

Abstract
Sox8, Sox9, and Sox10 constitute subgroup E within the Sox family of transcription factors. Many Sox proteins are essential regulators of development. Sox9, for instance, is required for chondrogenesis and male sex determination; Sox10 plays key roles in neural crest development and peripheral gliogenesis. The function of Sox8 has not been studied so far. Here, we generated mice deficient in this third member of subgroup E. In analogy to the case for the related Sox9 and Sox10, we expected severe developmental defects in these mice. Despite strong expression of Sox8 in many tissues, including neural crest, nervous system, muscle, cartilage, adrenal gland, kidney, and testis, homozygous mice developed normally in utero, were born at Mendelian frequencies, and were viable. A substantial reduction in weight was observed in these mice; however, this reduction was not attributable to significant structural deficits in any of the Sox8-expressing tissues. Because of frequent coexpression with either Sox9 or Sox10, the mild phenotype of Sox8-deficient mice might at least in part be due to functional redundancy between group E Sox proteins.
The Sox protein family constitutes a group of transcription factors with an already large but still increasing number of family members. Its occurrence is confined to the animal kingdom, where Sox proteins have diverse functions both during development and in the adult. These functions range from roles in early embryogenesis to functions in lineage specification and terminal differentiation events. Processes known to rely on Sox proteins include endoderm formation, neural induction, neural crest and lens development, gliogenesis, chondrogenesis, hemopoiesis, and sex determination (for reviews, see references 5, 22, and 32). All family members are characterized by possession of a specific type of DNA-binding domain, the minor groove-interacting high-mobility-group domain. Sequence similarities outside this domain are found only between subsets of Sox proteins and provide criteria which further subdivide this protein family into subgroups A to G. These subgroups are present in organisms from Caenorhabditis elegans and Drosophila to humans (5, 32). Genes coding for Sox proteins of the same subgroup tend to have similar genomic organizations.
One of the well-characterized groups of Sox proteins is subgroup E. It consists of the three members Sox8, Sox9, and Sox10, with Sox8 being the most recently identified (23, 25). Inactivation of a single Sox9 allele in humans is the cause of a severe skeletal malformation syndrome called campomelic dysplasia (7, 31). In male patients, campomelic dysplasia is often associated with XY sex reversal. In agreement with the observed phenotype, Sox9 expression is highest in chondrocytes and Sertoli cells of the testis (13, 17, 33). Other expression domains of Sox9 include brain, otic and nasal placode, lung, and kidney. These tissues are only rarely affected in campomelic dysplasia patients. The severity of the phenotype already observed in the heterozygous state might also explain why standard gene disruption techniques in mice have proven unsuccessful for Sox9. When homozygous Sox9-deficient ES cells were used to generate chimeras, Sox9-deficient ES cells failed to contribute to the chondrocyte population in these chimeric mice, impressively proving the essential role of Sox9 in this cell type (4). Many chondrocyte-specific genes are furthermore under direct control of Sox9, including the genes for type II collagen, type XI collagen, aggrecan, and cartilage-derived retinoic acid-sensitive protein genes (1, 15, 16, 26, 34).
Sox10 on the other hand, is expressed first in the early neural crest, then throughout the forming peripheral nervous system (PNS), and finally in glial cells of the PNS and central nervous system (CNS) (14). As in the case of Sox9, mutation or loss of a single Sox10 allele is already phenotypically apparent. Sox10 haploinsufficiency causes disturbances of neural crest development that are visible as partial pigmentation defects and aganglionosis of the distal colon in mice and humans (6, 9, 24, 28). In humans, this defect is known as Waardenburg-Hirschsprung disease. Peripheral neuropathies are often associated with Sox10-dependent Waardenburg-Hirschsprung disease (27, 29), correlating with the strong expression of Sox10 at later times in peripheral glia (14). Central myelinopathies present a further, less frequent complication (11), in agreement with Sox10 expression in myelinating glia of the CNS (14). Inactivation or deletion of both Sox10 alleles in mice leads to a complete loss of neural crest-derived melanocytes and enteric nervous system and proves that Sox10 is an essential factor for all gliogenesis of the PNS (6, 21). Target genes of Sox10 include genes important for glial development (ErbB3 gene) and identity (protein zero gene) (6, 21).
Recently, Sox8 was identified as the third group E Sox protein in mice, humans, and chickens (2, 23, 25). Existing reports on Sox8 expression are preliminary and partially contradictory, but they hint at expression during development in many tissues and organs, including branchial arches, nervous system, eye, male gonad, kidney, and limbs. Prominent places of expression in the adult were brain and testis. Chromosomal localization of human SOX8 to 16p13.3 placed it in a region often deleted in patients with ATR-16 syndrome (characterized by a combination of α-thalassemia, facial malformations, and mental retardation) and targeted in a Japanese family by a translocation event causing microphthalmia and congenital cataract (microphthalmia-cataract syndrome [CATM]). Localization to the syntenic region on mouse chromosome 17 places Sox8 in proximity to the tw18 mutation which causes abnormal mesodermal cell migration and is lethal prior to organogenesis.
In analogy to the case for Sox9 and Sox10, it appeared reasonable to assume that inactivation or deletion of Sox8 in mice should cause severe developmental defects in some of the tissues that express it. The phenotype could then be instrumental in identifying Sox8-dependent disease phenotypes in humans. Here, we deleted the Sox8 gene by homologous recombination in ES cells and subsequently generated Sox8-deficient mice. The simultaneous replacement of the Sox8 gene by a lacZ marker allowed a detailed analysis of Sox8 expression and should have facilitated detection of developmental defects in these mice. Surprisingly, homozygous Sox8-deficient mice failed to exhibit a major developmental defect in any of the Sox8-specific expression domains. Despite a significant weight reduction, they were viable and fertile. Possible reasons for and implications of this unexpected finding are discussed.
MATERIALS AND METHODS
Construction of targeting vector.
Genomic sequence from the Sox8 locus of 129/Sv mice was obtained by screening a bacterial artificial chromosome library. A 2.8-kb XbaI fragment ending at bp 56 of exon 1 was used as a 5′ homology region. Using an NruI linker, the fragment was extended to the start codon of Sox8, enabling us to place the start codon of the lacZ marker gene exactly over the Sox8 start codon. A 1.9-kb BglII/MscI fragment immediately downstream of the open reading frame was used as a 3′ homology region. Both the combination of the 5′ homology region and lacZ and the 3′ homology region were inserted into pPNT (30) on either side of the neomycin resistance cassette (Fig. 1A). The targeting vector thus replaced the complete open reading frame of Sox8 by a lacZ marker gene. The construct was linearized with NotI before electroporation.
FIG. 1.

Targeted disruption of Sox8 in mice. (A) Schematic representation of the targeting vector (top), the Sox8 wild-type locus (middle), and the mutant locus (bottom). The three known Sox8 exons are shown as boxes; introns and flanking regions are shown as bars. Regions of homology between wild-type locus and targeting vector are in black. Restriction sites for BamHI (B), BglII (Bg), EcoRI (E), MscI (M), and XbaI (X) are shown, as well as the localization of 5′ and 3′ probes. (B) Southern blot analysis of DNAs from adult wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice digested with EcoRI for use of the 5′ probe. (C) Southern blot analysis of DNAs from mice of all three genotypes digested with BamHI for use of the 3′ probe. The sizes of bands corresponding to the wild-type and the targeted alleles are indicated. (D) Analysis of Sox8 expression in various tissues of wild-type (+/+) and homozygous (−/−) adult mice by RT-PCR using primer pairs specific for the coding regions of the Sox8, lacZ, and GAPDH genes.
Gene targeting and generation of mouse mutants.
The linearized construct was electroporated into R1 ES cells (129 X1 × 129 S1), which were then selected with G418 (200 μg per ml) and ganciclovir (2 μM). Selected ES cell clones were screened by Southern blotting with a 1.8-kb 5′ probe, which recognized a 9.2-kb fragment of the wild-type allele and a 7.7-kb fragment of the targeted allele in genomic DNA digested with EcoRI (Fig. 1A and B). Appropriate integration of the 3′ end of the targeting construct was verified using a 0.7-kb 3′ probe on ES cell DNA digested with BamHI. This probe hybridized to a 6.7-kb fragment in the targeted allele, as opposed to a 7.5-kb fragment in the wild-type allele (Fig. 1A and C). Hybridization with a neo probe indicated that only a single integration event had occurred in all positive ES cell clones. Targeted ES cells were injected into C57BL/6J blastocysts to generate chimeras, and chimeric males transmitted the targeted allele to their offspring. Homozygous mutant mice were generated by heterozygote intercrosses. A probe corresponding to the Sox8 open reading frame failed to hybridize to genomic DNA from homozygous mice (data not shown). Genotyping was routinely performed by PCR analysis using a common upper primer located at bp 82 to 101 upstream of the start codon (5′-GTC CTG CGT GGC AAC CTT GG-3′) and two lower primers located at bp 308 to 327 (5′-GCC CAC ACC ATG AAG GCA TTC-3′) and 494 to 513 (5′-TAA AAA TGC GCT CAG GTC AA-3′) downstream of the start codon in Sox8 and lacZ, respectively. DNA was obtained from tail tips or, in the case of embryos, from yolk sacs. PCR was performed in 20-μl reaction mixtures containing standard buffer, 10% dimethyl sulfoxide, and a 0.25 μM concentration of each primer. The cycling conditions consisted of an initial 1-min denaturing step at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 56°C, and 30 s at 72°C. A 430-bp fragment was indicative of the wild-type allele, and a 617-bp fragment was indicative of the targeted allele. All analyses described in this paper were carried out with littermates of the F2, F3, or F4 generation.
Histological staining procedures, in situ hybridization, and X-ray and RT-PCR analyses.
Embryos were isolated at 9.5 to 18.5 days postcoitum (dpc) from staged pregnancies. Detection of β-galactosidase activity in whole-mount embryos and organs of adult mice was by standard procedures (10). After overnight fixation in 1% paraformaldehyde, embryos were incubated for several hours at 37°C in 1% X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) until blue precipitates were detectable. Prior to vibratome sectioning (100 μm), embryos were embedded in 2% agarose–phosphate-buffered saline. In situ hybridizations on paraformaldehyde-fixed vibratome sections using a digoxigenin-labeled antisense probe corresponding to the 3′ half of the Sox8 open reading frame and the 3′ untranslated region were as described previously (21).
For staining of bones and cartilage, skeletons were incubated at room temperature in Alizarin Red S–1% KOH (overnight) and then in Alcian Blue–20% acetic acid–80% ethanol (1 to 2 days). After bleaching in 10% KOH and fixation in ethanol, skeletons were stored in glycerol. X-ray pictures of adult mice were taken at 40 kV during a 1-min exposure.
RNAs from several organs of adult males were prepared using Trizol reagent (GibcoBRL), transcribed into cDNA using Superscript reverse transcriptase (GibcoBRL), and then used for reverse transcription-PCR (RT-PCR) analyses with primer pairs specific for the Sox8, lacZ, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) genes essentially as described previously (21). The following primer pairs were used: for the Sox8 gene, 5′-GTC CTG CGT GGC AAC CTT GG-3′ and 5′-GCC CAC ACC ATG AAG GCA TTC-3′, yielding a 0.43-kb product; for lacZ, 5′-GGT CGG CTT ACG GCG GTG ATT T-3′ and 5′-AGC GGC GTC AGC AGT TGT TTT T-3′, yielding a 0.59-kb product; and for the GAPDH gene, 5′-GCCATCAA(C/T)GACCCCTTCATT-3′ and 5′-CGCCTGCTTCACCACCTTCTT-3′, yielding a 0.7-kb product. Amplification products obtained after 23 cycles (GAPDH) or 28 cycles (Sox8 and lacZ) were separated for each gene on 2% agarose gels.
RESULTS
Targeted mutagenesis of Sox8.
The mouse Sox8 gene is localized within the t-complex on mouse chromosome 17 (23, 25). Its genomic organization is very similar to those of Sox9 and Sox10, with introns at conserved positions and all coding sequences confined to three exons (Fig. 1A). Exon 1 carries a short 5′ untranslated leader, and exon 3 carries a significantly longer 3′ untranslated region. To generate Sox8-deficient mice, we replaced the complete open reading frame and both introns by the lacZ marker gene and a neo cassette. The start codon of the lacZ marker was placed exactly over the start codon of the endogenous Sox8 gene. Transcription of the lacZ marker and transcription of the neo cassette were in the same direction. Detection of homologous recombination events in ES cell clones followed positive and negative selection procedures and was achieved by Southern blotting with a probe localized immediately upstream of the 5′ homology flank present in the targeting construct (Fig. 1A and B). After confirmation of the homologous recombination event with a probe taken from the region just outside the 3′ homology flank (Fig. 1A and C), a neo probe, and a probe corresponding to the Sox8 open reading frame (data not shown), chimeras were generated by blastocyst injection and germ line transmission was achieved (Fig. 1B and C). Backcrosses of heterozygous mice were performed to bring the knockout allele on a C57BL/6J background. All results described in this report were obtained with progeny of heterozygous mice belonging to generations N1 to N3.
In a large number of crosses between heterozygous and wild-type mice, approximately half of the progeny were heterozygous for the Sox8 deletion (Table 1), indicating that, contrary to observations with mice carrying a Sox10 deletion (6), there was no significant loss of heterozygous mice during the first postnatal weeks. Mice with a single intact copy of the Sox8 gene were indistinguishable from their wild-type littermates on gross anatomical and behavioral levels.
TABLE 1.
Genotype distribution in matings involving Sox8lacZ/+ micea
| Mating | % of total littermates
|
||
|---|---|---|---|
| Sox8+/+ | Sox8lacZ/+ | Sox8lacZ/lacZ | |
| Sox8lacZ/+ × Sox8+/+ | 49 | 45 | |
| Sox8lacZ/+ × Sox8lacZ/+ | 24 | 51 | 22 |
More than 15 litters were counted for both mating types. Mice that died before weaning and could not be genotyped account for the difference from 100%.
Heterozygous intercrosses were performed for the analysis of homozygous mice. Here, too, mice of all three genotypes were obtained in the expected Mendelian ratios (Table 1). Thus, mice homozygous for the Sox8 deletion complete embryonic development, are born, and are viable. They have normal life spans under standard housing conditions. Homozygous mice of both genders are fertile and can be used to establish a colony of homozygous Sox8-deficient mice.
Sox8 expression was checked in adult males of all three genotypes by RT-PCR (Fig. 1D). Among the tested tissues, we detected Sox8 expression in brain, spinal cord, and testis of wild-type mice, in agreement with previous analyses (25). There was no Sox8 expression in any tissue of homozygous mice, and there was no compensatory increase in expression levels of either Sox9 or Sox10 (Fig. 1D and data not shown). Instead, tissues that are positive for Sox8 in wild-type mice specifically expressed the lacZ marker, indicating that expression of the lacZ marker replaces Sox8 expression in our mouse model. Heterozygous littermates exhibited expression of both Sox8 and lacZ (data not shown).
Expression of the lacZ marker gene during embryonic development of Sox8-deficient mice.
As judged by comparison of in situ hybridization with a Sox8-specific antisense probe and detection of β-galactosidase activity using X-Gal staining, expression of the lacZ marker and Sox8 were highly similar during development (Fig. 2D and G and data not shown). Due to the high sensitivity of the X-Gal staining technique, we were therefore able to closely monitor Sox8 expression during development (Fig. 2).
FIG. 2.
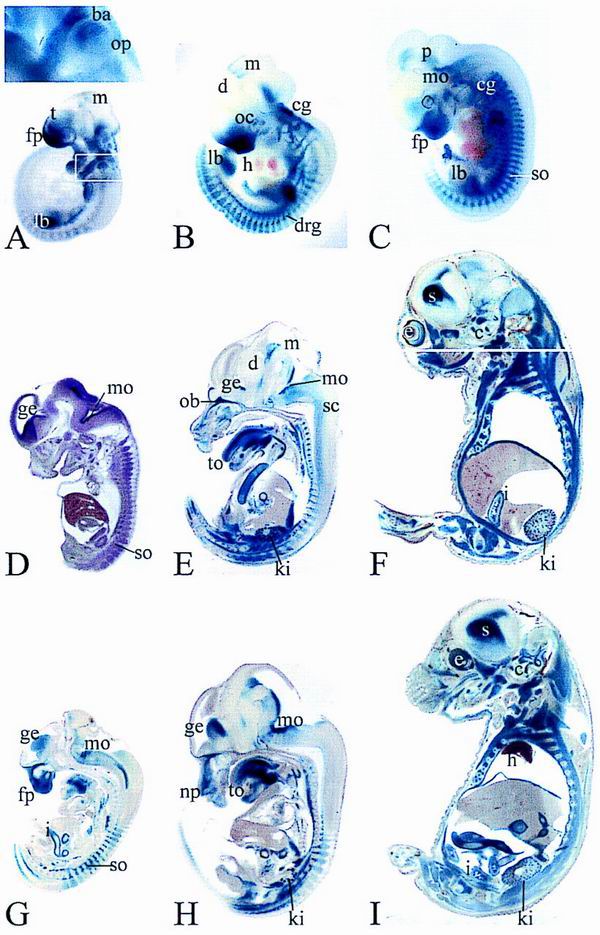
Developmental expression of Sox8 and Sox8lacZ. β-Galactosidase activity was detected colorimetrically using X-Gal substrate in age-matched whole-mount (A to C) and vibratome-sectioned (E to I) heterozygous (A to F) and homozygous (G to I) embryos at 9.5 dpc (A), 10.5 dpc (B), 11.5 dpc (C), 12.5 dpc (G), 14.5 dpc (E and H), and 16.5 dpc (F and I). Age-matched heterozygous and homozygous embryos were stained in parallel for identical times. No lacZ staining was detected in wild-type littermates under the conditions used. In panel A, the magnified area corresponds to the boxed region of the embryo. Panel F is a composite of two adjacent sections. Panel D shows in situ hybridization of a vibratome-sectioned embryo at 12.5 dpc with a Sox8-specific antisense riboprobe. The ventral surface is always to the left. Abbreviations: ba, branchial arches; c, cochlea; cg, cranial ganglia; d, diencephalon; drg, dorsal root ganglion; e, eye; fp, facial process; ge, ganglionic eminence; h, heart; i, intestine; k, kidney; l, limb bud; np, nasal pit; mo, medulla oblongata; m, mesencephalon; ob, olfactory bulb; oc, optic cup; op, otic placode; p, pons; so, somite; sc, spinal cord; s, striatum; t, telencephalon; to, tongue.
At 9.5 dpc, Sox8 is already present in various parts of the embryo (Fig. 2A). Expression levels are high in the facial process and the branchial arches. Additional Sox8-positive sites include the otic placode, cranial ganglia, and limb buds. Faint X-Gal staining in certain areas of the CNS and the forming somites is indicative of beginning Sox8 expression. Sox8 is thus found in cells of ectodermal and mesodermal origin, with particularly high levels in several neural crest derivatives. It is not restricted to derivatives of a single germ layer.
Through 10.5 and 11.5 dpc, expression in the facial mesenchyme recedes to the ventro-rostral region including the nasal invagination, thereby revealing the optic cup as a Sox8-positive site (Fig. 2B and C). Within the brain, there are several main areas of Sox8 expression. In addition to the pontine region of the hindbrain, there is a Sox8-specific signal in the ventricular zone at the border between diencephalon and mesencephalon. In the telencephalon, the olfactory bulb exhibits β-galactosidase activity (Fig. 2B and C). Fainter staining is observed in the basal telencephalon in a region that corresponds to the ganglionic eminence. Taking staining of the otic vesicle into account, Sox8 is found in all of the developing sensory organs (Fig. 2B and C). Sox8 is also detected in two parallel stripes of cells in the ventral region of the spinal cord (data not shown). Significant β-galactosidase activity is also detected in the sympathetic chain and in the enteric nervous system, showing that Sox8 is widely expressed throughout the PNS at this time of development (Fig. 2B and C). Intense staining in cranial ganglia and dorsal root ganglia masks the myotomes as a site of weaker expression at 10.5 dpc (Fig. 2B). At 11.5 dpc, expression levels in myotomes have caught up (Fig. 2C). With ongoing limb formation, Sox8 becomes restricted to the base of the limb buds. The heart is consistently negative for Sox8 throughout development, as are liver and adipose tissue.
Through 14.5 dpc, Sox8 expression remains strong in the enteric nervous system, whereas it slowly fades in other parts of the PNS (Fig. 2D to I). Nevertheless, at later times of development, residual β-galactosidase activity is still detectable in various nerves, such as the trigeminal nerve (Fig. 3D). In contrast to expression in the PNS, expression in distinct regions of the CNS continues to increase (Fig. 2D to I). Neuronal occurrence of Sox8 is evident in some nuclei of the pons and the medulla oblongata (Fig. 2D to I). In the forebrain, the lateral olfactory tract is heavily stained (Fig. 2D, E, G, and H). In the ganglionic eminence, Sox8 expression is detected in the ventricular and subventricular zones and in the mantle zone, indicating that it correlates with birth and terminal differentiation of striatal neurons (Fig. 2D, E, G, and H).
FIG. 3.
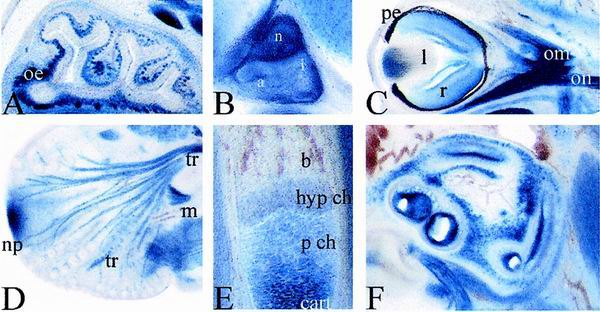
Developmental lacZ expression in various tissues of Sox8-deficient embryos. Higher magnifications of X-Gal-stained transverse (A and C) and sagittal (B, D, E, and F) vibratome sections of homozygous embryos at 15.5 dpc (A to D) and 16.5 dpc (E and F), showing nasal epithelium (A), pituitary (B), eye (C), trigeminus (D), humerus (E), and cochlea (F), are presented. Abbreviations: a, pars anterior of pituitary; b, bone; drg, dorsal root ganglia; cart, hyaline cartilage; hyp ch, hypertrophic chondrocytes; i, pars intermedia of pituitary; l, lens; m, muscle; n, pars nervosa of the pituitary; np, nasal pit; oe, olfactory epithelium; on; optic nerve; om, ocular muscle; p ch, proliferating chondrocytes; pe, pigment epithelium; r, retina; sc, spinal cord; tr, trigeminal nerve.
At 16.5 dpc, there is prominent Sox8 expression in all layers of the striatum, which by this time has developed from the ganglionic eminence (Fig. 2F and I). Additionally, Sox8 becomes strongly expressed throughout the ventricular zone of the brain. This is at a time when the ventricular zone switches from generation of mainly neuronal to primarily glial progenitors. Regions of the developing cerebellum also stain positive for Sox8. Within the eye, β-galactosidase activity is found in retina as well as the lens (Fig. 3C). Other sites of Sox8 expression in the head include nasal epithelium and innervation (Fig. 3A), cochlear epithelium (Fig. 3F), pituitary gland (Fig. 3B), and tooth buds. Throughout the body and limbs, skeletal muscles are major sites of Sox8 expression (e.g., the diaphragm, intercostal muscles, and ribs [Fig. 2E, F, H, and I]). Heart muscle and smooth muscle, in contrast, remain Sox8 negative. There is also strong Sox8 expression in cartilaginous and endochondral skeletal structures (e.g., the cartilage of nose and ear, thyroid and hyoid cartilage, cartilage of ribs, vertebrae, and limbs [Fig. 2E, F, and I]). In the forelimbs of 16.5-dpc-old embryos, β-galactosidase activity is high in the cartilagenous templates of humerus, radius, ulna, carpals, and metacarpals and in proliferating chondrocytes of growth plates (Fig. 3E). When chondrocytes become hypertrophic, X-Gal staining fades rapidly. The residual β-galactosidase activity is most likely due to the long half-life of the protein.
Expression in the kidney starts at the tip of the ureteric tree. From 14.5 dpc onwards it appears to be restricted to glomeruli and proximal tubules (Fig. 2E, F, H, and I; see Fig. 5A). Within the adrenal gland, Sox8 expression is restricted to the medulla (see Fig. 5A); within the male gonad, Sox8 occurs primarily within the testis cords (data not shown).
FIG. 5.
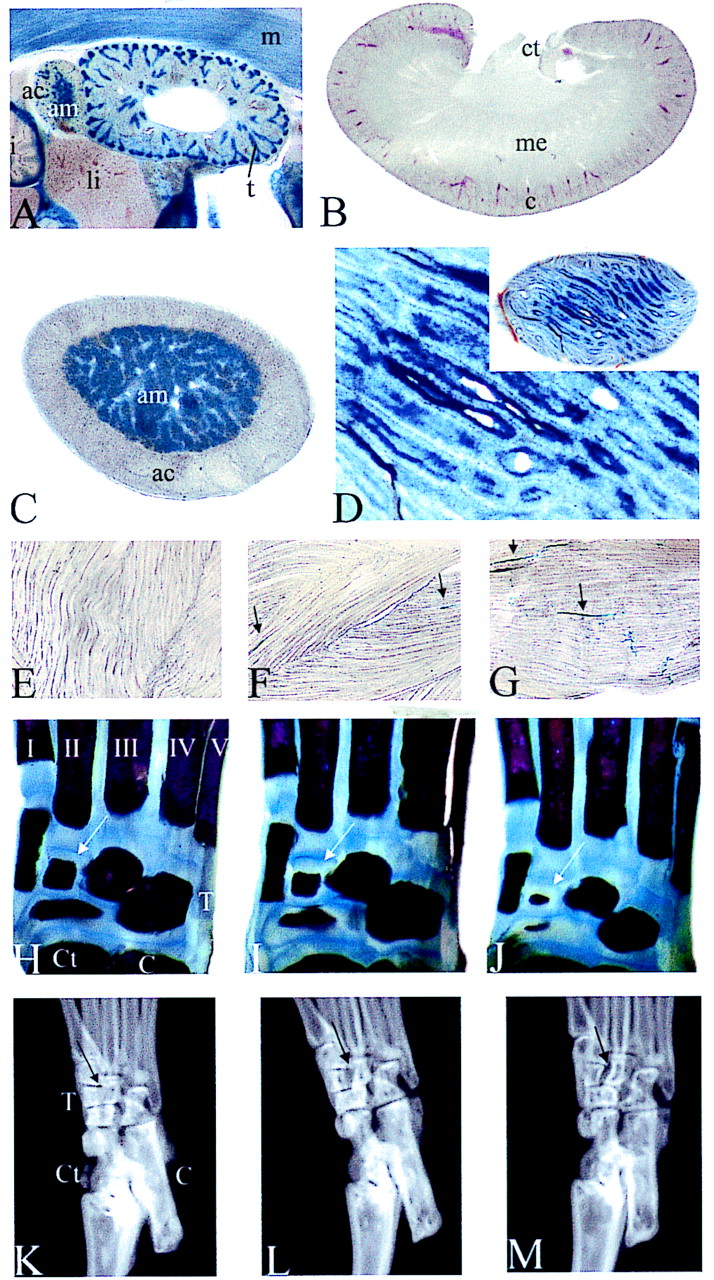
Tissue analyses in Sox8-deficient mice. (A to G) β-Galactosidase activity was detected colorimetrically using X-Gal substrate in vibratome-sectioned kidneys (A and B), adrenal gland (A and C), testis (D), and skeletal muscle (E to G) of 16.5-dpc-old embryos (A) and adult mice (B to G). Tissues were from homozygous (A to D and G), heterozygous (F), and wild-type (E) mice. Arrows in panels F and G point to X-Gal-stained cells or myotubes. (H to J) Alizarin Red S and Alcian Blue staining of bones and cartilage in the hind feet of 7-day-old wild-type (H), heterozygous (I), and homozygous (J) mice. Tarsals with genotype-dependent differing sizes are marked by arrows. (K to M) X-ray autoradiograms of hindfeet of adult wild-type (K), heterozygous (L), and homozygous (M) littermates. The arrows mark an area of differential ossification. Abbreviations: ac, adrenal cortex; am, adrenal medulla; C, calcaneus; c, cortex of the kidney; Ct, caput tali; ct, collecting tubule; li, liver; m, skeletal muscle; me, medulla of the kidney; T, tarsals; t, tubules; I to V, metatarsals.
Having established this detailed expression pattern, we used side-by-side comparison to detect differences of Sox8 expression in the homozygous Sox8-deficient embryos. However, expression patterns were identical with regard to both topology and chronology (compare Fig. 2D to F with Fig. 2G to I). Histological examination of Sox8-deficient embryos likewise failed to detect major developmental or structural alterations in Sox8-expressing areas (data not shown). Thus, we conclude that there is no major developmental defect in the absence of Sox8 expression.
Postnatal expression of Sox8.
The initial sites of Sox8 expression in the CNS correlate with regional generation and differentiation of neurons, arguing for an early role of Sox8 in regional specification of neuronal development. At later stages of embryonic development, Sox8 becomes prominently expressed in the ventricular zone. This is at a time when the ventricular zone switches from generation of mainly neuronal to primarily glial progenitors, arguing that Sox8 might have an additional role in gliogenesis. As gliogenesis in the CNS continues and actually peaks after birth, we monitored Sox8 expression in the CNS into adulthood (Fig. 4). During the first postnatal weeks, Sox8 is still detectable in neurons of striatum and hindbrain nuclei (Fig. 4A to C and data not shown). However, this expression is gradually reduced. At the same time, Sox8-positive cells from the ventricular zone spread throughout the brain. These cells appear to express Sox8 permanently. By morphology and location, these cells are primarily glia and include oligodendrocytes. In the adult striatum, there is strong Sox8 expression in oligodendrocytes of the fiber tracts and in isolated cells with astrocytic rather than neuronal morphology (Fig. 4J). Thus, in contrast to the case for the embryonic and early postnatal CNS, there is no evidence for significant neuronal expression of Sox8 in the adult striatum. In effect, the neuronal expression appears to be replaced by a primarily glial expression throughout the brain. Other examples of potentially astrocytic expression are visible in the external granular layer of the cerebellum, the septum or the dentate gyrus, and other parts of the hippocampus (Fig. 4K and L and data not shown). Intense X-Gal staining in the myelinated fiber tracts of the cerebrum (including the corpus callosum and anterior commissure) and the neuropil of the cerebellum are indicative of the presence of Sox8 in oligodendrocytes (Fig. 4E, F, K, and L). The pattern of Sox8-expressing cells in the adult spinal cord is also most compatible with a mixed glial expression (Fig. 4G to I). Comparative analysis of heterozygous and homozygous Sox8-deficient mice did not reveal significant differences in the type, number, or distribution of Sox8-expressing cells (compare Fig. 4B, E, and H with Fig. 4C, F, and I), arguing that there is no major loss or misorganization of these cells in the absence of Sox8.
FIG. 4.
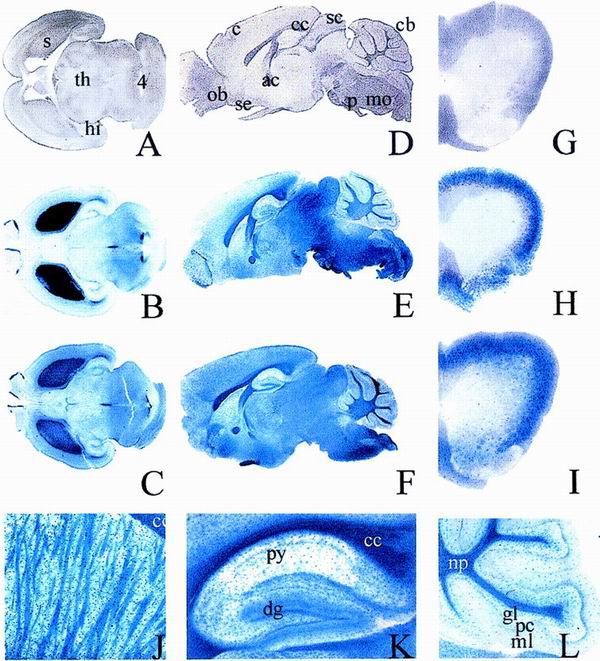
Postnatal CNS expression of the lacZ marker in Sox8-deficient mice. β-Galactosidase activity was detected colorimetrically using X-Gal substrate in age-matched vibratome-sectioned brains (A to F and J to L) and spinal cords (G to I) of wild-type (A, D, and G), heterozygous (B, E, and H), and homozygous (C, F, and I to L) littermates at birth (A to C) or at 4 months of age (D to L). Higher magnifications of homozygous adult brain include striatum (J), hippocampus (K), and cerebellum (L). (A to C and G to I) Transverse sections; (D to F, J, and K) sagittal sections. Abbreviations: ac, anterior commissure; c, cerebral cortex; cb, cerebrum; cc, corpus callosum; dg, dentate gyrus; gl, granular cell layer; hi, hippocampus; mo, medulla oblongata; ml, molecular layer; np, neuropil; ob, olfactory bulb; p, pons; pc, purkinje cell layer; s, striatum; sc, superior colliculus; se, septum; th, thalamus; 4, fourth ventricle.
Because of the strong expression in the developing kidney, adrenal gland, and testis, we also monitored Sox8 expression in these organs (Fig. 5A to D). With the conclusion of kidney morphogenesis, Sox8 expression was rapidly extinguished in both glomeruli and proximal tubuli so that there was no detectable Sox8-specific signal in the adult kidney (compare Fig. 5A and B). Again, there was no difference between heterozygous and homozygous mutant littermates (data not shown). In contrast to that in kidney, Sox8 expression in adrenal gland and testis persisted into adulthood. Sox8 expression in the adrenal gland was confined to the medulla at all times (Fig. 5A and C). The medulla was present in both the adult homozygous and adult heterozygous mice. In the adult testis, Sox8 expression was restricted to the interior lining of the testicular tubules (Fig. 5D). High-resolution analysis revealed localization to Sertoli cells and not to spermatogonia. Sertoli cells are present in homozygous Sox8-deficient males, and spermatogenesis proceeds normally, as expected from the normal fertility of these mice.
The muscle is another place of intense X-Gal staining and therefore of prominent Sox8 expression during late embryogenesis. However, in the adult skeletal muscle no significant amount of either Sox8 or lacZ transcripts was detected by RT-PCR (Fig. 1D). To reconcile these findings, we monitored Sox8 expression in muscle during postnatal development. At 1 week after birth, there is still significant Sox8 expression throughout the skeletal muscle. After 2 weeks, however, Sox8 expression has faded dramatically. In the heterozygous adult only very few cells exhibit X-Gal staining (Fig. 5F). Littermates of all genotypes exhibited comparable muscle morphologies at this level of resolution (Fig. 5E to G). Interestingly, there are approximately twice as many LacZ-positive cells in the adult homozygous skeletal muscle as in the heterozygous skeletal muscle (Fig. 5F and G). In addition, we observed selective X-Gal staining in the endplates of neuromuscular junctions from homozygous Sox-8-deficient mice (Fig. 5G).
Skeletons of wild-type and homozygous Sox8-deficient littermates do not exhibit conspicuous differences (Fig. 6B and data not shown). All major bones appear to be present in normal size and shape. However, closer inspection of the hind foot revealed a significant reduction in the size of several tarsals at postnatal day 7 (Fig. 5H to J) and a resulting failure of these tarsals to fully fuse in the adult (Fig. 5K to M). Interestingly, heterozygotes display an intermediate phenotype at both time points, arguing that the effect of Sox8 on this phenotype is dose dependent.
FIG. 6.

Growth parameters of Sox8-deficient mice after birth. (A) Body length of age-matched adult wild-type (+/+) and homozygous (−/−) female (f) and male (m) mice measured from nose to anus (n ≥ 10 for each genotype and sex). (B) X-ray autoradiograms of age-matched adult female mice. (C to F) Absolute weight (C and E) and weight gains (D and F) of wild-type (open symbols) and homozygous Sox8-deficient (filled symbols) littermates (n ≥ 6 for each genotype and sex) were monitored for the first 100 days after birth for both males (C and D) and females (E and F). Data are means ± standard errors of the means; statistically significant differences from wild-type controls were observed in weight measurements from postnatal day 19 onwards (P < 0.05 for males and P < 0.001 for females by Student's t test).
Weight reduction of Sox8-deficient mice.
Analysis of postnatal development also failed to reveal major developmental alterations, apparent losses, or obvious defects in organ systems. Nevertheless, adult homozygous mice exhibit a severe weight reduction relative to their wild-type littermates (Fig. 6C to F). In both sexes, homozygous mice are approximately 30% lighter than their wild-type littermates (Fig. 6C and E). This weight difference was not apparent during the first two postnatal weeks. In the following weeks, however, homozygous mice gained consistently less weight than wild-type mice (Fig. 6D and F). The reduction in body weight was not paralleled by a shorter length, measured from nose to anus (Fig. 6A). Homozygous Sox8-deficient mice were on average less than 10% shorter than their wild-type littermates, and the difference was not statistically significant. The difference in weight was due to smaller adipose stores (Fig. 6B). Together with our analyses of developmental processes in these mice, this finding seems to indicate that the Sox8-specific defect is based on physiological or metabolic differences rather than on a gross anatomical change. The exact cause of this defect will have to be determined in future studies.
DISCUSSION
Using homologous recombination, we have deleted the gene for Sox8 in mouse. We expected a profound developmental defect in at least one of the tissues and organs that exhibit transient or permanent Sox8 expression, for two reasons. Strong Sox8 expression coincides in many organs with essential phases of their development, and defects in these organs have been shown to cause severe phenotypes in other mouse models. Additionally, inactivation or deletion of the highly related Sox9 and Sox10 genes has serious consequences for embryonic development, with whole cell lineages being eliminated. Chondrocytes fail to form in the absence of Sox9 (4), whereas melanocytes, peripheral glia, and the enteric nervous system are missing in the absence of Sox10 (6, 9, 12, 28). The importance of Sox9 and Sox10 is further documented by the occurrence of haploinsufficiency. Inactivation of only one Sox9 or Sox10 allele manifests itself in such syndromes as campomelic dysplasia and Waardenburg-Hirschsprung syndrome (8, 24, 31). Thus, it came as a complete surprise that even homozygous Sox8 mutant mice are viable and show only a mild defect that manifests itself mainly in reduced weight from postnatal week 3. We were unable to detect gross developmental defects in any of the Sox8-expressing tissues and organs, including CNS, neural crest derivatives, myotomes, skeletal muscles, cartilage, kidney, adrenal gland, testis, eye, ear, and nose.
How can we explain the relatively mild phenotype in Sox8-deficient mice? It is noteworthy that expression of Sox8 overlaps very strongly with expression of either Sox9, Sox10, or both. Thus, all three Sox proteins are coexpressed in various derivatives of the neural crest. One prominent example is the facial mesenchyme (6, 17, 33). Cranial ganglia, dorsal root ganglia, and sympathetic ganglia also express Sox10, and to a lesser degree Sox9, in addition to Sox8 (6, 14, 17, 33). In the enteric nervous system, expression is well documented for both Sox8 and Sox10 (14). There are also sites of coexpression outside the neural crest. The otic vesicle and cochlea, for instance, express all three members of group E of Sox proteins (6, 17, 33). Sox8 expression in kidney and testis coincides exactly with expression of Sox9 in these tissues (13, 33). Chondrocytes express substantial amounts of Sox9 and low levels of Sox10 in addition to Sox8 (6, 17, 33). This pattern of coexpression also holds true for the adult CNS. Sox8 appears to be expressed in astrocytes and oligodendrocytes, whereas Sox9 is found primarily in astrocytes and Sox10 is found primarily in oligodendrocytes (14; M. Wegner, unpublished data).
Sox8 expression is thus not only broader than expression of either Sox9 (33) or Sox10 (14). Its pattern also corresponds to a large extent to the composite expression pattern of Sox9 and Sox10. Thus, one would have to argue that these three group E Sox proteins function redundantly and that in most tissues the loss of Sox8 can be compensated for by either one of the closely related Sox proteins. At least in the adult, functional compensation does not require a compensatory increase of Sox9 or Sox10 expression levels as indicated by our RT-PCR analyses.
Neither Sox9 nor Sox10 has so far been reported in muscle, lens, or striatal neurons, indicating that expression in these cells might be fairly unique to Sox8 among group E Sox proteins. Redundancy therefore appears to be an unlikely explanation for the lack of a phenotype in these particular cell types, unless one wants to invoke functional compensation by more distantly related Sox proteins expressed in these tissues (3, 18).
Furthermore, if redundancy explains the lack of a phenotype in Sox8-deficient animals, why does the same redundancy fail to provoke a similar lack of phenotype after inactivation of Sox9 or Sox10? After all, there is a loss of enteric neural crest in Sox10-deficient mice, and there is a block in chondrocyte development in the absence of Sox9 despite expression of Sox8 in these cells (4, 9, 28). Thus, these three Sox proteins do not seem capable of compensating for each other's loss in every coexpressing tissue. Either group E Sox proteins have diverged in function in a tissue-specific manner, or what appears to be a simple coexpression is instead a complex cross-regulatory network between these Sox proteins. One may regulate expression of the other in a manner that varies from tissue to tissue and that might even be different in a given tissue at various times of development. Deletion of the upstream Sox would then simultaneously lead to changed expression of the downstream Sox. There are precedents for the existence of such regulatory networks. Phox2a and Phox2b, for instance, are paired-type homeodomain proteins with strongly overlapping expression (19). Depending on the tissue, one activates expression of the other. In Phox2a- or Phox2b-knockout mice, developmental defects in a given tissue are usually more pronounced if the deleted gene represents the upstream effector in this tissue (19, 20). Detailed comparative expression analyses and the generation of double or triple knockouts will be needed to clarify this issue.
In light of the mild consequences of Sox8 deficiency and the severe phenotypes associated with Sox9 or Sox10 mutations, it also deserves to be mentioned that conservation between species is more pronounced for Sox9 and Sox10 than for Sox8. For Sox9 and Sox10, the amino acid identities between human and mouse proteins are 95 and 98%, respectively; that for Sox8 is only 84%. This lower degree of conservation might be indicative of a less fundamental role in development.
Also, one has to assess our results in light of the previously formulated hypotheses that Sox8 might be involved in the ATR-16 syndrome, which is characterized by α-thalassemia, facial malformations, and mental retardation, or in CATM (23, 25). ATR-16 syndrome is a deletion syndrome with usually more than 1 Mb deleted. The α-thalassemia appears to be due to loss of one or two α-globin genes. Thus, if anything, loss of Sox8 would have to be causative of mental retardation or facial malformations. Taking the phenotype of Sox8-deficient mice into account, loss of Sox8 appears to be an unlikely cause for the facial malformations observed in ATR-16 syndrome. In light of the transient expression of Sox8 in neuronal CNS populations (especially in the striatum and hindbrain), a contribution to mental retardation cannot be ruled out. However, the lack of cytoarchitectural defects in Sox8-expressing CNS regions and the almost complete switch of Sox8 expression to glia in the adult CNS do not suggest a strong link to mental retardation.
With regard to an involvement of Sox8 mutations in CATM, Sox8 expression in the developing eye might be relevant. However, the Sox8-deficient mice do not develop any signs of microphthalmia or cataract. It has to be noted, though, that in the reported case, CATM resulted from a balanced translocation (25). We cannot exclude a translocation event that altered the expression or functional characteristics of Sox8. Straightforward inactivation of Sox8 by translocation, however, would in all likelihood not suffice to cause CATM.
Given the phenotype of Sox8-deficient mice, the Sox8 gene also appears to be an unlikely candidate for the gene responsible for the mesodermal cell migration defects and early lethality of the tw18 mouse mutant, despite the close chromosomal localization (25).
Finally, we have shown that Sox8-deficient mice exhibit a significant weight reduction, most likely due to a significant decrease in fat tissue. As indicated above, however, adipose tissue does not express Sox8, and there is no major histological abnormality in this tissue. Its reduction is therefore most likely due to changes in energy homeostasis and its regulation, such as reduced efficacy of food utilization, increased energy expenditure through decreased sleep or increased locomotor activity during wakefulness, and disproportionate energy allocation. There is no reduction in food intake (data not shown). Causes for these physiological changes may lie in disturbances affecting various hormonal systems at different levels, from CNS and pituitary to endocrine organ and target tissues. Good candidates include the growth hormone-IGF1 axis, the leptin-melanocortin pathway, and various steroid and thyroid hormones. Because of these many possibilities, the extensive cross-regulation between hormonal systems, and the broad expression of Sox8, it is difficult to favor a particular hypothesis at this time. Only in-depth physiological analysis of these mice will clarify this issue in the future.
ACKNOWLEDGMENTS
We thank C. Haas for help with X-ray pictures, P. Lommes for in situ hybridization, and T. Mordhorst for expert technical assistance.
REFERENCES
- 1.Bell D M, Leung K K H, Wheatley S C, Ng L J, Zhou S, Ling K W, Sham M H, Koopman P, Tam P P L, Cheah K S E. Sox9 directly regulates the type-II collagen gene. Nat Genet. 1997;16:174–178. doi: 10.1038/ng0697-174. [DOI] [PubMed] [Google Scholar]
- 2.Bell K M, Western P S, Sinclair A H. Sox8 expression during chick embryogenesis. Mech Dev. 2000;94:257–260. doi: 10.1016/s0925-4773(00)00296-3. [DOI] [PubMed] [Google Scholar]
- 3.Beranger F, Mejean C, Moniot B, Berta P, Vandromme M. Muscle differentiation is antagonized by SOX15, a new member of the SOX protein family. J Biol Chem. 2000;275:16103–16109. doi: 10.1074/jbc.275.21.16103. [DOI] [PubMed] [Google Scholar]
- 4.Bi W, Deng J M, Zhang Z, Behringer R R, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 5.Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol. 2000;227:239–255. doi: 10.1006/dbio.2000.9883. [DOI] [PubMed] [Google Scholar]
- 6.Britsch S, Goerich D E, Riethmacher D, Peirano R I, Rossner M, Nave K A, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster J W, Dominguez-Steglich M A, Guioli S, Kwok C, Weller P A, Stevanovic M, Weissenbach J, Mansour S, Young I D, Goodfellow P N, Brook J D, Schafer A J. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525–530. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 8.Foster J W, Graves J A. An SRY-related sequence on the marsupial X chromosome: implications for the evolution of the mammalian testis-determining gene. Proc Natl Acad Sci USA. 1994;91:1927–1931. doi: 10.1073/pnas.91.5.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herbarth B, Pingault V, Bondurand N, Kuhlbrodt K, Hermans-Borgmeyer I, Puliti A, Lemort N, Goossens M, Wegner M. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc Natl Acad Sci USA. 1998;95:5161–5165. doi: 10.1073/pnas.95.9.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 11.Inoue K, Tanabe Y, Lupski J R. Myelin deficiencies in both the central and peripheral nervous system associated with a SOX10 mutation. Ann Neurol. 1999;46:313–318. doi: 10.1002/1531-8249(199909)46:3<313::aid-ana6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Kapur R P. Early death of neural crest cells is responsible for total enteric aganglionosis in Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatr Dev Pathol. 1999;2:559–569. doi: 10.1007/s100249900162. [DOI] [PubMed] [Google Scholar]
- 13.Kent J, Wheatley S C, Andrews J E, Sinclair A H, Koopman P. A male-specific role for SOX9 in vertebrate sex determination. Development. 1996;122:2813–2822. doi: 10.1242/dev.122.9.2813. [DOI] [PubMed] [Google Scholar]
- 14.Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18:237–250. doi: 10.1523/JNEUROSCI.18-01-00237.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefebvre V, Huang W, Harley V R, Goodfellow P N, DeCrombrugghe B. Sox9 is a potent activator of the chondrocyte-specific enhancer of the proα1(II) collagen gene. Mol Cell Biol. 1997;17:2336–2346. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Li H, Tanaka K, Tsumaki N, Yamada Y. Identification of an enhancer sequence within the first intron required for cartilage-specific transcription of the alpha2(XI) collagen gene. J Biol Chem. 2000;275:12712–12718. doi: 10.1074/jbc.275.17.12712. [DOI] [PubMed] [Google Scholar]
- 17.Ng L-J, Wheatley S, Muscat G E O, Conway-Campbell J, Bowles J, Wright E, Bell D M, Tam P P L, Cheah K S E, Koopman P. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108–121. doi: 10.1006/dbio.1996.8487. [DOI] [PubMed] [Google Scholar]
- 18.Nishiguchi S, Wood H, Kondoh H, Lovell-Badge R, Episkopou V. Sox1 directly regulates the γ-crystallin genes and is essential for lens development in mice. Genes Dev. 1998;12:776–781. doi: 10.1101/gad.12.6.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pattyn A, Morin X, Cremer H, Goridis C, Brunet J F. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065–4075. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- 20.Pattyn A, Morin X, Cremer H, Goridis C, Brunet J F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 21.Peirano R I, Goerich D E, Riethmacher D, Wegner M. Protein zero expression is regulated by the glial transcription factor Sox10. Mol Cell Biol. 2000;20:3198–3209. doi: 10.1128/mcb.20.9.3198-3209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pevny L H, Lovell-Badge R. Sox genes find their feet. Curr Opin Genet Dev. 1997;7:338–344. doi: 10.1016/s0959-437x(97)80147-5. [DOI] [PubMed] [Google Scholar]
- 23.Pfeifer D, Poulat F, Holinski-Feder E, Kooy F, Scherer G. The SOX8 gene is located within 700 kb of the tip of chromosome 16p and is deleted in a patient with ATR-16 syndrome. Genomics. 2000;63:108–116. doi: 10.1006/geno.1999.6060. [DOI] [PubMed] [Google Scholar]
- 24.Pingault V, Bondurand N, Kuhlbrodt K, Goerich D E, Prehu M-O, Puliti A, Herbarth B, Hermans-Borgmeyer I, Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I, Romeo G, Smith J C, Read A P, Wegner M, Goossens M. Sox10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 25.Schepers G E, Bullejos M, Hosking B M, Koopman P. Cloning and characterisation of the sry-related transcription factor gene sox8. Nucleic Acids Res. 2000;28:1473–1480. doi: 10.1093/nar/28.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekiya I, Tsuji K, Koopman P, Watanabe H, Yamada Y, Shinomiya K, Nifuji A, Noda M. SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem. 2000;275:10738–10744. doi: 10.1074/jbc.275.15.10738. [DOI] [PubMed] [Google Scholar]
- 27.Southard-Smith E M, Angrist M, Ellison J S, Agarwala R, Baxevanis A D, Chakravarti A, Pavan W J. The Sox10(Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 1999;9:215–225. [PubMed] [Google Scholar]
- 28.Southard-Smith E M, Kos L, Pavan W J. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat Genet. 1998;18:60–64. doi: 10.1038/ng0198-60. [DOI] [PubMed] [Google Scholar]
- 29.Touraine R L, Attie-Bitach T, Manceau E, Korsch E, Sarda P, Pingault V, Encha-Razavi F, Pelet A, Auge J, Nivelon-Chevallier A, Holschneider A M, Munnes M, Doerfler W, Goossens M, Munnich A, Vekemans M, Lyonnet S. Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am J Hum Genet. 2000;66:1496–1503. doi: 10.1086/302895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tybulewicz V L, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 31.Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli F D, Keutel J, Hustert E, Wolf U, Tommerup N, Schempp W, Scherer G. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene Sox9. Cell. 1994;79:1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 32.Wegner M. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 1999;27:1409–1420. doi: 10.1093/nar/27.6.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright E, Hargrave M R, Christiansen J, Cooper L, Kun J, Evans T, Gangadharan U, Greenfield A, Koopman P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat Genet. 1995;9:15–20. doi: 10.1038/ng0195-15. [DOI] [PubMed] [Google Scholar]
- 34.Xie W F, Zhang X, Sakano S, Lefebvre V, Sandell L J. Trans-activation of the mouse cartilage-derived retinoic acid-sensitive protein gene by Sox9. J Bone Miner Res. 1999;14:757–763. doi: 10.1359/jbmr.1999.14.5.757. [DOI] [PubMed] [Google Scholar]