

Abstract
We have isolated a cDNA homologous to known dual-specificity phosphatases from a mouse macrophage cDNA library and termed it MKP-M (for mitogen-activated protein kinase phosphatase isolated from macrophages). Three other presumed splice variant isoforms have also been identified for MKP-M. The longest and most abundant mRNA contains an open reading frame corresponding to 677 amino acids and produces an 80-kDa protein. The deduced amino acid sequence of MKP-M is most similar to those of hVH-5 (or mouse M3/6) and VHP1, a Caenorhabditis elegans tyrosine phosphatase. It includes an N-terminal rhodanase homology domain, the extended active-site sequence motif (V/L)X(V/I)HCXAG(I/V)SRSXT(I/V)XXAY(L/I)M (where X is any amino acid), and a C-terminal PEST sequence. Northern blot analysis revealed a dominant MKP-M mRNA species of approximately 5.5 kb detected ubiquitously among all tissues examined. MKP-M was constitutively expressed in mouse macrophage cell lines, and its expression levels were rapidly increased by lipopolysaccharide (LPS) stimulation but not by tumor necrosis factor alpha (TNF-α), gamma interferon, interleukin-2 (IL-2), or IL-15 stimulation. Immunocytochemical analysis showed MKP-M to be present within cytosol. When expressed in COS7 cells, MKP-M blocks activation of mitogen-activated protein kinases with the selectivity c-Jun N-terminal kinase (JNK) ≫ p38 = extracellular signal-regulated kinase. Furthermore, expression of a catalytically inactive form of MKP-M in a mouse macrophage cell line increased the intensity and duration of JNK activation and TNF-α secretion after LPS stimulation, suggesting that MKP-M is at least partially responsible for the desensitization of LPS-mediated JNK activation and cytokine secretion in macrophages.
Mitogen-activated protein kinases (MAPKs) are activated by phosphorylation of threonine and tyrosine residues within a signature sequence of T-X-Y by dual-specificity MAPK kinases (MKKs). MKKs are in turn phosphorylated and activated by a family of serine/threonine MKK kinases (47). Extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) (also called stress-activated protein kinase), and p38 (also called RK or CSBP) are distinct classes of MAPKs playing important roles in various cellular events. JNKs are activated by diverse stimuli, including DNA damage, heat shock, bacterial components, inflammatory cytokines, and Fas (31). Activated JNKs play an essential role in the activation of transcriptional factors, such as c-Jun (21), ATF-2 (16), Elk-1 (57), and ets-2 (50). In macrophages, activated JNKs mediate the expression of inducible nitric oxide synthase (6), cyclooxygenase-2 (56), chemokines such as RANTES (23), and cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and IL-6 (39), all of which not only potently activate host defense mechanisms but also often lead to excessive inflammatory responses in microbial infection.
MAPK activation is a reversible process, and an emerging family of dual-specificity protein phosphatases (DSPs) have been shown to inactivate MAPKs through dephosphorylation of both threonine and tyrosine residues that are essential for the enzymatic activity (4). DSPs share two common features: a catalytic domain with significant amino acid sequence homology to a vaccinia virus DSP, VH-1, and an N-terminal region homologous to the catalytic domain of the cdc25 phosphatase (rhodanase homology domain). Among DSP family members, some show highly selective substrate specificity while others efficiently inactivate all three classes of MAPKs. Interestingly, gene expression of many DSPs is significantly induced following stimulation with growth factors, cytokines, or cell stresses, and this induction of MAPK phosphatases (MKPs) may function as a negative feedback mechanism of MAPK activity.
In this study, in order to elucidate the regulatory mechanisms of MAPK pathways in macrophages, we screened a cDNA library from a mouse macrophage cell line with a cDNA probe of MKP-1, a known MKP. We isolated a partial cDNA containing the extended active-site sequence motif, (V/L)X(V/I)HCXAG(I/V)SRSXT(I/V)XXAY(L/I)M (where X is any amino acid), conserved in all DSPs (27, 35, 41). By rescreening the library and extending the cDNA by the method of 5′ rapid amplification of cDNA ends (5′-RACE), we have isolated a full-length cDNA, which we named MKP-M (for MKP isolated from macrophages). It includes an N-terminal rhodanase homology domain, the extended active-site sequence motif mentioned above, and a C-terminal PEST sequence. MKP-M was constitutively expressed in mouse macrophage cell lines, and its mRNA expression levels were rapidly induced by stimulation with lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria, but not by TNF-α, gamma interferon (IFN-γ), IL-2, or IL-15 stimulation. When expressed in COS7 cells, MKP-M inactivated JNK more significantly than p38 or ERK. Expression of a dominant-negative form of MKP-M in a mouse macrophage cell line increased the levels and duration of JNK activation and TNF-α secretion after LPS stimulation. These results indicated that MKP-M is at least partially responsible for the downregulation of LPS-mediated JNK activation and TNF-α secretion in macrophages, possibly preventing excessive inflammatory responses.
MATERIALS AND METHODS
Antibodies.
A polyclonal antibody against MKP-M was prepared by immunizing rabbits with a glutathione S-transferase (GST)–MKP-M fusion protein encompassing amino acids 297 to 373 of MKP-M. This region of MKP-M is not homologous to any other known phosphatases as determined by a database search. Affinity purification was achieved by passing the crude serum through a column containing the immunogen immobilized on glutathione-Sepharose beads (Amersham Pharmacia Biotech, Inc.). An antiphosphotyrosine monoclonal antibody, 4G10, was purchased from Upstate Biotechnology Inc. (Lake Placid, N.Y.). An antihemagglutinin (anti-HA) monoclonal antibody, 12CA5, and a polyclonal anti-p38 antibody were obtained from Santa Cruz Biotech (Santa Cruz, Calif.). The anti-FLAG M2 monoclonal antibody was purchased from Sigma Chemical Co. (St. Louis, Mo.). A polyclonal anti-JNK1 antibody was purchased from Santa Cruz Biotechnology. A phospho-specific anti-p38 polyclonal antibody and a polyclonal anti-ERK antibody were purchased from New England Biolabs (Beverly, Mass.).
Reagents.
Recombinant mouse IL-2, IL-1β, TNF-α, and IFN-γ and human IL-15 were purchased from Peprotech Corporation (Seattle, Wash.). PD98059, a specific inhibitor of ERK kinase (MEK), and SB208530, a specific inhibitor of p38 MAPK, were purchased from Calbiochem (San Diego, Calif.). LPS from Escherichia coli serotype B6:026, anisomycin, phorbol myristate acetate (PMA), dicoumarol, and p-nitrophenyl phosphate (pNPP) were obtained from Sigma Chemical Co. RPMI 1640 medium and Dulbecco's modified Eagle medium were from Gibco BRL (Rockville, Md.). Synthetic E. coli-type lipid A (compound 506) was a generous gift from T. Ogawa and has been previously described (37, 46).
Cells.
All cell lines were grown in tissue culture flasks at 37°C in 5% CO2–95% air and passaged every 2 or 3 days to maintain logarithmic growth. A mouse macrophage cell line, RAW264.7, and a monkey kidney cell line, COS7, were obtained from the RIKEN cell bank (Tsukuba, Japan) and maintained in Dulbecco's modified Eagle medium with 10% fetal calf serum (Sigma Chemical Co.).
Isolation of the full-length cDNA clone encoding mouse MKP-M.
In an effort to clone the cDNA for a DSP expressed in macrophages, a reverse transcription-PCR (RT-PCR)-prepared cDNA encoding the N-terminal domain of the mouse MKP-1 was 32P labeled by random priming and used for screening a J774.1 cDNA library constructed in the Uni-Zap cloning vector (Stratagene, La Jolla, Calif.), kindly provided by H. Yagita (Juntendo University, Tokyo, Japan). After the transfer of the phage plaques, the nitrocellulose membranes were UV cross-linked and soaked in prehybridization solution (6× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 5× Denhardt's reagent, 0.5% sodium dodecyl sulfate [SDS], 100 mg of denatured salmon sperm DNA per ml, and 50% formamide) for 3 h at 42°C, followed by incubation with 32P-labeled probe in hybridization solution (6× SSC, 0.5% SDS, 100 mg of denatured salmon sperm DNA per ml, and 50% formamide) for 14 h at 42°C. The membranes were washed in 2× SSC–0.1% SDS for 10 min twice at room temperature and in 1× SSC–0.1% SDS for 10 min twice at room temperature and were exposed to RX-U films (Fuji Film, Tokyo, Japan). The inserts of the positive phage clones were excised using the Rapid Excision Kit (Stratagene) according to the manufacturer's instructions to generate subclones in the pBK-CMV plasmid (Stratagene). DNA sequence analysis was performed on these plasmid clones using a DNA sequencer (model 373A) and a Thermo Sequence cycle sequencing kit (PE Biosystems, Foster City, Calif.). The 4.2-kb insert of one positive clone seemed to encode a protein containing the extended active-site sequence motif but not identical with any known phosphatase sequences. This cDNA insert was recloned into the pBluescript II vector, yielding pBS-MKP-M-1. A set of oligonucleotides was prepared based on the nucleotide sequence of pBS-MKP-M-1 and used for 5′ extension with J774.1 total cDNA as a template and a 5′-RACE kit from Gibco BRL. The 5′-RACE DNA fragment was cloned into pBS-MKP-M-1 to generate a plasmid containing the full-length MKP-M cDNA, which was named pBS-MKP-M full.
Mammalian expression plasmids.
The coding region of the mouse MKP-M cDNA was amplified by PCR using a pair of primers (CGACGCGTATGGCCCATGAGATGATTGGAA and CGACGCGTCTATTTTTTGTGAACAGGAAAC) from pBS-MKP-M full and inserted into the MluI site of the expression plasmid pEFBOS-FLAG or pEFBOS-Myc (36), which encode an N-terminal FLAG or Myc epitope, respectively, to yield pEFBOS-FLAG/MKP-M or pEFBOS-Myc/MKP-M, respectively.
The C244S mutation in the phosphatase catalytic domain was constructed by recombinant PCR (24) using four primers: CGACGCGTATGGCCCATGAGATGATTGGAA, CGACGCGTCTATTTTTTGTGAACAGGAAAC, GTGCTTATCCACAGCTTA, and GATCCCAGCTAAGCTGTG. The PCR product carrying the nucleotide substitution was cloned into the MluI site of pEFBOS-FLAG or pEFBOS-Myc to yield pEFBOS-FLAG/MKP-M-CS or pEFBOS-Myc/MKP-M-CS, respectively. The one-nucleotide substitution (from TGC to AGC) was confirmed by nucleotide sequencing.
The cDNA for the C-terminally truncated MKP-M (encoding amino acids 1 to 435 of MKP-M A1 and excluding the C-terminal PEST sequence) was constructed by PCR with the primer pair CGACGCGTATGGCCCATGAGATGATTGGAA and CGACGCGTCTAGTTGGTCCCATCCAGTGTG, using pBS-MKP-M full or pEFBOSFLAG-MKP-M-CS as a template. The 1.3-kb PCR product was digested with MluI and cloned into pEFBOS-FLAG or pEFBOS-Myc to yield expression plasmids pEFBOS-FLAG-MKP-M-trunc, pEFBOS-FLAG-MKP-M-CS-trunc, pEFBOS-Myc-MKP-M-trunc, and pEFBOS-Myc-MKP-M-CS-trunc. The NheI-NheI insert (including FLAG tag cDNA) of pEFBOS-FLAG-MKP-M-trunc or pEFBOS-FLAG-MKP-M-CS-trunc was introduced into the NheI site of pcDNA3.1(+). The plasmid encoding MKP-M in the right direction was termed pcDNA3.1(+)-FLAG-MKP-M-trunc or pcDNA3.1(+)-FLAG-MKP-M-CS-trunc, respectively.
For the expression plasmid of the mouse MKP-1, the coding region of the mouse MKP-1 cDNA was amplified by RT-PCR using the primer pair CGACGCGTATGGTGATGGAGGTGGGCATCC and CGACGCGTTAAATAAATAAGGACCAGCTCC from total RNA isolated from J774.1 cells and inserted into the MluI site of the expression plasmid pEFBOS-FLAG to yield pEFBOS-FLAG/MKP-1.
The C258S mutation in the phosphatase catalytic domain was constructed by recombinant PCR using four primers: CGACGCGTATGGTGATGGAGGTGGGCATCC, GGAGATGCCGGCCTGGCTATGAAC, CGACGCGTTAAATAAATAAGGACCAGCTCC, and AGAGTGTTTGTTCATAGCCAGGCC. The PCR product carrying the nucleotide substitution was cloned into the MluI site of pEFBOS-Myc to yield pEFBOS-Myc/MKP-1-CS. The one-nucleotide substitution (from TGC to AGC) was confirmed by nucleotide sequencing.
The Myc-tagged expression plasmids for M3/6, pcDNA3-M3/6, and its CS mutant, cDNA3-M3/6-CS, have been previously described (25).
For the expression plasmids of MAPKs, the coding regions of mouse ERK2, JNK2, and p38α were amplified by RT-PCR from total RNA isolated from J774.1 and inserted with the N-terminal HA tag into a mammalian expression vector, pcDNA3 (Invitrogen, Carlsbad, Calif.), to yield pcDNA3-HA/ERK2, pcDNA3-HA/JNK2, and pcDNA3-HA/p38α, respectively.
The structures of all plasmid constructs were verified by restriction enzyme mapping and nucleotide sequencing.
MKP-M expression in E. coli.
For bacterial expression of MKP-M, open reading frames (ORFs) of MKP-M were subcloned into pGEX5X2 (Amersham Pharmacia Biotech, Inc.) as follows. For the generation of pGEX5X2-MKP-M full WT and pGEX5X2-MKP-M full CS, full-length MKP-M cDNA was amplified by PCR using the primer pair CGGGATCCCCATGGCCCATGAGAT GATTGG and GCGTCGACTATTTTTTGTGAACAGGAAACA with pBS-MKP-M full or pEFBOSFLAG-MKP-M-CS as a template, respectively. The PCR products were inserted into the BamHI and SalI sites of pGEX5X2. For the generation of pGEX5X2-MKP-M trunc WT and pGEX5X2-MKP-M trunc CS, PCR was performed using the primer pair CGGGATCCCCATGGCCCATGAGATGATTGG and GCGTCGACTAGTTGGTCCCATCCAGTGTGG with pBS-MKP-M full or pEFBOSFLAG-MKP-M-CS as a template. The PCR products were inserted into the BamHI and SalI sites of pGEX5X2. The four plasmids generated, pGEX5X2-MKP-M full WT, pGEX5X2-MKP-M full CS, pGEX5X2-MKP-M trunc WT, and pGEX5X2-MKP-M trunc CS, were used to transform E. coli strain BL21 (Stratagene). Preparation of the GST fusion proteins was performed as previously described (38).
Phosphatase activity.
Each GST–MKP-M fusion protein was assayed for intrinsic phosphatase activity as previously described (28) with minor modifications. Briefly, various amounts of GST–MKP-M fusion proteins were incubated for 1 h at 30°C in a reaction volume of 200 μl containing 20 mM pNPP and 50 mM imidazole (pH 7.5) (both from Sigma Chemical Co.). The reaction was stopped by the addition of 800 μl of 0.25N NaOH, and pNPP hydrolysis was measured by absorbance at 410 nm.
Transient transfection.
Cells were plated onto 60-mm-diameter plates at 106 cells/plate on the day before transfection. Combinations of expression plasmid DNAs (6 μg total per plate) were transfected using Lipofectamine (Gibco BRL) according to the manufacturer's instructions. Cells were harvested 48 h later with phosphate-buffered saline and used for further analyses.
Northern blot analysis.
Total cellular RNA was extracted using Trizol reagent (Gibco BRL) according to the manufacturer's instructions. Aliquots (20 μg) of the total RNA were fractionated on a 1% agarose gel containing 20 mM morpholinepropanesulfonic acid (MOPS), 5 mM sodium acetate, 1 mM EDTA (pH 7.0), and 6% (vol/vol) formaldehyde and transferred to a nylon membrane. After UV cross-linking, membranes were soaked in prehybridization solution (6× SSC, 5× Denhardt's reagent, 0.5% SDS, 100 mg of denatured salmon sperm DNA per ml, and 50% formamide) for 3 h at 42°C, followed by incubation with a 32P-labeled probe in hybridization solution (6× SSC, 0.5% SDS, 100 mg of denatured salmon sperm DNA per ml, and 50% formamide) for 14 h at 42°C. The membranes were washed in 2× SSC–0.1% SDS for 10 min twice at room temperature and in 0.1× SSC–0.1% SDS for 10 min twice at 50°C and were exposed to RX-U films (Fuji Film). For MKP-M gene expression, a 220-bp MKP-M cDNA fragment encoding amino acids 297 to 373, which is not homologous with any reported protein, was prepared by PCR from pBS-MKP-M full and used as a probe. For MKP-1 gene expression, a 679-bp mouse MKP-1 cDNA fragment encoding amino acids 272 to 367 and including 392 bp of the 3′ untranslated region was prepared by RT-PCR from J774.1 mRNA and used as a probe. For PAC-1 gene expression, a 564-bp mouse PAC-1 cDNA fragment encoding amino acids 290 to 318 and including 495 bp of the 3′ untranslated region was prepared by RT-PCR from J774.1 mRNA and used as a probe. For M3/6 gene expression, the 2.0-kb insert of pcDNA3-M3/6 containing the whole ORF was cut out with KpnI and BamHI and used as a probe. For β-actin mRNA detection, mouse β-actin cDNA containing the whole ORF was prepared by RT-PCR and used as a probe.
Immunoblotting.
Cells were lysed in PLC buffer (50 mM HEPES [pH 7.0], 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM NaPPi, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 10 μg of aprotinin per ml, 10 μg of leupeptin per ml) at 108 cells/ml. The lysates were separated on SDS-polyacrylamide gels and electrotransferred to Immobilon polyvinylidene difluoride membranes (Millipore Corporation, Bedford, Mass.). The membranes were blocked for 2 h in 2% bovine serum albumin–TBST (TBST is 20 mM Tris-HCl [pH 7.6], 0.15 M sodium chloride, and 0.1% Tween 20), incubated for 1 h with primary antibodies in TBST, washed three times with TBST, and incubated for 1 h with horseradish peroxidase-conjugated anti-mouse or -rabbit immunoglobulin (Amersham Pharmacia Biotech) diluted 1:10,000 in TBST. After three washes in TBST, the blot was developed with the enhanced chemiluminescence system (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
Immunoprecipitation.
The cell lysates were incubated with the indicated antibody for 2 h at 4°C, followed by incubation with protein A-Sepharose beads (Amersham Pharmacia Biotech) for an additional 1 h. The beads were washed three times in PLC lysis buffer and then suspended in SDS sample buffer heated at 95°C for 5 min. The eluted proteins were applied to SDS-polyacrylamide gels, and proteins were detected by Western blotting.
Immune complex kinase assay.
Cell lysates (107 cells/sample) were incubated with 0.4 μg of antibody for 2 h at 4°C, followed by incubation with protein A-Sepharose beads for additional 1 h. The beads were washed three times in PLC lysis buffer and then once in kinase buffer (20 mM Tris-HCl [pH 7.4], 20 mM MgCl2, 2 mM EGTA, 0.5 mM sodium vanadate, 10 mM β-glycerophosphate, and 1 mM dithiothreitol). The kinase reaction was initiated by the addition of 40 μl of kinase buffer with 20 μM ATP, 5 μCi of [γ-32P]ATP, and 0.5 μg of myelin basic protein (Sigma Chemical Co.) for ERK, GST–c-Jun(5–89) (37) for JNK, or GST–ATF2(1–109) (37) for p38 MAPK and was allowed to proceed for 20 min at 30°C. The reaction was terminated by the addition of 2× SDS sample buffer. Samples were boiled and resolved by SDS-polyacrylamide gel electrophoresis, and the fixed gel was exposed to an X-ray film.
Immunocytochemical analysis.
COS7 cells were transfected with pEFBOS-FLAG/MKP-M or pEFBOS-FLAG-MKP-M-trunc using Lipofectamine as described above. After the cells were fixed with formaldehyde, immunohistochemistry was performed using monoclonal anti-FLAG M2 antibody followed by fluorescein isothiocyanate-conjugated rat anti-mouse immunoglobulin secondary antibody as previously described (37). For RAW264.7 cells, analysis was performed using affinity-purified anti-MKP-M antibody followed by fluorescein isothiocyanate-conjugated goat anti-rabbit immunoglobulin secondary antibody. Confocal microscopic analyses were performed using MRC-1024 (Bio-Rad Laboratories, Hercules, Calif.).
ELISA.
RAW264.7 cells (5 × 105 cells/ml) were incubated for 8 h with or without 1 ng of LPS per ml. Cell culture supernatants were collected, and levels of murine TNF-α were quantitated using a commercial enzyme-linked immunosorbent assay (ELISA) kit (TECHNE Corporation, Minneapolis, Minn.) according to the manufacturer's instructions.
Nucleotide sequence accession numbers.
The GenBank/EMBL accession numbers for the sequences determined in this study are as follow: MKP-M A1, AF345951; MKP-M A2, AF345952; MKP-M B1, AF345953; MKP-M B2, AF345954.
RESULTS
Identification of the cDNA encoding a novel DSP.
In an attempt to identify a new MKP in mouse macrophages, we screened a cDNA library prepared from a mouse macrophage cell line, J774.1, using the mouse MKP-1 cDNA encoding the N-terminal domain as the probe by washing under nonstringent conditions (see Materials and Methods). Preliminary sequence analyses of isolated clones indicated that one partial cDNA clone contained the consensus sequence for the DSPs. Full-length cDNA clones were subsequently obtained by 5′-RACE. We refer to the protein encoded by this cDNA as MKP-M, for MKP isolated from macrophages.
In the full-length cDNA clones obtained, an in-frame termination codon was identified in the predicted 5′ untranslated sequence, and a termination codon was found prior to the predicted 3′ untranslated region. The ORF encodes a 677-amino-acid protein. Computer-assisted sequence analysis of the MKP-M protein demonstrated the presence of an N-terminal rhodanase homology domain, the extended active-site sequence motif (V/L)X(V/I)HCXAG(I/V)SRSXT(I/V)XXAY (L/I)M (where X is any amino acid) that is conserved in DSPs, and a C-terminal PEST sequence (Fig. 1A). Comparison of this sequence with the GenBank/EMBL data bank revealed significant homology with other members of the DSP family. Direct comparison between MKP-M and its closest homologue, M3/6, showed 53% identity.
FIG. 1.

Molecular cloning of MKP-M. (A) Deduced amino acid sequence of MKP-M (A1 mRNA isoform). The extended active site conserved among DSPs is underlined. The rhodanase homology domain is marked by a dotted line. The putative PEST sequence is double underlined. (B) Schematic presentation of the MKP-M mRNA isoforms. The ORFs are indicated by open squares. The 5′ and 3′ untranslated regions are indicated by black lines. RHOD, rhodanase homology domain; aa, amino acids. (C) Protein expression of MKP-M mRNA isoforms. The coding regions of four MKP-M mRNA isoforms were cloned into the pEFBOS-FLAG vector and transiently expressed in COS7 cells. Western blotting was performed by using anti-FLAG monoclonal antibody. Protein bands A and C represent full-length products of the A1 and B1 mRNA isoforms, respectively. Band B is a possible degradation product of A1. Numbers on the left are molecular masses in kilodaltons. (D) In vitro measurement of MKP-M phosphatase activity. Full-length or C-terminally truncated wild-type or catalytically inactive mutant MKP-M was subcloned into pGEX5X2. GST fusion proteins were expressed in E. coli, and purified proteins were analyzed for their phosphatase activities as described in Materials and Methods. OD, optical density.
During the course of cloning, we additionally identified three presumed splice variant isoforms of MKP-M (Fig. 1B) and subsequently named the originally isolated isoform MKP-M A1. A variant mRNA isoform termed A2 has a 165-base deletion in the N-terminal region and is presumed to encode a 622-amino-acid protein. Another variant, termed B1, has a 966-base deletion and encodes a 355-amino-acid protein. The B2 isoform includes both deletions and encodes a 300-amino-acid protein (Fig. 1B). All three of these mRNA variants encode the extended active-site sequence motif. When the cDNAs encompassing these mRNA isoforms were cloned in frame into a FLAG-tagged expression vector, only the A1 and B1 isoforms expressed detectable amounts of proteins in transient-expression assays (Fig. 1C).
To establish that MKP-M possesses endogenous catalytic activity, we expressed the full-length MKP-M A1 isoform as a GST fusion protein in E. coli and tested its phosphatase activity using pNPP as a substrate (Fig. 1D). Increasing the concentration of GST–MKP-M resulted in a linear rise in pNPP hydrolysis, indicating that MKP-M has endogenous phosphatase activity.
Tissue distribution of MKP-M gene expression.
Northern blot analysis was performed on total RNAs isolated from various mouse tissues using an MKP-M probe (Fig. 2). A dominant transcript of ≈5.5 kb was detected in every tissue examined, with higher expression observed in kidney, heart, and testis. Using various parts of the MKP-M cDNA as probes, we confirmed that the dominant transcript represents the A1 mRNA isoform (data not shown).
FIG. 2.

Tissue distribution of MKP-M. Total RNAs (20 μg each) from various mouse tissues were hybridized with an MKP-M cDNA probe as described in Materials and Methods. The membrane was stripped and reprobed with a mouse β-actin cDNA.
Regulation of MKP-M gene expression in a mouse macrophage cell line, RAW264.7.
RAW264.7 is a well-established mouse macrophage cell line known to respond to various stimuli (37). To determine the effects of LPS and various cytokines on the MKP-M mRNA expression, RAW264.7 cells were treated for 2 h with LPS, IL-15, IL-2, IL-1β, IFN-γ, TNF-α, or synthetic lipid A and total RNAs were isolated. Using a probe from the cDNA sequence coding the C-terminal domain of MKP-M, which is not homologous to any other MKP family proteins, we detected an MKP-M mRNA increase after LPS or synthetic lipid A stimulation but not after IL-2, IL-1β, IL-15, IFN-γ, or TNF-α stimulation (Fig. 3). The same RNA samples were also analyzed for the expression of the genes for other MKP-M members, i.e., MKP-1, PAC-1, and M3/6. The MKP-1 mRNA level was increased in response to IL-1β, TNF-α, IFN-γ, IL-15, IL-2, LPS, and synthetic lipid A. PAC-1 gene expression was induced by IL-1β, IFN-γ, LPS, and lipid A. Gene expression of M3/6, which is most homologous to MKP-M among the DSP members, was barely detectable in RAW264.7 cells, consistent with a previous report that M3/6 mRNA expression is rather specific to neuronal cells (54). M3/6 mRNA, however, was increased only with LPS and lipid A, similar to the case for MKP-M gene expression. We used synthetic lipid A to rule out possible contamination in the commercial grade of LPS, as has been previously reported (22). These data indicated that unlike the case for some other MKPs, gene expression of MKP-M, along with that of its homologue M3/6, is specifically induced by LPS in the RAW264.7 cell line. We observed very similar patterns of MKP-M gene expression in another mouse macrophage cell line, J774.1, and in mouse peritoneal macrophages (data not shown). In LPS-stimulated RAW264.7 cells, the MKP-M mRNA increase was first detected at 1 h and rapidly reached the maximal level by 2 h (data not shown). MKP-M expression stayed highly elevated for up to 24 h.
FIG. 3.

LPS induces MKP-M gene expression in a mouse macrophage cell line. RAW264.7 cells were left untreated (unstim) or treated for 2 h with LPS (1 μg/ml), IL-15 (10 ng/ml), IL-2 (10 ng/ml), IL-1β (10 ng/ml), IFN-γ (10 ng/ml), TNF-α (10 ng/ml), or synthetic lipid A (1 μg/ml). Total RNA was extracted for Northern blot analysis using a 32P-labeled MKP-M cDNA probe. The same blot was stripped and rehybridized with a mouse MKP-1, PAC-1, or M3/6 cDNA probe. A picture of the ethidium bromide (Et-Br)-stained gel is also shown.
Activation of p38 MAPK, but not ERK, is necessary for LPS-mediated MKP-M mRNA induction.
LPS stimulation of macrophages is known to activate MAPK pathways, including those of ERK, JNK, and p38 MAPK (17–19, 32, 45, 51). To investigate the mechanisms of MKP-M mRNA upregulation, RAW264.7 cells were pretreated with a specific inhibitor of the ERK (PD98059) or p38 MAPK (SB208530) pathway, followed by stimulation with LPS. We confirmed that pretreatment with PD98059 and SB208530 effectively suppressed the LPS-mediated activation of ERK and p38 MAPK, respectively, in a dose-dependent manner (Fig. 4A). Pretreatment with SB208530 inhibited the MKP-M mRNA increase in a dose-dependent manner (Fig. 4A), suggesting that p38 activation is involved in MKP-M mRNA upregulation by LPS. Unexpectedly, PD98059 treatment enhanced the increase in MKP-M mRNA mediated by LPS (Fig. 4A). On the other hand, pretreatment with dicoumarol, a known JNK inhibitor (10, 34), did not inhibit the LPS-mediated MKP-M mRNA increase (Fig. 4B), indicating that JNK activation may not be involved in the MKP-M mRNA upregulation by LPS. However, as dicoumarol could not completely inhibit the JNK activation (Fig. 4B), we could not rule out the possibility that a low-level activation of JNK is involved in the MKP-M mRNA increase mediated by LPS.
FIG. 4.

The p38 MAPK pathway, but not the ERK pathway, is important for LPS-mediated MKP-M mRNA upregulation in RAW264.7 cells. (A) RAW264.7 cells were pretreated with a series of concentrations of either PD98059 or SB208530 for 30 min, followed by 2 h of stimulation with 1 μg of LPS per ml. Total RNAs (20 μg each) were extracted for Northern blot analysis using a 32P-labeled MKP-M cDNA probe. A picture of the ethidium bromide (EtBr)-stained gel is also shown. For in vitro kinase assays for ERK and p38, RAW264.7 cells were pretreated with a series of concentrations of either PD98059 or SB208530 for 30 min, followed by 30 min of stimulation with 1 μg of LPS per ml. ERK or p38 was immunoprecipitated with specific antibodies and assayed for kinase activity as described in Materials and Methods. (B) RAW264.7 cells were pretreated with a series of concentrations of dicoumarol for 30 min, followed by 2 h of stimulation with 1 μg of LPS per ml. Total RNAs (20 μg each) were extracted for Northern blot analysis using a 32P-labeled MKP-M cDNA probe. A picture of the ethidium bromide-stained gel is also shown. For the in vitro kinase assay for JNK1, RAW264.7 cells were pretreated with a series of concentrations of dicoumarol for 30 min, followed by 30 min of stimulation with 1 μg of LPS per ml. JNK1 was immunoprecipitated with its specific antibody and assayed for kinase activity as described in Materials and Methods. (C) RAW264.7 cells were untreated (unstim) or treated with LPS (1 μg/ml), anisomycin (Aniso) (1 μg/ml), or PMA (100 nM) for 2 h. Extracted total RNAs were analyzed as described for panel A.
To further elucidate the role of MAPK pathways in MKP-M gene expression in macrophages, we treated RAW264.7 cells with anisomycin, a potent activator of JNK and p38 MAPK (20), or with PMA, which activates ERKs but not JNK or p38 MAPK in RAW264.7 cells (1, 11). As shown in Fig. 4C, the MKP-M mRNA level was significantly increased in response to anisomycin stimulation, whereas the MKP-M mRNA increase induced by PMA was marginal. In contrast, PMA potently increased the gene expression of both MKP-1 and PAC-1 (Fig. 4C). These results indicate that p38 MAPKs, but not ERKs, play important roles in the gene induction of MKP-M in macrophages.
Analysis of MKP-M protein expression levels in RAW264.7 cells.
A polyclonal antibody against MKP-M was prepared by immunizing rabbits with a GST–MKP-M fusion protein encompassing amino acids 297 to 373 of MKP-M. This region of MKP-M is not homologous to any other known phosphatases as determined by a database search. The antiserum was affinity purified through an antigen-conjugated column and used for Western blot analyses. RAW264.7 cells constitutively expressed an 80-kDa MKP-M protein (Fig. 5). After LPS stimulation, the MKP-M protein level started to increase at 2 h and became significantly elevated by 24 h. This result was consistent with the mRNA data and demonstrated that LPS could increase MKP-M expression at both the mRNA and protein levels in RAW264.7 cells.
FIG. 5.

Analysis of MKP-M protein expression levels in RAW264.7 cells after LPS treatment. RAW264.7 cells were stimulated with LPS (1 μg/ml) for the indicated times. Whole-cell lysates (50 μg each) were prepared and analyzed for MKP-M protein expression by using the affinity-purified anti-MKP-M polyclonal antibody. As a control, the same set of cell lysates were examined for JNK1 expression levels using the anti-JNK1 polyclonal antibody.
MKP-M selectively inactivates JNK.
As most DSPs specifically dephosphorylate MAPKs, we presumed that MKP-M functions as an MKP. In order to examine whether MKP-M could inactivate MAPKs in intact cells, we cotransfected the expression plasmid for each HA-tagged MAPK (p42ERK2, p54-JNK2, or p38α) together with pEFBOS-FLAG-MKP-M in COS7 cells. To obtain a clear impression of the relative effectiveness of MKP-M in inactivation of each MAPK, cells were transfected with a range of plasmid concentrations (0.01 to 1.0 μg/plate) of pEFBOS-FLAG-MKP-M. These concentrations were chosen to give a reproducible dose-dependent increase in the levels of immunodetectable MKP-M protein, while the expression of each MAPK was unchanged (Fig. 6A). Epidermal growth factor (EGF) stimulation was used for ERK2 and p38α. As EGF did not effectively activate JNK2 in COS7 cells, UV irradiation was used for the JNK2 assay. Using this approach, EGF-stimulated ERK2 and p38α activation was inhibited by ≈50% when cells were transfected with 1 μg of MKP-M plasmid (Fig. 6A). On the other hand, the same amount of MKP-M plasmid reduced UV-stimulated JNK2 activation by ≈90%, and 50% inactivation of JNK2 could be achieved with less than 0.1 μg of MKP-M plasmid.
FIG. 6.

MKP-M selectively inactivates JNK. (A) COS7 cells were transiently transfected with 1 μg of pcDNA3-HA-ERK2, -JNK2, or -p38α, along with increasing amounts of pEFBOS-FLAG-MKPM. An empty vector, pEFBOS-FLAG, was also added so that the total amount of plasmids was 2 μg for each transfection. At 48 h after transfection, the cells were treated with either 100 ng of EGF per ml or 400 J of UV per m2 for 30 min. Total cell lysates were prepared, and MAPK was immunoprecipitated with anti-HA antibody. MAPK enzymatic activity was assessed by phosphorylation of 0.5 μg of myelin basic protein (ERK2), GST–c-Jun(5–89), or GST–ATF(21–109). The autoradiogram shows phosphorylated substrate proteins following separation by SDS-polyacrylamide gel electrophoresis and is representative of three identical experiments. The amounts of expressed proteins were assessed by Western blotting with either anti-FLAG or anti-HA antibody. Following drying of the gel, substrate bands were excised for counting with a scintillation counter. The relative MAPK activity with increasing amounts of MKP-M expression plasmid is shown as a graph. The stimulated kinase activity without MKP-M expression is defined as 100. (B) The same experiment as for panel A was performed for M3/6 using increasing amounts of pcDNA3-M3/6 and the empty vector, pcDNA3. The amount of exogenously expressed M3/6 was detected using anti-Myc antibody.
As a control, we also examined the substrate specificity of M3/6, the closest homologue of MKP-M, in the same cell system (Fig. 6B). Consistent with previous reports (25, 44), M3/6 effectively inactivated both p38α and JNK2 (slightly more effectively for p38α), whereas it did not effectively inactivate ERK2. Together, these data suggested that MKP-M displayed significant selectivity toward JNK2 compared with ERK2 or p38α.
MKP-M subcellular localization.
In order to study the subcellular localization of exogenously expressed MKP-M, FLAG-tagged MKP-M was transiently expressed in COS7 cells and immunostaining was performed with anti-FLAG antibody. As shown in Fig. 7A, FLAG-tagged MKP-M was detected only in the cell cytoplasm. No staining was obtained with the same antibody when the cells were transfected with the vector alone, confirming the specificity of the immunostaining. Also, no cell compartment was stained with the control antibody. When the C-terminally truncated mutant was expressed in COS7 cells, it was also detected in the cell cytoplasm, suggesting that the C-terminal domain was not important for the cytoplasmic localization of MKP-M (Fig. 7A). We further studied the localization of the endogenous MKP-M by using the affinity-purified anti-MKP-M polyclonal antibody. As shown in Fig. 7B, this antibody specifically detected the endogenous MKP-M in the cytoplasm of RAW264.7 cells. The cytoplasmic distribution of MKP-M did not change after LPS treatment of the cells (data not shown).
FIG. 7.
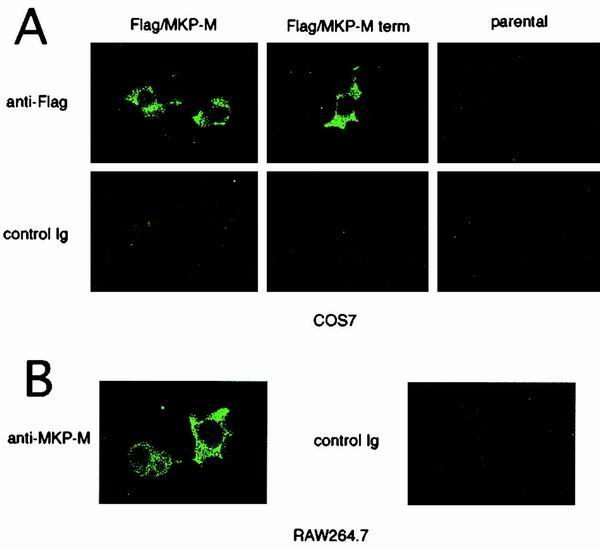
MKP-M localizes to the cell cytoplasm. (A) The localization of the exogenously expressed FLAG-tagged MKP-M or C-terminally truncated version of MKP-M (MKP-M term) in COS7 cells was visualized by immunofluorescence with anti-FLAG or control antibody. (B) The localization of endogenous MKP-M in RAW264.7 cells was visualized by immunofluorescence with anti-MKP-M or preimmune antibody. Ig, immunoglobulin.
MKP-M is involved in the regulation of LPS-stimulated JNK activity.
It has been reported that the cysteine in the extended active-site sequence is absolutely necessary for the enzymatic activity of DSPs (43). Thus, in order to make a catalytically inactive mutant of MKP-M, we replaced cysteine 244 in the extended active-site sequence of MKP-M with serine and overexpressed this mutant along with HA-tagged JNK2 in RAW264.7 cells. The full-length MKP-M containing this CS substitution did not contain endogenous phosphatase activity (Fig. 1D). Although the overexpression of this mutant did not significantly change the basal JNK activity, LPS-mediated JNK activity was moderately increased at 30 min and remained significantly elevated at 6 h in the presence of the mutant MKP-M, indicating that this C244S mutant of MKP-M functioned in a dominant-negative fashion (Fig. 8A). We also overexpressed CS mutants of M3/6 and MKP-1 in RAW264.7 cells. These mutants were also confirmed to be catalytically inactive by in vitro phosphatase assays (data not shown). In contrast to the MKP-M CS mutant, these mutant phosphatases did not significantly affect the LPS-induced JNK2 activation, although their expression levels were comparable to that of the MKP-M CS mutant (Fig. 8A).
FIG. 8.

Expression of a catalytically inactive MKP-M enhances the magnitude and the duration of LPS-mediated JNK activation in RAW264.7 cells. (A) COS7 cells were transiently transfected with 1 μg of pEFBOS-FLAG/MKP-M CS, 1 μg of pcDNA3-M3/6, 0.2 μg of pEFBOS-Myc/MKP-1 CS (to adjust the amount of protein expression) plus 0.8 μg of pEFBOS-Myc, or 1 μg of the empty vector (EV) (pEFBOS-Myc), along with 1 μg of pcDNA3-HA/JNK2. At 48 h after the transfection, protein expression of MKPs and HA-JNK2 was examined with anti-Myc or anti-HA antibody, respectively. At 48 h after the transfection, the cells were left untreated or stimulated with 1 μg of LPS per ml for 30 min or 6 h. Cell lysates were then prepared, and HA-JNK2 was immunoprecipitated with anti-HA antibody. JNK enzymatic activity was measured by using GST–c-Jun(5–89) as the substrate. The experiment was done with three independent clones for each group, and a typical result is shown. (B) RAW264.7 cells stably transfected with pcDNA3.1(+)-MKP-M CS trunc, pcDNA3.1(+)-MKP-M WT trunc, or pcDNA3.1(+) were untreated or stimulated with 1 μg of LPS per ml for 30 min. Endogenous JNK1 was immunoprecipitated with anti-JNK1 antibody, and the JNK activity was measured as described for panel A. Protein expression of the exogenous MKP-M was measured with anti-FLAG antibody. As a control, tyrosine phosphorylation of p38 was measured with the phospho-specific anti-p38 antibody. (C) RAW264.7 cells stably transfected with pcDNA3.1(+)-MKP-M CS trunc or pcDNA3.1 were untreated or stimulated with 1 μg of LPS per ml for the indicated times. Endogenous JNK1 was immunoprecipitated with anti-JNK1 antibody, and the JNK1 kinase activity was measured as described for panel A. (D) RAW264.7 cells stably transfected with pcDNA3.1(+)-MKP-M CS term, pcDNA3.1(+)-MKP-M wild type trunc, or pcDNA3.1 were untreated or stimulated with 1 ng of LPS per ml for 12 h. The TNF-α concentrations in the culture supernatants were measured by ELISA. Three independent clones were examined for each group. The assay of each clone was done in triplicate, and the averages and standard deviations are shown.
We further tried to establish a RAW264.7 cell line stably expressing the wild-type MKP-M and its dominant-negative mutant. We found it difficult to obtain a cell line stably expressing the full-length MKP-M protein, probably due to the instability of the protein caused by the presence of the C-terminal PEST sequence. Thus, we made an expression plasmid of the C-terminally truncated versions of both the wild type and the C244S mutant of MKP-M. We confirmed that the C-terminally truncated wild-type MKP-M contained endogenous phosphatase activity in vitro, while its CS mutant did not (Fig. 1D). Using these expression plasmids, we obtained RAW264.7 cells stably expressing the C-terminally truncated MKP-Ms. We analyzed three independent clones for each transfectant, and the result for a typical clone is shown in Fig. 8B. The wild-type MKP-M transfectants showed significantly lower JNK activity both before and after LPS stimulation. On the other hand, the catalytically inactive MKP-M transfectants showed basal JNK activity similar to that of the empty vector transfectants. However, at 30 min after LPS stimulation, JNK activity in these transfectants was approximately 2 times higher than that in the empty vector-transfected cell lines.
Furthermore, we examined the effects of the MKP-M CS mutant on the kinetics of JNK activation in LPS-stimulated macrophages (Fig. 8C). In the empty vector-transfected RAW264.7 cells, JNK activity started to decrease by 4 h and was down to the basal level by 6 h after the LPS stimulation. In contrast, when the cells stably expressing the C244S MKP-M mutant were treated with LPS, the JNK activity remained highly elevated at least until 6 h after the LPS stimulation. Together, these data revealed that MKP-M may not be involved in maintaining the basal JNK activity but is involved in downregulating the extent and, more significantly, the duration of JNK activity after LPS stimulation.
Expression of a catalytically inactive MKP-M mutant enhances LPS-mediated TNF-α secretion from RAW264.7 cells.
It has been reported that JNK activation plays an important role in the production of cytokines such as TNF-α, IL-1, and IL-6 (39), all of which are potent activators of inflammation caused by LPS. To examine the roles of MKP-M in LPS-mediated cytokine production, TNF-α production from RAW264.7 cell lines stably transfected with vector alone, C-terminally truncated wild-type MKP-M, or the C244S mutant was measured by ELISA after 12 h of LPS stimulation. Three independent clones were examined for each group. As shown in Fig. 8D, a significant increase of TNF-α production was detected in the culture supernatants of the MKP-M CS cell lines compared with that of the empty vector transfectants. In contrast, TNF-α production from wild-type MKP-M transfectants was significantly lower than that from the empty vector transfectants. These results indicate that MKP-M negatively regulates TNF-α production in macrophages by inactivating JNK after LPS stimulation.
DISCUSSION
In this paper, we have reported the molecular cloning and characterization of a novel DSP termed MKP-M from a mouse macrophage cDNA library. MKP-M is localized in the cytoplasm and preferentially deactivates JNK among the three subsets of MAPKs. In macrophages, LPS stimulation induced MKP-M mRNA expression at least partly through p38 MAPK activation. The expression of a dominant-negative mutant of MKP-M in macrophages enhanced LPS-stimulated JNK activation in both transient and stable expression systems, suggesting that MKP-M is involved in the downregulation of JNK activity in macrophages after LPS stimulation.
DSPs form a family of proteins characterized by a highly homologous catalytic domain and selectively dephosphorylate MAPKs (27). They play important roles in the maintenance of cellular functions, because dysregulation of MAPKs can be detrimental to cells. Comparison of the primary sequence of MKP-M with those of related DSPs revealed an extended active-site sequence motif, (V/L)X(V/I)HCXAG(I/V)SRSXT(I/V)XXAY(L/I)M (where X is any amino acid) that appeared to be a hallmark of this family of proteins. Additional areas of homology included a rhodanase homology domain in the N-terminal region, which may play a role in defining substrate specificity or serve as a recruiting site for other proteins (29). In addition, the presence of a proline-rich carboxyl extension in MKP-M is interesting. Although it is not conserved among most DSP family proteins, this extension is homologous with those of a mammalian DSP, hVH-5 (or M3/6 for its mouse version) (35, 54), and a Caenorhabditis elegans tyrosine phosphatase, VHP-1. Although the biological significance of this extension in MKP-M remains obscure, it contains a putative PEST sequence, KLCQFSPVQEVSEQSPETSPD (Fig. 1A). An abundance of proline (P), glutamate (E), serine (S), and threonine (T) residues is commonly found in the most rapidly degraded eukaryotic proteins, and the half-lives of proteins containing PEST sequences are frequently less than 1 h (48). Thus, the putative PEST sequence may be utilized as signals for rapid degradation of MKP-M. As the MKP-M mRNA level in macrophages is rapidly increased by LPS stimulation but remains elevated for at least 24 h (data not shown), the temporal abundance of MKP-M may be regulated at both the mRNA and protein levels. Consistently, a C-terminally truncated version of MKP-M without the PEST sequence was easily expressed at much higher levels than the full-length version in stable transfection of macrophages. Interestingly, hVH-5 also contains PEST sequences in the homologous proline-rich extension (35).
Other than the predominantly expressed mRNA isoform (A1), we identified three additional mRNA variants (A2, B1, and B2), which are probably produced by alternative splicing. The mRNA levels for these variants were significantly lower than that for the major mRNA isoform (A1), as Northern blot analysis revealed a dominant mRNA band that was confirmed to be the A1 isoform. In transient-transfection assays, only one of the three variants (B1) produced a stable protein of 45 kDa. As this putative short form of MKP-M protein does not contain the portion of the protein used to make the polyclonal antibody, we could not confirm the endogenous expression level of this 45-kDa protein. Although the MKP-M B1 protein lacks the C-terminal portion of the phosphatase domain and may not encode an active phosphatase, it contains an extended active-site sequence motif. The significance of these alternative spliced forms of MKP-M remains unknown.
The major form of MKP-M mRNA (A1) encodes an active MKP of 80 kDa. In cells, MKP-M displayed substrate selectivity for JNK2 compared with ERK2 and p38α. This substrate selectivity of MKP-M (JNK ≫ ERK = p38) is unique and is different from those of MKP-1 (also called hVH1 or CL100) (p38 > JNK > ERK) (13), MKP-3 (also called PYST1) (14, 41) and MKP-4 (ERK > JNK = p38) (42), MKP-2 (also called hVH-2 or TYP-1) (ERK = JNK > p38) (9), MKP-5 (p38 = JNK > ERK) (52, 53), or PAC-1 (ERK = p38 > JNK) (9). Interestingly, it is somewhat similar to that of hVH-5 and M3/6 (p38 = JNK ≫ ERK) (25), to which MKP-M is most homologous. It has recently been reported that the RILPHLYL sequence in the N-terminal domains of hVH-5 and M3/6, which shares significant homology with the JNK-binding site of c-Jun protein (called the delta domain), is important for the ability of these phosphatases to dephosphorylate JNK (25). Although the ILP and L(Y/F)LG elements of this motif occur in all known mammalian DSPs, the presence of the N-terminal arginine, which is unique to hVH-5 and M3/6, makes this sequence conform to the delta domain consensus known to be critical for binding to JNK. Like hVH5 and M3/6, MKP-M contains arginine in the N-terminal position of this motif. As substrate binding to some DSPs is accompanied by catalytic activation through the stabilization of the active phosphatase conformation (5, 12), this delta domain-like motif may contribute to the selective JNK dephosphorylation by MKP-M.
It seems unreasonable that both hVH-5 (or M3/6) and MKP-M are localized mainly in the cell cytoplasm, considering that JNK efficiently phosphorylates several transcription factors and was reported to translocate to the nucleus in response to UV or hypoxia (26, 40). However, others have found that JNK constitutively exists in both cytoplasm and nuclei and can be activated without evident nuclear translocation at least by gamma irradiation (8). More recently it has been reported that activated JNK translocates to mitochondria and interacts with an antiapoptotic protein, Bcl-xL (30). Therefore, it seems reasonable to speculate that controlling JNK activity in the cytoplasm is essential for some of the physiological functions of JNK.
An interesting feature of many DSPs is their tight and rapid transcriptional induction by growth factors and/or cellular stresses. Indeed, MKP-1 was first identified as an immediate-early gene rapidly induced by mitogens, heat shock, or oxidative stress (7, 28). Other DSP genes also undergo transcriptional upregulation in response to various stimuli (4). In many instances, induction of each DSP gene seems to be specific to given stimuli (4). In mouse macrophage cell lines, a small amount of MKP-M mRNA was constitutively expressed. The amount of MKP-M mRNA significantly increased in response to LPS stimulation but not in response to IL-1β, TNF-α, IFN-γ, IL-15, or IL-2 stimulation. In contrast, MKP-1 mRNA responded to all of these cytokines, and PAC-1 mRNA responded to IL-1β and IFN-γ stimulation in addition to LPS stimulation. Interestingly, the mRNA level of M3/6, the closest homologue of MKP-M, also increased in response to LPS and lipid A stimulation but not in response to cytokine stimulation, indicating that M3/6 and MKP-M may share similar transcriptional control elements. The mRNA expression level of M3/6, however, seemed rather low in macrophages.
LPS-stimulated MKP-M mRNA induction seemed to be independent of ERK activation and to be mediated at least partly by the activation of the p38 MAPK pathway, as treatment with SB208530, a specific inhibitor of p38 activation, successfully inhibited the MKP-M mRNA increase after LPS stimulation, suggesting that ERK activation may have an inhibitory effect on MKP-M gene expression. In RAW264.7 cells, p38 MAPK is strongly activated by LPS treatment but only weakly activated by IL-1β, TNF-α, IFN-γ, IL-2, or IL-15 treatment (data not shown). Thus, we speculate that this LPS-specific response of MKP-M transcription may be due to its dependence on p38 MAPK activation. We presume that significant involvement of JNK in MKP-M transcriptional induction is unlikely, as dicoumarol, which partially inhibited LPS-mediated JNK activation, did not significantly affect MKP-M mRNA induction (Fig. 4B). However, as dicoumarol could not inhibit the JNK activation completely and no other powerful and specific JNK inhibitor was available, we could not rule out the possibility that a low-level activation of JNK is involved in the MKP-M mRNA increase mediated by LPS. On the other hand, the expression of the MKP-1 gene is dependent on the activation of ERK in CCL39 cells (3) or of JNK in NIH 3T3 cells (2). PAC-1 gene expression was reported to be dependent on ERK activation in T cells (15). These reports are consistent with our finding that both MKP-1 and PAC-1 are responsive to different sets of stimuli than is MKP-M.
LPS, a cell wall component of gram-negative bacteria, is a complex glycolipid composed of a hydrophilic polysaccharide region and a hydrophobic domain known as lipid A that is responsible for most of the biological functions of LPS (49). LPS stimulates host cells to produce endogenous mediators, including bioactive lipids (e.g., platelet-activating factor and thromboxane A2), reduced oxygen species (e.g., NO), and, in particular, cytokines such as IL-1, IL-6, IFN-γ, and TNF-α (49). It activates a transcriptional factor, NF-κB (33, 55), and MAPKs, including ERKs (17, 32), JNKs (18, 51), and p38 MAPKs (19, 45). On the other hand, these responses have to be carefully regulated, as excessive and long-lasting inflammation often causes tissue damages. Thus, it is important to understand the mechanisms by which the JNK activity is regulated in macrophages in response to bacterial components such as LPS.
Proximally, JNKs are activated by the dual-specificity MKKs 4 and 7, which in turn are activated by the MEK kinases. MEK kinase 1 has recently been reported to be essential for JNK activation in embryonic stem cells (58). In spite of these upstream kinases, JNK activation in macrophages after stimulation with LPS is a temporary process even in the continued presence of activating stimuli, indicating that the duration of JNK activity also provides important control mechanisms for LPS-induced inflammatory responses. However, the exact mechanisms of JNK deactivation after LPS stimulation have remained unknown.
Our results showed that the expression of a dominant-negative form of MKP-M increased both the magnitude and duration of JNK activity induced by LPS stimulation in a mouse macrophage cell line. It also decreased TNF-α secretion induced by LPS (Fig. 8D). These findings indicate that MKP-M, the transcription of which is induced by LPS, plays an important role in the JNK deactivation process in LPS-treated macrophages. It was rather surprising, as other MAPKs (at least MKP-1 and PAC-1) are also induced by LPS in these types of cells. However, in contrast to MKP-M, most of the other MKP members dephosphorylate ERKs or p38 MAPKs more preferentially, and M3/6, which is structurally related to MKP-M and preferentially dephosphorylates JNKs, is specifically expressed in brain, lung, and heart in adult mice (54). In fact, the expression of the catalytically inactive mutants of MKP-1 or M3/6 in RAW264.7 cells did not have significant effects on LPS-mediated JNK activation (Fig. 8A).
In summary, these observations provide evidence that MKP-M, a newly identified MKP protein, specifically dephosphorylates JNK and plays an important role in regulating the LPS-mediated JNK activity and cytokine production in macrophages. MKP-M may become a new therapeutic target for the control of inflammatory responses caused by bacterial infection.
ACKNOWLEDGMENTS
We thank K. Itano and A. Nishikawa for their technical assistance.
This work was supported in part by grants from Ono Pharmaceutical Company, the Yokoyama Research Foundation for Clinical Pharmacology, and the Naito Foundation (to T.M.); the Ministry of Education, Science and Culture of the Japanese Government (JSPS-RFTF97L00703) and the Yakult Bioscience Foundation (to Y.Y.); and the National Cancer Institute (CA42533) (to A.S.K.).
REFERENCES
- 1.Barbour S E, Wong C, Rabah D, Kapur A, Carter A D. Mature macrophage cell lines exhibit variable responses to LPS. Mol Immunol. 1998;35:977–987. doi: 10.1016/s0161-5890(98)00070-4. [DOI] [PubMed] [Google Scholar]
- 2.Bokemeyer D, Sorokin A, Yan M, Ahn N G, Templeton D J, Dunn M J. Induction of mitogen-activated protein kinase phosphatase 1 by the stress-activated protein kinase signaling pathway but not by extracellular signal-regulated kinase in fibroblasts. J Biol Chem. 1996;271:639–642. doi: 10.1074/jbc.271.2.639. [DOI] [PubMed] [Google Scholar]
- 3.Brondello J M, Brunet A, Pouyssegur J, McKenzie F R. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J Biol Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- 4.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 5.Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- 6.Chan E D, Riches D W. Potential role of the JNK/SAPK signal transduction pathway in the induction of iNOS by TNF-alpha. Biochem Biophys Res Commun. 1998;253:790–796. doi: 10.1006/bbrc.1998.9857. [DOI] [PubMed] [Google Scholar]
- 7.Charles C H, Abler A S, Lau L F. cDNA sequence of a growth factor-inducible immediate early gene and characterization of its encoded protein. Oncogene. 1992;7:187–190. [PubMed] [Google Scholar]
- 8.Chen Y R, Meyer C F, Tan T H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 9.Chu Y, Solski P A, Khosravi-Far R, Der C J, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- 10.Cross J V, Deak J C, Rich E A, Qian Y, Lewis M, Parrott L A, Mochida K, Gustafson D, Vande Pol S, Templeton D J. Quinone reductase inhibitors block SAPK/JNK and NFkappaB pathways and potentiate apoptosis. J Biol Chem. 1999;274:31150–31154. doi: 10.1074/jbc.274.44.31150. [DOI] [PubMed] [Google Scholar]
- 11.Dziarski R, Jin Y P, Gupta D. Differential activation of extracellular signal-regulated kinase (ERK) 1, ERK2, p38, and c-Jun NH2-terminal kinase mitogen-activated protein kinases by bacterial peptidoglycan. J Infect Dis. 1996;174:777–785. doi: 10.1093/infdis/174.4.777. [DOI] [PubMed] [Google Scholar]
- 12.Fjeld C C, Rice A E, Kim Y, Gee K R, Denu J M. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J Biol Chem. 2000;275:6749–6757. doi: 10.1074/jbc.275.10.6749. [DOI] [PubMed] [Google Scholar]
- 13.Franklin C C, Kraft A S. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 14.Groom L A, Sneddon A A, Alessi D R, Dowd S, Keyse S M. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- 15.Grumont R J, Rasko J E, Strasser A, Gerondakis S. Activation of the mitogen-activated protein kinase pathway induces transcription of the PAC-1 phosphatase gene. Mol Cell Biol. 1996;16:2913–2921. doi: 10.1128/mcb.16.6.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta S, Campbell D, Derijard B, Davis R J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 17.Hambleton J, McMahon M, DeFranco A L. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-induced signaling events. J Exp Med. 1995;182:147–154. doi: 10.1084/jem.182.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hambleton J, Weinstein S L, Lem L, DeFranco A L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci USA. 1996;93:2774–2778. doi: 10.1073/pnas.93.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han J, Lee J D, Bibbs L, Ulevitch R J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 20.Hazzalin C A, Cano E, Cuenda A, Barratt M J, Cohen P, Mahadevan L C. p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr Biol. 1996;6:1028–1031. doi: 10.1016/s0960-9822(02)00649-8. [DOI] [PubMed] [Google Scholar]
- 21.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 22.Hirschfeld M, Ma Y, Weis J H, Vogel S N, Weis J J. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 23.Hiura T S, Kempiak S J, Nel A E. Activation of the human RANTES gene promoter in a macrophage cell line by lipopolysaccharide is dependent on stress-activated protein kinases and the IkappaB kinase cascade: implications for exacerbation of allergic inflammation by environmental pollutants. Clin Immunol. 1999;90:287–301. doi: 10.1006/clim.1998.4659. [DOI] [PubMed] [Google Scholar]
- 24.Ishizaka-Ikeda E, Fukunaga R, Wood W I, Goeddel D V, Nagata S. Signal transduction mediated by growth hormone receptor and its chimeric molecules with the granulocyte colony-stimulating factor receptor. Proc Natl Acad Sci USA. 1993;90:123–127. doi: 10.1073/pnas.90.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson T R, Biggs J R, Winbourn S E, Kraft A S. Regulation of dual-specificity phosphatases M3/6 and hVH5 by phorbol esters. Analysis of a delta-like domain. J Biol Chem. 2000;275:31755–31762. doi: 10.1074/jbc.M004182200. [DOI] [PubMed] [Google Scholar]
- 26.Kawasaki H, Moriguchi T, Matsuda S, Li H Z, Nakamura S, Shimohama S, Kimura J, Gotoh Y, Nishida E. Ras-dependent and Ras-independent activation pathways for the stress-activated-protein-kinase cascade. Eur J Biochem. 1996;241:315–321. doi: 10.1111/j.1432-1033.1996.00315.x. [DOI] [PubMed] [Google Scholar]
- 27.Keyse S M. An emerging family of dual specificity MAP kinase phosphatases. Biochim Biophys Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- 28.Keyse S M, Emslie E A. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature. 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 29.Keyse S M, Ginsburg M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem Sci. 1993;18:377–378. doi: 10.1016/0968-0004(93)90092-2. [DOI] [PubMed] [Google Scholar]
- 30.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen Y N, Campbell A, Sudha T, Yuan Z M, Narula J, Weichselbaum R, Nalin C, Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. . (Erratum, 275:19433.) [DOI] [PubMed] [Google Scholar]
- 31.Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 32.Liu M K, Herrera-Velit P, Brownsey R W, Reiner N E. CD14-dependent activation of protein kinase C and mitogen-activated protein kinases (p42 and p44) in human monocytes treated with bacterial lipopolysaccharide. J Immunol. 1994;153:2642–2652. [PubMed] [Google Scholar]
- 33.Mackman N, Brand K, Edgington T S. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J Exp Med. 1991;174:1517–1526. doi: 10.1084/jem.174.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangelus M, Kroyter A, Galron R, Sokolovsky M. Reactive oxygen species regulate signaling pathways induced by M1 muscarinic receptors in PC12M1 cells. J Neurochem. 2001;76:1701–1711. doi: 10.1046/j.1471-4159.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 35.Martell K J, Seasholtz A F, Kwak S P, Clemens K K, Dixon J E. hVH-5: a protein tyrosine phosphatase abundant in brain that inactivates mitogen-activated protein kinase. J Neurochem. 1995;65:1823–1833. doi: 10.1046/j.1471-4159.1995.65041823.x. [DOI] [PubMed] [Google Scholar]
- 36.Matsuguchi T, Kraft A S. Regulation of myeloid cell growth by distinct effectors of Ras. Oncogene. 1998;17:2701–2709. doi: 10.1038/sj.onc.1202201. [DOI] [PubMed] [Google Scholar]
- 37.Matsuguchi T, Musikacharoen T, Ogawa T, Yoshikai Y. Gene expression of toll-like receptor 2, but not toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. J Immunol. 2000;165:5767–5772. doi: 10.4049/jimmunol.165.10.5767. [DOI] [PubMed] [Google Scholar]
- 38.Matsuguchi T, Salgia R, Hallek M, Eder M, Druker B, Ernst T J, Griffin J D. Shc phosphorylation in myeloid cells is regulated by granulocyte macrophage colony-stimulating factor, interleukin-3, and steel factor and is constitutively increased by p210BCR/ABL. J Biol Chem. 1994;269:5016–5021. [PubMed] [Google Scholar]
- 39.Meng F, Lowell C A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizukami Y, Yoshioka K, Morimoto S, Yoshida K. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J Biol Chem. 1997;272:16657–16662. doi: 10.1074/jbc.272.26.16657. [DOI] [PubMed] [Google Scholar]
- 41.Muda M, Boschert U, Dickinson R, Martinou J C, Martinou I, Camps M, Schlegel W, Arkinstall S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996;271:4319–4326. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- 42.Muda M, Boschert U, Smith A, Antonsson B, Gillieron C, Chabert C, Camps M, Martinou I, Ashworth A, Arkinstall S. Molecular cloning and functional characterization of a novel mitogen-activated protein kinase phosphatase, MKP-4. J Biol Chem. 1997;272:5141–5151. doi: 10.1074/jbc.272.8.5141. [DOI] [PubMed] [Google Scholar]
- 43.Muda M, Theodosiou A, Gillieron C, Smith A, Chabert C, Camps M, Boschert U, Rodrigues N, Davies K, Ashworth A, Arkinstall S. The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J Biol Chem. 1998;273:9323–9329. doi: 10.1074/jbc.273.15.9323. [DOI] [PubMed] [Google Scholar]
- 44.Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. 1996;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- 45.Nick J A, Avdi N J, Gerwins P, Johnson G L, Worthen G S. Activation of a p38 mitogen-activated protein kinase in human neutrophils by lipopolysaccharide. J Immunol. 1996;156:4867–4875. [PubMed] [Google Scholar]
- 46.Ogawa T, Suda Y, Kashihara W, Hayashi T, Shimoyama T, Kusumoto S, Tamura T. Immunobiological activities of chemically defined lipid A from Helicobacter pylori LPS in comparison with Porphyromonas gingivalis lipid A and Escherichia coli-type synthetic lipid A (compound 506) Vaccine. 1997;15:1598–1605. doi: 10.1016/s0264-410x(97)00102-3. [DOI] [PubMed] [Google Scholar]
- 47.Robinson M J, Cobb M H. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 48.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 49.Schletter J, Heine H, Ulmer A J, Rietschel E T. Molecular mechanisms of endotoxin activity. Arch Microbiol. 1995;164:383–389. doi: 10.1007/BF02529735. [DOI] [PubMed] [Google Scholar]
- 50.Smith J L, Schaffner A E, Hofmeister J K, Hartman M, Wei G, Forsthoefel D, Hume D A, Ostrowski M C. Ets-2 is a target for an Akt (protein kinase B)/Jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol Cell Biol. 2000;20:8026–8034. doi: 10.1128/mcb.20.21.8026-8034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swantek J L, Cobb M H, Geppert T D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanoue T, Moriguchi T, Nishida E. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J Biol Chem. 1999;274:19949–19956. doi: 10.1074/jbc.274.28.19949. [DOI] [PubMed] [Google Scholar]
- 53.Theodosiou A, Smith A, Gillieron C, Arkinstall S, Ashworth A. MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene. 1999;18:6981–6988. doi: 10.1038/sj.onc.1203185. [DOI] [PubMed] [Google Scholar]
- 54.Theodosiou A M, Rodrigues N R, Nesbit M A, Ambrose H J, Paterson H, McLellan-Arnold E, Boyd Y, Leversha M A, Owen N, Blake D J, Ashworth A, Davies K E. A member of the MAP kinase phosphatase gene family in mouse containing a complex trinucleotide repeat in the coding region. Hum Mol Genet. 1996;5:675–684. doi: 10.1093/hmg/5.5.675. [DOI] [PubMed] [Google Scholar]
- 55.Vincenti M P, Burrell T A, Taffet S M. Regulation of NF-kappa B activity in murine macrophages: effect of bacterial lipopolysaccharide and phorbol ester. J Cell Physiol. 1992;150:204–213. doi: 10.1002/jcp.1041500127. [DOI] [PubMed] [Google Scholar]
- 56.Wadleigh D J, Reddy S T, Kopp E, Ghosh S, Herschman H R. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem. 2000;275:6259–6266. doi: 10.1074/jbc.275.9.6259. [DOI] [PubMed] [Google Scholar]
- 57.Whitmarsh A J, Shore P, Sharrocks A D, Davis R J. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 58.Xia Y, Makris C, Su B, Li E, Yang J, Nemerow G R, Karin M. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA. 2000;97:5243–5248. doi: 10.1073/pnas.97.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]