

Abstract
N 6‐Methyladenosine (m6A) is an important RNA modification catalyzed by methyltransferase‐like 3 (METTL3) and METTL14. m6A homeostasis mediated by the methyltransferase (MTase) complex plays key roles in various biological processes. However, the mechanism underlying METTL14 protein stability and its role in m6A homeostasis remain elusive. Here, we show that METTL14 stability is regulated by the competitive interaction of METTL3 with the E3 ligase STUB1. STUB1 directly interacts with METTL14 to mediate its ubiquitination at lysine residues K148, K156, and K162 for subsequent degradation, resulting in a significant decrease in total m6A levels. The amino acid regions 450–454 and 464–480 of METTL3 are essential to promote METTL14 stabilization. Changes in STUB1 expression affect METTL14 protein levels, m6A modification and tumorigenesis. Collectively, our findings uncover an ubiquitination mechanism controlling METTL14 protein levels to fine‐tune m6A homeostasis. Finally, we present evidence that modulating STUB1 expression to degrade METTL14 could represent a promising therapeutic strategy against cancer.
Keywords: m6A homeostasis, METTL14, METTL3, STUB1, ubiquitination‐mediated degradation
Subject Categories: Cancer, Post-translational Modifications & Proteolysis, RNA Biology
The competitive interaction of METTL3 with STUB1 controls METTL14 levels, thereby fine‐tuning m6A homeostasis. Enforced STUB1 expression delays the progression of cholangiocarcinoma by reducing METTL14 and m6A levels in the tumor.
Introduction
N 6‐Methyladenosine (m6A) is one of the most prevalent RNA modifications in eukaryotic cells (Fu et al, 2014; Wang et al, 2016b; Huang et al, 2021; Ren et al, 2021). m6A is deposited reversibly and at specific sites on RNA transcripts by “writers” (Wang et al, 2016b). The core components of the m6A complex are the methyltransferases METTL3 and METTL14 (Liu et al, 2014; Wang et al, 2016a, 2016b) and Wilms’ tumor 1‐associating protein (WTAP; Ping et al, 2014), forming the methyltransferase (MTase) complex. A portion of m6A modifications can be identified by “reader” proteins that generally exact the functions of this modification, which can be removed by “erasers”, including the demethylases Fat mass and obesity‐associated protein (FTO; Jia et al, 2011) and AlkB homolog 5 (ALKBH5; Zheng et al, 2013). Due to its ability to bind directly to S‐adenosylmethionine (SAM), METTL3 was identified as a major methyltransferase responsible for installing m6A modifications. METTL3 interacts with METTL14 to form a heterodimeric complex with a methyltransferase domain; METTL14 plays a structural role in this complex critical for substrate recognition (Wang et al, 2016a, 2016b; Huang et al, 2019). Increasing evidence suggests that m6A homeostasis is important for normal cellular processes. Its dysregulation triggered by the MTase complex, including impaired expression of m6A writers, readers, and erasers, has been linked to many human diseases, including neurological disorders (Livneh et al, 2020), leukemia (Vu et al, 2019), and diverse types of solid cancers (Deng et al, 2018; Gu et al, 2020; Huang et al, 2020).
In addition to the self‐regulatory properties of m6A writers, several studies have also shown that post‐translational modifications (PTMs) are critical for the dynamic regulation of RNA methyltransferase activity. Several PTMs of METTL3 have been identified, including phosphorylation (Scholler et al, 2018; Sun et al, 2020a), SUMOylation (Du et al, 2018; Xu et al, 2020), and ubiquitination (Du et al, 2018). For example, extracellular signal‐regulated kinases (ERKs) mediate the phosphorylation of METTL3, promoting the nuclear localization of the complex and decreasing its ubiquitination via recruitment of the deubiquitinase ubiquitin‐specific protease 5 (USP5). Interestingly, ERK‐mediated interactions between METTL3 and other complex members appear to affect only WTAP, as METTL14 was not obviously affected by treatment with an ERK inhibitor (Sun et al, 2020a). Moreover, endoplasmic reticulum (ER) stress induces METTL14 expression by suppressing HMG‐CoA reductase degradation 1 (HRD1)‐mediated ubiquitination to promote mRNA decay induced by C/EBP‐homologous protein (CHOP) and to inhibit CHOP‐induced apoptosis under stress conditions (Wei et al, 2021). These findings indicate that PTMs indeed play important roles in regulating m6A modification.
Protein ubiquitination, one of the most common PTMs, mainly controls protein degradation and homeostasis (Pohl & Dikic, 2019; Pla‐Prats & Thomä, 2022). Ubiquitination is an evolutionarily conserved modification that tags proteins for degradation and is mediated by a cascade of enzymatic reactions sequentially implemented by ubiquitin‐activating (E1), −conjugating (E2), and ‐ligating (E3) enzymes. The E3 ubiquitin (Ub) ligase transfers the Ub molecule from the E2 enzyme to substrate proteins, triggering the subsequent degradation of the ubiquitinated protein by the 26S proteasome (Morreale & Walden, 2016; Collins & Goldberg, 2017; Zheng & Shabek, 2017). The abundance of core components of the m6A complex, METTL3 (Du et al, 2018), METTL14 (Wei et al, 2021), and WTAP (Bansal et al, 2014), can be modulated by ubiquitination, suggesting that ubiquitination plays an important role in controlling the protein levels of m6A writers and modifications; however, the molecular mechanism underlying this type of posttranslational regulation of m6A writers remains elusive. In addition, although several hundred genes encoding E3 ligases have been identified in the human genome based on characteristic structural motifs, specific E3 ligases involved in regulating the ubiquitination‐mediated degradation of m6A writers still need to be identified.
METTL3 and METTL14 can reciprocally stabilize each other at the protein level; however, METTL14 is unstable when expressed by itself, and co‐expression with METTL3 greatly stabilizes METLL14 (Wang et al, 2014; Yang et al, 2017). These observations have led to the speculation that METTL14 might be protected by METTL3 to evade degradation and maintain m6A homeostasis. Therefore, in this study, we aimed to determine the specific degradation pathway that METTL14 is involved in, along with its underlying molecular mechanisms and physiological roles. We discovered that the ubiquitin–proteasome system controls METTL14 stability and that METTL14 is specifically modified by ubiquitin at the major lysine residues K148, K156, and K162. Using liquid chromatography tandem mass spectrometry (LC–MS/MS), we determined that STIP1 homology and U‐box‐containing protein 1 (STUB1) serves as an E3 Ub ligase that specifically targets and ubiquitinates METTL14, leading to its proteasomal degradation. STUB1 deficiency elevates METTL14 protein levels, inducing an increase in m6A modifications and resulting in cancer cell growth and tumorigenesis in vivo and in vitro. Interestingly, METTL3 directly binds to the ubiquitin domain of METTL14 and protects its ubiquitinated sites from STUB1‐induced ubiquitination‐mediated degradation. Therefore, we elucidated the molecular mechanism underlying the ubiquitin–proteasome system regulating METTL14 and the role of METTL3 in the stability of METTL14 and uncovered a direct link between ubiquitination and RNA m6A methylation. Our results suggest that targeting METTL14 by increasing STUB1 levels could help to suppress tumorigenesis.
Results
METTL14 requires METTL3 for its stabilization
METTL14 by itself is unstable, whereas co‐expression of METTL3 stabilizes METLL14 protein abundance (Wang et al, 2014; Yang et al, 2017), raising the possibility that METTL14 might require METTL3 to maintain its stability. To explore this notion, we investigated METTL14 abundance with or without METTL3. METTL3 knockdown cells had significantly lower METTL14 protein levels than control cells, despite showing only a slight change in METTL14 mRNA levels (Figs 1A and B, and EV1A). Moreover, when we attempted to overexpress METTL14 alone, we obtained only very low levels for the encoded protein, whereas METTL3 could be stably overexpressed by itself, as evidenced by the accumulation of METTL3 (Figs 1C and EV1B). WATP, a core member of the MTase complex, did not affect the protein levels of METTL14, suggesting its dispensable role in METTL14 protein stability (Fig EV1C and D).
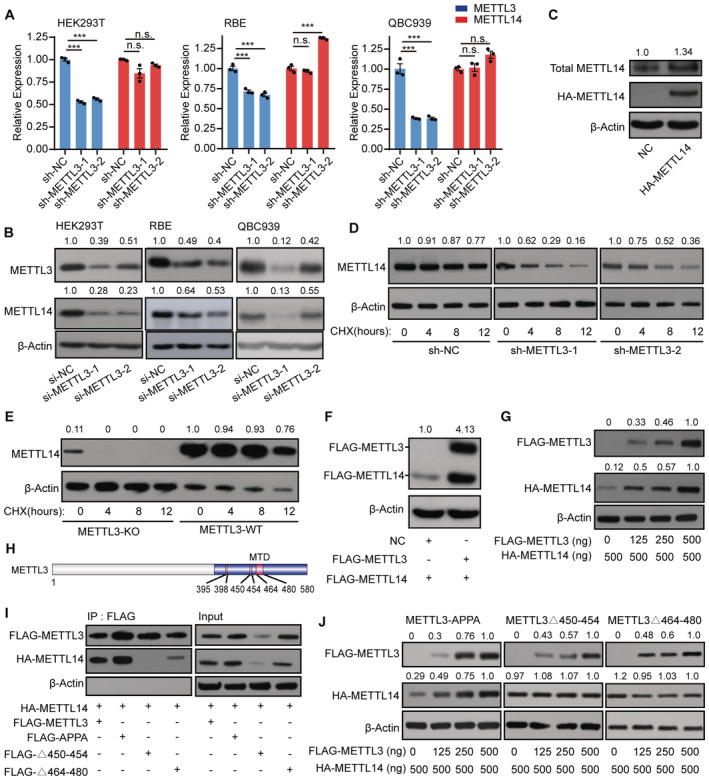
Figure 1. METTL3 protects METTL14 from degradation.
-
A, BRT‐qPCR analysis of METTL3 and METTL14 expression (A) and immunoblot analysis of METTL3 and METTL14 protein levels (B) in shRNA knockdown METTL3 cells. Data are mean ± SEM from three biological replicates and were analyzed by two‐tailed unpaired t‐test. ***P < 0.001; n.s., not significant. β‐actin was used as the loading control in the immunoblots. METTL3 or METTL14 /β‐actin densitometric ratio was recorded by ImageJ.
-
CImmunoblots showing total METTL14 protein levels in HA‐tagged METTL14‐overexpressing cells. The total METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
DImmunoblots showing the effects of cycloheximide (CHX) treatment on SK‐Cha‐1 cells. Cells with shRNA‐mediated knockdown of METTL3 (sh‐METTL3) and negative control (sh‐NC) SK‐Cha‐1 cells were exposed to 100 μg/ml CHX. The METTL14 levels dramatically decreased in sh‐METTL3 cells exposed to CHX compared with the negative control. β‐actin was used as the loading control. The METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
EImmunoblots showing the protein levels in the CHX assay in the METTL3 knockout cell line E14TG2a. After 4 h of exposure to CHX, METTL14 levels dramatically decreased in METTL3 knockout (METTL3‐KO) cells, whereas those in METTL3 wild‐type (METTL3‐WT) cells showed only a slight change. β‐actin was used as the loading control. The METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
FImmunoblots showing the protein level of METTL14 after transfection with the FLAG‐METTL3 plasmid. β‐actin was used as the loading control. The FLAG‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
GMETTL14 protein levels detected under a concentration gradient of FLAG‐METTL3 plasmids. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
HSchematic diagram of METTL3 showing the different domains. MTD, methyltransferase domain.
-
IHA‐METTL14 Co‐IP with FLAG‐tagged METTL3 domain‐deletion mutants. β‐actin was used as the loading control.
-
JHA‐METTL14 protein levels detected under a concentration gradient of plasmids encoding FLAG‐tagged METTL3 domain‐deletion mutants. β‐actin was used as the loading control. The HA‐METTL14 or FLAG‐METTL3 mutants /β‐actin densitometric ratio was recorded by ImageJ.
Source data are available online for this figure.
Figure EV1. METTL3 but not WTAP protects METTL14 protein.
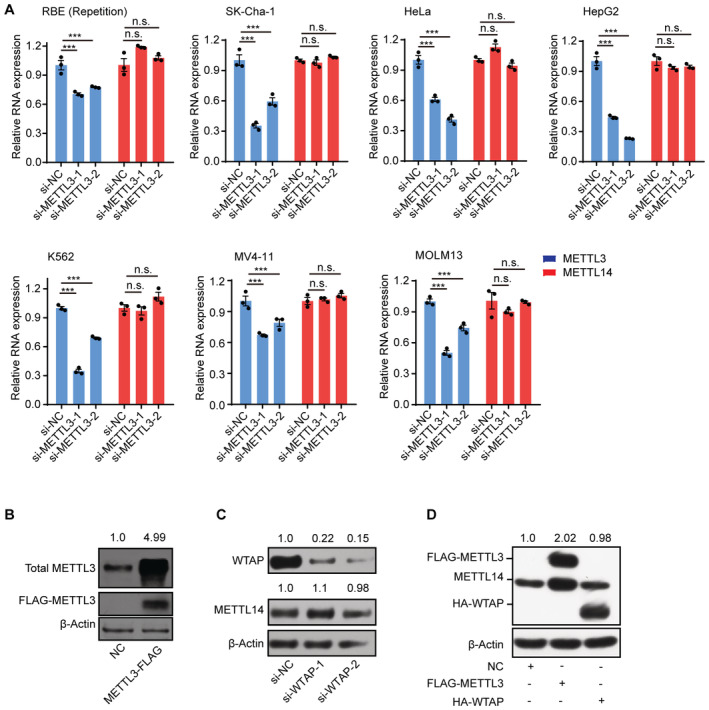
- RT‐qPCR analysis of the expression levels of METTL3 and METTL14 in siRNA knockdown METTL3 cells. Data are mean ± SEM of three biological replicates and were analyzed by two‐tailed unpaired t‐test. ***P < 0.001; n.s., not significant.
- Immunoblots showing total METTL3 protein levels in FLAG‐tagged METTL3‐overexpressing cells. β‐actin was used as the loading control. The total METTL3/β‐actin densitometric ratio was recorded by ImageJ.
- Immunoblots showing METTL14 and WTAP protein levels in WTAP knockdown cells. β‐actin was used as the loading control. The METTL14 or WTAP/β‐actin densitometric ratio was recorded by ImageJ.
- Immunoblots showing METTL14 protein levels in FLAG‐tagged METTL3 or HA‐tagged WTAP‐overexpressing cells. β‐actin was used as the loading control. The METTL14/ β‐actin densitometric ratio was recorded by ImageJ.
Source data are available online for this figure.
We performed an assay using cycloheximide (CHX), a specific inhibitor of protein synthesis (Wang et al, 2014; Han et al, 2022), to investigate whether METTL3 influences METTL14 protein stability. Accordingly, we applied CHX to block protein synthesis in both short hairpin RNA (shRNA)‐mediated METTL3 knockdown (sh‐METTL3) cells and control cells and measured the degradation rate of METTL14. In the control group, CHX reduced METTL14 protein abundance only slightly; however, in sh‐METTL3 cells, CHX dramatically decreased METTL14 protein levels (Fig 1D). We observed a similar pattern in a METTL3 knockout cell line (E14TG2a; Fig 1E). These results support the idea that METTL3 may contribute to the stability of METTL14. Notably, when we transfected HEK293T cells with FLAG‐METTL3 plasmids, the level of METTL14 increased significantly (Fig 1F). To validate this observation, we transfected cells with progressively increasing concentrations of METTL3 plasmids but with a constant amount of METTL14 plasmid. METTL14 protein levels clearly increased with increasing METTL3 level, suggesting that METTL3 maintains METTL14 stabilization in a protein level‐dependent manner (Fig 1G).
To assess the effects of METTL3 on METTL14 stability more directly, we investigated the METTL3 domains involved in METTL14 stabilization. Previous studies demonstrated that regions from amino acids (aa) 450–454 and aa 464–480 of METTL3 comprise its binding regions with METTL14 (Wang et al, 2016b). Thus, we asked whether these domains might be responsible for METTL14 stabilization. Immunoprecipitation assays revealed that both the aa 450–454 and aa 464–480 domains of METTL3, but not its catalytic site, interact with METTL14 to regulate its stability (Fig 1H and I). Notably, METTL14 protein levels increased with increasing concentrations of plasmids encoding wild‐type METTL3 (METTL3‐WT) and METTL3‐APPA (in which the DPPW motif was mutated to APPA), but not plasmids encoding versions of METTL3 with the aa 450–454 and 464–480 regions deleted (Fig 1J). These results suggest that these two regions are required for the ability of METTL3 to stabilize METTL14 in a protein level‐dependent manner (Fig 1J). These data confirm the notion that METTL14 protein stabilization requires METTL3 to assemble into a tight asymmetric heterodimer.
METTL14 is degraded by the ubiquitin–proteasome system
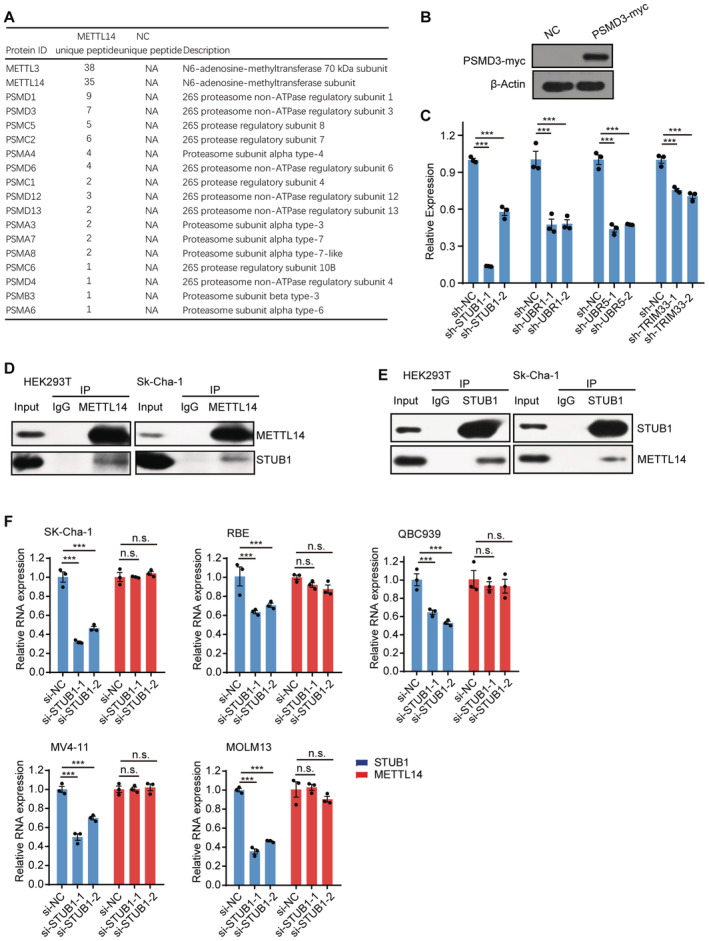
The above results confirmed that METTL3 helps to stabilize METTL14. We then investigated the underlying mechanism. To this end, we looked for potential co‐factors of METTL14 via a co‐immunoprecipitation (Co‐IP) assay in HEK293T cells transfected with a construct encoding METTL14‐GFP (a fusion between green fluorescent protein [GFP] and METTL14). METTL14‐GFP was effectively enriched in the IP assay (Fig 2A). Through silver staining followed by liquid chromatography tandem mass spectrometry (LC–MS/MS) analysis and gene ontology (GO) pathway analysis, we observed that many of the proteins that interacted with METTL14 are significantly enriched in the GO “Proteasome” category (Fig 2B and Dataset EV1). In particular, several proteasome subunits were found in the METTL14‐GFP group, including five proteasome subunit alpha (PSMAs), one PSMBs, four PSMCs, and eight PSMDs (Fig EV2A), suggesting that METTL14 may undergo proteolysis via the proteasome system. We further validated the interaction between METTL14 and the proteasome subunits, such as proteasome 26S subunit, non‐ATPase 3 (PSMD3; Figs 2C and EV2B).
Figure 2. The ubiquitin–proteasome system regulates METTL14 protein stability.
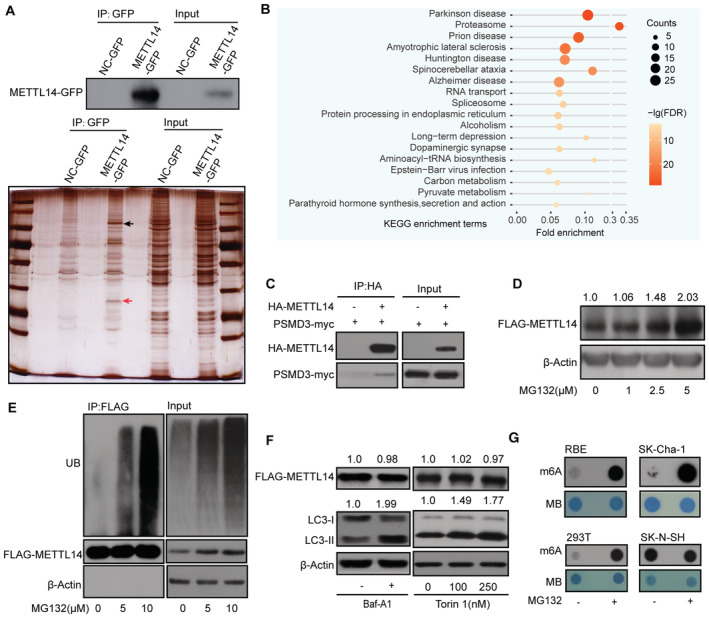
- METTL14‐GFP Co‐IP assay in HEK293T cells. Silver staining identified the specific bands (red arrow) for METTL14‐IP (black arrow).
- GO analysis of the METTL14 interactome.
- Co‐IP of HA‐METTL14 with the proteasome subunit PSMD3‐myc in HEK293T cells.
- METTL14 protein levels increase significantly with increasing MG132 concentration. The FLAG‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
- METTL14 ubiquitination, as detected by IP of METTL14 with anti‐FLAG antibody and immunoblotting with anti‐Ub antibody. The accumulation of Ub and METTL14 was confirmed in whole‐cell lysates. β‐actin was used as the loading control.
- Immunoblot analysis of FLAG‐METTL14, LC3‐II, and β‐actin in HEK293T cells exposed to the autophagy inhibitor Baf‐A1 and activator Torin 1, respectively. The FLAG‐METTL14 or LC3‐II /β‐actin densitometric ratio was recorded by ImageJ.
- Dot blots of m6A levels in the presence of MG132 in 293 T, RBE, SK‐Cha‐1, and SK‐N‐SH cells.
Source data are available online for this figure.
Figure EV2. STUB1 interacts with METTL14 but is dispensable for regulating METTL14 mRNA levels.
-
AList of the “Proteasome” category in the METTL14 interactome identified by MS, including five PSMAs, one PSMBs, four PSMCs, and eight PSMDs.
-
BImmunoblots showing PSMD3‐myc protein levels in myc‐tagged PSMD3‐ overexpressing cells. β‐actin was used as the loading control.
-
CRT‐qPCR analysis of the efficiency of shRNA‐knockdown for STUB1, UBR1, UBR5, and TRIM33. β‐actin was used as the reference. Data are mean ± SEM of three biological replicates and were analyzed by two‐tailed unpaired t‐test. (n.s., not significant; ***P < 0.001).
-
D, ECo‐IP of METTL14 and STUB1 in HEK293T and SK‐Cha‐1 cells.
-
FRT‐qPCR analysis of expression levels of STUB1 and METTL14 in STUB1 knockdown cells. Data are mean ± SEM of three biological replicates and were analyzed by two‐tailed unpaired t‐test. ***P < 0.001; n.s., not significant.
Source data are available online for this figure.
To explore whether METTL14 is degraded by the ubiquitin–proteasome system, we treated HEK293T cells with the proteasome inhibitor MG132 and observed that METTL14 protein levels increase significantly (Fig 2D). Co‐IP experiments revealed a smear signal of ubiquitination in Flag‐METTL14 samples (Fig 2E). Importantly, both METTL14 protein levels and ubiquitination increased in an MG132‐concentration‐dependent manner, suggesting that the ubiquitin–proteasome system could be responsible for METTL14 degradation (Fig 2D and E). A previous study showed that ultraviolet B (UVB) irradiation downregulated METTL14 protein levels via selective autophagy (Yang et al, 2021); however, we did not detect significant effects of the autophagy inhibitors (Klionsky et al, 2021), including Baf‐A1 (Klionsky et al, 2021), or of the activator Torin 1 (Klionsky et al, 2021), on METTL14 protein levels in cells (Fig 2F). Moreover, m6A levels greatly increased in the presence of MG132, which is consistent with the greater stability of METTL14 (Fig 2G). These results indicate that METTL14 can be degraded by the ubiquitin–proteasome system under normal physiological conditions.
The E3 ubiquitin ligase STUB1 positively regulates METTL14 ubiquitination
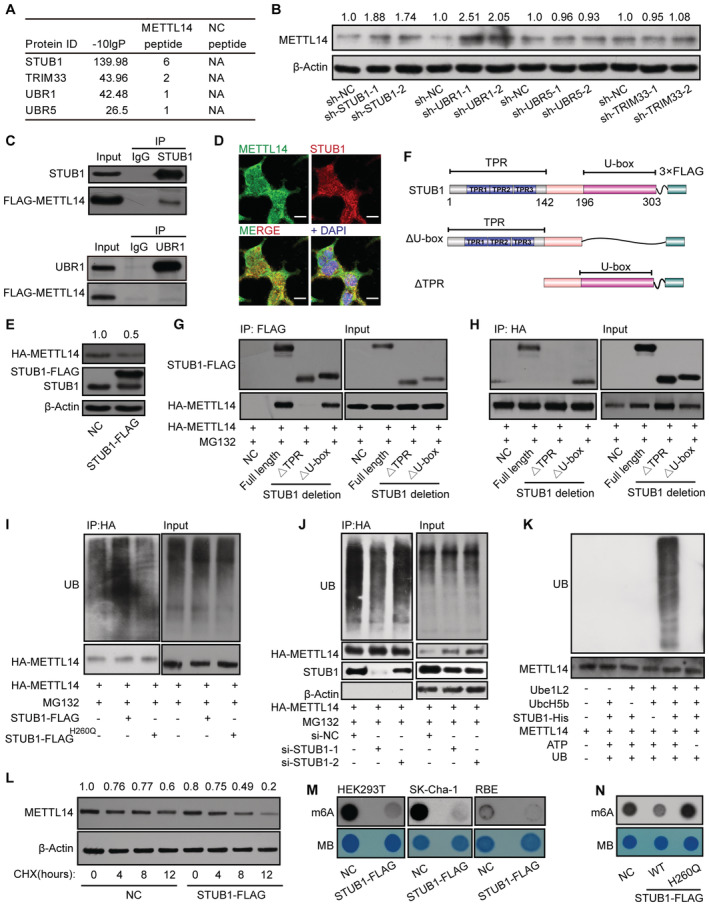
We further investigated how METTL14 is degraded by the ubiquitin–proteasome system. The transfer of Ub by an E3 ligase is the last step in ubiquitination; the E3 ligase serves as both a Ub transferase and a matchmaker that specifically recognizes the substrate (Zheng & Shabek, 2017; Clague et al, 2019). To identify the E3 ligase that specifically modulates the ubiquitination and degradation of METTL14, we took a closer look at METTL14 interactors and identified several previously reported E3 ligases, including the STIP1 homology and U‐box‐containing protein 1 (STUB1), Ubiquitin Protein Ligase E3 Component N‐recognin 1 (UBR1), Tripartite Motif‐containing 33 (TRIM33), and UBR5 (Fig 3A). We knocked down each of these candidates in HEK293T cells using two shRNAs (Fig EV2C). Endogenous METTL14 protein levels significantly increased after the knockdown of STUB1 and UBR1, but not UBR5 or TRIM33, compared with the levels in control cells (Fig 3B).
Figure 3. STUB1 is the E3 ubiquitin ligase to promote METTL14 ubiquitination.
-
AList of E3 ligases in the METTL14 interactome identified by MS.
-
BImmunoblots showing METTL14 protein levels in cells with shRNA knockdown of UBR1, STUB1, UBR5, or TRIM33. The METTL14 /β‐actin densitometric ratio was recorded by ImageJ.
-
CCo‐IP assays between FLAG‐METTL14 and STUB1 or URB1.
-
DImmunofluorescence of METTL14 (green) colocalized with that of STUB1 (red) in HEK293T cells. Nuclei were stained with DAPI (blue). Scale bar, 20 μm.
-
EImmunoblots showing the protein level of METTL14 after transfection with a plasmid encoding STUB1. β‐actin was used as the loading control. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
FSchematic diagram of STUB1‐domain‐deletion mutants.
-
G, HHA‐METTL14 Co‐IP with FLAG‐tagged STUB1‐domain‐deletion mutants.
-
IImmunoblots showing METTL14 ubiquitination assays under ectopic expression of STUB1 or the STUB1 H260Q mutant, which encodes a protein that lacks E3 ligase activity.
-
JImmunoblots showing METTL14 ubiquitination assays under STUB1 siRNA knockdown.
-
KIn vitro ubiquitination assays showing that purified METLL14 is ubiquitinated by purified STUB1‐His. Ube1L2 and UbcH5b were the E1 and E2 enzymes, respectively, in the ubiquitination system. ATP and ubiquitin (UB) were also added to the assay.
-
LImmunoblots showing CHX assay in HEK293T cells. Negative control (NC; no transfected cells) and STUB1‐FLAG plasmid‐transfected HEK293T cells were exposed to 100 μg/ml CHX for 0, 4, 8, or 12 h. β‐actin was used as the loading control. The METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
M, NDot blot assays showing that enforced expression of wild‐type STUB1, but not the STUB1 H260Q mutant, significantly decreases total m6A levels.
Source data are available online for this figure.
To further investigate whether METTL14 is a novel substrate of STUB1 or UBR1, we conducted a Co‐IP assay. In this assay, METTL14 interacted only with STUB1, suggesting a possible direct interaction between STUB1 and METTL14; however, UBR1 may regulate the stability of METTL14 independently of ubiquitination‐mediated degradation (Fig 3C). We further validated the interaction of endogenous STUB1 and METTL14 in SK‐Cha‐1 and HEK293T cells (Fig EV2D and E). Consistent with this finding, immunofluorescence (IF) experiments also showed that METTL14 colocalizes with STUB1 (Fig 3D). Moreover, METTL14 protein levels were significantly reduced upon STUB1 upregulation (Fig 3E), whereas METTL14 mRNA levels showed no significant changes in STUB1 knockdown cells, suggesting that STUB1 does not regulate the transcription of METTL14 (Fig EV2F). Together, these results indicate that STUB1 is the E3 ubiquitin ligase that positively regulates the ubiquitination of METTL14.
To further test this hypothesis, we delineated the possible interacting domains of STUB1 and METTL14 by truncation and/or deletion analysis (Fig 3F). Immunoprecipitation analysis revealed that the tetratricopeptide repeat (TPR) domain of STUB1 interacts with METTL14 (Fig 3G and H). We also created a series of GFP‐tagged METTL14 truncations and determined that the domain from aa 286 to 456 of METTL14 interacts with STUB1 (Fig EV3A and B). Subsequently, we investigated whether the ubiquitination of METTL14 is regulated by STUB1. Ectopic expression of STUB1 markedly increased METTL14 ubiquitination compared with control cells; however, a similar increase was not observed in STUB1H260Q mutants (Chen et al, 2013; Cho et al, 2018), which lack E3 ligase activity (Fig 3I). Furthermore, knockdown of STUB1 dramatically decreased the ubiquitination of METTL14 compared with control cells (Fig 3J). To further confirm these findings, we performed in vitro ubiquitination assays and showed that purified METLL14 is ubiquitinated by STUB1‐His (Figs 3K and EV3C). These results indicate that METTL14 is a substrate of STUB1.
Figure EV3. Possible ubiquitination sites in METTL14.
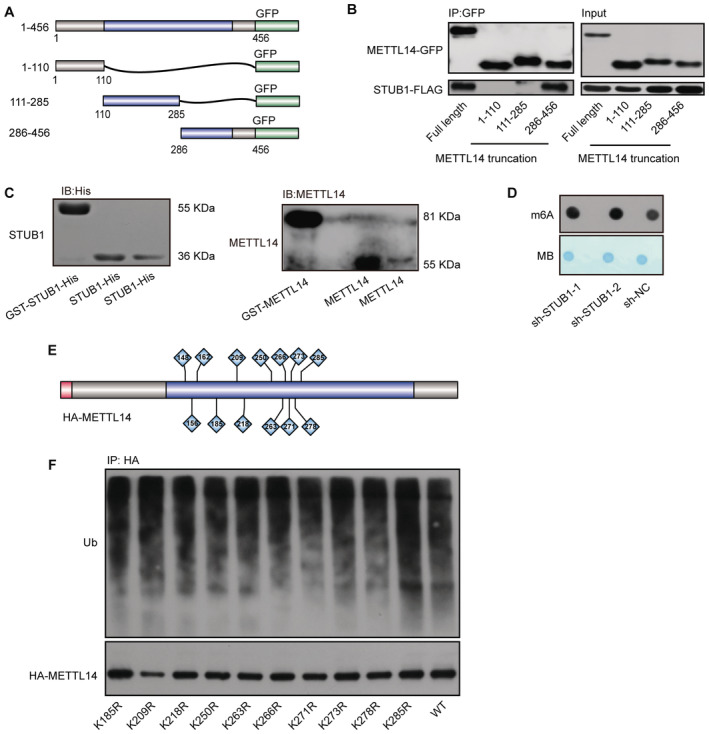
- Schematic diagram of METTL14‐domain‐deletion mutants. All mutants were GFP‐tagged.
- Co‐IP of GFP‐tagged METTL14‐domain‐deletion mutant with FLAG‐tagged STUB1.
- Immunoblots showing purified GST‐STUB1‐His, STUB1‐His, GST‐METTL14, and METTL14.
- Dot blot assays showing the role of sh‐STUB1 in regulating m6A levels.
- Schematic diagram of the lysine sites located at aa 111–285 of METTL14.
- METTL14 ubiquitination, as detected by IP of a series of lysine (K)‐to‐arginine (R) mutations in aa 111–285. The accumulation of Ub and METTL14 was confirmed in whole‐cell lysates. WT, wild type.
Source data are available online for this figure.
We performed CHX chase assays to confirm that ubiquitination mediated by STUB1 promotes the instability of METTL14 protein. Ectopic expression of wild‐type STUB1, but not the STUB1 mutant, with no E3 ligase activity, promoted the degradation of METTL14 in HEK293T or SK‐Cha‐1 cells that were treated with CHX (Fig 3L). We also investigated the function of STUB1 in regulating m6A levels in vitro. m6A is installed by the METTL3–METTL14 complex. Indeed, overexpressing wild‐type STUB1 significantly decreased the total m6A levels in HEK293T cells and two other cholangiocarcinoma cell lines, whereas knocking down STUB1 dramatically increased m6A levels (Figs 3M and EV3D). Consistent with this finding, the ectopic expression of STUB1 H260Q did not affect global m6A levels (Fig 3N). Together, these results demonstrate that STUB1, an E3 ubiquitin ligase, is responsible for interaction with and ubiquitination of METTL14.
STUB1 mediates METTL14 ubiquitination at K148, K156, and K162
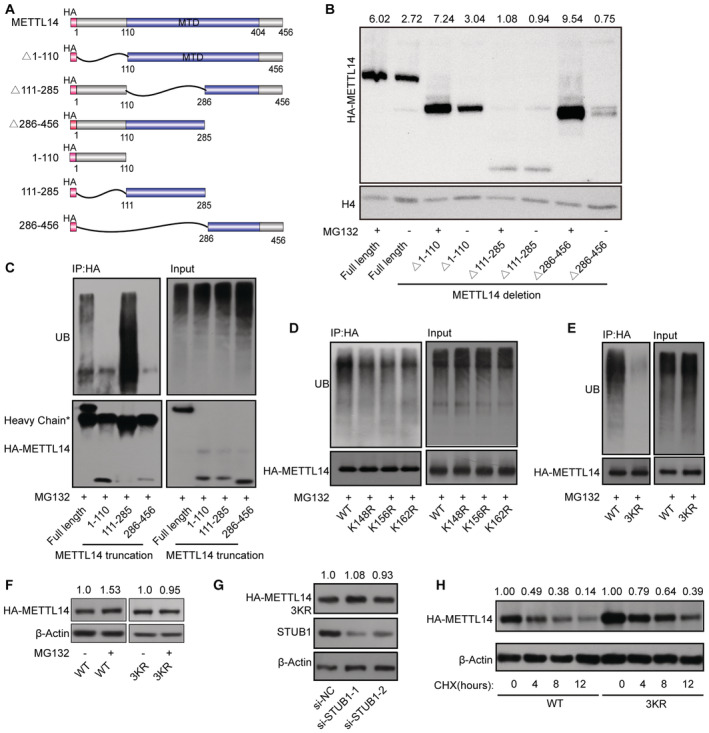
We then explored the molecular mechanism by which STUB1 mediates the ubiquitination‐induced degradation of METTL14. To identify the lysine poly‐ubiquitin sites in METTL14, we used truncated METTL14 fragments (tagged with the hemagglutinin epitope, HA; Fig 4A). In degradation suppression assays, MG132 treatment did not affect METTL14Δ111–285, indicating that ubiquitination of lysine may occur in the aa 111–285 region of METTL14, which is located within the MTase domain (Fig 4B). A ubiquitin tagging assay further demonstrated that this portion of METTL14 is significantly ubiquitinated, whereas the aa 1–110 and aa 286–456 regions were not (Fig 4C). To further uncover the ubiquitinated lysine site(s), we performed ubiquitination assays after introducing a series of lysine (K)‐to‐arginine (R) mutations in aa 111–285 region of METTL14, either individually or in various combinations (Fig EV3E). Only mutations at K148, K156, and K162 significantly decreased METTL14 ubiquitination, and this decrease was dramatically enhanced (by ∼90%) in a mutant with all three residues replaced with arginine (3KR), suggesting that these three sites are the major ubiquitinated residues of METTL14 (Figs 4D and E, and EV3F). Both MG132 treatment and STUB1 knockdown had little effect on METTL14‐3KR accumulation (Fig 4F and G). We then investigated whether ubiquitination at K148, K156, and K162 is responsible for the stability of METTL14. Mutating all three residues to R dramatically inhibited METTL14 degradation in CHX chase assays (Fig 4H). Collectively, these data indicate that the ubiquitination of the K148, K156, and K162 residues of METTL14 is responsible for its STUB1‐mediated degradation.
Figure 4. K148, K156, and K162 are the main sites of METTL14 ubiquitination by STUB1.
-
ASchematic diagram of METTL14‐domain‐deletion mutants. All mutants were HA‐tagged.
-
BImmunoblots showing the abundance of METTL14 truncated mutants with or without 10 μM MG132 treatment. Histone H4 was used as the loading control. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
CMETTL14 ubiquitination, as detected by IP of METTL14 truncated mutants with anti‐HA antibody and immunoblotting with anti‐ubiquitin (anti‐Ub) antibody. Accumulation of Ub and METTL14 was confirmed in whole‐cell lysates.
-
D, EMETTL14 ubiquitination, as detected by IP of a series of mutants with lysine (K)‐to‐arginine (R) substitutions in the aa 111–285 portion of the protein. The accumulation of Ub and METTL14 was confirmed in the whole‐cell lysates. WT, wild type; 3KR, K148, K156, and K162 all mutated to R.
-
FImmunoblots showing the levels of METTL14 protein after transfection with METTL14‐3KR mutant plasmids with or without 10 μM MG132. β‐actin was used as the loading control. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
GImmunoblots showing the protein level of METTL14 after transfection with METTL14‐3KR mutant plasmids under STUB1 knockdown. β‐actin was used as the loading control. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
HImmunoblots showing HEK293T cells in response to CHX treatment. HA‐METTL14 and HA‐METTL14‐3KR plasmid‐transfected HEK293T cells were exposed to 100 μg/ml CHX for 0, 4, 8, or 12 h. β‐actin was used as the loading control. The HA‐METTL14/β‐actin densitometric ratio was recorded by ImageJ.
Source data are available online for this figure.
METTL3 protects METTL14 from STUB1‐mediated ubiquitination to maintain m6A homeostasis
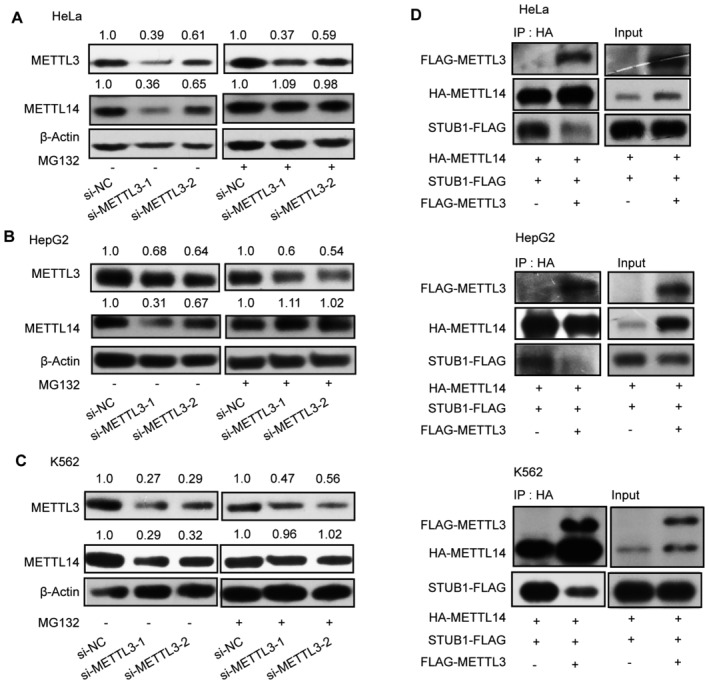
The above results revealed that STUB1 regulates the ubiquitination‐mediated degradation of METTL14. We thus investigated whether METTL3 protects METTL14 from STUB1‐mediated ubiquitination, thereby maintaining cellular m6A homeostasis. The decrease in METTL14 protein levels triggered by METTL3 knockdown could be significantly reduced by MG132 treatment, suggesting that METTL3 protects METTL14 from degradation (Figs 5A and EV4A–C). Additionally, the METTL14 ubiquitination level was lower in cells transfected with ectopic METTL3‐wt and METTL3‐APPA compared with the METTL3 Δ450–454 and Δ464–480 mutants (Fig 5B). These results suggest that both the aa 450–454 and aa 464–480 domains of METTL3, but not its catalytic site, are responsible for protecting METTL14 from ubiquitination‐mediated degradation. To gain further insights into how METTL3 decreases METTL14 ubiquitination, we examined whether METTL3 could weaken the interaction between METTL14 and STUB1. We determined that the interaction between METTL14 and STUB1 is indeed affected by the overexpression of METLL3 (Figs 5C and EV4D). Moreover, the METTL3 Δ450–454 and Δ464–480 mutants did not affect the interaction between METTL14 and STUB1 (Fig 5C).
Figure 5. METTL3 protects METTL14 from STUB1‐mediated ubiquitination to maintain m6A homeostasis.
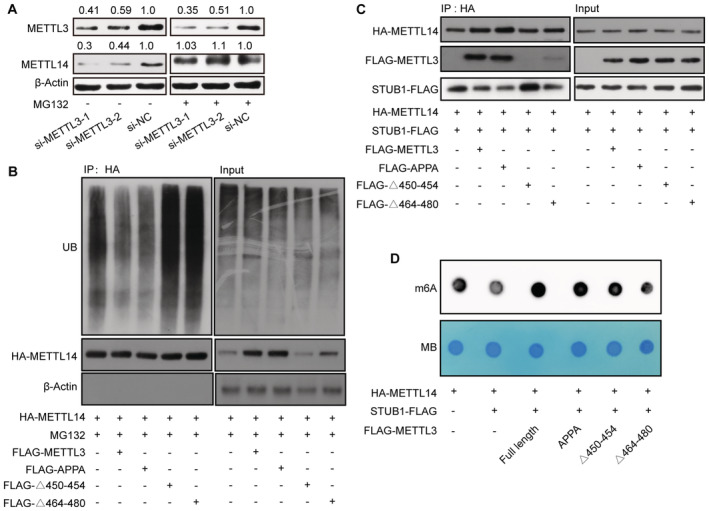
- Immunoblots showing METTL14 protein levels under METTL3 knockdown with or without 10 μM MG132 treatment. β‐actin was used as the loading control. The METTL3 or METTL14/β‐actin densitometric ratio was recorded by ImageJ.
- METTL14 ubiquitination, as detected by IP of METTL3 truncated mutants with anti‐HA antibody. The accumulation of Ub and METTL14 was confirmed in whole‐cell lysates. β‐actin was used as the loading control.
- Immunoblots showing METTL14 protein levels, as detected by IP in response to METTL3 truncated mutants and STUB1‐FLAG. β‐actin was used as the loading control.
- Dot blot assays showing the activities of METTL3 truncated mutants and STUB1‐FLAG in regulating m6A levels.
Source data are available online for this figure.
Figure EV4. METTL3 protects METTL14 from STUB1‐mediated ubiquitination.
-
A–CImmunoblots showing METTL14 protein levels under METTL3 knockdown with or without 10 μM MG132 treatment in HeLa (A), HepG2 (B), and K562 (C) cells. β‐actin was used as the loading control. The METTL3 or METTL14/β‐actin densitometric ratio was recorded by ImageJ.
-
DImmunoblots showing the interaction between METTL14 and STUB1 detected by IP with or without FLAG‐METTL3 in HeLa, HepG2, and K562 cells.
Source data are available online for this figure.
We also investigated the function of the METTL3 Δ450–454 and Δ464–480 mutants and STUB1 in regulating m6A levels in vitro. Compared with the control, enforced wild‐type STUB1 expression significantly decreased total m6A levels; the reduced m6A levels were rescued by introducing FLAG‐METTL3 and METTL3‐APPA, but not the METTL3 Δ450–454 or Δ464–480 mutants (Fig 5D). Together, these data support the possibility that METTL14 protein stabilization primarily requires the METTL3 450–454 and 464–480 domains to assemble into a tight asymmetric heterodimer to protect it from STUB1‐mediated ubiquitination; the coordination of the protection and degradation systems regulates METTL14 levels and m6A homeostasis.
Ectopic STUB1‐mediated degradation of METTL14 suppresses tumorigenesis: clinical relevance
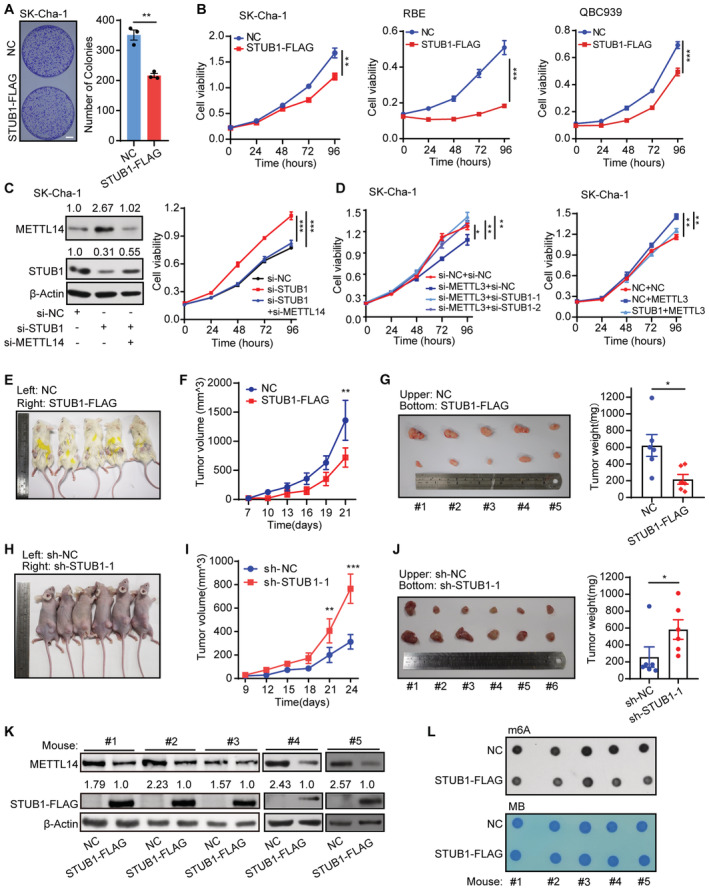
High levels of METTL14 increase m6A modifications, which may play a critical role in tumorigenesis (Weng et al, 2018; Sun et al, 2020b; Yang et al, 2021; Guan et al, 2022). Therefore, we investigated whether increasing STUB1‐induced ubiquitination‐mediated degradation of METTL14 could be a promising strategy for cancer treatment. Specifically, we investigated its effect on cholangiocarcinoma (CCA; Rizvi et al, 2018). CCA is a common primary hepatobiliary malignancy with a poor prognosis, whose incidence and mortality rate have been increasing worldwide over the past two decades, especially in China (Rizvi et al, 2018; Chen et al, 2020; Brindley et al, 2021). Due to its largely unknown pathogenetic mechanism, the major challenge associated with CCA is the lack of efficient tools for early diagnosis and clinical treatment. CCK‐8 and colony formation assays showed that ectopic STUB1 expression greatly reduced cancer cell growth, whereas STUB1 knockdown promoted cancer cell growth (Figs 6A and B, and EV5A–D). These results suggest that STUB1 can inhibit tumorigenesis.
Figure 6. STUB1‐mediated degradation of METTL14 suppresses tumorigenesis.
-
AMorphology of colonies of SK‐Cha‐1 cells overexpressing STUB1‐FLAG. Data are mean ± SEM of three biological replicates and were analyzed by two‐tailed unpaired t‐test. (**P < 0.01).
-
BCCK‐8 assays showing cell viability in SK‐Cha‐1, RBE, and QBC939 cells overexpressing STUB1‐FLAG. Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA (**P < 0.01, ***P < 0.001).
-
CImmunoblots showing METTL14 protein levels in SK‐Cha‐1 cells with STUB1 knockdown and METTL14 knockdown (left panel). β‐actin was used as the loading control. The METTL14 or STUB1 /β‐actin densitometric ratio was recorded by ImageJ. CCK‐8 assays showing cell viability upon the function of METTL14 activity in STUB1 knockdown cells (right panel). Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA (***P < 0.001).
-
DCCK‐8 assays showing cell viability upon the function of METTL3 in STUB1 knockdown or overexpression cells. Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA (*P < 0.05, **P < 0.01).
-
E–GFollowing the subcutaneous inoculation of SK‐Cha‐1‐NC (left) and SK‐Cha‐1‐STUB1‐FLAG (right) cells into the flanks of male NOD‐SCID mice (E), overexpression of STUB1‐FLAG inhibited malignant cell proliferation (F) and reduced subsequent tumor size and growth (G) in vivo. Data are mean ± SEM. Statistical significance was analyzed by two‐way ANOVA (F) or two‐tailed unpaired t‐test (G) (Five mice, *P < 0.05; **P < 0.01).
-
H–JFollowing the subcutaneous inoculation of SK‐Cha‐1‐sh‐NC (left) and SK‐Cha‐1‐sh‐STUB1‐1 (right) cells into the flanks of male nude mice (H), STUB1 knockdown promoted malignant cell proliferation (I) and increased subsequent tumor size and growth (J). Data are mean ± SEM. Statistical significance was analyzed by two‐way ANOVA (I) or two‐tailed unpaired t‐test (J). (Six mice, *P < 0.05; **P < 0.01; ***P < 0.001).
-
K, LImmunoblots (K) and dot blots (L) showing METTL14 protein levels and m6A levels under STUB1‐FLAG overexpression in five pairs of mouse tumors. β‐actin was used as the loading control. The METTL14 or STUB1‐FLAG/β‐actin densitometric ratio was recorded by ImageJ.
Source data are available online for this figure.
Figure EV5. STUB1‐mediated degradation of METTL14 can suppress CCA progression and has potential clinical relevance.
-
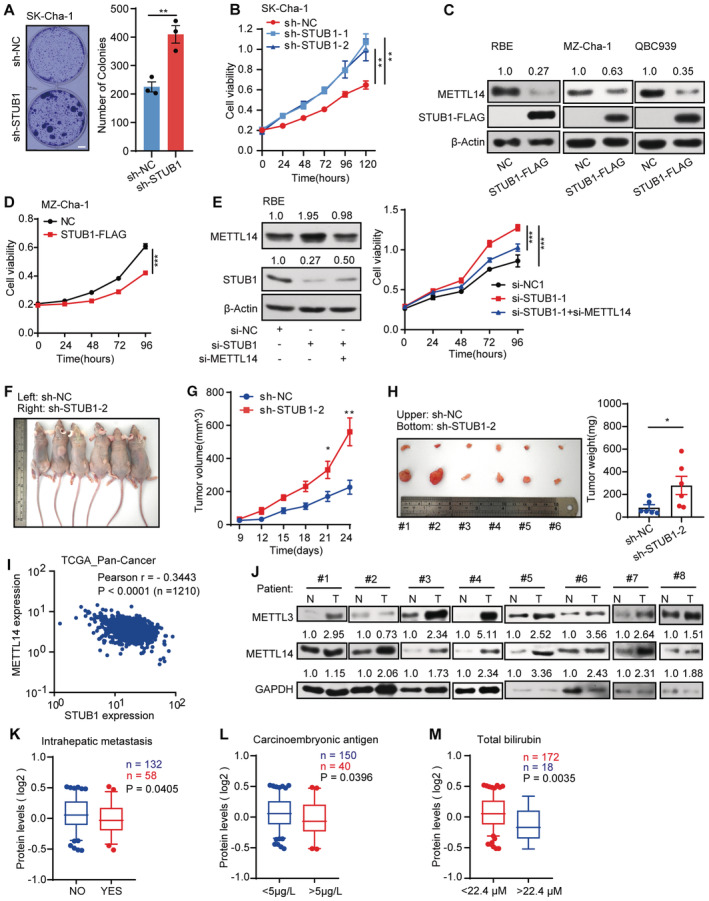
AMorphology of colonies of Sk‐Cha‐1 cells upon shRNA‐mediated STUB1 knockdown. Data are mean ± SEM of three biological replicates and were analyzed by two‐tailed unpaired t‐test. (**P < 0.01).
-
BCCK‐8 assays showing cell viability after shRNA‐mediated STUB1 knockdown. Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA. (**P < 0.01).
-
CImmunoblots showing METTL14 protein levels in CCA cells overexpressing STUB1‐FLAG. β‐actin was used as the loading control. The METTL14 /β‐actin densitometric ratio was recorded by ImageJ.
-
DCCK‐8 assays showing cell viability in MZ‐Cha‐1 cells overexpressing STUB1. Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA (***P < 0.001).
-
EImmunoblots showing METTL14 protein levels under STUB1 knockdown and METTL14 knockdown (left panel) in RBE cells. β‐actin was used as the loading control. The METTL14 or STUB1/β‐actin densitometric ratio was recorded by ImageJ. CCK‐8 assays showing cell viability upon the function of METTL14 in STUB1 knockdown cells (right panel). Data are mean ± SEM of three biological replicates and were analyzed by two‐way ANOVA (***P < 0.001).
-
F–HFollowing the subcutaneous inoculation of SK‐Cha‐1‐sh‐NC (left) and SK‐Cha‐1‐sh‐STUB1‐2 (right) cells into the flanks of male nude mice (F), STUB1 knockdown promoted the proliferation of malignant cells (G) and increased subsequent tumor size and growth (H). Data are mean ± SEM. Statistical significance was analyzed by two‐way ANOVA (G) and two‐tailed unpaired t‐test (H) (Six mice, *P < 0.05; **P < 0.01).
-
IPan‐cancer analysis using TGCA data sets consisting of 1,210 patient samples showing that METTL14 and STUB1 expression is negatively correlated (Pearson's correlation coefficient − 0.3443, P < 0.001).
-
JImmunoblot analysis showing METTL3 and METTL14 protein levels in eight pairs of CCA patient samples. N, adjacent non‐tumor tissue; T, tumor tissue. GAPDH was used as the loading control. The METTL3 or METTL14 /GAPDH densitometric ratio was recorded by ImageJ.
-
K–MSTUB1 protein levels are low in patient samples with high carcinoembryonic antigen and total bilirubin, and intrahepatic metastasis in the Fudan University intrahepatic cholangiocarcinoma (FU‐iCCA) cohort (using patient samples from the 5–95% bin according to STUB1 protein levels; a two‐tailed unpaired t‐test was used). In the boxplot, the central band indicates the median, the box indicates the interquartile range, and the whiskers indicate the 5–95% percentile.
Source data are available online for this figure.
To investigate whether the effect of STUB1 on CCA tumorigenesis is METTL14‐dependent, we performed rescue experiments. We knocked down METTL14 in STUB1 knockdown cells and found that both the increases in METTL14 protein levels and cell proliferation caused by the knockdown of STUB1 by small interfering RNAs (siRNA) are reversed (Figs 6C and EV5E). These results suggest that the role of STUB1 in CCA tumorigenesis is at least partially METTL14‐dependent. Notably, the ectopic expression of METTL3 partially rescued the anti‐cancer effects of STUB1 upregulation with regard to cell growth and colony formation in CCA cells, suggesting that METTL3 is important for METTL14‐dependent oncogenic activity (Fig 6D). These results indicate that the ectopic expression of STUB1 can undercut the protection from METTL3 to competitively mediate METTL14 degradation and suppress tumorigenesis.
To further explore the potential of STUB1 as a cancer treatment strategy, we injected mice with SK‐Cha‐1 cells ectopically expressing STUB1 loading enforced STUB1 in vivo. Compared with the negative control, cells ectopically expressing STUB1 significantly suppressed the growth rate, volume, and weight of tumors (Fig 6E–G). We also injected cohorts of animals with STUB1‐knockdown SK‐Cha‐1 cells. Strikingly, these mice showed substantial increases in tumor size, weight, and volume (Figs 6H–J and EV5F–H). Consistent with these findings, the ectopic expression of STUB1 resulted in the ubiquitination and degradation of METTL14 and reduced m6A levels in tumor samples (Fig 6K and L). Together, these data suggest that enforced STUB1 expression can delay the progression of tumorigenesis by causing the degradation of METTL14 in vivo.
Based on the mechanism and function of STUB1 in mediating METTL14 degradation identified above, we explored the clinical relevance of this process in cancer. A pan‐cancer analysis using The Cancer Genome Atlas (TCGA) pan‐cancer dataset, which consists of 1,210 patient samples, showed that most cancers present a strong negative correlation between METTL14 and STUB1 expression, especially CCA (Figs 7A and EV5I). This observation suggests that STUB1‐mediated regulation of METTL14 is common among many cancers. Using immunoblot analysis, we determined that STUB1 protein levels are significantly lower in CCA tumor tissues than in adjacent non‐tumor tissues (Fig 7B), while both METTL3 and METTL14 protein levels were significantly higher in CCA tumor tissues (Fig EV5J), pointing to the possible diagnostic utility of STUB1 and METTL14 for CCA.
Figure 7. STUB1‐mediated degradation of METTL14 has potential clinical relevance.
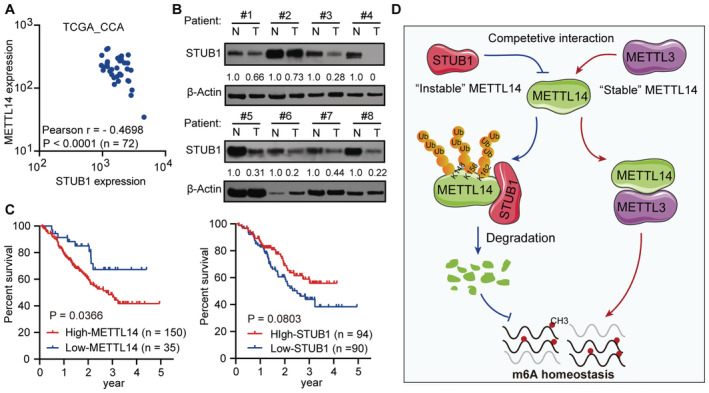
- METTL14 and STUB1 expression show a strong negative correlation in the TGCA data sets consisting of 72 CCA patient samples (Pearson coefficient: −0.4698, P < 0.001).
- Immunoblot analysis showing STUB1 protein levels in eight pairs of CCA patient samples. N, adjacent non‐tumor tissue; T, tumor tissue. β‐actin was used as the loading control. The STUB1/β‐actin densitometric ratio was recorded by ImageJ.
- The 5‐year disease‐free survival of patients with high expression levels of METTL14 (P = 0.0366) and low expression levels of STUB1 (P = 0.0803) in the Fudan University intrahepatic cholangiocarcinoma (FU‐iCCA) cohort among patient samples within the 5–95% ranges of STUB1 and METTL14 protein levels.
- Working model for the role of the competitive interaction of METTL3 and STUB1 in maintaining METTL14 levels and the subsequent regulation of m6A homeostasis.
Source data are available online for this figure.
We then reanalyzed the data on STUB1 and METTL14 in an existing cancer proteomics dataset, the quantitative proteomics dataset from the Fudan University intrahepatic cholangiocarcinoma (FU‐iCCA) cohort (Dong et al, 2022). To ensure the accuracy of our analysis, we selected patients within the 5–95% ranges of STUB1 and METTL14 levels to exclude outliers. Patient samples with signatures of high carcinoembryonic antigen and total bilirubin, and intrahepatic metastasis, which contribute to CCA progression, showed significantly lower STUB1 levels than the others (Fig EV5K–M), suggesting that there is a clinically relevant relationship between STUB1 protein levels and CCA carcinogenicity. Notably, lower levels of STUB1 (P = 0.0803) and higher levels of METTL14 (P = 0.0366) may lead to reduced 5‐year disease‐free survival (Fig 7C). Moreover, high levels of STUB1 were significantly associated with good patient outcomes, suggesting that STUB1 could serve as a biomarker for cancer prognosis and that improving STUB1 levels could represent a promising strategy for cancer treatment in the future.
In summary, we propose a competitive interaction model in which METTL3 serves as a “bodyguard” for METTL14, protecting it from STUB1‐mediated degradation (Fig 7D). We showed that K148, K156, and K162 are the ubiquitination sites of METTL14 for STUB1‐mediated degradation. The METTL3 domains comprising residues 450–454 and 464–480 help control the stability of METTL14 and protect it from STUB1‐mediated ubiquitination. The coordination of the protection and degradation systems precisely controls METTL14 levels and m6A homeostasis. Thus, the genetic upregulation of STUB1 effectively releases METTL14 from its defense system, resulting in the degradation of METTL14, which suppresses m6A modification and hence tumorigenesis.
Discussion
The homeostasis of RNA m6A modifications is important for various physiological and pathological processes, and its dysregulation has been linked to many human diseases (Vu et al, 2019; Huang et al, 2020, 2021; Livneh et al, 2020). Recently, several studies have reported that both PTMs and the self‐regulatory properties of m6A writers can promote RNA N 6‐methyladenosine modification (Du et al, 2018; Scholler et al, 2018; Xu et al, 2020; Sun et al, 2020a). However, the roles of PTMs in regulating the methyltransferase complex and RNA m6A modifications remain poorly understood. PTMs of METTL3, including SUMOylation and phosphorylation, increase the abundance of m6A RNA modifications (Du et al, 2018; Sun et al, 2020a). For instance, ERK can phosphorylate METTL3 at S43/S50/S525 and WTAP at S306/S341, thereby stabilizing the m6A methyltransferase complex to guarantee m6A homeostasis (Du et al, 2018). METTL3 is modified by SUMO1 mainly at the lysine residues K177, K211, K212 and K215 to regulate m6A RNA methyltransferase activity (Du et al, 2018). Additionally, protein arginine methyltransferase 1 (PRMT1) can mediate the arginine methylation of METTL14 at different sites to maintain m6A deposition (Liu et al, 2021; Wang et al, 2021). In the current study, we demonstrate that the PTM of METTL14 can suppress m6A modification. Ubiquitination of the K148, K156, and K162 residues of METTL14 by the E3 ligase STUB1 can significantly suppress METTL14 stability and inhibit RNA m6A deposition, providing a direct link between ubiquitination and RNA m6A methylation.
During ER stress, METTL14 expression can be induced by suppressing HRD1‐mediated ubiquitination to promote CHOP mRNA m6A modification and decay in liver disease (Wei et al, 2021). Interestingly, we showed that STUB1‐mediated K148, K156 and K162 ubiquitination of METTL14 can weaken the METTL3–METTL14 complex. It is also worth noting that the aa 450–454 and 464–480 regions in the MTase domains of METTL3not only execute m6A methylation activity but also serve as bodyguards of METTL14. It appears that PTMs on m6A writers can regulate the interactions of m6A regulators in the methyltransferase complex. These findings suggest that different PTMs of m6A writers can result in distinct functions in m6A modification. However, further investigations of the complex regulation of PTMs among m6A writers are needed.
The crystal structure of the MTase domains in the METTL3–METTL14 complex has already been analyzed (Wang et al, 2016a, 2016b; Huang et al, 2019); however, the underlying molecular mechanism by which METTL3 regulates METTL14 protein stability is largely unknown. We showed for the first time that the aa 450–454 and aa 464–480 regions in the MTase domains of METTL3 not only exhibit m6A methylation activity but also serve as bodyguards of METTL14. Importantly, the K148, K156 and K162 sites, which have been recognized in the MTase domains of METTL14, are considerably protected by METTL3 away from E3 ligase STUB1, pointing to a spatial competition close to the ubiquitination sites. This result also indicates the binding site for STUB1 on METTL14 as well as the ubiquitination sites of the METTL14–METTL3 structure.
A previous study demonstrated that METTL14 is degraded by Neighbor of Brca1 (NBR1)‐mediated selective autophagy under UVB irradiation in skin tumorigenesis (Yang et al, 2021). However, in the current study, METTL14 protein levels were only slightly altered in cells exposed to autophagy inhibitors (Klionsky et al, 2021), including Baf‐A1 (Klionsky et al, 2021), and the activator Torin 1 (Klionsky et al, 2021); instead, METTL14 levels were mainly regulated by ubiquitination‐mediated degradation. These contrasting results may be due to the different conditions used in these studies. UVB irradiation induces autophagy in skin cells and regulates METTL14 degradation (Yang et al, 2021); however, in the current study, we investigated METTL14 stability under normal physiological conditions. These observations suggest that METTL14 protein levels are mainly regulated by ubiquitination‐mediated degradation under normal physiological conditions but is modulated by other pathways including autophagy in response to different stresses. Indeed, ER stress can upregulate METTL14 expression by competing against HRD1‐ER‐associated ubiquitination‐mediated degradation in liver disease (Wei et al, 2021). HRD1 is an ER membrane protein that triggers ER‐located METTL14 degradation in ER proteotoxic liver disease, whereas STUB1 can serve as a potential E3 ligase to regulate whole‐cell METTL14 levels via the ubiquitination of K148, K156, and K162 to maintain m6A homeostasis under normal physiological conditions. These observations suggest that the posttranslational regulation of METTL14 is complex and may involve different operators under distinct cellular conditions.
RNA N 6‐methyladenosine modification and m6A writers are generally upregulated in cancers (Gu et al, 2020; Huang et al, 2021). Targeting the mRNAs of these m6A writers, including METTL3 and METTL14, could be a promising strategy for cancer therapy. For instance, knockdown of METTL14 can significantly suppress the progression of acute myeloid leukemia (Weng et al, 2018), CCA (Ye et al, 2022), and breast cancer (Sun et al, 2020b). In the current study, we investigated another approach: to downregulate m6A writers and RNA N 6‐methyladenosine modification in cancer cells. We demonstrated that the E3 ligase STUB1 directly regulated the ubiquitination‐mediated degradation of METTL14. Compared with the targeting of mRNAs by siRNAs or shRNAs, the direct targeting of proteins may be more efficient. To date, several proteasome inhibitors, such as MG132, bortezomib, and carfilzomib, have been used to inhibit antioncogenes in both clinical and pre‐clinical studies (Lee & Goldberg, 1998; Dimopoulos et al, 2015; Cowan et al, 2022).
Several recent reports propose that improving ubiquitination‐mediated degradation by specific E3 ligases targeting critical oncoproteins represents another promising approach to cancer therapy (Burslem & Crews, 2020; Li & Crews, 2022). For instance, upregulated E3‐ligase Smad ubiquitination regulatory factor 2 (SMURF2) specifically mediates Sirtuin‐1 (SIRT1)‐regulated ubiquitination‐mediated degradation, thereby suppressing the progression of colorectal cancer (Yu et al, 2020). In this study, we demonstrated that enforced STUB1 expression can significantly improve K148‐, K156‐, and K162‐linked polyubiquitination of METTL14 and degrade this protein, resulting in low global m6A modification levels and an antitumor effect. A recent study also showed that elvitegravir can suppress metastasis by targeting METTL3 and enhancing its STUB1‐mediated proteasomal degradation in esophageal squamous cell carcinoma (ESCC), suggesting that STUB1 has different regulatory functions during the treatment of some cancers with different drugs (Liao et al, 2022). Interestingly, METTL3 has many other functions apart from m6A modification. For example, METTL3, independently of METTL14, associates with chromatin and localizes to promoters to activate oncogenic genes in order to maintain a leukemic state (Barbieri et al, 2017). Notably, METTL14 has not been found to have other functions beyond m6A modifications to date, suggesting that it may have more specific effects than METTL3. Thus, STUB1‐induced ubiquitination‐mediated degradation of METTL14 may represent an excellent strategy for cancer treatment.
Taken together, our results show that STUB1 is an E3 Ub ligase that mediates the K148‐, K156‐, and K162‐linked polyubiquitination of METTL14, thereby inhibiting m6A deposition. METTL3 can directly bind to the ubiquitin domain of METTL14, thereby protecting it from STUB1‐dependent ubiquitination. These findings highlight the importance of polyubiquitination for METTL14 protein stability, suggesting that increasing STUB1 levels could be a promising strategy for suppressing tumorigenesis.
Materials and Methods
Patients
Human CCA tumor (designated as T) and peritumoral (designated as N) tissues were obtained with informed consent from Sun Yat‐sen Memorial Hospital and approved by the Hospital's Protection of Human Subjects Committee (Approval No. 2017126). Informed consent was obtained from all subjects; the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. The detailed clinicopathological characteristics of the CCA patients are summarized in Appendix Table S1. The samples were stored in liquid nitrogen until use.
Cell cultures, treatment and transfection
HEK293T, HepG2, Hela, and SK‐N‐SH cells were cultured in DMEM (Gibico, USA) with 10% fetal bovine serum (FBS; Gibico); MV4‐11 cells were cultured in IMDM (Gibico) with 10% FBS (Gibico); and SK‐CHA‐1, RBE, QBC939, MOLM‐13 and K562 cells were maintained in RPMI‐1640 (Hyclone, USA) with 10% FBS (Gibico) at 37°C and 5% CO2. Unless otherwise indicated, the cells were treated with proteasome inhibitor MG132 (5 μM, #S1748‐5 mg, Beyondtime, China) for 6 h, cycloheximide (100 μg/ml, Sigma, USA). Transient transfections of overexpression vector or siRNA were performed using Lipofectamine 2000 reagent or Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's instructions. To generate lentivirus for constructing stable cell lines, pGreenPuro vector for knockdown with shRNA or pCDH‐CMV‐MCS‐EF1‐Puro vector for constitutive gene overexpression was packaged in Lentivector Expression Systems (System Biosciences, Germany) consisting of pVSV‐G, pPACKH1‐GAG and pPACKH1‐REV. Virus in culture supernatant was harvested after 48 h transfection and then enriched and precipitated virus using Lentivirus Precipitation Solution (#EMB810A‐1, ExCell Bio, China). The lentivirus was resuspended in fresh complete culture medium. All lentiviruses were stored at −80°C before usage. 3 × 105 cells were resuspended in 300 μl virus suspension, followed by incubation at 37°C and 5% CO2. After 6 h, sufficient complete culture medium should be supplemented. 48 h later, cells were centrifuged, washed, and resuspended in fresh medium containing 1 μg/ml puromycin (#S7417, Selleck, USA). To confirm target knockdown or overexpression, cells were collected for RT‐qPCR and immunoblotting.
Plasmid construction
Wild‐type METTL3, METTL14, STUB1, and UBR1 gene were amplified from HEK293T mRNA and cloned into pCDH1‐MSCV‐MCS‐EF1‐copGFP‐T2A‐Puro or pCDH1‐CMV‐EF1‐Puro (#C510B‐1, ADDgene, USA). Stable RNA interference was performed using small hairpin RNA (shRNA) which was cloned into pGreenPuro shRNA cloning (System Biosciences). Transient RNA interference was performed using small interfering (siRNA; Genepharma, China). Primers listed in Appendix Table S2.
Co‐IP and immunoprecipitation (IP) assay
For co‐IP, HEK293T cells were collected by centrifugation at 100 g for 3 min after transfected with the indicated plasmids for 48 h. The cell pellet was lysed in Co‐IP lysis buffer (50 mM Tris–HCl pH 7.4, 150 nM NaCl, 1 mM EDTA, 1% Triton‐X100 and protease inhibitor cocktail [Thermo Fisher Scientific, USA]) and rotated at 4°C for 1 h. The cell debris was removed by centrifugation at 21,130 g for 15 min. The supernatant was incubated with 1 μg anti‐FLAG (Sigma, F2555) or anti‐HA (Cell Signal Technology, 3724s, USA) for 16 h at 4°C. Subsequently, magnetic beads were added into the mixture to incubate for 2 h. Subsequently, the beads were washed with 1 ml wash buffer (50 mM Tris–HCl pH 7.4, 150 nM NaCl, 1 mM EDTA) three times. Finally, protein‐bound beads were mixed with 5 × loading buffer (Fdbio Science, China) to the final concentration of 1 × loading buffer and boiled for 10 min at 100°C, 5 min.
IP was used for detecting ubiquitin. For 6‐cm dish, cells were lysed on ice for 15 min in 100 μl Ub‐IP Buffer 1 (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8.0, protease inhibitor cocktail [Thermo Fisher Scientific]). Subsequently, 900 μl Ub‐IP Buffer 2 (0.01% SDS, 10 mM EDTA, 1% Triton‐X100, 150 mM NaCl, 50 mM Tris–HCl pH 8.0, protease inhibitor cocktail [Thermo Fisher Scientific]) was added and rotated at 4°C for 30 min. The insoluble matter (chromatin and lipids) was broken by sonication. The cell debris was removed by centrifugation at 15,000 rpm for 15 min. The following steps were as described above for the Co‐IP (Sun et al, 2021; Han et al, 2022).
Protein purification and in vitro ubiquitination assays
HA‐METTL14 or STUB1‐6 × His cDNA was cloned into pET‐N‐GST‐Thrombin‐C‐His vector (Beyotime Biotechnology, China) and transformed into DE3 Transetta (DE3) Chemically Competent cells (Han et al, 2022; TransGen Biotechnology, China). Protein production was induced in optimal conditions: 16°C, 0.5 mM IPTG (TransGen Biotechnology), 2 h for GST‐HA‐METTL14; 25°C, 0.5 mM IPTG, 4 h for GST‐STUB1‐6 × His. The following steps were maintained at 4°C. The pelleted bacteria were resuspended in phosphate buffered saline (PBS) with lysozyme and supplemented with 1 mM PMSF (Beyotime Biotechnology) and then lysed by sonication. The cell debris was removed by centrifugation at 15,000 rpm for 15 min and 0.45 μm filter. GST‐tag proteins were purified using ProteinIso® GST Resin (TransGen Biotechnology). The purified protein was removed GST‐tag by Thrombin (Sigma Aldrich, USA) for purified HA‐METTL14 or STUB1‐6 × His. The proteins were stored at −80°C.
For in vitro complete ubiquitination assays, prepare the reactions in total 30 μl, including the reaction buffer (50 mM Tris–HCl pH 7.5, 2 mM ATP, 5 nM MgCl2, 2 mM DTT, 0.1 U PPIase), 5 μg monoUb (R&D Systems, U‐100H‐10 M, USA), 50 ng E1 Ube1L2 (R&D Systems, E302‐025), 200 ng E2 UBcH5b (R&D Systems, E23‐622‐100), 500 ng E3 STUB1‐6 × His, and 500 ng substrate HA‐METTL14. The reaction was placed at 30°C for 2 h. Finally, stop the reaction by adding protein loading buffer and boiling for 10 min at 100°C, 5 min for immunoblotting.
Immunoblotting analysis
Total protein was extracted by RIPA (Beyotime Biotechnology) supplemented with 1 × complete ULTRA protease inhibitor (Roche, Switzerland), adding 5 × loading buffer (Fdbio Science, China) to the final concentration of 1 × loading buffer and boiling for 10 min at 100°C, 5 min. The tumor samples from patients and mice were first grinded by Freezing Tissuelyser (LUKYM‐II, China) and then extracted by RIPA (Beyotime Biotechnology). The protein samples were resolved by SDS–PAGE and then transferred onto a PVDF membrane (EMD Millipore, USA) by electrophoretical system. The transferred PVDF membrane was incubated in blocking buffer (1 × TBST containing 5% BSA) for 1 h at room temperature, and then was incubated with the appropriate primary antibody (diluting by 1 × TBST containing 1% BSA) at 4°C overnight. The primary antibodies were as follow: anti‐METTL3 (1:2,000, Proteintech, 15073‐1‐AP, China), anti‐METTL14 (1:2,000, Sigma‐Aldrich, HPA038002), anti‐STUB1 (1:5,000, Abcam, ab134064, USA); anti‐UBR1 (1:5,000, Proteintech, 26,069‐1‐AP), anti‐β‐ACTIN (1:10,000, Sigma‐Aldrich, A2228), anti‐Ub (1:2,000, Cell Signal Technology, 3936s), anti‐GFP (1:2,000, Abcam, ab290), anti‐FLAG (1:10,000, Sigma, F2555), and anti‐HA(1:10,000, Cell Signal Technology, 3,724 s). After three washes with 1 × TBST, the membranes were incubated for 1 h at room temperature with horseradish peroxidase (HPR)‐conjugated secondary antibody (Cell Signal Technology). All incubations steps were on a plate shaker. Membrane was visualized by the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore).
RNA isolation and reverse transcription quantitative PCR (RT‐qPCR)
Total RNA was extracted using RNAiso Plus (Takara, Japan) according to the manufacturer's instructions. All RNA was stored at −80°C before reverse transcription and quantitative PCR. RNA was reverse transcribed into cDNA with an RT reagent Kit RR047A (Takara) and following quantitative PCR (qPCR) analysis was performed using TB Green premix ExTaq Real‐Time PCR kit (Takara) on an ABI QuantStudio 6 Flex PCR system (Applied Biosystems, USA) with primers listed in Appendix Table S2. All data were normalized to ACTB expression as a control. Relative expression levels were calculated using the method.
Cell proliferation assay
Cell proliferation assays using Cell Counting Kit‐8 (Dojindo Molecular Technologies, Japan) were performed as previously described (Fang et al, 2020; Wang et al, 2020). Briefly, 3 × 104 SK‐Cha‐1 cells were seeded in triplicate in 96‐well plates with 100 μl of the appropriate complete medium per well. At 0, 24, 48, 72, and 96 h, the CCK‐8 reagent (10 μl) was added to each well and incubated for another 2.5 h prior to measurement. Absorbance at 480 and 630 nm was measured using VICTORTM X5 Multilabel Plate Reader (PerkinElmer, USA).
The m6A dot blot
RNA samples extracted from cell were spotted to a nylon membrane (Fisher) followed by UV crosslinking at UV 254 nm, 0.12 J/cm2. The equal amounts of RNA were confirmed using 0.02% methylene blue in 0.3 M sodium acetate. And then methylene blue staining was removed by 0.5% SDS in PBS. The SDS should be washed off thoroughly five times using TBST before blocking by TBST containing 5% non‐fat milk. The membrane was incubated with 1:5,000 diluted anti‐m6A antibody (Synaptic System, 202003) overnight at 4°C. The membrane was incubated with HRP‐conjugated anti‐rabbit IgG (1:10,000 dilution) for 1 h and visualized by Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore).
Colony forming unit (CFU) assay
For SK‐Cha‐1 cells, 3,000 cells were seeded in a 6‐cm dish and continued to grow in fresh medium for 14 days at 37°C and 5% CO2 (Ye et al, 2022). One experiment group included at least three replicates. The colonies were fixed by cold methanol for 10 min and visualized using 0.5% crystal violet staining solution. The number of colonies were calculated by ImageJ.
Immunofluorescence
Cell was plated in 35‐mm glass bottom culture dishes (MetTek, #P35G‐1.0‐14‐C) before 48 h. After washing with PBS, cells were fixed on ice for 10 min in 4% PFA and permeabilized and blocked in 1% Triton X‐100, QuickBlock™ Blocking Buffer for Immunol Staining (Beyondtime, #P0260) in PBS for 30 min at room temperature. The samples were then incubated with the appropriate primary antibody overnight at 4°C. The next day, dishes were washed three times with PBS and then incubated with fluorescent secondary antibody in the dark for 1 h at room temperature. The secondary antibodies used were: anti‐Mouse IgG (H + L) Alexa Fluor® 488 1:500 (A‐11001, Life Technologies, USA), anti‐Mouse IgG (H + L) Alexa Fluor® 594 conjugate 1:500 (A‐11005, Life Technologies), and anti‐rabbit IgG (H + L) Alexa Fluor® 594 1:500 (A‐11012, Life Technologies). Dishes were then washed three times with PBS and dyed with DAPI to stain nuclei. The dishes were examined in 404, 488, and 594 nm laser system by Zeiss LSM880 with Fast airy scan.
Xenotransplantation model
Six‐week‐old male BALB/c nude mice and 6‐week‐old male NOD‐SCID mice (Beijing Vital River Laboratory Animal Technology Co., Ltd., China) were maintained under specific pathogen‐free conditions in the Laboratory Animal Center of Sun Yat‐sen University, and all procedures on mice were performed according to the Institutional Animal Ethical Guidelines (Approval No. SYSU‐IACUC‐2022‐B1637). In each mouse, stable knockdown STUB1 SK‐Cha‐1 cells (3 × 106) were subcutaneously injected into the dorsal right flanks of the mice, and NC into the dorsal left flanks. The mice were monitored every 3 days for tumor growth 9 days after xenotransplantation. The neoplasms were dissected from mice 24 days from transplant and weighed for statistical analysis (Ye et al, 2022).
Silver staining and MS analysis
HEK293T cells expressing METTL14‐GFP or control were collected for coimmunoprecipitation using GFP‐crosslinked beads. The input and Co‐IP samples were subjected silver staining to detect the specific bands. The whole METTL14‐GFP and control co‐IP lysis were subjected to MS analysis.
Statistical analysis
Pearson's correlation coefficients were calculated to determine the correlation between METTL14 and STUB1 mRNA levels. Two‐tailed Student's t test was used to analyze STUB1 protein levels between patients with different clinical index. Disease‐free survival was calculated from the date of complete remission (CR) until either relapse or death in remission. Disease‐free survival was analyzed using the Kaplan–Meier method with a log‐rank test. Two‐tailed tests were used for univariate comparisons. Student's two‐tailed unpaired t‐test was used for statistical comparisons, and the data are expressed as the mean ± SEM of three independent experiments. P < 0.05 was considered statistically significant.
Author contributions
Zhan‐Cheng Zeng: Resources; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft. Qi Pan: Resources; formal analysis; investigation; visualization; methodology. Yu‐Meng Sun: Data curation; software; formal analysis; funding acquisition; investigation; visualization. Heng‐Jing Huang: Data curation; formal analysis; validation; investigation; visualization; methodology. Xiao‐Tong Chen: Resources; software; formal analysis; investigation; methodology. Tian‐Qi Chen: Resources; data curation; investigation; methodology. Bo He: Resources; investigation; methodology. Hua Ye: Resources; formal analysis; methodology. Shun‐Xin Zhu: Resources; software; formal analysis; methodology. Ke‐Jia Pu: Formal analysis; investigation; methodology. Ke Fang: Resources; data curation; formal analysis; investigation. Wei Huang: Formal analysis; investigation; methodology. Yue‐Qin Chen: Conceptualization; resources; supervision; writing – review and editing. Wen‐Tao Wang: Conceptualization; data curation; formal analysis; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This research was supported by the National Key R&D Program of China (No. 2021YFA1300502), the National Natural Science Foundation of China (Nos 32170570 and 81772621), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (No. 2021B1515020002), and the Scientific and Technological Planning Project of Guangzhou City (No. 202102020070).
EMBO reports (2023) 24: e55762
Data availability
No primary datasets have been generated and deposited for this study.
References
- Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB et al (2014) WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28: 1171–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan‐Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N et al (2017) Promoter‐bound METTL3 maintains myeloid leukaemia by m(6)A‐dependent translation control. Nature 552: 126–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley PJ, Bachini M, Ilyas SI, Khan SA, Loukas A, Sirica AE, Teh BT, Wongkham S, Gores GJ (2021) Cholangiocarcinoma. Nat Rev Dis Primers 7: 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, Crews CM (2020) Proteolysis‐targeting chimeras as therapeutics and tools for biological discovery. Cell 181: 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, Jinasena D, Fu J, Lin F, Chen C et al (2013) The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39: 272–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu Y, Zhang Y (2020) Current status of laparoscopic radical hilar cholangiocarcinoma in mainland China. Biosci Trends 14: 168–173 [DOI] [PubMed] [Google Scholar]
- Cho Y, Kang HG, Kim SJ, Lee S, Jee S, Ahn SG, Kang MJ, Song JS, Chung JY, Yi EC et al (2018) Post‐translational modification of OCT4 in breast cancer tumorigenesis. Cell Death Differ 25: 1781–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, Komander D (2019) Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol 20: 338–352 [DOI] [PubMed] [Google Scholar]
- Collins GA, Goldberg AL (2017) The logic of the 26S proteasome. Cell 169: 792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Green D, Kwok M, Lee S, Coffey D, Holmberg L, Tuazon S, Gopal A, Libby E (2022) Diagnosis and management of multiple myeloma: a review. JAMA 327: 464–477 [DOI] [PubMed] [Google Scholar]
- Deng X, Su R, Weng H, Huang H, Li Z, Chen J (2018) RNA N‐methyladenosine modification in cancers: current status and perspectives. Cell Res 28: 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos M, Richardson P, Moreau P, Anderson K (2015) Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol 12: 42–54 [DOI] [PubMed] [Google Scholar]
- Dong L, Lu D, Chen R, Lin Y, Zhu H, Zhang Z, Cai S, Cui P, Song G, Rao D et al (2022) Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 40: 70–87 [DOI] [PubMed] [Google Scholar]
- Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R et al (2018) SUMOylation of the m6A‐RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res 46: 5195–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K, Huang W, Sun YM, Chen TQ, Zeng ZC, Yang QQ, Pan Q, Han C, Sun LY, Luo XQ et al (2020) Cis‐acting lnc‐eRNA SEELA directly binds histone H4 to promote histone recognition and leukemia progression. Genome Biol 21: 269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C (2014) Gene expression regulation mediated through reversible m(6)a RNA methylation. Nat Rev Genet 15: 293–306 [DOI] [PubMed] [Google Scholar]
- Gu C, Shi X, Dai C, Shen F, Rocco G, Chen J, Huang Z, Chen C, He C, Huang T et al (2020) RNA m6A modification in cancers: molecular mechanisms and potential clinical applications. Innovation (Camb) 1: 100066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q, Lin H, Miao L, Guo H, Chen Y, Zhuo Z, He J (2022) Functions, mechanisms, and therapeutic implications of METTL14 in human cancer. J Hematol Oncol 15: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Sun LY, Luo XQ, Pan Q, Sun YM, Zeng ZC, Chen TQ, Huang W, Fang K, Wang WT et al (2022) Chromatin‐associated orphan snoRNA regulates DNA damage‐mediated differentiation via a non‐canonical complex. Cell Rep 38: 110421 [DOI] [PubMed] [Google Scholar]
- Huang J, Dong X, Gong Z, Qin L, Yang S, Zhu Y, Wang X, Zhang D, Zou T, Yin P et al (2019) Solution structure of the RNA recognition domain of METTL3‐METTL14 N‐methyladenosine methyltransferase. Protein Cell 10: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Chen J (2020) m6A modification in coding and non‐coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37: 270–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Chen TQ, Fang K, Zeng ZC, Ye H, Chen YQ (2021) N6‐methyladenosine methyltransferases: functions, regulation, and clinical potential. J Hematol Oncol 14: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y et al (2011) N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdel‐Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo‐Arozena A et al (2021) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17: 1–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Goldberg A (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8: 397–403 [DOI] [PubMed] [Google Scholar]
- Li K, Crews C (2022) PROTACs: past, present and future. Chem Soc Rev 51: 5214–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, He Y, Li SJ, Zhang GG, Yu W, Yang J, Huang ZJ, Zheng CC, He QY, Li Y et al (2022) Anti‐HIV drug elvitegravir suppresses cancer metastasis via increased proteasomal degradation of m6A methyltransferase METTL3. Cancer Res 82: 2444–2457 [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X et al (2014) A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10: 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang H, Zhao X, Luo Q, Wang Q, Tan K, Wang Z, Jiang J, Cui J, Du E et al (2021) Arginine methylation of METTL14 promotes RNA N(6)‐methyladenosine modification and endoderm differentiation of mouse embryonic stem cells. Nat Commun 12: 3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh I, Moshitch‐Moshkovitz S, Amariglio N, Rechavi G, Dominissini D (2020) The mA epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci 21: 36–51 [DOI] [PubMed] [Google Scholar]
- Morreale FE, Walden H (2016) Types of ubiquitin ligases. Cell 165: 248 [DOI] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6‐methyladenosine methyltransferase. Cell Res 24: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla‐Prats C, Thomä N (2022) Quality control of protein complex assembly by the ubiquitin‐proteasome system. Trends Cell Biol 32: 696–706 [DOI] [PubMed] [Google Scholar]
- Pohl C, Dikic I (2019) Cellular quality control by the ubiquitin‐proteasome system and autophagy. Science 366: 818–822 [DOI] [PubMed] [Google Scholar]
- Ren F, Miao R, Xiao R, Mei J (2021) m6A reader Igf2bp3 enables germ plasm assembly by m6A‐dependent regulation of gene expression in zebrafish. Sci Bull 66: 1119–1128 [DOI] [PubMed] [Google Scholar]
- Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ (2018) Cholangiocarcinoma – evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 15: 95–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, Meister G (2018) Interactions, localization, and phosphorylation of the m(6)a generating METTL3‐METTL14‐WTAP complex. RNA 24: 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HL, Zhu AC, Gao Y, Terajima H, Fei Q, Liu S, Zhang L, Zhang Z, Harada BT, He YY et al (2020a) Stabilization of ERK‐phosphorylated METTL3 by USP5 increases m(6)a methylation. Mol Cell 80: 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Wu Z, Wang X, Wang Y, Hu X, Qin W, Lu S, Xu D, Wu Y, Chen Q et al (2020b) LNC942 promoting METTL14‐mediated mA methylation in breast cancer cell proliferation and progression. Oncogene 39: 5358–5372 [DOI] [PubMed] [Google Scholar]
- Sun L, Wang W, Han C, Huang W, Sun Y, Fang K, Zeng Z, Yang Q, Pan Q, Chen T et al (2021) The oncomicropeptide APPLE promotes hematopoietic malignancy by enhancing translation initiation. Mol Cell 81: 4493–4508 [DOI] [PubMed] [Google Scholar]
- Vu L, Cheng Y, Kharas M (2019) The biology of mA RNA methylation in Normal and malignant hematopoiesis. Cancer Discov 9: 25–33 [DOI] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC (2014) N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, Nam Y (2016a) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63: 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C et al (2016b) Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature 534: 575–578 [DOI] [PubMed] [Google Scholar]
- Wang WT, Chen TQ, Zeng ZC, Pan Q, Huang W, Han C, Fang K, Sun LY, Yang QQ, Wang D et al (2020) The lncRNA LAMP5‐AS1 drives leukemia cell stemness by directly modulating DOT1L methyltransferase activity in MLL leukemia. J Hematol Oncol 13: 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Pan Z, Adhikari S, Harada BT, Shen L, Yuan W, Abeywardana T, Al‐Hadid Q, Stark JM, He C et al (2021) M(6) a deposition is regulated by PRMT1‐mediated arginine methylation of METTL14 in its disordered C‐terminal region. EMBO J 40: e106309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Harada BT, Lu D, Ma R, Gao B, Xu Y, Montauti E, Mani N, Chaudhuri SM, Gregory S et al (2021) HRD1‐mediated METTL14 degradation regulates m(6)a mRNA modification to suppress ER proteotoxic liver disease. Mol Cell 81: 5052–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)a modification. Cell Stem Cell 22: 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W, Xin Y, Li W, Yang C, Bai Y et al (2020) SUMO1 modification of methyltransferase‐like 3 promotes tumor progression via regulating snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 10: 5671–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y et al (2017) Extensive translation of circular RNAs driven by N(6)‐methyladenosine. Cell Res 27: 626–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yang S, Cui YH, Wei J, Shah P, Park G, Cui X, He C, He YY (2021) METTL14 facilitates global genome repair and suppresses skin tumorigenesis. Proc Natl Acad Sci USA 118: e2025948118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Chen T, Zeng Z, He B, Yang Q, Pan Q, Chen Y, Wang W (2022) The m6A writers regulated by the IL‐6/STAT3 inflammatory pathway facilitate cancer cell stemness in cholangiocarcinoma. Cancer Biol Med 19: 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Dong L, Li H, Liu Z, Luo Z, Duan G, Dai X, Lin Z (2020) Ubiquitination‐mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene 39: 4450–4464 [DOI] [PubMed] [Google Scholar]
- Zheng N, Shabek N (2017) Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem 86: 129–157 [DOI] [PubMed] [Google Scholar]
- Zheng G, Dahl J, Niu Y, Fedorcsak P, Huang C, Li C, Vågbø C, Shi Y, Wang W, Song S et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]