

Abstract
The extracellular matrix protein fibulin-1 is a distinct component of vessel walls and can be associated with other ligands present in basement membranes, microfibrils, and elastic fibers. Its biological role was investigated by the targeted inactivation of the fibulin-1 gene in mice. This led to massive hemorrhages in several tissues starting at midgestation, ultimately resulting in the death of almost all homozygous embryos upon birth. Histological analysis demonstrated dilation and ruptures in the endothelial lining of various small vessels but not in that of larger vessels. Kidneys displayed a distinct malformation of glomeruli and disorganization of podocytes. A delayed development of lung alveoli suggested impairment in lung inflation. Immunohistology demonstrated the absence of fibulin-1 in its typical localizations but no aberrant patterns for several other extracellular matrix proteins. Electron microscopy revealed intact basement membranes but very irregular cytoplasmic processes of capillary endothelial cells in the organs that were most severely affected. Absence of fibulin-1 caused considerable blood loss but did not compromise blood clotting. The data indicate a strong but restricted abnormality in some endothelial compartments which, together with some kidney and lung defects, may be responsible for early death.
The cardiovascular system is the first complex organ to appear during embryonic development; it depends to a large part on the formation of numerous blood vessels by a process known as angiogenesis. This process is initiated by endothelial cells, has a distinct plasticity, and in the end leads to a considerable heterogeneity of the endothelium and the vessel walls in different organs (19, 44). Angiogenesis is controlled by various cytokines including vascular endothelial growth factors (VEGF), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), and transforming growth factor β, which transmit their signals through several receptor kinases (7, 12, 22, 45). Later stages include the recruitment of mesenchymal cells by the endothelium and the deposition of an extracellular matrix under the control of transforming growth factor β and PDGF, converting the vessel walls into a stable functional unit (14, 19, 45).
Gene targeting in mice has been used to show that several of these cytokines and receptors are essential in early development, and most null mutants died on embryonic days 8.5 to 14.5 (7, 22). For VEGF, even haploinsufficiency caused midgestation death (11, 18). Other deficiencies, such as for PDGF (30, 32) and several factors involved in blood coagulation (13, 25, 55, 56, 60), often showed an incomplete block of embryogenesis, and mice which survived until their neonatal death exhibited massive hemorrhaging involving several organs. This suggested that these components play a major role in the promotion of vessel wall stability and that their absence causes death by blood loss.
Integrity of vessel walls is also determined by their extracellular matrix, which includes basement membranes, elastic and collagenous fibers, and other interstitial structures. A substantial number of receptors involved in cell-cell and cell-matrix interactions and their extracellular ligands have been examined by gene targeting, and some mutants showed a phenotype involving defects in the heart and/or vessels (3, 17, 27). Absence of the fibril-forming collagen type I caused aortic ruptures at embryonic day 14 (33), and fibronectin deficiency led to even earlier death with severe defects in heart and vessel organization (20). Lack of elastin impaired late-gestation arterial morphogenesis, and the mutants showed a disorganized accumulation of smooth muscle cells (31). On the other hand, mutations in the elastin-associated fibrillins cause Marfan syndrome and related disorders in humans and experimental animals (40, 42). Moderate to fatal hemorrhage was observed in the absence of integrin receptor genes including the subunits α5 (61), αV (4), and β3 (24). Involvement of other integrins may have escaped detection because β1 subunit-deficient mice die prior to angiogenesis (16). A role of these integrins in vessel formation was, however, indicated from studies with β1-integrin-deficient embryonic stem (ES) cells that formed teratomas and embryoid bodies with a vasculature of poor quality (9).
There are many more known extracellular matrix proteins which could contribute to vessel wall stability but have not yet been examined by genetic elimination. They include the fibulins, which were initially characterized as two isoforms (fibulin-1 and fibulin-2) located in various vessel walls, basement membranes, and microfibrillar structures (38, 43, 46, 49). They are particularly prominent during heart valve development (10, 35, 62), and fibulin-1 is expressed in the developing aorta prior to elastogenesis (26). Fibulin-1 of 90 kDa was shown to bind fibrinogen (59); fibronectin, nidogen, and several other basement membrane proteins (5, 48); aggrecan and versican (2); and the angiogenesis inhibitor endostatin (50). The biological consequences of these interactions are not yet understood.
In the present study we have used homologous recombination in ES cells to generate transgenic mice which lack fibulin-1. These mice develop massive bleeding in the cranial mesenchyme, skin, and skeletal muscles, and most of them die shortly after birth. They also show a reduced loop formation in renal glomeruli and a delay in the proper formation of lung alveoli. Since these phenotypes are accompanied by irregular cell shape in some endothelial compartments, it indicates that fibulin-1 may interact directly or indirectly with endothelial cells. This has set the stage for a more precise analysis of the underlying molecular mechanisms.
MATERIALS AND METHODS
Construction of targeting vector, ES cell transfection, and generation of fibulin-1-deficient mice.
A 12-kb genomic mouse clone was isolated from a 129SV genomic phage library (Stratagene) using the 5′ part of fibulin-1 cDNA as probe (39). A neomycin cassette under the control of a phosphoglycerol kinase promoter and poly (A)+ signal was inserted into the XhoI site of the ATG-containing exon. A thymidine kinase cassette was added at the 3′ end of the construct. ES cell culture, electroporation of the targeting constructs into R1 ES cells, and isolation and analysis of neomycin-resistant ES cell clones were carried out as previously described (16). Three ES cell clones which had undergone homologous recombination were used to generate germ line chimeric mice as described in a previous study (16). Chimeric males were mated with either C57BL/6 females or 129SV females to obtain outbred or inbred lines, respectively.
Outbred homozygous (−/−) mice obtained from two independent ES cell clones were used for all subsequent examinations. They were compared to wild-type and heterozygous mice, both of which were, except in the fibulin-1 expression levels (mRNA and concentration in serum), indistinguishable in all other assays.
Southern, Northern, and RT-PCR analysis.
Genomic DNA was isolated from ES cells or tail biopsy specimens, digested with EcoRI, separated on a 0.7% agarose gel, and transferred to a Zeta-probe nylon membrane (Bio-Rad) by capillary blotting. The membrane was hybridized with a random-primed 32P-labeled external probe (Fig. 1) by using standard methods. For Northern analysis, total RNA was extracted from kidneys of 6-week-old mice using Trizol reagent as specified by the manufacturer (Gibco/BRL). A 10-μg portion of total RNA was glyoxylated, run through a 1% agarose gel, and subsequently blotted to a Zeta-probe nylon membrane and probed with random-primed 32P-labeled full-length cDNA probes specific for fibulin-1 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control.
FIG. 1.

Targeted disruption of the mouse fibulin-1 gene. (A) Partial structure of the mouse fibulin-1 gene (39). the targeting vector, and the homologous recombinant. The pgk-neo cassette was cloned in the direction of transcription into the XhoI site of exon-1, and the pgk-tk cassette was cloned into the 3 end of the genomic DNA. Restriction sites are shown for EcoRI (E), HindIII (H), NotI (N), PstI (P), SalI (S), and XhoI (X). (B) Southern blot analysis of EcoRI-digested genomic DNA isolated from 4-week-old F2 mice hybridized to an external 0.6-kb PstI fragment (probe A). The 10-kb fragment represents the wild-type allele, and the 3.8-kb fragment represents the targeted allele. (C and D) Analysis of mouse kidney total RNA for fibulin-1 by Northern-blot analysis (C) and RT-PCR (D). Northern analysis shows two fibulin-1 mRNA transcripts of 2.3 and 2.7 kb detectable in wild-type and heterozygous mice. A control hybridization was performed with a probe for GAPDH (lower panel). For RT-PCR, two fibulin-1 fragments of 400 bp (lanes 1) and 800 bp (lanes 2) and a GAPDH fragment of 200 bp (lanes 3) were amplified with specific primers. (E and F) Analysis of fibulin-1 protein expression by immunoblotting of plasma proteins after sodium dodecyl sulfate-polyacrylamide gel electrophoresis under nonreducing conditions (E) and quantitation of plasma fibulin-1 by radioimmunoinhibition assay shown as mean and standard deviations (F). The numbers of animals analyzed are indicated in parentheses.
Reverse transcriptase PCR (RT-PCR) was performed as follows. First-strand cDNA was synthesized using Superscript reverse transcriptase (Gibco/BRL) as specified by the supplier. The reaction mixture was supplemented with hexamer random primers and 5 μg of total kidney RNA as the template. Subsequently, PCR was carried out with two specific primer pairs: for fibulin-1, 400-bp (primer 1, 5′-CCATAACTGCCGGCTGGGAG-3′; primer 2, 5′-GCTGGTGGGGCGCACTCATC-3′) or 800-bp (primer 1 and primer 3, 5′-CGGTAGGAGCAGATGTGAC-3′) products were amplified, and for the control GAPDH, a 200-bp product was amplified using primers 4 (5′-CTGCCAAGTATGATGACATCA-3′) and 5 (5′-TACTCCTTGGAGGCCATGTAG-3′).
Histological analysis and immunohistochemistry.
Whole embryos or dissected organs were either fixed in phosphate-buffered saline (pH 7.4) (PBS)–4% paraformaldehyde, dehydrated, and embedded into paraffin or directly embedded in tissue-freezing medium (Tissue Tech; Leitz Industries), frozen on dry ice, and stored at −80°C. For paraffin-embedded tissues, 5-μm sections were cut with a Leitz microtome and collected onto polylysine-coated slides (Shandon). Cryosections (8 μm thick) were cut on a Leitz cryostat and further processed on Superfrost slides (Menzel). Staining with haematoxylin and eosin (HE) or methylene blue was performed by standard procedures. Sections (30 and 7 μm) were stained with antibodies against caveolin (Transduction Lab.) and analyzed by fluorescence micrography.
For the immunolocalization of different proteins, affinity-purified polyclonal rabbit antibodies against fibulin-1, fibulin-2 (38), collagen IV, perlecan (52), laminin α2 chain (57), fibronectin (Sigma), integrin α8 (23), ZO-1, and claudin-1 (Zymed) or rat monoclonal antibodies against nidogen-1 (JF3; Chemicon), cytokeratin (lu-5; Dianova), PECAM-1 (MEC13.3; Pharminogen), synaptopodin (G1D4; Progen) (37), and occludin (Z-T22; Zymed) were used together with the appropriate Cy-3-labeled secondary antibodies (Dianova) for detection. Biotinylated lectin BS-1 was used together with Cy-3-labeled streptavidin (Sigma). Tissue sections were blocked with 5% goat serum in PBS. After sequential incubation with the primary antibody for 2 h and the secondary antibody for 1 h, both diluted in 5% goat serum–PBS, and intermediate washes with PBS, sections were mounted in Fluorosave (Calbiochem) and analyzed under an Axiophot fluorescence microscope (Zeiss). Immunodetection of proteins after Western blotting and radioimmunoinhibition assays (28) were performed by standard procedures.
Electron microscopic analysis.
Small pieces of dissected organs were fixed in 4% buffered paraformaldehyde, rinsed in cacodylate buffer three times, and treated with 1% uranyl acetate in 70% ethanol (8 h) for contrast enhancement. They were subsequently dehydrated in a graded series of ethanol, and specimens were then embedded in Araldite (Serva). Semithin sections of plastic-embedded specimens were cut with a glass knife on an ultramicrotome (Reichert, Bensheim, Germany) and stained with methylene blue. Ultrathin sections (30 to 60 nm) for electron microscopic observation were processed on the same microtome with a diamond knife and placed on copper grids. Transmission electron microscopy was performed using a 902A electron microscope (Zeiss, Oberkochem, Germany).
Hematological analysis and determination of bleeding time.
After amputation of the tip of the tail, the bleeding time was monitored by gently absorbing the blood drop without touching the wound at 30-s intervals until the bleeding stopped. A modification of the standard Lee-White clotting test allowed the determination of blood coagulation times in small volumes. Glass capillaries (50 μl) were filled with 5 μl of blood using an Eppendorf pipette tip connected to a silicon tube. The blood column was moved every 30 s until a blood clot attached to the capillary wall. Blood cell counts and hematocrit measurements were done by standard methods. Platelet aggregation initiated by collagen I (10 μg/ml) or thrombin (10 μg/ml) was analyzed by fluorescence-activated cell sorting (Becton-Dickenson) using antibodies against P-selectin (Pharmingen) and fibrinogen (Sigma).
RESULTS
Targeted disruption of the fibulin-1 gene eliminates expression in homozygous mice.
To engineer a targeted deletion of fibulin-1, a mouse strain 129Sv genomic phage library was screened with the 5′ part of the mouse fibulin-1 cDNA to identify clones covering the 5′ region of the gene. The fibulin-1 gene was disrupted by insertion of a pgk-neo cassette, which was introduced in the direction of transcription into a 10.9-kb genomic construct at a single XhoI site in the first exon (Fig. 1A). In addition, a pgk-TK cassette was inserted at the 3′ site of the targeting construct for negative selection. ES cells (R1) were transfected with this construct, and G418-resistant clones were analyzed by Southern blotting with the external probe A, which detects a 10-kb EcoRI fragment in the wild-type allele and a 3.8-kb fragment from the targeted allele. Of 450 ES cell clones tested 19 were found to be positive for homologous recombination. Targeted ES cell clones were further tested for single integrations using internal probes (data not shown).
Three of the targeted ES cell clones were injected into 3.5-day-old C57BL/6 blastocysts and transferred to foster mice. Heterozygous offspring of chimeric F1 males mated with C57BL/6 females, as identified by Southern blotting (Fig. 1B), developed normally and were phenotypically indistinguishable from wild-type littermates. A reduced fibulin-1 mRNA level in heterozygous compared to wild-type mice was shown by Northern blotting (Fig. 1C) and RT-PCR analysis of adult kidney (Fig. 1D). Homozygous fibulin-1-deficient mice displayed a complete loss of expression of the two splice variants of fibulin-1 mRNA (39), as shown in both assays. Analysis of fibulin-1 levels in blood plasma by immunoblotting (Fig. 1E) confirmed that fibulin-1 protein is absent in homozygous mice. The mean concentration of fibulin-1 in plasma (3.5 ± 0.75 μg/ml) in wild-type animals, as determined by radioimmunoassay, was reduced to about half (1.62 ± 0.35 μg/ml) in heterozygotes, which was a statistically significant drop (independent t test: = −9.049, P < 10−9). Fibulin-1 was no longer detectable (<0.04 μg/ml) in the plasma of homozygous mice (Fig. 1F).
Postnatal lethality of fibulin-1 deficiency.
Analysis of 4-week-old offspring of heterozygous crossings showed that the proportion of fibulin-1-deficient mice was only 2% and thus was 14-fold lower than expected (Table 1). The frequency of heterozygous animals was about twice that of the wild type and thus was not affected by the inactivation of one fibulin-1 allele. The low frequency of fibulin-1-deficient mice raised the possibility that the fibulin-1 gene disruption might cause embryonic death. However, genotyping at embryonic stages E9.5 and E18.5 followed a normal Mendelian distribution (Table 1). After birth, almost all of the fibulin-1-deficient mice died during the first 24 to 48 h. Both their size and weight were more than 20% lower than those of their littermates. A larger number of surviving homozygotes could be obtained by intensive animal care and extensive breeding. Almost all fibulin-1-deficient animals which survived the first 2 days after birth developed normally but showed a 10 to 30% reduction in size and weight for several months. Most of the adults regained weight and were not longer distinguishable from heterozygotes. Mating of homozygotes with heterozygote or homozygote partners resulted in normal-sized litters. Fibulin-1-deficient mice were born in the expected Mendelian ratio and showed the same severity of the phenotype.
TABLE 1.
Genotype frequency of offspring from heterozygous matings of fibulin-1-deficient mice
| Stage | No. of animals of genotype:
|
Total no. | ||
|---|---|---|---|---|
| +/+ | +/− | −/− | ||
| 9.5-day embryo | 6 | 12 | 7 | 25 |
| 18.5-day embryo | 17 | 35 | 19 | 71 |
| Adult (4 wk)a | 60 | 106 | 3 | 169 |
+/+ mice made up 35%, +/− mice made up 63%, and −/− mice made up 2% of adult mice.
Spontaneous bleeding of fibulin-1-deficient mice.
Examination of the gross appearance of embryos at different stages of gestation revealed spontaneous bleeding of fibulin-1-deficient animals starting at day E12.5 (Fig. 2A). Although not all fibulin-1-deficient mice were macroscopically affected at this early stage, histological analysis demonstrated bleeding events found predominantly in the region of the telencephalon and beneath the spinal cord for almost every fibulin-1-deficient embryo. In addition, several embryos displayed bleeding of a typical petechial phenotype, which followed the track of the blood vessels under the skin (Fig. 2A). Although the severity of the bleeding increased during gestation, it did not have a fatal effect on embryonic development until birth. At the perinatal stage, every homozygote could be visually detected by the severe bleeding found predominantly in the regions of the snout and the hind limbs (Fig. 2B). Mice which were delivered at day E18.5 by cesarean section showed the same severe bleeding, indicating that this is not provoked by birth stress. A few neonates exhibited strong bleeding into the cranial mesenchyme, which caused compression of the brain (data not shown). No bleeding was found in the inner organs or into the abdominal cavity. The gross appearance of the few surviving fibulin-1-deficient mice changed by day 2 after birth. Subcutaneous blood was cleared, and spontaneous bleeding was no longer observed.
FIG. 2.
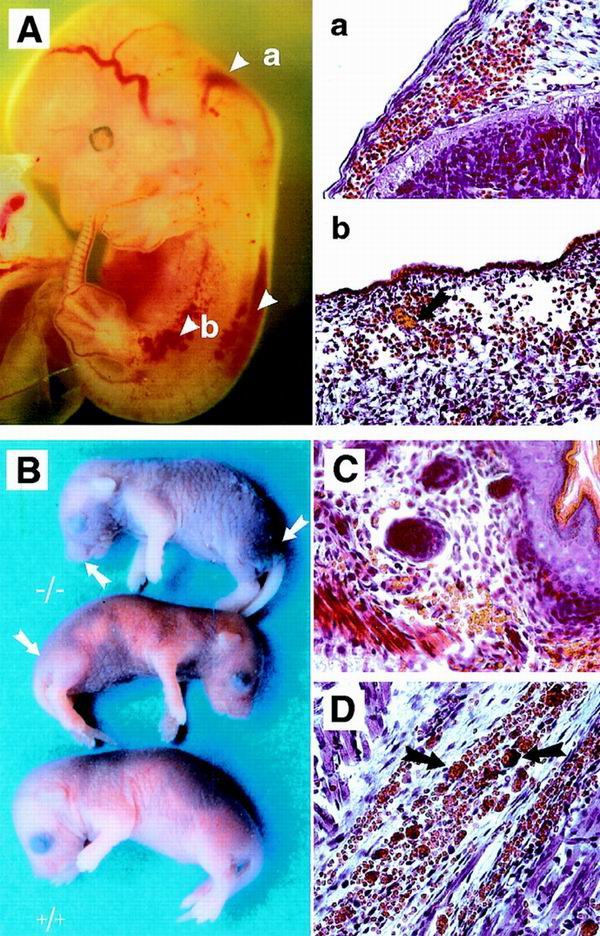
Spontaneous bleeding in fibulin-1-deficient embryos and neonates. (A) Severe bleeding in the cranial mesenchyme in the region of the myelencephalon and the spinal cord in an E12.5 embryo. Petechial bleeding is seen along blood vessels under the skin. Transverse sections through two regions (a and b) show a large number of erythrocytes outside the blood vessels, some of which are aggregated (arrow in b). (B) Newborn fibulin-1-deficient mice (−/−) with severe bleeding under the skin and in muscle predominantly of the snout, hind legs, and abdominal regions. (C and D) Histological analysis of sections from skin (C) and muscle (D). Note the large number of aggregated erythrocytes outside the blood vessels (arrows). Bar, 50μm.
Histological analysis of day E12.5 embryos demonstrated extravasal blood in the mesenchyme close to the telencephalon and the spinal cord and under the epidermis (Fig. 2A). For day E18.5 embryos, extravasal blood was found in the reticular layer of the skin and to a great extent in the subdermal space. In the skeletal muscle, erythrocytes were located both between the muscle fibers and in the perimysium (Fig. 2C and D). Fibulin-1-deficient embryos at day 13.5 of gestation with massive bleeding into the cranial mesenchyme (Fig. 3A) were further examined by immunostaining. Staining for the endothelial marker PECAM-1 (Fig. 3B) and the basement membrane protein perlecan (Fig. 3C) showed that the continuous endothelial cell basement membrane layer was interrupted at several positions while the surrounding mesenchyme appeared intact, excluding sectioning artefacts. As a consequence, this leads to leakage of erythrocytes into the extravasal space.
FIG. 3.
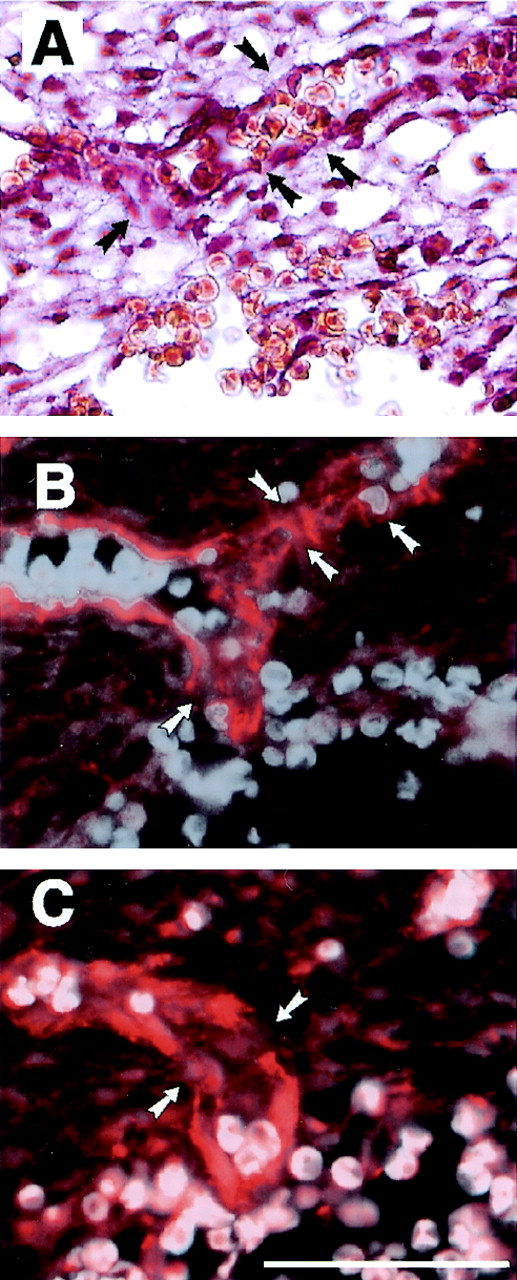
Severe bleeding in the cranial mesenchyme of fibulin-1-deficient E13.5 embryos with leaking venous plexus. (A) A sinus venosus, next to the rupture, is leaking erythrocytes, as shown by HE staining. (B and C) Immunofluorescence of serial sections with antibodies against PECAM-1 (B) and perlecan (C) revealed multiply disrupted endothelial cell layers (arrows) in the same region. Erythrocytes were made visible in panels B and C by superimposing their autofluorescence. Bar, 50 μm.
The massive aggregates of erythrocytes outside the blood vessels (Fig. 2A and D) could be stained by antibodies to fibrinogen, indicating a normal function of the coagulation system (data not shown). Hematological analyses of bleeding and coagulation time were not different for fibulin-1-deficient newborn animals and heterozygous controls (Table 2). Furthermore, no difference could be detected in the aggregation and P-selectin expression of platelets after stimulation with either thrombin or collagen type I (data not shown). However, a remarkable reduction (about 70%) was observed in the hematocrit values, indicating severe bleeding which could affect the survival of the homozygous mutants (Table 2). A similar reduction (50 to 70%) was also detected in erythrocyte numbers and hemoglobin content in the homozygous E18.5 embryos and neonates.
TABLE 2.
Hematological analysis of heterozygous and homozygous mice with fibulin-1 deficiencya
| Mouse | Blood coagulation time (min) | Bleeding time (min) | Hematocrit (%) |
|---|---|---|---|
| 18.5-day embryo | |||
| +/− | 2.5–3.5 | 3.5–5 | 41–44 |
| −/− | 2–4 | 3.5–6 | 30–36 |
| Newborn | |||
| +/− | 2–4 | 2–4.5 | 46–49 |
| −/− | 2–2.5 | 2.5–4.5 | 10–20 |
Ranges were determined using groups of two to seven individual animals. Blood coagulation and bleeding times were determined in triplicate.
Delayed lung development caused by fibulin-1 deficiency.
After birth, only about 50% of the fibulin-1-deficient newborns were able to start spontaneous breathing. Examination of the lungs of E18.5 embryos delivered by cesarean section showed that the parenchymal septa containing extracellular matrix were thickened and the saculi were not properly expanded. Surviving homozygotes at neonatal day 1 showed enlarged saculi with the septa still thickened (Fig. 4A). To identify the cells involved in this delayed development, a double immunostaining was performed for the endothelial marker PECAM-1 (Fig. 4B) and the epithelial marker cytokeratin (Fig. 4C). This demonstrated the same regional restriction of epithelial cells within the developing alveoli for both heterozygous and homozygous animals. The endothelial compartments of homozygotes appeared regularly organized, but electron microscopy of the vessels often showed a dilated and irregular lumen, indicating lack of a proper spatial control in the formation of vessel walls (data not shown). A similar pattern could also be demonstrated by antibodies against the vessel wall components collagen IV and endostatin (data not shown).
FIG. 4.
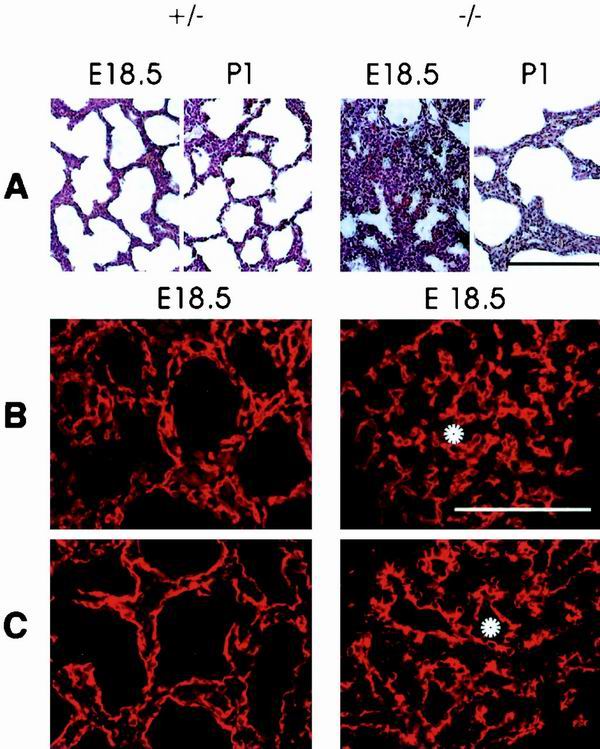
Delayed lung development in fibulin-1-deficient mice at stage E18.5 and 1 day after birth. (A and B) HE staining of fibulin-1-deficient animals shows improperly expanded sacculi in the lungs of spontaneous breathing E18.5 embryos and enlarged sacculi with thickened septa at perinatal day 1 compared to heterozygous controls. (B and C) Immunostaining for the endothelial marker PECAM-1 (B) and epithelial marker cytokeratin (C) demonstrates the proper arrangement of both cell types. Examples of sacculi that are not expanded in homozygous animals are indicated by white asterisks. Bar, 100 μm.
Abnormal formation of kidney glomeruli.
In 10 to 40% of kidney glomeruli of newborn fibulin-1-deficient animals, the organization of the endothelial cell layer and the podocytes was dramatically affected. The lumen of the capillary loop was dilated, and the number of its individual loops was reduced. In some cases a single capillary filled the complete glomerular space (Fig. 5A and B). Examinations at the ultrastructural level showed that all components of the glomeruli, the capillaries and endothelial cell layer, podocytes, and mesangial cells could be detected inside the Bowman capsule of homozygotes. The capillaries were completely covered by podocytes, and the glomerular basement membranes appeared normal. However, the number of foot processes was drastically reduced and their shapes were altered, leading to a decrease in the number of filtration slits (Fig. 5C and D), while the slit membranes appeared normal (Fig. 5D, inset). This alteration could be detected in all glomeruli of fibulin-1-deficient mice, including those which appeared normal at the light microscopic level. Renal tubules and collecting ducts appeared normal in both heterozygous and homozygous animals at the light microscopic (Fig. 5A and B) and electron microscopic levels. The urine showed no abnormalities in either protein concentration or composition (data not shown).
FIG. 5.

Light and electron microscopic analyses of kidneys from heterozygous (A and C) and homozygous (B and D) stage E18.5 embryos. (A and B) Methylene blue staining of thin kidney sections (bar, 15 μm) in the heterozygote shows a normal glomerular structure with small capillary lumen (l), mesangial cells (m), and podocytes (p) surrounded by Bowman's capsule and regular tubules (t). The homozygous kidney shows a single enlarged, glomerular capillary surrounded by podocytes and mesengial cells. (C and D) Electron microscopy (bar 0.7 μm) of heterozygotes reveals a regular array of podocyte (p) foot processes divided by small slits. The homozygotes show a decreased number of atypical foot processes (arrows) and an endothelium (e) with multiple luminal processes. However, a normal glomerular basement membrane (white arrowheads) is located between the capillary endothelium and podocytes in both cases. Regular formed slit membranes were found between the foot processes of podocytes (inset in panel D, black arrowheads).
Kidney glomeruli were further analyzed by immunohistochemical methods (Fig. 6). As expected, fibulin-1 was expressed prominently in both the mesangium and the basement membrane of the glomeruli of heterozygous mice but was missing in the homozygous animals (Fig. 6A and B). The weak fibulin-2 staining in heterozygous glomeruli was not increased in fibulin-1-deficient mice (Fig. 6C and D). Several potential fibulin-1 ligands such as fibronectin (Fig. 6I and J) and the basement membrane proteins nidogen-1 (Fig. 6E and F), laminin α2 chain (Fig. 6G and H), collagen IV, and perlecan (data not shown) were found at similar levels of staining intensity in the glomeruli of both heterozygous and homozygous fibulin-1-deficient mice. Nevertheless, the staining patterns of these proteins was different in the two groups, reflecting the disorder in morphological organization in fibulin-1-deficient mice. All three cell types of the glomerular cave, the endothelial cells, represented by PECAM 1 (Fig. 6K and L), the podocytes, represented by synaptopodin (37) (Fig. 6M and N), and the mesangial cells, represented by integrin α8 (23), were irregularly distributed for the homozygous animals. In mice with completely dilated capillaries, the mesangial cells and podocytes were lined up around the endthelium, forming clusters with almost no overlaps, as shown by double staining (Fig. 6N and P).
FIG. 6.

Immunofluorescence staining of kidney glomeruli from heterozygous (+/−) and fibulin-1-deficient (−/−) newborn mice. Protein expression around a single kidney glomerulus was analyzed by using antibodies against fibulin-1 (A and B), fibulin-2 (C and D), nidogen-1 (E and F), laminin α2 chain (G and H), fibronectin (I and J), and PECAM-1 (K and L) and by double staining for synaptopodin (M and N) and integrin α8 (O and P). Bar, 25 μm.
Embryonic heart and aorta development is almost normal.
Fibulin-1 is expressed at early stages of cardiovascular development and later becomes a prominent component of heart valves and the aortic media (10, 26, 62). Homozygous embryos, as identified by their hemorrhages, showed no obvious failure in heart function, however, and no gross histological abnormalities in the heart and aorta until birth. Some subtle changes could be revealed at the ultrastructural level. Cardiomyocytes frequently had an irregular shape and had cytoplasmic processes oriented toward endocardial and capillary endothelial cells (see Fig. 9D). These features could not be dected in normal and heterozygous controls (data not shown). Several abnormalities in the shape of endothelial cells lining myocardial capillaries were also observed (see below). Immunohistological examination of fibulin-1-deficient mice demonstrated the absence of fibulin-1 from heart valves, arteries, and capillaries within the myocardium. The expression of fibulin-2 was apparently not changed (Fig. 7). The examination of both tissues with all the other antibodies used in the experiment in Fig. 6 also did not reveal any difference between heterozygous and homozygous animals.
FIG. 9.

Electron microscopy of capillaries demonstrates unusual endothelial cell shapes in homozygous perinatal animals but not in adults. Ultrathin cross-sections of capillaries are shown for heterozygous (A) and homozygous (B to F) stage E18.5 embryos. (A) Representative heart capillary from a heterozygote with a single endothelial cell having a smooth inner surface to the capillary lumen (l) without cytoplasmic processes. (B) Heart capillary from a homozygote demonstrates multiple endothelial cytoplasmic processes. (C) Other heart capillaries show multilayered endothelial cells (arrows). (D) Endothelial cells (e) with vacuoles are seen together with their basement membranes (arrows) detached from an irregularly shaped cardiomyocyte (cm). (E) Enlarged renal capillaries show an irregular luminal surface with cytoplasmic processes (arrows), while a tubular epithelium cell (t) seems to be unaltered. (F) Multiple cytoplasmic processes (arrows) are recognizable in a brain capillary endothelial cell which is partially covered by a normal pericyte (p). (G and H) Heart capillaries from homozygous adults (H) do not show obvious alterations compared to heterozygotes (G). Bars: 4 μm (A), 1 μm (B, C, and E), 0.7 μm (D), 5 μm (F), 3 μm and (G and H).
FIG. 7.

Immunofluorescence staining for fibulin-1 and fibulin-2 of heart valve (A to D) and myocardium including a large artery (E to H). Staining was performed with equal concentrations of antibodies against fibulin-1 (A, B, E, and F) and fibulin-2 (C, D, G, and H). Heterozygous and homozygous embryos were from stage E18.5. Bar, 100 μm.
Abnormal morphogenesis in various capillaries in embryos and perinatal mice recovers in adults.
A distinct enlargement of the capillary lumen was the most common phenotype in organs such as the heart and kidney (Fig. 5B and 8B and D) in fibulin-1-deficient late embryos and perinatal mice. In addition, the capillary shape was more irregular than that in normal or heterozygous animals (Fig. 8A and C). Medium-sized vessels, which are surrounded by smooth muscle cells, showed no obvious differences in diameter and shape between heterozygous and homozygous embryos, however (Fig. 8E and F).
FIG. 8.

Light microscopy of capillaries and arteries. Antibody staining against caveolin was performed with 30-μm (A and B) and 7-μm (C and D) semithin sections of ventricular heart capillaries and small arteries of heterozygous and homozygous embryos at stage E18.5. (A and C) In the densely packed myocardium, heterozygotes show relatively homogenous and round capillaries (arrows) partially filled with erythrocytes. (B and D) Homozygous animals have mainly enlarged capillaries of irregular shape (arrows). (E and F) Methylene blue staining of small coronary arteries show no differences between the two groups of mice and are characterized by monolayers of smooth muscle cells (arrows) and endothelial cells (arrowheads) and no obvious alteration in the endothelial surface. Bar, 30 μm.
Several capillaries were more closely examined by electron microscopy. Normal capillaries usually show a single endothelial cell per cross-section, with a smooth surface towards the lumen, and this was also found in heterozygous animals (Fig. 9A). The most common observation for homozygotes, besides the irregular capillary shape, was the excessive development of many cytoplasmic processes of endothelial cells, which sometimes filled a substantial part of the vessel lumen (Fig. 9B, E, and F). Other alterations included the formation of multilayers of endothelial cells (Fig. 9C) or the disruption of endothelial cell-cell contacts (Fig. 9D) that could cause leaky vessel walls. Several endothelial cells also showed a distinct increase in the number of intracellular vacuoles (Fig. 9C to E). These changes are restricted to endothelial cells, since an adjacent tubular epithelium cell (Fig. 9E) and pericyte (Fig. 9F) have a normal shape. Furthermore, pericytes were found adjacent to the capillary endothelium at the same frequency as that in normal controls. None of these changes could be detected by electron microscopy of macrovascular endothelium. In the few surviving animals, all alterations in the capillary system recovered, with no obvious differences between heterozygotes and homozygtes (Fig. 9G and H).
The distribution of tight junction proteins is not affected in endothelial cells.
The morphology of the tight-junction contacts between endothelial cells were investigated at stage E13.5 and E18.5 for several tissues including the brain. Double staining with antibodies against VE-cadherin and with lectin BS-1 (Fig. 10A, B, E and F) or with antibodies against ZO-1 (Fig. 10C, D, G, and H) showed no differences in the distribution or intensity of the staining, as demonstrated for E18.5 brain capillaries. This was also found for the tight-junction proteins occludin and claudin-1 (data not shown).
FIG. 10.

Double-fluorescence staining of E18.5 brain capillaries from wild-type (A to D) and fibulin-1-deficient (E to H) mice either with antibodies against VE-cadherin (A and E) and with lectin BS-1 (B and F) or with antibodies against VE-cadherin (C and G) and ZO-1 (D and H). VE-cadherin staining is restricted in both heterozygous (A) and homozygous (E) animals to tight junctions of capillaries, stained by lectin BS-1 (B and F). VE-cadherin (C and G) and ZO-1 (D and H) expression fully overlaps in both wild-type and fibulin-1-deficient mice.
DISCUSSION
Fibulin-1 was the first member of a new family of calcium-binding extracellular matrix proteins characterized some 10 years ago (1, 28). Four more members are now known (21), and a fibulin-1 homologue has also been identified in the genome of Caenorhabditis elegans (6). Mammalian fibulin-1 was also shown to be a ligand for a diverse group of extracellular proteins (2, 5, 48, 50, 51, 59) but did not promote cell adhesion through integrin receptors (41). In the present study we have analyzed the biological consequences of these potential functions by deleting the fibulin-1 gene in mice. Heterozygous fibulin-1-deficient mice showed the expected decrease in serum fibulin-1 and tissue mRNA levels but were otherwise indistinguishable from normal animals. Most homozygous mice, however, died during the first 2 days after birth, due to a combination of blood loss and renal and respiratory impairments. This indicated that fibulin-1 plays a crucial role in vessel development, from midgestation until an early perinatal stage. However, a few homozygotes survived and developed to a fertile age. For the histological abnormalities in lung and kidney podocytes, these animals might be less severely affected and might attain normal functioning after birth with only minor residua. For the capillary phenotype, however, the changes in the vessel stability during the first 2 days after birth was correlated with a normalization of the morphology of the capillaries. This suggested a distinct switch in the requirement for fibulin-1, which might be correlated with an obvious reduction of fibulin-1 expression in adult animals.
The most obvious phenotype of fibulin-1-deficient mice were massive hemorrhages in skin, muscle, and perineurial tissue but not in most other organs. This blood leakage is presumably caused by imperfect endothelial cell linings in capillaries and, as a consequence, the disruption of continuous vessel wall structures (Fig. 3). The capillaries in nearly all the organs examined were abnormally widened and irregular, although in the heart, lungs, kidneys, and organs of the abdominal cavity this was not associated with hemorrhages. Since we did not observe an increased number of endothelial nuclei per capillary cross-section, it is likely that the increased capillary diameter is caused by enlargement of endothelial cells. Ultrastructural analyses support this interpretation, by showing many luminal cytoplasmic processes; an overlap of endothelial cells, which provides the impression of multilayered cells; and the increased formation of intracellular vacuoles. More limited changes in cellular shape were also observed for cardiomyocytes and podocytes, which showed a reduced number of foot processes and slits, while no obvious changes were detected for the slit membranes or for renal tubular epithelium and pericytes. This indicates that the primary impact of fibulin-1 deficiency is on endothelial cells in developing small capillaries, with a limited involvement of endothelial cells of larger vessels and other cell types.
There are several other genes whose elimination produced similar but not completely identical vascular phenotypes associated with late gestation or neonatal death. Absence of the PDGF B chain prevented glomerular tuft formation and at later stages caused fatal hemorrhages (30). Vessel instability was explained by the lack of attraction of pericytes by PDGF (32). Absence of the PDGF receptor β subunit also interfered with glomerular capillarization, while muscle capillaries had a narrow and irregular shape which may have caused microangiopathic hemolytic anemia (54). The knockout of the integrin α3 subunit also caused a reduced branching of glomerular loops and lung alveoli, with patterns similar to that of fibulin-1 deficiency, but was not associated with bleeding (29). Massive intracerebral and intestinal hemorrhages were, however, observed in about 20% of surviving mice lacking the αV integrin (4). Elimination of the laminin α5 chain, which is a potential ligand for the α3β1 integrin, also interferes with glomerular vascularization due to defects in the formation of the glomerular basement membrane (34). This basement membrane appears not to be affected by fibulin-1 deficiency.
A common vascular defect can apparently be generated by the elimination of quite diverse proteins which are involved primarily in cell signaling and cell-matrix interactions during angiogenesis (7, 22, 44, 45). Late stages include the investment with smooth muscle cells or pericytes and their communication with endothelial cells followed by matrix deposition for the formation of a stable, nonleaky vessel bed (14, 19). This stage is preceded by a plasticity window in vessel remodeling regulated by VEGF and PDGF (8), while angiopoietin-1 seems to be required for making vessels resistant to leakage (58). As shown here, fibulin-1 is also required for the stabilization of vessel walls, at least in certain tissues. A more general observation, however, is the bizarre changes in the shape of capillary endothelial cells, which have rarely been observed in other studies on malformed vessels. This could indicate that fibulin-1 controls the cytoskeletal organization of these endothelial cells directly or indirectly, arresting them at least transiently at a certain premature angiogenic stage. Such cytoskeletal changes as well as vacuole formation are known from other endothelial cell studies to be controlled by integrins and growth factors (15, 47, 53).
A molecular interpretation of our data is still difficult, but at least it allows some possibilities to be eliminated. The binding of fibulin-1 to fibrinogen was previously interpreted to indicate a role in blood coagulation (59). This seems not to be an essential function, since coagulation, platelet aggregation, and bleeding time are apparently normal in fibulin-1-deficient mice. A lack of other coagulation proteins such as fibrinogen, prothrombin, tissue factor inhibitor, and coagulation factor V causes more severe defects, including massive hemorrhages (13, 25, 55, 56). As shown previously with tumor cells (41) and more recently with endothelial cells (R. Timpl, unpublished data), fibulin-1C and -1D are not substrates for integrin-mediated cell adhesion. This indicates that fibulin-1 does not make a significant contribution to cell-matrix interactions, which have been implicated in playing a role in vessel stability from studies with fibronectin (20) and αV integrin (4) knockout mice. However, there is still the possibility that fibulin-1 may bind to other cellular receptors. In this context, it is of interest that fibulin-1 is also present at relatively high concentration in blood (1, 28), and it could therefore bind to endothelial cells via luminal receptors.
A major function of fibulin-1 could be involvement in the supramolecular organization of various extracellular structures such as basement membranes (28, 38, 62), microfibrils (35), and elastic fibers (46, 51). The basement membranes of fibulin-1-deficient mice, however, appeared normal by electron microscopy and immunohistology, indicating that its absence can be tolerated. Fibulin-1 is also an important component during heart development and switches from a localization in the cardiac jelly, presumably mediated by hyaluronan/versican (2, 35), to a microfibrillar association. Another major location is in the elastic fibers of the aorta and dermis (46, 51). Again, development of heart, aorta, and heart valves seems not to be severely compromised in the absence of fibulin-1, possibly because fibulin-2 could take over some functions. Fibulin-1 was also shown to bind to the angiogenesis inhibitor endostatin, an interaction considered to retain endostatin in tissues and vessel walls after proteolytic release from collagen XVIII (36, 50). This distribution also remains unchanged in fibulin-1-deficient mice, indicating efficient replacement by other extracellular ligands such as nidogen-2 and fibulin-2.
The distinct vessel wall and endothelial cell changes in homozygous mice indicate a particular role for fibulin-1 during angiogenesis, which is not yet understood at the molecular level. Several previous predictions of fibulin-1 functions based on its binding repertoire and tissue localization failed to provide satisfying answers. Further activities of fibulin-1, such as binding to endothelial cells and angiogenic growth factors, are now under investigation. A moderate but distinct calcium-dependent interaction of fibulin-1 with bFGF could be demonstrated by a solid-phase binding assay (unpublished data). We considered in this context that fibulin-1 may also modulate the expression or activity of other components involved in angiogenesis and thus may play a more indirect role within a particular signaling cascade. However, we observed no changes in the expression of the tyrosine kinase receptors flt, flk, tie, and tek, of the ligands VEGF-A, VEGF-B, Ang-1, Ang-2, and bFGF, and of VE-cadherin when examined by semiquantitative RT-PCR in E18.5 kidneys (unpublished data). The distribution of VE-cadherin as well as of other endothelial tight junction proteins like occludin, claudin-1, and ZO-1 was not altered for fibulin-1 homozygotes after immunostaining. While the present study has concentrated on developmental features, further studies based on adult fibulin-1-deficient mice could reveal other aspects of fibulin-1 functions. These animals can now be generated in sufficient numbers, and preliminary data indicate moderate to severe abnormalities in the aorta (G. Kostka and W. Bloch, unpublished data). Further studies are required to reveal whether such animals will become suitable models for studying vascular diseases.
ACKNOWLEDGMENTS
We are grateful for the expert technical assistance of Christa Hintz-Martini and Sabine Sass, and we thank S. Massberg (University of Munich) for the platelet assays. We also appreciate help from and discussion with U. Mayer, B. Bader, H. Gerhardt, and H. Wolburg.
This study was supported by EC grant BI04 CT960537, the Deutsche Forschungsgemeinschaft (SFB 266), the Swedish Medical Research Council, and NIH grant GM 55625.
REFERENCES
- 1.Argraves W S, Tran H, Burgess W H, Dickerson K. Fibulin is an extracellular matrix and plasma glycoprotein with repeated domain structure. J Cell Biol. 1990;111:3155–3164. doi: 10.1083/jcb.111.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aspberg A, Adam S, Kostka G, Timpl R, Heinegard D. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J Biol Chem. 1999;274:20444–20449. doi: 10.1074/jbc.274.29.20444. [DOI] [PubMed] [Google Scholar]
- 3.Aszódi A, Pfeiffer A, Wendel M, Hiripi L, Fässler R. Mouse models of extracellular matrix diseases. J Mol Med. 1998;76:238–252. doi: 10.1007/s001090050214. [DOI] [PubMed] [Google Scholar]
- 4.Bader B L, Rayburn H, Crowley D, Hynes R O. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all αV integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 5.Balbona K, Tran H, Godyna S, Ingham K C, Strickland D K, Argraves W S. Fibulin binds to itself and to the carboxyl-terminal heparin-binding region of fibronectin. J Biol Chem. 1992;267:20120–20125. [PubMed] [Google Scholar]
- 6.Barth J L, Argraves K M, Roark E F, Little C D, Argraves W S. Identification of chicken and C. elegans fibulin-1 homologs and characterization of the C. elegans fibulin-1 gene. Matrix Biol. 1998;17:635–646. doi: 10.1016/s0945-053x(98)90114-7. [DOI] [PubMed] [Google Scholar]
- 7.Beck L, D'Amore P A. Vascular development: cellular and molecular regulation. FASEB J. 1997;11:365–373. [PubMed] [Google Scholar]
- 8.Benjamin L E, Hemo I, Keshet E. A plasticity window for blood vessel remodeling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125:1591–1598. doi: 10.1242/dev.125.9.1591. [DOI] [PubMed] [Google Scholar]
- 9.Bloch W, Forsberg E, Lentini S, Brakebusch C, Martin K, Krell H W, Weidle U H, Addicks K, Fässler R. β1 integrin is essential for teratoma growth and angiogenesis. J Cell Biol. 1997;139:265–278. doi: 10.1083/jcb.139.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouchey D, Argraves W S, Little C D. Fibulin-1, vitronectin and fibronectin expression during avian cardiac valve and septa development. Anat Rec. 1996;244:540–551. doi: 10.1002/(SICI)1097-0185(199604)244:4<540::AID-AR12>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P, Jain R K. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 13.Cui J, O'Shea K S, Purkayastha A, Saunder T L, Ginsburg D. Fatal hemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384:66–68. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 14.Darland D C, D'Amore P A. Blood vessel maturation: Vascular development comes of age. J Clin Investig. 1999;103:157–158. doi: 10.1172/JCI6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis G E, Camarillo C W. An α2β1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res. 1996;224:39–51. doi: 10.1006/excr.1996.0109. [DOI] [PubMed] [Google Scholar]
- 16.Fässler R, Meyer M. Consequences of lack of β1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 17.Fässler R, Georges-Labouesse E, Hirsch E. Genetic analysis of integrin function in mice. Curr Opin Cell Biol. 1996;8:641–646. doi: 10.1016/s0955-0674(96)80105-0. [DOI] [PubMed] [Google Scholar]
- 18.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea K S, Powell-Braxton L, Hillan K J, Moore M W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 19.Folkman J, D'Amore P A. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- 20.George E L, Baldwin H S, Hynes R O. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of presursor cells. Blood. 1997;90:3073–3081. [PubMed] [Google Scholar]
- 21.Giltay R, Timpl R, Kostka G. Sequence, recombinant expression and tissue localization of two novel extracellular matrix proteins, fibulin-3 and fibulin-4. Matrix Biol. 1999;18:469–480. doi: 10.1016/s0945-053x(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 22.Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 23.Hartner A, Schöckelmann H, Pröls F, Müller U, Sterzel R B. α8 integrin in glomerular mesangial cells and in experimental glomerulonephritis. Kidney Int. 1999;56:1468–1480. doi: 10.1046/j.1523-1755.1999.00662.x. [DOI] [PubMed] [Google Scholar]
- 24.Hodivala-Dilke K M, McHugh K P, Tsakiris D A, Rayburn H, Crowley D, Ullman-Culeré M, Ross F P, Coller B S, Teitelbaum S, Hynes R O. β3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Investig. 1999;103:229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Z-F, Higuchi D, Lasky N, Broze G J. Tissue factor pathway inhibitor gene disruption produces intrauterine lethality in mice. Blood. 1997;90:944–951. [PubMed] [Google Scholar]
- 26.Hungerford J E, Owens G K, Argraves W S, Little C D. Development of aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev Biol. 1996;178:375–392. doi: 10.1006/dbio.1996.0225. [DOI] [PubMed] [Google Scholar]
- 27.Hynes R O. Targeted mutations in cell adhesion genes: what have we learned from them? Dev Biol. 1996;180:402–412. doi: 10.1006/dbio.1996.0314. [DOI] [PubMed] [Google Scholar]
- 28.Kluge M, Mann K, Dziadek M, Timpl R. Characterization of a novel calcium-binding 90-kDa glycoprotein (BM-90) shared by basement membranes and serum. Eur J Biochem. 1990;193:651–659. doi: 10.1111/j.1432-1033.1990.tb19383.x. [DOI] [PubMed] [Google Scholar]
- 29.Kreidberg J A, Donovan M J, Goldstein S L, Rennke H, Shepherd K, Jones R C, Jaenisch R. α3β1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 30.Levéen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 31.Li D Y, Brooke B, Davis E C, Mecham R P, Sörensen L K, Boak B B, Eichwald E, Keating M T. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl P, Johansson P R, Levéen P, Betsholtz C. Pericyte loss and microaneurism formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 33.Löhler J, Timpl R, Jaenisch R. Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell. 1984;38:597–606. doi: 10.1016/0092-8674(84)90514-2. [DOI] [PubMed] [Google Scholar]
- 34.Miner J H, Li C. Defective glomerulogenesis in the absence of laminin α5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217:278–289. doi: 10.1006/dbio.1999.9546. [DOI] [PubMed] [Google Scholar]
- 35.Miosge N, Sasaki T, Chu M L, Herken R, Timpl R. Ultrastructural localization of microfibrillar fibulin-1 and fibulin-2 during heart development indicates a switch in molecular associations. Cell Mol Life Sci. 1997;54:606–613. doi: 10.1007/s000180050188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miosge N, Sasaki T, Timpl R. Angiogenesis inhibitor endostatin is a distinct component of elatic fibers in vessel walls. FASEB J. 1999;13:1743–1750. doi: 10.1096/fasebj.13.13.1743. [DOI] [PubMed] [Google Scholar]
- 37.Mundel P, Reiser J, Kritz W. Introduction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8:697–705. doi: 10.1681/ASN.V85697. [DOI] [PubMed] [Google Scholar]
- 38.Pan T-C, Sasaki T, Zhang R-Z, Fässler R, Timpl R, Chu M-L. Structure and expression of fibulin-2, a novel extracellular matrix protein with multiple EGF-like repeats and consensus motifs for calcium-binding. J Cell Biol. 1993;123:1269–1277. doi: 10.1083/jcb.123.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan T-C, Kostka G, Zhang R-Z, Timpl R, Chu M-L. Complete exon-intron organization of the mouse fibulin-1 gene and its comparison with the human fibulin-1 gene. FEBS Lett. 1999;444:38–42. doi: 10.1016/s0014-5793(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 40.Pereira L, Andrikopoulos K, Tian J, Lee S Y, Keene D R, Ono R, Reinhardt D P, Sakai L Y, Biery N J, Bunton T, Dietz H C, Ramirez F. Targetting the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 41.Pfaff M, Sasaki T, Tangemann K, Chu M-L, Timpl R. Integrin-binding and cell-adhesion studies of fibulins reveal a particular affinity for αIIbβ3. Exp Cell Res. 1995;219:87–92. doi: 10.1006/excr.1995.1208. [DOI] [PubMed] [Google Scholar]
- 42.Ramirez F. Fibrillin mutations in Marfan syndrome and related phenotypes. Curr Opin Genet Dev. 1996;6:309–315. doi: 10.1016/s0959-437x(96)80007-4. [DOI] [PubMed] [Google Scholar]
- 43.Reinhardt D P, Sasaki T, Dzamba B J, Keene D R, Chu M-L, Göhring W, Timpl R, Sakai L Y. Fibrillin-1 and fibulin-2 interact and are colocalized in some tissues. J Biol Chem. 1996;271:19489–19496. doi: 10.1074/jbc.271.32.19489. [DOI] [PubMed] [Google Scholar]
- 44.Risau W. Differentiation of endothelium. FASEB J. 1995;9:926–933. [PubMed] [Google Scholar]
- 45.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 46.Roark E F, Keene D R, Haudenschild C C, Godyna S, Little C D, Argraves W S. The association of human fibulin-1 with elastic fibers: an immunohistological, ultrastructural, and RNA study. J Histochem Cytochem. 1995;43:401–411. doi: 10.1177/43.4.7534784. [DOI] [PubMed] [Google Scholar]
- 47.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki T, Kostka G, Göhring W, Wiedemann H, Mann K, Chu M-L, Timpl R. Structural characterization of two variants of fibulin-1 that differ in nidogen affinity. J Mol Biol. 1995;245:241–250. doi: 10.1006/jmbi.1994.0020. [DOI] [PubMed] [Google Scholar]
- 49.Sasaki T, Wiedemann H, Matzner M, Chu M L, Timpl R. Expression of fibulin-2 by fibroblasts and deposition with fibronectin into a fibrillar matrix. J Cell Sci. 1996;109:2895–2904. doi: 10.1242/jcs.109.12.2895. [DOI] [PubMed] [Google Scholar]
- 50.Sasaki T, Fukai N, Mann K, Göhring W, Olsen B R, Timpl R. Structure, function and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. EMBO J. 1998;17:4249–4256. doi: 10.1093/emboj/17.15.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sasaki T, Göhring W, Miosge N, Abrams W R, Rosenbloom J, Timpl R. Tropoelastin binding to fibulins, nidogen-2 and other extracellular matrix proteins. FEBS Lett. 1999;460:280–284. doi: 10.1016/s0014-5793(99)01362-9. [DOI] [PubMed] [Google Scholar]
- 52.Schulze B, Mann K, Battistutta R, Wiedemann H, Timpl R. Structural properties of recombinant domain III-3 of perlecan containing a globular domain inserted into an epidermal-growth-factor-like motif. Eur J Biochem. 1995;231:551–556. doi: 10.1111/j.1432-1033.1995.tb20731.x. [DOI] [PubMed] [Google Scholar]
- 53.Sims J R, Karp S, Ingber D E. Altering the cellular mechanical force balance results in integrated changes in cell, cytoskeletal and nuclear shape. J Cell Sci. 1992;103:1215–1222. doi: 10.1242/jcs.103.4.1215. [DOI] [PubMed] [Google Scholar]
- 54.Soriano P. Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes Dev. 1994;8:1888–1898. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 55.Suh T T, Holmbäck K, Jensen N J, Daugherty C C, Small K, Simon D I, Potter S S, Degen J L. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 56.Sun W Y, Witte D P, Degen J L, Colbert M C, Burkart M C, Holmbäck K, Xiao Q, Bugge T H, Degen S F J. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci USA. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Talts J F, Mann K, Yamada Y, Timpl R. Structural analysis and proteolytic processing of recombinant G domain of mouse laminin α2 chain. FEBS Lett. 1998;426:71–76. doi: 10.1016/s0014-5793(98)00312-3. [DOI] [PubMed] [Google Scholar]
- 58.Thurston G, Suri C, Smith K, McClain J, Sato T N, Yancopoulos G D, McDonald D M. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 59.Tran H, Tanaka A, Litvinovich S V, Medved L V, Haudenschild C C, Argraves W S. The interaction of fibulin-1 with fibrinogen. A potential role in hemostasis and thrombosis. J Biol Chem. 1995;270:19458–19464. doi: 10.1074/jbc.270.33.19458. [DOI] [PubMed] [Google Scholar]
- 60.Xue J, Wu Q, Westfield L A, Tuley E A, Lu D, Zhang Q, Shim K, Zheng X, Sadler J E. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci USA. 1998;95:7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang J T, Rayburn H, Hynes R O. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H Y, Kluge M, Timpl R, Chu M L, Ekblom P. The extracellular matrix glycoproteins BM-90 and tenascin are expressed in the mesenchyme at sites of endothelial-mesenchymal conversion in the embryonic mouse heart. Differentiation. 1993;52:211–220. doi: 10.1111/j.1432-0436.1993.tb00633.x. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H-Y, Chu M-L, Pan T-C, Sasaki T, Timpl R, Ekblom P. Extracellular matrix protein fibulin-2 is expressed in the embryonic endocardial cushion tissue and is a prominent component of valves in adult heart. Dev Biol. 1995;167:18–26. doi: 10.1006/dbio.1995.1003. [DOI] [PubMed] [Google Scholar]