

Abstract
Chronic alcohol consumption is associated with intestinal fungal dysbiosis, yet we understand little about how alterations of intestinal fungi (mycobiota) contribute to the pathogenesis of alcohol-associated liver disease. By reanalyzing internal transcribed spacer 2 amplicon sequencing of fecal samples from a cohort of 66 patients with alcohol use disorder for presence (as opposed to relative abundance) of fungal species, we observed that the presence of Malassezia restricta was associated with increased markers of liver injury. M. restricta exacerbates ethanol-induced liver injury both in acute binge and chronic ethanol-feeding models in mice. Using bone marrow chimeric mice, we found that the disease exacerbating effect by M. restricta was mediated by C-type lectin domain family 4, member N on bone marrow-derived cells. M. restricta induces inflammatory cytokines and chemokines in Kupffer cells through C-type lectin domain family 4, member N signaling. Targeting fungal pathobionts might be a therapeutic strategy for alcohol-associated liver disease.
INTRODUCTION
Alcohol-associated liver disease is one of the most prevalent liver diseases, and the leading chronic cause of alcohol-attributable deaths.1 Disturbances of the intestinal microbiota, which include bacteria, fungi, and viruses, have been linked to alcohol-associated liver disease.2,3 Despite most research focusing on the bacterial microbiota, the fungal microbiota (also called mycobiota) has been implicated in the pathogenesis of various liver diseases.4 Intestinal fungi contribute to the development of ethanol-induced liver disease in mice,5 correlate with disease severity in patients with alcohol-associated hepatitis,6 and predict the outcome of therapeutic interventions.7 Specifically, we found that the fecal proportion of the genus (Malassezia restricta M. restricta) was higher in alcohol use disorder (AUD) patients with the progressive liver disease compared with nonprogressive liver disease. Moreover, the relative proportion of M. restricta decreased after 2 weeks of alcohol abstinence.
M. restricta is a major component of human skin mycobiota and is associated with a spectrum of cutaneous diseases.8 In the human gut, Malassezia genus is the second most abundant fungal genus analyzed by internal transcribed spacer (ITS) sequencing9,10 with M. restricta identified as the most abundant Malassezia species.11 M. restricta is involved in the pathogenesis of gut inflammation and cancer.12 M. restricta is especially abundant in inflammatory bowel disease patients carrying loss-of-function mutations in caspase recruitment domain-containing protein 9 (Card9), which induces a strong inflammatory response from myeloid phagocytes. Colonization of mice with M. restricta exacerbates disease severity of dextran sulfate sodium-induced colitis in mice.13 Malassezia species infiltrate pancreatic ductal adenocarcinoma tumors both in humans and mice, and promote disease progression by driving the complement cascade through the activation of mannose-binding lectin.14
C-type lectin receptors (CLRs) are pattern recognition receptors for fungal pathogens,15 which include C-type lectin domain family 7, member a (also known as dectin-1), Clec4n (dectin-2), CD209 (DC-SIGN), Clec4e (mincle), and Mrc1 (mannose receptor). Clec4n is a FcRg-coupled CLR that is constitutively expressed on dendritic cells, tissue macrophages, and inflammatory monocytes, and it recognizes α-mannans and O-linked mannoproteins of M. restricta.16 After recognition, Clec4n activates various downstream kinases and pathways such as spleen-associated tyrosine kinase/Card9, spleen-associated tyrosine kinase-phospholipase C gamma 2 and the rapidly accelerated fibrosarcoma kinase to activate nuclear factor kappa B subunit 1 (Nfkb1) and the NLR family pyrin domain containing 3 inflammasome to produce inflammatory and anti-inflammatory cytokines and chemokines.17 In this study, we evaluated the role of M. restricta for the development of alcohol-associated liver injury.
PATIENTS AND METHODS
Human study
Patients with AUD (n=66) were enrolled from April 2017 until January 2019 at St. Luc University Hospital, Brussels, Belgium, as described before.7 The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institution’s human research and ethical committee (Université Catholique de Louvain, Brussels, Belgium; B403201422657). Written informed consent was obtained from all patients and healthy volunteers.
Standard biochemical serum studies, including measurement of aspartate and alanine aminotransferases (AST, ALT), gamma-glutamyltransferase (GGT), alkaline phosphatase, were performed at the clinical laboratory associated with St. Luc University Hospital, Brussels, Belgium.7 Serum caspase-cleaved and intact cytokeratin 18 (CK18-M65) was used to assess liver cell necrosis and apoptosis (CK18-M65 ELISA kit; TECO medical AG, Sissach, Switzerland).7,18,19
Fecal DNA extraction, sequencing, and analysis was performed as previously described and data presented in this study have been reanalyzed.7 Raw sequences from ITS2 gene sequencing were registered previously at NCBI under BioProject PRJNA703732 (refer to https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA703732), as described.7
Mouse models
C57BL/6 mice used in Figures 2 and 5 were bred in the animal facility at the University of California, San Diego (La Jolla, CA). C57BL/6 mice in Figures 3 and 4 were purchased from Charles River. Clec4n −/− mice were a generous gift from Dr. Yoichiro Iawakura of Tokyo University of Science, Japan.20
FIGURE 2.
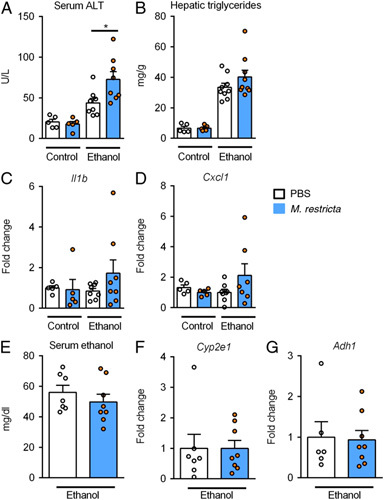
Malassezia restricta exacerbates acute ethanol-induced liver injury in mice. Wild type mice were gavaged once with M. restricta or PBS followed by a dose of ethanol (300 µL, 30% vol/vol) or an isocaloric amount of dextrose (300 μL, 0.42 g/mL) 1 hour later, and harvested after 9 hours. A, Serum levels of ALT. B, Hepatic triglyceride content. C and D, Hepatic levels of Il1b and Cxcl1 mRNAs. E, Serum ethanol. F and G, Hepatic levels of Cyp2e1 and Adh1 mRNAs. Fold changes were calculated relative to the mRNA expression of vehicle treated mice given an isocaloric amount of dextrose. Results are expressed as mean ± SEM (A–G). p values among groups of mice fed with control diet or ethanol diet in the presence or absence of M. restricta are determined by 1-way ANOVA with Tukey post hoc test (A–D). p values between groups of mice gavaged with M. restricta or PBS are determined by unpaired Student t test (E–G). Number of mice per group is indicated in each column, 3 technical replicates. *p < 0.05. Abbreviations: Adh1, alcohol dehydrogenase 1; ALT, alanine aminotransferase; Cxcl1, chemokine (C-X-C motif) ligand 1; Cyp2e1, cytochrome P450 family 2 subfamily E polypeptide 1; Il1b, interleukin 1 beta.
FIGURE 5.
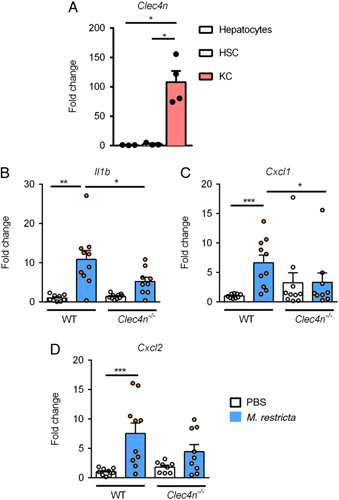
Malassezia restricta induces inflammatory genes in Kupffer cells. A, Primary mouse hepatocytes (Hep), Kupffer cells (KC), and hepatic stellate cells (HSC) were isolated from wild type mice. Expression of Clec4n was measured by quantitative PCR. Fold changes were calculated relative to Clec4n mRNA expression of hepatocytes. p values among groups are determined by 1-way ANOVA with Tukey post hoc test. B–D, Primary Kupffer cells were isolated from wild type (WT) and Clec4n –/– mice and stimulated with Malassezia restricta lysate (40 µg/mL) in culture for 8 hours (n = 4 independent experiments). Fold changes were calculated relative to mRNA expression of vehicle treated Kupffer cells isolated from WT mice. p values among groups of WT and Clec4n –/– in the presence or absence of M. restricta lysate are determined by 1-way ANOVA with Tukey post hoc test (B–D). *p<0.05. Abbreviations: Cxcl1, chemokine (C-X-C motif) ligand 1; Cxcl2, chemokine (C-X-C motif) ligand 2; Il1b, interleukin 1 beta.
FIGURE 3.
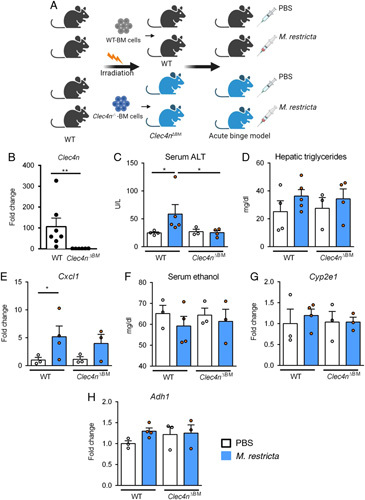
Malassezia restricta exacerbates acute ethanol-induced liver injury through Clec4n on bone marrow-derived cells. A, Wild type (WT) recipient mice underwent transplantation of WT or Clec4n −/− bone marrow (Clec4n ΔBM ). Chimeric mice were gavaged once with a dose of 108 CFUs M. restricta or PBS followed by a dose of ethanol (300 µL, 30% vol/vol) 1 hour later, and harvested after 9 hours. B, Hepatic Clec4n mRNA expression. Fold change was calculated relative to the Clec4n mRNA expression of vehicle treated Clec4n ΔBM mice. p value is determined by Mann-Whitney test. **p<0.01. C, Serum levels of ALT. D, Hepatic triglyceride content. E, Hepatic levels of Cxcl1 mRNA. F, Serum ethanol. G and H, Hepatic levels of Cyp2e1 and Adh1 mRNAs. Fold changes were calculated relative to mRNA expression of vehicle treated wild type mice transplanted with wild type bone marrow. Results are expressed as mean ± SEM (B–H). p values among groups of ethanol-fed WT mice and Clec4n ΔBM mice in the presence or absence of M. restricta are determined by Kruskal-Wallis test with Dunn multiple comparisons test (C–H). Number of mice per group is indicated in each column. *p <0.05, **p<0.01. Abbreviations: Adh1, alcohol dehydrogenase 1; ALT, alanine amino-transferase; Clec4n, C-type lectin domain family 4, member N; Cxcl1, chemokine (C-X-C motif) ligand 1; Cyp2e1, cytochrome P450 family 2 subfamily E polypeptide 1.
FIGURE 4.
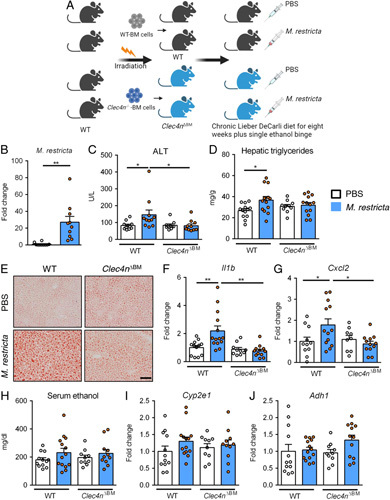
Malassezia restricta exacerbates chronic ethanol-induced steatohepatitis through Clec4n on bone marrow-derived cells. A, Wild type (WT) recipient mice underwent transplantation of WT or Clec4n −/− bone marrow (Clec4n ΔBM ) and subjected to the chronic Lieber DeCarli diet feeding model plus one single binge for 8 weeks. Mice were gavaged vehicle (PBS) or M. restricta (108 CFUs) every third day during the last 4 weeks. B, Relative abundance of M. restricta in fecal samples from wild type mice transplanted with wild type bone marrow supplemented with M. restricta or PBS. Fold change was calculated relative to PBS treated mice. C, Serum levels of ALT. D, Hepatic triglyceride content. E, Representative images of Oil Red O stained liver tissue. Scale bar=100 μm. F and G, Hepatic levels of Il1b and Cxcl2 mRNAs. H, Serum ethanol. I and J, Hepatic levels of Cyp2e1 and Adh1 mRNAs. Fold changes were calculated relative to mRNA expression of vehicle treated wild type mice transplanted with wild type bone marrow. Results are expressed as mean ± SEM. p values among groups of ethanol-fed WT mice and Clec4n ΔBM mice in the presence or absence of M. restricta are determined by 1-way ANOVA with Tukey post hoc test (C, D, F–J). Number of mice per group is indicated in each column, 3 technical replicates. *p<0.05, **p<0.01. Abbreviations: Adh1, alcohol dehydrogenase 1; ALT, alanine amino-transferase; Clec4n, C-type lectin domain family 4, member N; Cxcl2, chemokine (C-X-C motif) ligand 2; Cyp2e1, cytochrome P450 family 2 subfamily E polypeptide 1; Il1b, interleukin 1 beta.
To evaluate the effect of M. restricta on the acute ethanol-induced liver injury, female mice (age, 9–12 wk) were subjected to an acute binge model as described.21 Mice were gavaged with either vehicle (PBS) or live M. restricta [108 colony forming units (CFUs)]. One hour later, mice were given a single dose of 30% (vol/vol) ethanol at a dose of 3 g/kg in the morning. Pair-control mice were given an isocaloric amount of dextrose (0.42 g/mL) at a dose of 5.2 g/kg. Nine hours later, mice were harvested.
To assess the effect of M. restricta on chronic ethanol-induced liver disease, female mice (age, 10 wk) were placed on the chronic Lieber DeCarli diet model for 8 weeks as described.22 Mice were subjected to caloric intake from ethanol at 0 on day 1, 10% of total calories on days 2 and 3, 20% on days 4 and 5, 30% from day 6 until the end of 6 weeks, and 36% for the last 2 weeks. Mice were gavaged with either vehicle (PBS) or live M. restricta (108 CFUs) every third day during week 4–8. At the end, mice were given a single dose of 31.5% (vol/vol) ethanol (5 g/kg body weight) in the morning and sacrificed 9 hours later.
To generate bone marrow chimeric mice, C57BL/6 recipient female mice (age, 6 wks) received 5 Gy radiation twice using an x-ray irradiation system. Bone marrow cells were collected from either female C57BL/6 or Clec4n −/− donor mice and injected to recipient mice via the tail vein. Two weeks later, mice were injected i.p. with 200 μL of clodronate liposomes (5 mg/mL; Vrije Universiteit, Amsterdam, the Netherlands) to deplete radioresistant Kupffer cells.5 Mice were subjected to the acute binge model or chronic Lieber DeCarli model 2 weeks after bone marrow transplantation. For acute binge model, chimeric mice were gavaged with either vehicle (PBS) or live 108 CFUs of M. restricta. One hour later, mice were given a single dose of 30% (vol/vol) ethanol at a dose of 3 g/kg and then harvested after nine hours. For chronic Lieber DeCarli diet model, chimeric mice were gavaged with either vehicle (PBS) or live M. restricta (108 CFUs) every third day during weeks 4–8. At the end, mice were given a single dose of 31.5% (vol/vol) ethanol (5 g/kg body weight) in the morning and sacrificed 9 hours later.
All mice were bred and maintained under specific pathogen-free conditions in a standard environment with a 12-hour light–dark cycle at the animal facilities of University of California, San Diego under protocol S09042. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
M. restricta culture
M. restricta strain KCTC 27527 was used in this study.23 The fungal cells were grown in Leeming and Notman agar medium [0.5% (w/v) glucose, 0.01% (w/v) yeast extract, 1% (w/v) peptone, 0.8% (w/v) bile salt, 0.05% (w/v) glycerol monostearate, 0.1% (v/v) glycerol, 0.05% (v/v) Tween 60, 1.2% (w/v) agar, and 0.5% (v/v) whole fat cow milk] at 34 °C for 3 days, and used for mouse studies.23
Biochemical analysis
Mouse serum ALT levels were measured by ALT (SGPT) Kinetic (Teco Diagnostics). Mouse hepatic triglyceride levels were determined using Triglyceride Liquid Reagents kit (Pointe Scientific). Mouse serum levels of ethanol were measured using an Ethanol Assay kit (Abcam).
Real-time quantitative PCR (qPCR)
Mouse RNA was extracted from liver tissues using Trizol (Invitrogen) and cDNA was generated by reverse transcription using a High Capacity cDNA Reverse Transcription kit (ABI).24 Most of the primer sequences for mouse genes were originally obtained from the NIH qPrimerDepot and are as following:
| Mouse Adh1 | F: 5’-GGGTTCTCAACTGGCTATGG-3’ |
| R: 5’-ACAGACAGACCGACACCTCC-3’ | |
| Mouse Cyp2e1 | F: 5’-GGGACATTCCTGTGTTCCAG-3’ |
| R: 5’-CTTAGGGAAAACCTCCGCAC-3' | |
| Mouse Il1b | F: 5’-GGTCAAAGGTTTGGAAGCAG-3’ |
| R: 5’-TGTGAAATGCCACCTTTTGA-3' | |
| Mouse Cxcl1 | F: 5’-TGCACCCAAACCGAAGTC-3’ |
| R: 5’-GTCAGAAGCCAGCGTTCACC-3' | |
| Mouse Cxcl2 | F: 5’-AAAGTTTGCCTTGACCCTGAA-3’ |
| R: 5’-CTCAGACAGCGAGGCACATC-3' | |
| Mouse 18S | F: 5’-AGTCCCTGCCCTTTGTACACA-3’ |
| R: 5’-CGATCCGAGGGCCTCACTA-3' | |
| Mouse Clec4n | F: 5’-ATTTCATCACCCAGCAGC-3’ |
| R: 5’-AAAACATCATTCCAGCCCC-3' | |
| Mouse Cd4 | F: 5’-CAAGCGCCTAAGAGAGATGG-3’ |
| R: 5’-CACCTGTGCAAGAAGCAGAG-3’ | |
| Mouse Cd8 | F: 5’-ACGGGCATTGCTTCTTCTT-3’ |
| R: 5’-ACAGGGACGAAGCTGACTGT-3’ | |
| Mouse Cd160 | F: 5’-GGATTCTTTGCATGCTGTTG-3’ |
| R: 5’-AAGGAAGAACATCTGTCCTGAG-3’ | |
| Mouse Ly6g | F: 5’-ACACAACTACCTGCCCCTTC-3’ |
| R: 5’-CAGATGGGAAGGCAGAGATT-3’ | |
| Mouse F4/80 | F: 5’-GGATGTACAGATGGGGGATG-3’ |
| R: 5’-CATAAGCTGGGCAAGTGGTA-3’ | |
| Mouse Ptprc | F: 5’-AAGAGTTGTGAGGCTGGCAC-3’ |
| R: 5’-GCTCAAACTTCTGGCCTTTG-3’ |
Gene expression was determined with Sybr Green (Bio-Rad) using ABI StepOnePlus real-time PCR system. The qPCR values were normalized to 18S and expressed relative to the control mice or cells.
Histological analysis
To determine lipid accumulation, mouse liver sections were embedded in OCT compound. Five micrometer frozen sections were then cut and stained with Oil Red O (Sigma-Aldrich).
TUNEL analysis
The staining method for liver apoptosis was performed using In Situ Cell Death Detection Kit, TMR red (SIGMA) according to the manufacturer’s instructions. In brief, tissue sections were digested with proteinase K at room temperature for 30 minutes and incubated with a mixture of Label solution and Enzyme solution for 60 minutes at 37 °C. Nuclei were counter stained using DAPI (VECTASHIELD Antifade Mounting Medium with DAPI). The number of TUNEL positive nuclei were counted in three randomly selected regions on each slide. Quantification of apoptosis by TUNEL assay was expressed as a ratio of the number of positive nuclei (red) and the total number of nuclei (green) revealed by DAPI staining. Large positive nuclei were counted as apoptotic hepatocytes.
Mouse cell culture studies
Primary hepatic stellate cells, Kupffer cells and hepatocytes were isolated as previously described and expression of Clec4n was measured by real-time qPCR.25 Kupffer cells were isolated from C57BL/6 and Clec4n –/– mice and cultured with RPMI 1640 (Thermo Fisher Scientific) containing 10% FBS for 4 hours. After 12 hours of starvation in RPMI1640 (without FBS), Kupffer cells were stimulated with M. restricta lysate (40 µg/mL) or PBS (control) for 8 hours. M. restricta lysate was prepared by washing centrifugation pellets of M. restricta with PBS twice, dissolving in 1 mL PBS and then disrupting using 0.5 mm glass beads in a MINI-BEADBEATER BIOSPEC PRODUCTS twice for 30 seconds with 3 minutes cooling on ice between runs. The concentration of M. restricta lysate was 0.15 mg/mL, measured by bicinchoninic acid assay.
Statistical analysis
For human studies, the unpaired Mann-Whitney test/Wilcoxon rank-sum test was used for nonparametric data (eg, human data), and results are expressed as median and range for each continuous outcome, if not stated otherwise. Categorical variables were compared using the Pearson Chi-squared test. A p value ≤0.05 was considered statistically significant. Statistical analysis was performed using R statistical software, R version 2022.02.2 for Mac, 2022 the R Foundation for Statistical Computing.
For mouse studies, results are expressed as mean ± SEM (except when stated otherwise). Statistical tests were selected based on appropriate assumptions with respect to data distribution and variance characteristics. Significance of 2 groups were evaluated using an unpaired Student t test (parametric) or an unpaired Mann-Whitney test (nonparametric), respectively. Significance of multiple groups were evaluated using 1-way ANOVA with Tukey post hoc test (parametric) or Kruskal-Wallis test with Dunn multiple comparisons test (nonparametric), respectively. Statistical analyses were performed using GraphPad Prism v7.04. A p <0.05 was considered to be statistically significant.
RESULTS
Study population with AUD
The study population comprised 66 patients with AUD, as described.7 The cohort comprises 47 males and 19 females with a median age of 44 years and median body mass index of 24.2 kg/m2. The median serum values for the cohort were 47.5 IU/L and 37.5 IU/L for AST and ALT, respectively, 101.0 IU/L for GGT, 74.0 IU/L for alkaline phosphatase, and 352.5 U/L for the liver cell necrosis and apoptosis marker caspase-cleaved and intact cytokeratin 18 (CK18-M65).18
We previously analyzed the impact of relative abundance changes of fungi with regard to liver disease in AUD patients over time.7 We now aimed to evaluate whether the presence or absence of specific fungal species could predict the severity of liver disease in the same AUD cohort. We first determined the presence of fungal species for each subject using the UNITE database with ITS2 primers. We detected 35 fungal species in the feces of at least 1 AUD patient (Figure 1A). Saccharomyces cerevisiae and Candida albicans were the fungal species most frequently present in the stool samples of AUD patients. S. cerevisiae was detectable in all AUD subjects, whereas C. albicans could be identified in 62.1% of the patients. M. restricta was the third most common fungal species, present in less than half (45.5%) of all subjects. Candida zeylanoides was detected in 39.4%, Candida dubliniensis in 18.2%, Mucor lanceolatus in 10.6%, and Malassezia globosa in 9.1% of all AUD patients. Of note, control subjects from a prior publication are less frequently positive for M. restricta with 33.3% (p = 0.383 using Pearson Chi-squared test).7
FIGURE 1.
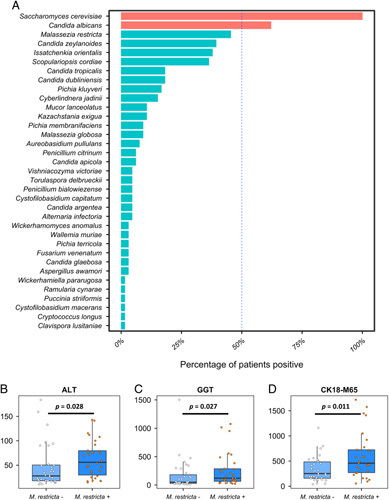
Malassezia restricta is one of the fungal species most frequently present in patients with alcohol use disorder (AUD) and its presence is associated with more severe alcohol-associated liver disease. A, Out of 69 tested fungal species, 35 fungal species were present in at least 1 AUD patient. Fungal species depicted in red are present in >50% of AUD patients, whereas species visualized in blue are more variably present in AUD patients with a positivity of <50% of all AUD patients. B, Serum ALT of M. restricta negative AUD patients (n=36) and M. restricta positive AUD patients (n=30). C, Serum GGT of M. restricta negative AUD patients (n=35) and M. restricta positive AUD patients (n=30). D, Serum CK18-M65 of M. restricta negative AUD patients (n=34) and M. restricta positive AUD patients (n=29). p Values were determined by Mann-Whitney test. A p value of ≤0.05 was considered statistically significant. Abbreviations: ALT, alanine aminotransferase; CK18-M65, caspase-cleaved and intact cytokeratin 18; GGT, gamma-glutamyltransferase.
Presence of fecal M. restricta associates with more severe liver disease in patients with AUD
Cytokeratin-18 (CK-18) is a major intermediate filament protein expressed in the liver. CK-18 cleavage generates a neoepitope, which can be detected by the monoclonal antibody M65 and allows the specific assessment of liver apoptosis. We and others have demonstrated the diagnostic value of CK18-M65 in liver disease, in particular alcohol-associated liver disease. CK18-M65 can differentiate progressive from non-progressive liver disease and has predictive value for mortality in patients with AUD.18,19 To assess whether the presence or absence of the above fungal species could predict the severity of liver disease, we performed multiple linear regression using the presence of fungal species to predict liver disease severity as indicated by CK18-M65 levels in our AUD cohort (Table 1). As we have demonstrated that the relative and absolute abundance of fecal C. albicans plays a major role in liver disease, particularly alcohol-associated liver disease,5,6,22 we therefore focused on less frequent fungal species (<50% presence of all AUD patients) to determine their predictive value for the degree of liver disease. The only 2 fungal species whose presence could predict liver disease severity in our model were M. restricta (363.33, 95% CI: 32.75–693.91, p = 0.03) and Mucor lanceolatus (672.60, 95% CI: 165.62–1179.57, p = 0.01, Table 1). The presence of other fungi, including C. zeylanoides, C. dubliniensis, or M. globosa did not significantly predict the degree of liver disease based on CK18-M65 levels.
TABLE 1.
Multiple linear regression using presence of fungal species to predict CK18-M65 values in patients with alcohol use disorder
| Species | Estimate | 95% CI | p |
|---|---|---|---|
| Alternaria infectoria | 31.55 | −663.65, 726.76 | 0.93 |
| Aspergillus awamori | −529.98 | −1266.71, 206.76 | 0.15 |
| Aureobasidium pullulans | 309.41 | −382.90, 1001.71 | 0.37 |
| Candida apicola | −170.29 | −789.98, 449.39 | 0.58 |
| Candida argentea | 39.59 | −648.94, 728.11 | 0.91 |
| Candida dubliniensis | −206.88 | −507.41, 93.65 | 0.17 |
| Candida glaebosa | 784.76 | −293.66, 1863.17 | 0.15 |
| Candida tropicalis | 169.84 | −235.74, 575.41 | 0.40 |
| Candida zeylanoides | −55.57 | −330.88, 219.74 | 0.68 |
| Clavispora lusitaniae | −567.58 | −1626.41, 491.26 | 0.28 |
| Cryptococcus longus | 924.81 | −760.49, 2610.10 | 0.27 |
| Cyberlindnera jadinii | −11.72 | −440.42, 416.98 | 0.96 |
| Cystofilobasidium capitatum | 525.76 | −96.82, 1148.35 | 0.09 |
| Cystofilobasidium macerans | 788.96 | −764.88, 2342.80 | 0.31 |
| Fusarium venenatum | −588.26 | −1282.09, 105.56 | 0.09 |
| Issatchenkia orientalis | −180.82 | −535.84, 174.21 | 0.31 |
| Kazachstania exigua | 121.00 | −261.98, 503.98 | 0.52 |
| Malassezia globosa | −341.77 | −854.23, 170.69 | 0.18 |
| Malassezia restricta | 363.33 | 32.75–693.91 | 0.03 |
| Mucor lanceolatus | 672.60 | 165.62–1179.57 | 0.01 |
| Penicillium bialowiezense | −151.05 | −725.79, 423.69 | 0.60 |
| Penicillium citrinum | −349.60 | −870.24, 171.03 | 0.18 |
| Pichia kluyveri | −48.44 | −525.33, 428.46 | 0.84 |
| Pichia membranifaciens | −161.25 | −716.71, 394.20 | 0.56 |
| Pichia terricola | 145.45 | −579.56, 870.47 | 0.68 |
| Puccinia striiformis | −585.64 | −1774.33, 603.06 | 0.32 |
| Ramularia cynarae | −125.07 | −1053.89, 803.76 | 0.79 |
| Scopulariopsis cordiae | −55.91 | −325.86, 214.04 | 0.68 |
| Torulaspora delbrueckii | −133.34 | −627.43, 360.74 | 0.59 |
| Vishniacozyma victoriae | −1072.42 | −2448.59, 303.75 | 0.12 |
| Wallemia muriae | 689.87 | −166.26, 1546.00 | 0.11 |
| Wickerhamomyces anomalus | −30.79 | −728.34, 666.75 | 0.93 |
Note: Bold font indicates statistical significance (p <0.05).
Based on the results of this multiple regression analysis, we opted to divide our AUD patient cohort into M. restricta positive and M. restricta negative subjects (Table 2). The 2 groups were not statistically different regarding sex, age, or body mass index. However, the M. restricta positive group had significantly more severe liver disease, as indicated by higher ALT (medians 56 vs. 28, p = 0.028), GGT (120.5 vs. 49, p = 0.027), and CK18-M65 levels (458.4 vs. 251.0, p = 0.011) compared with their M. restricta negative counterpart (Table 2, Figure 1B–D).
TABLE 2.
Baseline demographic and laboratory markers of the study population
| Alcohol use disorder—M. restricta negative (n=36) | Alcohol use disorder—M. restricta positive (n=30) | p | |
|---|---|---|---|
| Sex (male), n (%) | 24 (67) | 23 (77) | 0.37 |
| Age (y), n=66 | 45.0 (19.5) | 42.0 (15.0) | 0.83 |
| Body mass index (kg/m2), n=66 | 24.3 (4.9) | 23.7 (6.2) | 0.72 |
| AST (IU/L), n=66 | 33 (37.25) | 59 (75.5) | 0.18 |
| ALT (IU/L), n=66 | 28 (32.5) | 56 (50.0) | 0.028 |
| GGT (IU/L), n=65 | 49 (154) | 120.5 (222.5) | 0.027 |
| AP (IU/L), n=64 | 66 (31.5) | 76 (20) | 0.11 |
| Bilirubin (mg/dL), n=66 | 0.50 (0.25) | 0.45 (0.30) | 0.31 |
| Albumin (g/dL), n=63 | 4.6 (0.63) | 4.6 (0.5) | 0.80 |
| INR, n=65 | 1.0 (0.1) | 1.0 (0.2) | 0.94 |
| Creatinine (mg/dL), n=66 | 0.80 (0.16) | 0.82 (0.15) | 0.38 |
| Platelets (109/L), n=66 | 213 (85) | 238 (107) | 0.35 |
| CK18-M65 (U/L), n=63 | 251.0 (328.2) | 458.4 (456.2) | 0.011 |
Notes: Values are presented as median and interquartile range in parentheses. The number of subjects for which data were available is indicated in the first column. Continuous variables were compared using the Mann-Whitney test. Categorical variables were compared using the Pearson Chi-squared test. Bold font indicates statistical significance (p <0.05).
Abbreviations: ALT, alanine aminotransferase; AP, alkaline phosphatase; AST, aspartate aminotransferase; CK18-M65, caspase-cleaved and intact cytokeratin 18; GGT, gamma-glutamyltransferase; INR, international normalized ratio; M. restricta, Malassezia restricta.
M. restricta exacerbates acute ethanol-induced liver injury
Since the presence of M. restricta was associated with increased markers of liver injury, we initially tested whether M. restricta exacerbates acute ethanol-induced liver injury in mice. Wild type mice were gavaged once with M. restricta or vehicle (PBS) followed by a dose of ethanol or an isocaloric dose of dextrose 1 hour later, and then harvested after 9 hours. Mice gavaged with M. restricta displayed more severe acute ethanol-induced liver injury as indicated by elevated levels of serum ALT as compared with vehicle treated mice (Figure 2A). Hepatic triglycerides (Figure 2B) and inflammatory gene expression in the liver including Interleukin 1 beta (Il1b) and Chemokine (C-X-C motif) ligand 1 (Cxcl1) were slightly increased in M. restricta administered mice following acute binge of ethanol (Figure 2C, D). Treatment with M. restricta did not affect intestinal absorption or hepatic metabolism of ethanol, as serum levels of ethanol as well as expression of hepatic alcohol dehydrogenase 1 (Adh1) and cytochrome P450 family 2 subfamily E polypeptide 1 (Cyp2e1) (2 main hepatic enzymes that metabolize ethanol) did not differ significantly between mouse groups (Figure 2E–G). Immune cell infiltration plays an important role in the occurrence and progression of liver injury. The hepatic mRNA expression of CD4+ T cells (Cd4), CD8+ T cells (Cd8), natural killer cells (Cd160), macrophages (F4/80), neutrophils (Ly6g), and CD45+ leukocytes (Ptprc) was measured, but no significant difference was observed between the groups (Supplemental Figure S1, http://links.lww.com/HC9/A82).
M. restricta exacerbates acute ethanol-induced liver injury through Clec4n on bone marrow-derived cells
Clec4n (also known as dectin-2) is a pattern recognition receptor that plays a central role in immunity to fungal pathogens including Malassezia species. To investigate whether M. restricta-exacerbated acute ethanol-induced liver injury is mediated by Clec4n signaling, we generated Clec4n bone marrow chimeric mice (Clec4n ΔBM ) using a combination of clodronate-mediated Kupffer cell depletion, irradiation, and bone marrow transplantation. Wild type recipient mice were transplanted with bone marrow cells from either Clec4n −/− mice or wild type mice. Chimeric mice were gavaged with M. restricta or vehicle (PBS) and subjected to binge ethanol 1 hour later, and then harvested after 9 hours (Figure 3A). We confirmed reduced levels of Clec4n mRNA in whole livers of mice that received Clec4n −/− bone marrow, indicating successful bone marrow transplantation (Figure 3B).
Consistent with our initial observation, chimeric mice that received bone marrow from wild type mice and were gavaged with M. restricta, showed more severe acute ethanol-induced liver injury than vehicle gavaged mice. M. restricta-induced liver injury was reduced in chimeric mice that received bone marrow from Clec4n −/− mice (Figure 3C). Hepatic triglycerides (Figure 3D) and hepatic Cxcl1 in mice transplanted with Clec4n −/− bone marrow following acute ethanol binge were not decreased (Figure 3E). No significant difference in serum ethanol, hepatic expression of Cyp2e1, Adh1, and immune cell infiltration were observed among all groups (Figure 3F–H, Supplemental Figure S2, http://links.lww.com/HC9/A82).
M. restricta increases chronic ethanol-induced steatohepatitis through Clec4n on bone marrow-derived cells
To further investigate whether M. restricta exacerbates chronic ethanol-induced steatohepatitis and whether this is mediated by Clec4n signaling, we gavaged wild type and Clec4n ΔBM chimeric mice with M. restricta (108 CFUs every third day) during the last 4 weeks of chronic ethanol feeding (Figure 4A). M. restricta was hardly detectable in the wild type mice transplanted with wild type bone marrow and subjected to the chronic Lieber-DeCarli ethanol feeding model for 8 weeks with PBS gavage, indicating that M. restricta does not colonize the gastrointestinal tract of mice, or that the level of M. restricta was below the detection limit of nucleic acid amplification. Repeated oral gavage of mice with M. restricta during the ethanol feeding period, resulted in significantly higher proportions of M. restricta in fecal samples (Figure 4B). M. restricta increased ethanol-induced liver injury, steatosis and inflammation the wild type mice transplanted with wild type bone marrow (Figure 4C–G). M. restricta-exacerbated liver disease was reduced in ethanol-fed Clec4n ΔBM chimeric mice as compared with ethanol-fed wild type mice gavaged with M. restricta (Figure 4C–G). No significant difference in serum ethanol, hepatic expression of Cyp2e1 and Adh1 was observed among all groups (Figure 4H–J). Serum levels of translocated lipopolysaccharide did not differ between vehicle and M. restricta treated mice after ethanol feeding, suggesting that M. restricta does not alter intestinal permeability (Supplemental Figure S3, http://links.lww.com/HC9/A82). The mRNA expressions of natural killer cells (Cd160) and CD45 leukocytes (Ptprc) were increased in M. restricta-treated wild type mice, but reduced in the M. restricta-treated Clec4n ΔBM chimeric mice following chronic ethanol feeding (Supplemental Figure S3, http://links.lww.com/HC9/A82). Compared with vehicle-treated group, M. restricta induced total liver cell apoptosis and hepatocyte apoptosis in wild type mice transplanted with wild type bone marrow after ethanol feeding, as determined by TUNEL staining of liver sections. Apoptosis was reduced in M. restricta-treated ethanol-fed chimeric Clec4n ΔBM mice as compared with M. restricta-treated ethanol-fed wild type mice (Supplemental Figure S4, http://links.lww.com/HC9/A82). These results indicate that Clec4n on bone marrow-derived cells contributes to M. restricta-exacerbated ethanol-induced liver injury in mice.
M. restricta induces an inflammatory response in Kupffer cells
To determine the expression of Clec4n in different liver cell types, parenchymal and nonparenchymal hepatic cells were isolated from wild type mice. Clec4n mRNA was highly expressed in Kupffer cells as compared with hepatocytes or hepatic stellate cells (Figure 5A). To further define a mechanism by which M. restricta enhances ethanol-induced liver disease, Kupffer cells were isolated from wild type and Clec4n deficient mice and stimulated with whole M. restricta lysate. M. restricta induced the expression of Il1b, Cxcl1, Cxcl2 mRNA expression in Kupffer cells isolated from wild type mice (Figure 5B–D) after stimulation with M. restricta lysate. M. restricta lysate-stimulated Kupffer cells lacking Clec4n showed significantly decreased levels of Il1b and Cxcl1 mRNA, and lower levels of Cxcl2 mRNA than M. restricta lysate-stimulated Kupffer cells isolated from wild type mice (Figure 5B–D).
DISCUSSION
Intestinal fungal dysbiosis contributes to the development of alcohol-associated liver disease, yet the potential cause/effect relationship in the pathogenesis of disease remains largely unknown. The relative and absolute abundance of Candida species and in particular C. albicans, associates with more severe liver disease in patients with AUD and alcohol-associated hepatitis.6,7 Our current data links the presence of the specific fungus M. restricta with more severe alcohol-associated liver disease in patients with AUD, similar to a higher relative abundance of M. restricta in progressive liver disease versus nonprogressive liver disease.7 M. restricta exacerbates ethanol-induced liver injury both in acute binge and chronic ethanol feeding models, which was mediated by the pattern recognition of Clec4n on Kupffer cells and possibly other bone marrow-derived cells.
In our study, we have shown that wild type mice gavaged once with M. restricta followed by an ethanol binge, developed more severe acute ethanol-induced liver injury than the vehicle group. We suspect that M. restricta may promote acute liver injury by a rapid increase in M. restricta derived antigens translocating to the liver and together with other microbial products (such as endotoxin) affects the inflammation cascade and contributes to ethanol-induced liver injury.26 It is important to note that not all fungal species exacerbate ethanol-induced liver disease when gavaged (eg, gavage of the yeast Saccharomyces cerevisiae improves serum ALT levels and hepatic steatosis in a mouse model27).
In addition to Clec4n, other CLRs such as Clec4e (also known as Mincle) function as pattern recognition receptor in response to Malassezia species.16 Prior reports show that Malassezia pachydermatis and Malassezia furfur can be recognized by Clec4n and Mincle receptors and activate Card9 for the inflammatory response of mouse bone marrow-derived dendritic cells and neutrophils.16,28 Clec4n was especially important for responses to M. restricta in the development of colitis in mouse models, while Clec7a (also known as dectin-1) and Clec4e did not contribute significantly.13 In our study, Clec4n chimeric mice colonized with M. restricta developed less severe ethanol-induced liver disease than control wild type mice colonized with M. restricta, as determined by serum ALT levels and inflammatory markers. However, elimination of Clec4n on bone marrow–derived cells is not sufficient to completely prevent M. restricta-exacerbated ethanol-induced liver disease. For example, hepatic steatosis was not significantly different between these two groups following chronic ethanol feeding. Future studies are required to clarify whether Clec4e or other CLRs contribute to the effect of M. restricta on hepatic steatosis.
Despite the fact that M. restricta DNA could not be detected in the liver by qPCR (data not shown), M. restricta-derived antigens might translocate to the liver from the gut, and promote alcohol-related liver injury. Future studies are needed to identify specific M. restricta-derived antigens that contribute to alcohol-associated liver disease. Increased fungal cell wall polysaccharide-β-glucan activates Clec7a to induce liver inflammation through upregulation of Il1b.5 Clec4n may recognize Malassezia through a mannose-related structure. An O-linked mannobiose rich glycoprotein was identified as Malassezia ligand for Clec4n, which can directly activate dendritic cells to produce inflammatory cytokines.16 In addition, fungi derived metabolites also participate in the pathogenesis of alcohol-associated liver disease. For example, candidalysin, an exotoxin peptide produced by C. albicans exacerbates ethanol-induced liver disease by causing direct hepatocyte damage.22 Malassezia species are lipophilic and their growth is dependent on lipids produced by the environment. Malassezia produce a variety of enzymes including lipases and phospholipases, which could hydrolyze triglycerides, freeing specific saturated fatty acids and potentially affect fatty acid metabolism in humans.29 Therefore, it is interesting to speculate that M. restricta related lipid metabolism might contribute to alcohol-associated steatosis and liver disease. Further studies are required to address this notion and to identify the specific ligand of M. restricta that activates Clec4n.
CONCLUSIONS
In summary, our study showed that the presence of M. restricta associated with increased severity of alcohol-associated liver disease in patients and promoted ethanol-induced liver injury in mice. Manipulation of the gut mycobiota by targeting M. restricta may serve as a potential therapeutic strategy to prevent or treat liver disease.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Suling Zeng was responsible for acquisition, analysis and interpretation of data, and drafting of the manuscript. Phillipp Hartmann was responsible for data analysis of human cohorts. Minji Park and Won Hee Jung provided cultures of Malassezia restricta. Yi Duan provided assistance on culture of Malassezia restricta and bone marrow transplant. Cristina Llorente and Yanhan Wang were responsible for Kupffer cell experiments. Sonja Lang, Yanhan Wang, and Noemí Cabré provided assistance in animal experiments. Petra Bacher participated in study design. Derrick E. Fouts analyzed sequencing data. Peter Stärkel was responsible for collection of human samples. Bernd Schnabl was responsible for the study concept and design, study supervision and editing the manuscript. All authors edited and approved the manuscript.
ACKNOWLEDGEMENT
This study was supported in part by National Institutes of Health (NIH) grant K12 HD85036, Pinnacle Research Award in Liver Diseases Grant #PNC22-159963 from the American Association for the Study of Liver Diseases Foundation (to Phillipp Hartmann), NIH grants R01 AA24726, R37 AA020703, U01 AA026939, U01 AA026939-04S1, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and a Harrington Discovery Institute Foundation Grant (to Bernd Schnabl) and services provided by NIH centers P30 DK120515 and P50 AA011999. Peter Stärkel received grants from Fond National de Recherche Scientifique, Belgium (J.0146.17 and T.0217.18) and Région Wallonne, Belgium (Action de Recherche Concertée (ARC) 2018). Won Hee Jung received grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning 2021M3A9I4021431. Sonja Lang was supported by a DFG fellowship (LA 4286/1-1) and the “Clinical and Translational Research Fellowship in Liver Disease” by the American Association for the Study of Liver Diseases (AASLD) Foundation. Cristina Llorente was supported by the American Association for the Study of Liver Diseases (AASLD) Pinnacle Research Award in Liver Disease (8998GA), by the Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (ALPD) and Cirrhosis (P50 AA011999) funded by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and its Animal Core facilities, the Isenberg Endowed Fellowship jointly awarded by the Pilot/Feasibility Program of the San Diego Digestive Diseases Research Center (SDDRC) and the Hellman Family Foundation (P30 DK120515), the UC San Diego’s Hispanic Center of Excellence Program (D34HP31027) and NIH grant R01AA029106. Graphic abstract is created with a licence from BioRender.com.
CONFLICT OF INTEREST
Bernd Schnabl has been consulting for Ambys Medicines, Ferring Research Institute, Gelesis, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, Patara Pharmaceuticals and Takeda. He is founder of Nterica Bio. UC San Diego has filed several patents with Yi Duan, Cristina Llorente, and Bernd Schnabl as inventors related to this work. Bernd Schnabl’s institution UC San Diego has received research support from Artizan Biosciences, Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, Prodigy Biotech and Synlogic Operating Company. Peter Stärkel received grant support from Gilead Sciences, Belgium. The remaining authors declare no conflict of interest.
Footnotes
Current affiliation: Minji Park, CutisBio, Nonhyeon-Ro, Gangnam-gu, Seoul, Korea.
Abbreviations: Adh1, alcohol dehydrogenase 1; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUD, alcohol use disorder; CFUs, colony forming units; CK18-M65, caspase-cleaved and intact cytokeratin 18; Clec4n, C-type lectin domain family 4, member N; Clec7a, C-type lectin domain family 7, member A; CLRs, C-type lectin receptors; Cxcl, chemokine (C-X-C motif) ligand; Cyp2e1, cytochrome P450 family 2 subfamily E polypeptide 1; GGT, gamma-glutamyltransferase; Il1b, interleukin 1 beta; ITS, internal transcribed spacer; M. restricta, Malassezia restricta; qPCR, quantitative PCR.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.hepcommjournal.com.
Contributor Information
Suling Zeng, Email: szeng@ucsd.edu.
Phillipp Hartmann, Email: phhartmann@health.ucsd.edu.
Minji Park, Email: mjpark@cutisbio.com.
Yi Duan, Email: yid003@health.ucsd.edu.
Sonja Lang, Email: sonja.lang@uk-koeln.de.
Cristina Llorente, Email: allorenteizquierdo@health.ucsd.edu.
Yanhan Wang, Email: yaw015@health.ucsd.edu.
Noemí Cabré, Email: ncabrecasares@health.ucsd.edu.
Derrick E. Fouts, Email: dfouts@jcvi.org.
Petra Bacher, Email: Petra.Bacher@uksh.de.
Won Hee Jung, Email: whjung@cau.ac.kr.
Peter Stärkel, Email: peter.starkel@uclouvain.be.
Bernd Schnabl, Email: beschnabl@ucsd.edu.
REFERENCES
- 1.Gustot T, Jalan R. Acute-on-chronic liver failure in patients with alcohol-related liver disease. J Hepatol. 2019;70:319–27. [DOI] [PubMed] [Google Scholar]
- 2.Stärkel P, Schnabl B. Bidirectional communication between liver and gut during alcoholic liver disease. Semin Liver Dis. 2016;36:331–9. [DOI] [PubMed] [Google Scholar]
- 3.Chu H, Williams B, Schnabl B. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2018;2:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng S, Schnabl B. Roles for the mycobiome in liver disease. Liver Int. 2022;42:729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Investig. 2017;127:2829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology. 2020;71:522–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartmann P, Lang S, Zeng S, Duan Y, Zhang X, Wang Y, et al. Dynamic changes of the fungal microbiome in alcohol use disorder. Front Physiol. 2021;12:699253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vijaya Chandra SH, Srinivas R, Dawson TL, Jr, Common JE. Cutaneous Malassezia: commensal, pathogen, or protector? Front Cell Infect Microbiol. 2020;10:614446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suhr MJ, Banjara N, Hallen-Adams HE. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett Appl Microbiol. 2016;62:209–15. [DOI] [PubMed] [Google Scholar]
- 10.Raimondi S, Amaretti A, Gozzoli C, Simone M, Righini L, Candeliere F, et al. Longitudinal survey of fungi in the human gut: ITS profiling, phenotyping, and colonization. Front Microbiol. 2019;10:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gouba N, Raoult D, Drancourt M. Plant and fungal diversity in gut microbiota as revealed by molecular and culture investigations. PloS One. 2013;8:e59474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spatz M, Richard ML. Overview of the potential role of Malassezia in gut health and disease. Front Cell Infect Microbiol. 2020;10:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, Funari V, et al. Malassezia is associated with Crohn’s disease and exacerbates Colitis in mouse models. Cell Host Microbe. 2019;25:377–88.e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patin EC, Thompson A, Orr SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol. 2019;89:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa T, Itoh F, Yoshida S, Saijo S, Matsuzawa T, Gonoi T, et al. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe. 2013;13:477–88. [DOI] [PubMed] [Google Scholar]
- 17.Glocker EO, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller S, Nahon P, Rausch V, Peccerella T, Silva I, Yagmur E, et al. Caspase-cleaved keratin-18 fragments increase during alcohol withdrawal and predict liver-related death in patients with alcoholic liver disease. Hepatology. 2017;66:96–107. [DOI] [PubMed] [Google Scholar]
- 19.Maccioni L, Gao B, Leclercq S, Pirlot B, Horsmans Y, De Timary P, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut microbes. 2020;12:1782157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–91. [DOI] [PubMed] [Google Scholar]
- 21.Chen P, Miyamoto Y, Mazagova M, Lee KC, Eckmann L, Schnabl B. Microbiota protects mice against acute alcohol-induced liver injury. Alcohol Clin Exp Res. 2015;39:2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu H, Duan Y, Lang S, Jiang L, Wang Y, Llorente C, et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J Hepatol. 2020;72:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park M, Cho YJ, Lee YW, Jung WH. Whole genome sequencing analysis of the cutaneous pathogenic yeast Malassezia restricta and identification of the major lipase expressed on the scalp of patients with dandruff. Mycoses. 2017;60:188–97. [DOI] [PubMed] [Google Scholar]
- 24.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwaisako K, Haimerl M, Paik YH, Taura K, Kodama Y, Sirlin C, et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc Natl Acad Sci USA. 2012;109:E1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bala S, Marcos M, Gattu A, Catalano D, Szabo G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PloS One. 2014;9:e96864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izu H, Shobayashi M, Manabe Y, Goto K, Iefuji H. Sake yeast suppresses acute alcohol-induced liver injury in mice. Biosci Biotechnol Biochem. 2006;70:2488–93. [DOI] [PubMed] [Google Scholar]
- 28.Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, Sakuma M, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci USA. 2009;106:1897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Triana S, de Cock H, Ohm RA, Danies G, Wösten HAB, Restrepo S, et al. Lipid metabolic versatility in Malassezia spp. Yeasts studied through metabolic modeling. Front Microbiol. 2017;8:1772. [DOI] [PMC free article] [PubMed] [Google Scholar]