by the National Scleroderma Foundation (D.R.W.); the American Lung Association (D.R.W.); the Respiratory Health Association (P.A.R.); the American Thoracic Society (ATS Foundation/Boeringher Ingelheim Research Fello [P.A.R.]), (ATS/Mallinckrodt Research Grant in Sarcoidosis [D.R.W.]); the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR073270 [M.H.]); and the National Heart, Lung, and Blood Institute (HL146943 [P.A.R.] and R01HL134800 [C.J.G.]). This research was supported in part through the computational resources and staff contributions provided by the Genomics Compute Cluster, which is jointly supported by the Feinberg School of Medicine, the Center for Genetic Medicine, and Feinberg's Department of Biochemistry and Molecular Genetics, the Office of the Provost, the Office for Research, and Northwestern Information Technology. The Genomics Compute Cluster is part of Quest, Northwestern University’s high-performance computing facility, with the purpose of advancing research in genomics. D.R.W. is also funded by the ATS.
Author Contributions: P.A.R. and D.R.W. conceived and designed the study. E.S.M., M.A.C., K.A., and I.A.G. recruited volunteers and coordinated sample collection. A.P.L. and C.J.G. oversaw the IRB protocol and managed the study. E.S.M. and N.J. processed samples with the help of S.K. and A.V.M. A.S.F. performed RNA-ISH and immunofluorescence experiments under the supervision of C.J.G. G.G. transferred, processed, and stored the data. P.A.R. and B.K. performed the analyses and generated the figures. D.R.W. supervised the analyses. G.R.S.B., A.V.M., P.H.S.S., M.A., E.S.M., M.H., and C.J.G. assisted with experimental design and provided critical feedback throughout the study. P.A.R. and D.R.W. wrote the initial and final drafts. M.H. and C.J.G. edited the manuscript and provided critical review.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0123OC on September 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Franks TJ, Colby TV, Travis WD, Tuder RM, Reynolds HY, Brody AR, et al. Resident cellular components of the human lung: current knowledge and goals for research on cell phenotyping and function. Proc Am Thorac Soc . 2008;5:763–766. doi: 10.1513/pats.200803-025HR. [DOI] [PubMed] [Google Scholar]
- 2. Ardain A, Marakalala MJ, Leslie A. Tissue-resident innate immunity in the lung. Immunology . 2020;159:245–256. doi: 10.1111/imm.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agostini C, Chilosi M, Zambello R, Trentin L, Semenzato G. Pulmonary immune cells in health and disease: lymphocytes. Eur Respir J . 1993;6:1378–1401. [PubMed] [Google Scholar]
- 4. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol . 2014;14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 5. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med . 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity . 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scott CL, Henri S, Guilliams M. Mononuclear phagocytes of the intestine, the skin, and the lung. Immunol Rev . 2014;262:9–24. doi: 10.1111/imr.12220. [DOI] [PubMed] [Google Scholar]
- 8. McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest . 2021;131:140299. doi: 10.1172/JCI140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med . 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am J Respir Cell Mol Biol . 2017;57:294–306. doi: 10.1165/rcmb.2017-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J . 2020;55:1900646. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. NU SCRIPT Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature . 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol . 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol . 2017;17:451–460. doi: 10.1038/nri.2017.42. [DOI] [PubMed] [Google Scholar]
- 15. Coates BM, Staricha KL, Koch CM, Cheng Y, Shumaker DK, Budinger GRS, et al. Inflammatory monocytes drive influenza A virus-mediated lung injury in juvenile mice. J Immunol . 2018;200:2391–2404. doi: 10.4049/jimmunol.1701543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch CM, Anekalla KR, Hu Y-S, Davis JM, Ciesielski M, Gadhvi G, et al. 2020. https://www.biorxiv.org/content/10.1101/2020.06.08.141309v3
- 17. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol . 2020;21:145–157. doi: 10.1038/s41590-019-0568-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv . 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv . 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis . 2019;78:1379–1387. doi: 10.1136/annrheumdis-2018-214865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruhnke M, Böhme A, Buchheidt D, Cornely O, Donhuijsen K, Einsele H, et al. Infectious Diseases Working Party in Haematology and Oncology of the German Society for Haematology and Oncology Diagnosis of invasive fungal infections in hematology and oncology—guidelines from the Infectious Diseases Working Party in Haematology and Oncology of the German Society for Haematology and Oncology (AGIHO) Ann Oncol . 2012;23:823–833. doi: 10.1093/annonc/mdr407. [DOI] [PubMed] [Google Scholar]
- 23. Gharsalli H, Mlika M, Sahnoun I, Maalej S, Douik El Gharbi L, Mezni FE. The utility of bronchoalveolar lavage in the evaluation of interstitial lung diseases: a clinicopathological perspective. Semin Diagn Pathol . 2018;35:280–287. doi: 10.1053/j.semdp.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 24. Gibelin A, Parrot A, Fartoukh M, de Prost N. Rare respiratory diseases in the ICU: when to suspect them and specific approaches. Curr Opin Crit Care . 2019;25:29–36. doi: 10.1097/MCC.0000000000000572. [DOI] [PubMed] [Google Scholar]
- 25. Lachant DJ, Croft DP, McGrane Minton H, Hardy DJ, Prasad P, Kottmann RM. The clinical impact of pneumocystis and viral PCR testing on bronchoalveolar lavage in immunosuppressed patients. Respir Med . 2018;145:35–40. doi: 10.1016/j.rmed.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol . 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol . 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McInnes L, Healy J, Melville J.2018. https://arxiv.org/abs/1802.03426
- 29. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA . 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang S, Corbett SE, Koga Y, Wang Z, Johnson WE, Yajima M, et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol . 2020;21:57. doi: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience . 2020;9:giaa151. doi: 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DePasquale EAK, Schnell DJ, Van Camp PJ, Valiente-Alandí Í, Blaxall BC, Grimes HL, et al. DoubletDecon: deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep . 2019;29:1718–1727.e8. doi: 10.1016/j.celrep.2019.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, III, et al. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol . 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature . 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mould KJ, Moore CM, McManus SA, McCubbrey AL, McClendon JD, Griesmer CL, et al. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am J Respir Crit Care Med . 2021;203:946–956. doi: 10.1164/rccm.202005-1989OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell . 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell . 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell . 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 39. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun . 2016;7:10321. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity. 2019;51:655–670.e8. doi: 10.1016/j.immuni.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science . 2019;363:eaau0964. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 42. Sajti E, Link VM, Ouyang Z, Spann NJ, Westin E, Romanoski CE, et al. Transcriptomic and epigenetic mechanisms underlying myeloid diversity in the lung. Nat Immunol . 2020;21:221–231. doi: 10.1038/s41590-019-0582-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun . 2019;10:3964. doi: 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leach SM, Gibbings SL, Tewari AD, Atif SM, Vestal B, Danhorn T, et al. Human and mouse transcriptome profiling identifies cross-species homology in pulmonary and lymph node mononuclear phagocytes. Cell Rep . 2020;33:108337. doi: 10.1016/j.celrep.2020.108337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med . 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]