

ABSTRACT
The urokinase-type plasminogen activator receptor (uPAR) is an essential regulator for cell signaling in tumor cell proliferation, adhesion, and metastasis. The ubiquitous nature of uPAR in many aggressive cancer types makes uPAR an attractive target for immunotherapy. Here, we present a rapid and successful workflow for developing cross-reactive anti-uPAR recombinant antibodies (rAbs) using high-throughput optofluidic screening of single B-cells from human uPAR-immunized mice. A total of 80 human and cynomolgus uPAR cross-reactive plasma cells were identified, and selected mouse VH/VL domains were linked to the trastuzumab (Herceptin®) constant domains for the expression of mouse-human chimeric antibodies. The resulting rAbs were characterized by their tumor-cell recognition, binding activity, and cell adhesion inhibition on triple-negative breast cancer cells. In addition, the rAbs were shown to enact antibody-dependent cellular cytotoxicity (ADCC) in the presence of either human natural killer cells or peripheral blood mononuclear cells, and were evaluated for the potential use of uPAR-targeting antibody-drug conjugates (ADCs). Three lead antibodies (11857, 8163, and 3159) were evaluated for their therapeutic efficacy in vivo and were shown to suppress tumor growth. Finally, the binding epitopes of the lead antibodies were characterized, providing information on their unique binding modes to uPAR. Altogether, the strategy identified unique cross-reactive antibodies with ADCC, ADC, and functional inhibitory effects by targeting cell-surface uPAR, that can be tested in safety studies and serve as potential immunotherapeutics.
KEYWORDS: Antibody-dependent cellular cytotoxicity (ADCC), cancer therapeutics, cross-reactive antibody, single B-cell screening, Urokinase-type plasminogen activator receptor (uPAR)
Introduction
A key feature of tumor cells is their enhanced ability to degrade extracellular matrix (ECM), allowing tumor cell motility, invasion, and metastasis. The urokinase-type plasminogen activator receptor (uPAR) is an integral membrane protein tethered to the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor. This well-studied receptor is involved in the binding of various partners, such as urokinase-type plasminogen activator (uPA), vitronectin (VN), and transmembrane receptors, to regulate a wide variety of cellular processes including extracellular proteolysis, angiogenesis, cell adhesion, migration, and downstream signaling events.1 Several studies have demonstrated that the overexpression of uPAR is tumor-specific,2,3 making it a prominent biomarker for identifying tumor aggressiveness4–6 and an attractive target for cancer treatment,7 particularly breast cancer.8–11
Growing evidence suggests that uPAR and human epidermal growth factor receptor-2 (HER2) are co-amplified in both in situ and metastatic breast cancer, and they work cooperatively for tumor progression toward the onset of a metastatic phenotype.12,13 Moreover, the downregulation of uPAR using RNAi with an anti-HER2 antibody induces synergistic effects in inhibiting breast cancer cell growth, highlighting the potential of a combined therapy as an effective treatment for breast cancer.14 Although clinical outcomes have shown that US Food and Drug Administration-approved anti-HER2 antibodies are effective in metastatic HER2-positive breast cancer, several mechanisms of resistance to anti-HER2 therapy have been identified.15,16 In addition, HER2 is not an effective target for triple-negative breast cancer (TNBC) patients because of the absence of HER2 expression,9 so development of novel treatment strategies is needed. Several groups have developed a series of antagonists, such as recombinant antibodies (rAbs), small molecules, and peptides, to block the interaction of uPAR with its partners.17–22 Some of these uPAR-targeted agents have also been designed as novel preclinical immunotherapeutics,17,23,24 diagnostic imaging tools,17,25,26 and drug delivery vehicles,24 validating uPAR as a potential therapeutic target.
Despite the important role of uPAR as an anti-cancer target, no species cross-reactive anti-uPAR antibodies with efficient antitumor effect in vivo have been developed. Previously, fully human rAbs, 2G10 and 3C6,27 were identified from a human naïve Fab library using phage display technology and were shown to be effective against human TNBC cells in xenograft models,17 but they lacked cross-reactivity. This lack of species cross-reactivity of the inhibitors has hindered human efficacy studies and, in particular, safety studies in non-human primates. Since cynomolgus monkeys (cyno) are genetically similar to human compared to other species, they are the most relevant non-human primate model for conducting preclinical studies of antibody drugs.28 We, therefore, established a high-throughput discovery approach to develop novel human and cyno cross-reactive rAbs by using a microfluidic platform and opto-electropositioning (OEP) technology to screen uPAR-primed mouse B lymphocytes. Selected cross-reactive rAbs were shown to exhibit antibody-dependent cellular cytotoxicity (ADCC), antibody-drug conjugate (ADC) cytotoxicity, and inhibitory effects on cell adhesion against human breast cancer cells. Furthermore, lead antibodies showed their therapeutic efficacy in reducing tumor growth in an orthotopic animal model of human breast cancer, providing promising antibodies for further study. Finally, a binding model of the lead antibodies was proposed showing their binding epitopes that contribute to their unique activities against uPAR.
Results
High-throughput B-cell screening for human and cyno uPAR cross-reactive antibodies
Swiss Jim Lambert (SJL/J) mice (n = 8) were subjected to a 60-day immunization campaign by using a recombinant human soluble uPAR (suPAR), which lacks a GPI anchor, as an immunogen with seven buffering days between bleeds and boosts (Figure 1(a)). The suPAR antigen was prepared by endotoxin removal to reduce nonspecific pyrogenic reactions to immunized animals, and further characterization was performed using SDS-PAGE, immunoblot, and liquid chromatography-mass spectrometry (LC-MS)/MS (Suplementary Figure S1). Immunized mice were monitored with bi-weekly bleeds, followed by the determination of their antibody titers. Antisera binding curves demonstrated increased production of anti-uPAR antibodies within the first week of immunization, and sustained antibody production was maintained throughout the campaign with an antibody titer saturating at a 1 × 107 dilution of mouse antisera (Supplementary Figure S2). Although the titer from some of the mice dropped at the third bleed, later time points permitted in vivo affinity maturation and the generation of anti-uPAR antibodies with increased affinity and selectivity. With the confirmed maturation of uPAR-primed plasma B cells, spleens, and bone marrow from each animal were harvested to allow the isolation of CD45R(B220)-/CD138high antibody-secreting cells (ASCs) using magnetic beads and fluorescence-activated cell sorting (FACS).
Figure 1.
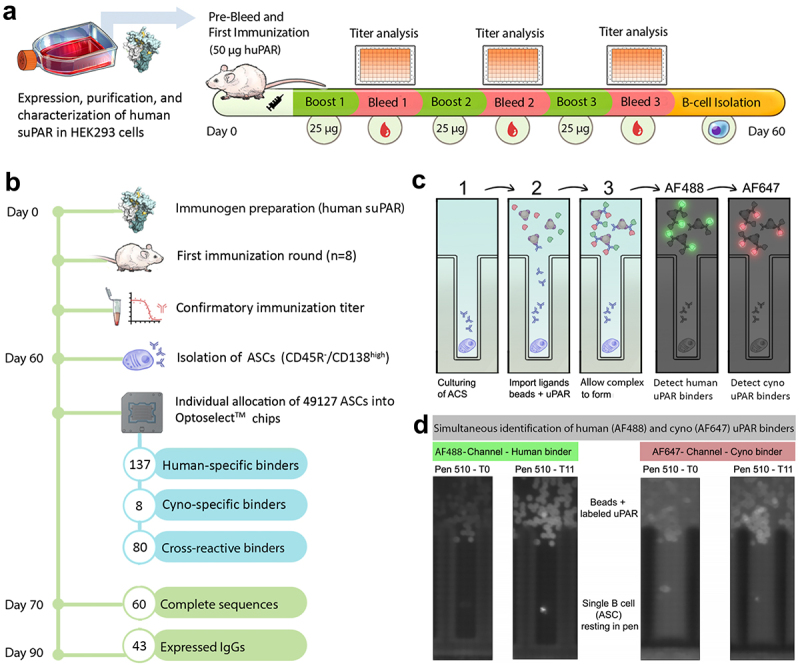
(a) Mouse immunization campaign pipeline. (b) A workflow for high-throughput discovery of cross-reactive anti-uPAR antibodies using the bacon platform. (c) Each ASC was cultured in individual nanopens to allow secreted antibodies to be accumulated. After culturing cells for 1–2 hours, anti-mouse IgG (H+L)-coated beads are imported into the channel along with fluorescently labeled uPAR (AF488-human suPAR, green; and AF647-cyno uPAR, red). As the secreted antibodies are immobilized on the beads, antigen recognition allows for a time-dependent accumulation of fluorescent signals immediately above each nanopen. (d) The increased signal in AF488 and AF647 channels was observed between T0 and T11, suggesting the antibodies were able to recognize human/cyno uPAR.
To screen and select cross-reactive antibodies against human and cyno uPAR, we performed a high-throughput screening of single B cells on the Beacon platform,29 which uses microfluidics combined with OEP technology to screen thousands of B cells from immunized animals.30 A total of 49,127 mouse ASCs were imported into nanopens on OptoSelect™ 3500 and 14k chips, and screened against human, mouse, and cyno uPAR. Overall, 225 binders were identified against human uPAR, from which 80 were cross-reactive to cyno uPAR, and no cells were able to produce reactive binders to mouse uPAR (Figure 1(b–d)). Interestingly, eight ASCs produced specific binders to cyno uPAR, and 137 ASCs were specific to human uPAR. It is possible the eight cyno-uPAR specific antibodies are false-positive. However, it is possible that these antibodies recognize an epitope where even a single amino acid difference affects the antigen binding and leads to more cyno uPAR-specific binding than human uPAR binding.
Human and cyno uPAR cross-reactive rAbs were generated and characterized with the trastuzumab constant region
VH/VL sequencing, cloning, and recombinant IgG expression
A total of 225 individual mouse B cells were exported from the Beacon, and 80 pairs of VH and VL sequences from human and cyno uPAR cross-reactive binders were then recovered using the rapid amplification of cDNA ends (RACE) protocol.31 A total of 60 clones showed the amplicons within 500–700 bp with 78% recovery and these amplicons were sequenced using next-generation sequencing (NGS), resulting in 46 pairs of VH and VL sequences.
Previous studies have shown that trastuzumab (Herceptin®), a humanized IgG1 monoclonal antibody that targets the HER2 protein, is able to promote tumor cell death by evoking ADCC through the interaction with IgG1 Fc and Fcγ receptors on human immune cells.32–34 In order to convey this effector function to our anti-uPAR antibodies, mouse VH and VL domains were fused to the constant domains of trastuzumab IgG1 to produce rAbs in a mouse/human chimeric antibody format. From the recovery pool, 43 initial rAbs were successfully expressed. A phylogenetic tree constructed for their complementarity-determining region (CDR) sequences indicated that they have>1% amino acid difference (Suplementary Figure S3).
Cell-surface uPAR recognition and binding affinity of antibody candidates
Since suPAR lacks the cell surface anchoring motif, antibodies generated by immunizing animals with suPAR can target protein regions that are not accessible for membrane-bound uPAR. Nonetheless, effective targeting of cell surface receptors benefits from recognizing both solvent-exposed epitopes and also native conformational states displayed on the cell surface.35 Although the Beacon platform allows on-cell screening, we preferred a more quantitative methodology. Thus, we applied FACS to evaluate the cell-surface uPAR recognition by each antibody in the triple-negative breast cancer cell line, MDA-MB-231, that expresses uPAR endogenously (Figure 2). Human isotype IgG1 (hIgG1) served as a negative control, and all antibodies were benchmarked against 2G10, 3C6, and trastuzumab (Tras). The binding curves demonstrated 11 antibodies recognized cell-surface uPAR in a dose-dependent manner and are comparable to trastuzumab binding to HER2 (EC50 = 3.6 nM) (Table 1 and Figure 2),36,37 showing a more potent ability to recognize cell-surface uPAR than 2G10 and 3C6.
Figure 2.
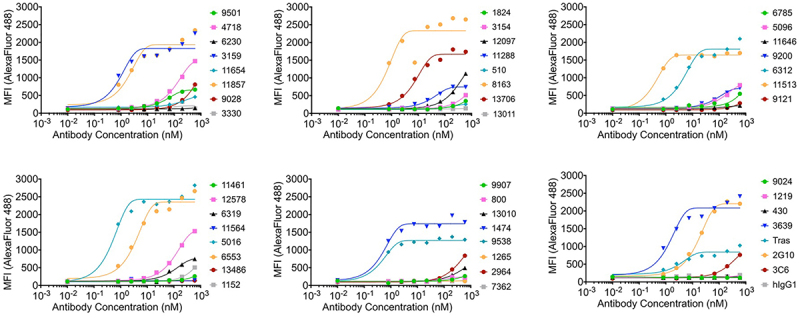
Binding curves for initial antibodies to MDA-MB-231 cells expressing human uPAR on the cell surface. MFI indicates median fluorescence intensity. Tras: trastuzumab. hIgG1: human isotype control.
Table 1.
In vitro characterization of novel anti-uPAR antibodies. Tras: Trastuzumab. hIgG1: Human isotype control. Dash (-): Not determined. NA: Not applicable.
| Anti-uPAR antibody |
Cellular recognition |
Human uPAR binding | Cyno uPAR binding | ADCC NK-92 |
ADCC PBMCs |
ADC Cytotoxicity |
Adhesion blocking | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID number | Max MFI | EC50 (95% CI, nM) | EC50 (95% CI, nM) | EC50 (95% CI, nM) | Max% | EC50 (nM) | Max% | EC50 (nM) | Max% | EC50 (nM) | IC50 (95% CI, µM) |
| 1474 | 1740 | 0.51 (0.22–1.07) | 0.14 (0.11–0.21) | 0.20 (0.15–0.29) | 15.56 | 0.73 | NA | NA | NA | NA | NA |
| 3159 | 1830 | 0.92 (0.20–5.60) | 0.53 (0.38–0.87) | 0.27 (0.20–0.42) | 28.18 | 0.89 | 47.41 | 0.11 | 32.02 | 0.75 | 1.61 (0.71–3.45) |
| 3639 | 2053 | 1.42 (0.50–5.32) | 0.52 (0.39–0.80) | 0.19 (0.17–0.21) | 34.09 | 5.95 | 57.06 | 0.53 | 2.32 | - | >10 |
| 5016 | 2429 | 0.51 (0.16–1.27) | 0.75 (0.58–1.08) | 0.11 (0.10–0.15) | 23.73 | 3.12 | 49.56 | 0.12 | 0 | - | >10 |
| 6312 | 1809 | 4.53 (2.07–11.22) | 0.28 (0.22–0.40) | 0.21 (0.18–0.27) | 16.44 | 4.03 | NA | NA | NA | NA | NA |
| 6553 | 2353 | 3.76 (1.74–9.17) | 0.33 (0.26–0.46) | 0.21 (0.16–0.29) | 17.44 | 5.6 | NA | NA | NA | NA | NA |
| 8163 | 2330 | 0.69 (<53.38) | 0.41 (0.28–0.79) | 0.15 (0.13–0.19) | 18.81 | 1.14 | 57.55 | 0.09 | 28.7 | 0.57 | 5.40 (2.11–31.5) |
| 9538 | 1270 | 0.46 (0.25–0.80) | 0.67 (0.59–0.78) | 1.12 (0.96–1.37) | 29.71 | 50.2 | 45.64 | 13.01 | 4.26 | - | 3.51 (1.80–8.40) |
| 11513 | 1642 | 0.39 (0.29–0.51) | 0.75 (0.58–1.08) | 0.28 (0.22–0.39) | 21.60 | - | NA | NA | NA | NA | NA |
| 11857 | 1936 | 2.07 (0.52–16.00) | 0.52 (0.39–0.80) | 0.18 (0.16–0.22) | 27.16 | 5.59 | 52.04 | 0.35 | 35.16 | 0.76 | 0.94 (0.17–76.9) |
| 13706 | 1669 | 7.61 (4.09–14.66) | 0.53 (0.38–0.87) | 0.30 (0.26–0.39) | 54.56 | 4.85 | 65.66 | 0.79 | 3.24 | - | >10 |
| 2G10 | 2204 | 16.65 (12.87–21.56) | 0.26 (0.19–0.46) | - | 33.92 | 22.53 | 65.45 | 8.65 | 4.9 | - | 3.46 (0.96–33.0) |
| 3C6 | 971 | - | 0.28 (0.22–0.40) | - | NA | NA | NA | NA | 1.42 | - | 0.81 (0.43–1.50) |
| hIgG1 | 155.3 | - | NA | NA | 0.47 | - | 4.8 | - | NA | NA | NA |
| Tras | 841.8 | 3.65 (0.67–70.55) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
The binding activity of 11 antibodies to human uPAR was further characterized using biolayer interferometry (BLI). All antibodies were prepared in IgG format to provide avidity in binding and showed equilibrium dissociation constant (KD) values in the pM range. One thing to be noted is that 2G10 and 3C6 IgGs displayed KD values in the double-digit nanomolar range (Suplementary Table S1).17 These results demonstrate the advantage of screening B cells that produce affinity matured antibodies capable of binding uPAR tightly and exhibit slow off rates.
Cross-reactivity profiles were confirmed by ELISA
To ensure the cross-reactivity of the 11 antibodies was maintained in the mouse/human chimeric format with the trastuzumab constant domains, we evaluated their binding reactivity to human and cyno uPAR by ELISA (Figure 3(a)). Our results demonstrated that all antibodies displayed cross-reactivity to both human and cyno uPAR with EC50 values ranging from 0.1–0.8 nM and 0.2–1.1 nM, respectively. The binding curves also revealed that 2G10 and 3C6 exhibited specificity of binding only for human uPAR, but not cyno uPAR (Figure 3(a) and Table 1).
Figure 3.
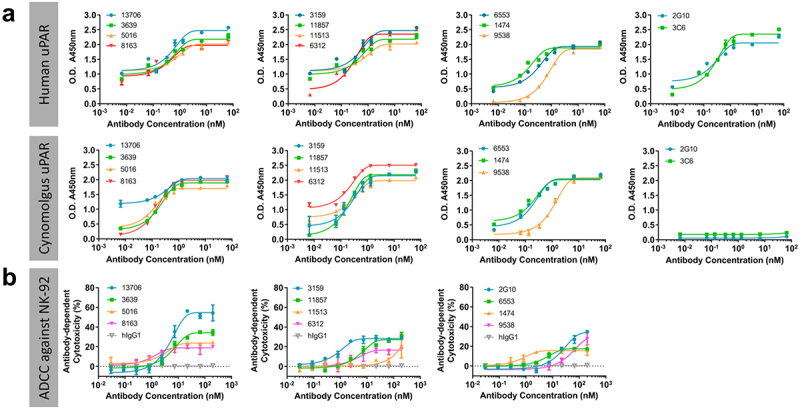
(a) Cross-reactivity profile of 11 antibodies evaluated by ELISA, showing they are cross-reactive to human and cyno uPAR, and 2G10 and 3C6 are specific binders for human uPAR. (b) ADCC activity of 11 antibodies and 2G10 in the presence of NK-92® MI CD16a effector cells against MDA-MB-231 cells. Two-way ANOVA followed by posthoc tukey test reveals significant activity over human isotype IgG1 (hIgG1). * = p≤0.001.
Antibody-dependent cellular cytotoxicity
With the confirmation of cell-surface uPAR recognition and cross-reactivity of the antibodies, 11 antibodies identified with >5% amino acid difference in CDR sequences were evaluated for their ADCC activity as a uPAR-targeted immunotherapy approach. The ADCC assay for the 11 antibodies was first performed using MDA-MB-231 cells as target cells in the presence of NK-92 MI CD16a effector cells. The resulting dose-response curves demonstrated that, except for 11513, 10 antibodies were able to induce ADCC with better or comparable EC50 values compared to 2G10, and no cytotoxicity was observed for the hIgG1 (Figure 3(b)). Seven antibodies (3159, 3639, 5016, 8163, 9538, 11857, 13706), which exerted effective ADCC response in NK-92 cells, were further tested for ADCC activity in the presence of human peripheral blood mononuclear cells (PBMCs) from three different healthy donors. Due to the intrinsic characteristics of effector cells from each PBMC donor,38 the inherent donor-to-donor variability was observed as expected between the seven antibodies, and the results showed they all facilitated effector cell function against MDA-MB-231 cells in a dose-dependent manner in the presence of healthy PBMCs (Figure 4). The mean maximum percentage of ADCC response for seven antibodies ranges from 46 to 66%, and six of them are more potent than 2G10 with 11 to 96-fold lower EC50 values from 0.1 to 0.8 nM (Table 1).
Figure 4.
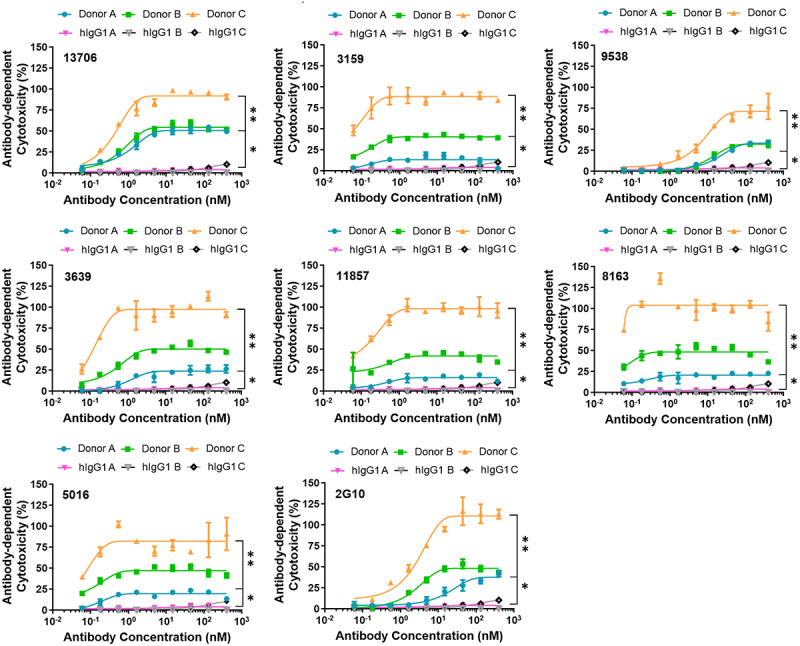
Selected seven antibodies were able to induce dose-dependent cell death in MBA-MB-231 cells in the presence of human PBMCs from three healthy donors. Two-way ANOVA followed by posthoc tukey test reveals significant activity over human isotype IgG1 (huIgG1). * =p≤0.05, ** = p≤0.001.
Cytotoxicity as ADCs and antibody internalization
In addition to Fc-mediated ADCC as a strategy for providing antitumor cytotoxicity, ADCs have been developed rapidly in recent decades to selectively deliver cytotoxic payloads directly to the targeted cancer cells.39 To determine if the ADC approach can be applied to the selected seven antibodies, ADC efficacy was assessed in vitro against the MDA-MB-231 cells with Fab-HFc-CL-MMAE, which recognizes the trastuzumab Fc moiety and has a cathepsin-cleavable linker connecting to monomethyl auristatin E (MMAE). Although all selected antibodies recognized cell-surface uPAR, only three of them (3159, 8163, and 11857) exhibited a concentration-dependent increase in ADC cytotoxicity in the presence of Fab-HFc-CL-MMAE with low EC50 values from 0.57 to 0.76 nM. (Figure 5(a)). The control experiment without treatment of Fab-HFc-CL-MMAE showed low cytotoxicity (Figure 5(b)).
Figure 5.
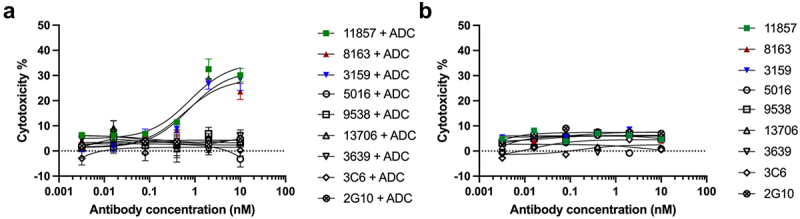
(a) Dose-dependent cytotoxicity was observed for antibodies (3159, 8163, and 11857) in MDA-MB-231 cells in the presence of anti-human Fc Fab conjugated to cytotoxic MMAE through a cathepsin-cleavable linker. (b) Low cytotoxicity was observed for all antibodies without the treatment of secondary ADC.
To investigate whether the ADC cytotoxicity was due to the internalization of the uPAR-antibody complex, confocal microscopy and colocalization analysis were performed. MDA-MB-231 cells were treated with Alexa Fluor 568-conjugated antibody and co-stained with LysotrackerTM Green, a pH-dependent fluorescent dye that selectively stains lysosomes. The internalization of the three antibodies (3159, 8163, and 11857) and their localization into lysosomes were observed by overlaying the images from their respective fluorescence channels and brightfield microscopy (Figure 6(a)). Negative controls were conducted with mouse and human non-relevant isotype IgG, and no internalization was observed (Supplementary Figure S4). In addition, the colocalization of antibodies and lysosomes was analyzed quantitatively using the Manders’ coefficient to determine the percentage of overlap between the red and green fluorescent signals, revealing that 53–60.5% of the three antibodies were internalized to the lysosomal compartment (Figure 6(b)). These results indicated that three antibodies (3159, 8163, and 11857) were able to target uPAR in a distinct complex and induce efficient internalization.
Figure 6.
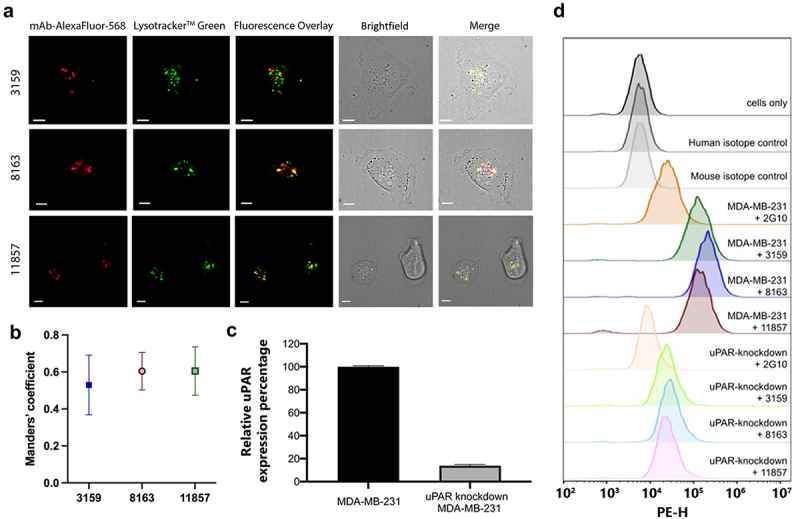
(a) Lead antibodies were internalized by MDA-MB-231 cells and colocalized with lysosomes. Scale bars = 10 µm. (b) Fluorescent signal overlap between antibodies and lysosomes is represented quantitatively using the manders’ coefficient. A high degree of overlap is observed for all lead antibodies, suggesting active lysosomal uptake. (c) Quantification of relative uPAR expression in MDA-MB-231 and uPAR-knockdown MDA-MB-231 cells from the immunoblot result. (d) On-target binding of uPAR on MDA-MB-231 and uPAR-knockdown MDA-MB-231 cells by flow cytometry.
Validation of antibody’s specificity using small interfering RNA knockdown and flow cytometry
To validate the specific binding of three antibodies (3159, 8163, 11857) to uPAR in MDA-MB-231 cells, uPAR expression was knocked down in the MDA-MB-231 cell line using small interfering RNA (siRNA). A significant reduction (~86%) in the uPAR expression was shown by immunoblot (Figure 6(c) and Supplementary Figure S5A). Flow cytometry was further performed for these three antibodies and 2G10, which served as a positive control, against MDA-MB-231 and uPAR-knockdown MDA-MB-231 cells (Figure 6(d)). The results indicated 14% of relative mean fluorescent intensity (MFI) was observed from uPAR-knockdown MDA-MB-231 cells and is consistent with the immunoblot results (Supplementary Figure S5B-C). The non-relevant isotype control antibodies (P2B2,40 human IgG1; P3.6.2.8.1, mouse IgG1) showed no significant shift in flow cytometry, suggesting that the 14% of relative MFI is from the residual uPAR expression from the uPAR knockdown experiment. These data show that our anti-uPAR antibodies displayed on-target binding of uPAR in MDA-MB-231 cells.
Inhibition of cell adhesion to vitronectin
The binding of VN to uPAR is known to induce intracellular signaling events that activate integrins to promote cancer cell adhesion and communication to the extracellular matrix.41 To investigate whether the seven antibodies have any functional inhibition on the tumor cell by targeting cell-surface uPAR, we evaluated their ability to block uPAR-mediated cell adhesion to VN. The results demonstrated that the seven antibodies were able to inhibit the adhesion of MDA-MB-231 cells to VN-coated plates in a dose-dependent manner (Table 1 and Supplementary Figure S6). Antibody 3159 showed the strongest inhibitory effect and is comparable to 3C6, which was identified as an inhibitor to abrogate uPAR-mediated cell adhesion in our previous study.42 Overall, the in vitro characterization of the antibodies highlighted three lead antibodies (3159, 8163, and 11857) are the most promising antibodies with ADCC activity, ADC cytotoxicity by inducing efficient uPAR-antibody internalization, on-target binding, and the functional inhibition on cell adhesion (Table 1), leading us to investigate their therapeutic efficacy in an orthotopic animal model of breast cancer. The CDR sequences of the three lead antibodies are shown in Supplementary Table S2.
Therapeutic efficacy in an orthotopic animal model of human breast cancer
To determine the in vivo therapeutic efficacy of the three lead antibodies, MDA-MB-231 cells were orthotopically implanted in the mammary fat pads of Foxn1nu nude mice, and animals with tumors (75–100 mm3 in volume) were treated weekly via intravenous injection with each antibody (30 mg/kg). Close monitoring of tumor growth across treatment groups revealed that all antibodies were able to reduce tumor burden relative to the untreated control (Figure 7(a)). Our data demonstrated 11857 exhibited the most potent efficacy with a 3.1 ± 0.4-fold smaller tumor burden on day 21 (p = 0.0039) compared to the untreated control. Such activity was maintained on Days 25 and 28 with a 3.3 ± 0.3 (p = 0.0058) and a 3.6 ± 0.6 (p = 0.0125) fold smaller tumor burden than that of the untreated control group, respectively (Figure 7(a)). Although 3159 and 8163 had little effect in reducing tumor volume in comparison with 11857, they were nonetheless effective in reducing tumor growth rates relative to the untreated control (Figure 7(b)). The superior antitumor activity of 11857 was also reflected in its ability to impair tumor growth rates in comparison to the untreated control (p = 0.0002), which was far superior to that of 3159 (p = 0.0141) and 8163 (p = 0.0267) (Figure 7(b)).
Figure 7.
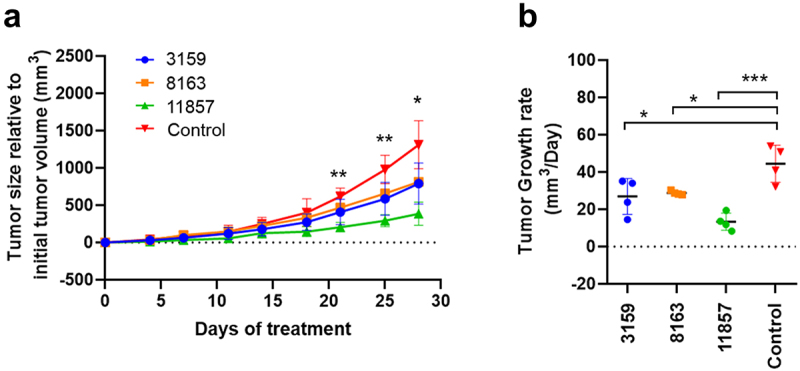
(a) The therapeutic efficacy of novel antibodies was determined in an orthotopic mice model of human breast cancer using MDA-MB-231 cells. Animals showed reduced tumor size in comparison to untreated controls, and significant tumor growth suppression was observed in animals treated with 11857 after 21 days of treatment. (b) A significant reduction in tumor growth rates was observed for antibody-treated animals throughout the 30-day treatment, whereas 11857 was the most active agent. Data are shown as mean ± standard deviation. Statistical analysis as two-way ANOVA, with a posthoc multiple comparisons using dunnett’s test. * p<0.05; ** p<0.01; *** p<0.001.
Epitope binning using biolayer interferometry
The decrease in tumor growth rate along with the ability of the three lead antibodies (3159, 8163, and 11857) to impair cell adhesion led us to investigate their binding epitopes on uPAR. The epitope binning was first performed to test whether the three lead antibodies could inhibit the binding of VN to uPAR by BLI. uPAR was immobilized on a biosensor, and the first association was performed with each lead antibody or no antibody, followed by the second association of VN. Compared with the binding response of VN to uPAR alone, the BLI measurements showed that 3159 completely abolished subsequent VN binding, 11857 significantly reduced the binding of VN to uPAR, and 8163 displayed a weaker inhibitory effect (Figure 8(a)). This is consistent with the result from our adhesion assay showing 3159 exhibited the strongest inhibition on VN-mediated cell adhesion, suggesting 3159 interfered with the interaction of VN and uPAR by blocking the VN-binding site. A previous report has shown that the binding affinity of VN to uPAR is in the 1 μM range.43 By changing the order of adding 3159 and VN, 3159 with stronger binding affinity and avidity competed with VN for binding to uPAR (Supplementary Figure S7). An epitope competition assay was also performed in different pairs of antibodies to identify if they have distinct binding epitopes. Interestingly, both 3159 and 8163 could bind to uPAR at the same time, but neither one of them was able to interact with uPAR once an antibody-uPAR complex was formed with 11857, suggesting 11857 has a partially overlapping epitope with 3159 and 8163 (Figure 8(b)). In addition, the BLI measurements also showed the binding epitopes of these three antibodies are distinct from 2G10 and 3C6, which are known to block uPA and integrin αVβ1 binding to uPAR, respectively (Supplementary Figure S8).
Figure 8.
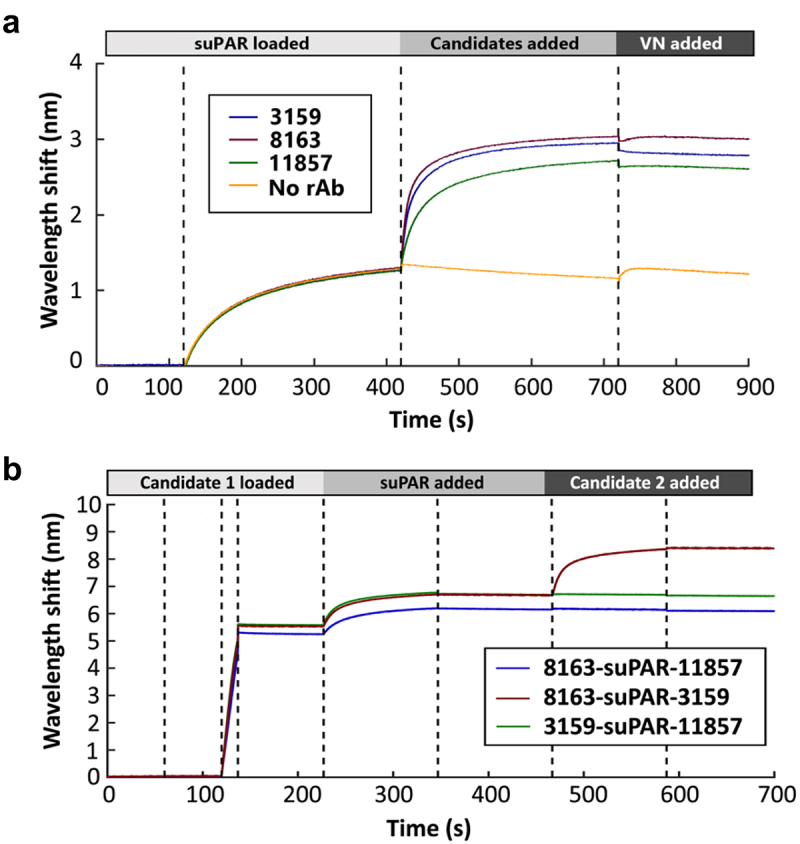
(a) BLI epitope binning of three lead antibodies and vitronectin to uPAR. The binding of each antibody to uPAR resulted in a decreased wavelength shift in the last association step (VN-added) demonstrating the competitive blocking of VN binding by each antibody. (b) BLI competition assay reveals 8163 and 3159 have distinct binding sites, and 11857 has a partially overlapping epitope with 8163 and 3159.
Discussion
The growing understanding of uPAR and its molecular partners in tumorigenesis, cancer progression, and metastasis has provided the basis for developing uPAR-targeted diagnostic, prognostic, and therapeutic strategies to treat a wide variety of tumors.44,45 Currently, no cross-reactive anti-uPAR antibodies with potential as a clinical candidate have been reported, preventing the evaluation of uPAR in non-human primates such as cynomolgus monkeys, which serve as a valuable model to provide the most relevant information on the safety, efficacy, and pharmacokinetic profiles of translational therapeutics for human use.46,47 Thus, this study presents a high-throughput antibody discovery pipeline allowing the identification of cross-reactive antibodies against human and cyno uPAR. The 60-day immunization campaign using recombinant human suPAR enabled in vivo affinity maturation in SJL mice to generate uPAR-primed B cells, and the Beacon platform allowed the culture, manipulation, and screening of single B cells in one day. A similar approach has recently been used for the successful development of neutralizing antibodies against SARS-CoV and SARS-CoV-2.48 Our approach provides an example of using immunization to bias the immune response coupled with the screening of antigen-primed B cells for identifying cross-reactive antibodies with strong binding affinity and antitumor activity, showing the power of in vivo development and affinity maturation in B cells for antibody selection.
Effective tumor-targeting antibodies induce direct and indirect effects on tumor cells, mediated by their Fab variable and Fc constant domains, respectively.49 The targeted therapies for HER2-positive breast cancer in clinical use (i.e., trastuzumab (Herceptin®) and pertuzumab (Perjeta®)) involve ADCC by targeting HER2 and recruiting immune effector cells through the Fc domain as part of their mechanism for killing tumors.50 To confer the ADCC activity to the mouse antibodies identified from the Beacon platform, we fused their Fab variable domains to the trastuzumab constant domains for expressing antibodies in a mouse/human chimeric format. Interestingly, different amplitudes of ADCC were observed in the presence of NK-92 cells, suggesting the epitope recognition of antibodies is critical to modulate ADCC activity. These findings corroborated previous studies that antigen binding altered an IgG conformation and affected the recognition of the Fc region by the FcγRIIIa and FcγRIIIb receptors on the surface of NK cells and PBMCs.51–53 In addition, the angle of its Fc domain relative to the target cell surface could govern the accessibility of the Fc region for the interaction with effector cells to induce ADCC.54,55
Although ADCC is one of the primary mechanisms for most antitumor mAbs currently in the clinic, recent findings suggested having antibodies with both ADCC-based and direct biological effects would benefit an effective anti-tumor response.56,57 Previous studies have shown that VN deficiency strongly impairs tumor growth in an orthotopic xenograft model of breast cancer.58 In addition, the binding of uPAR to VN has been shown to regulate cell adhesion and further trigger changes in cell morphology, migration, and signaling.59–61 A reported mAb 8B12 was found to inhibit the VN binding to uPAR and effectively reduce uPAR-mediated cell migration on the VN-coated surface.18 These studies revealed how inhibiting the interaction of cancer cells with the ECM can affect their pro-proliferative communication in the tumor microenvironment and overall tumorigenesis, supporting the idea that the inhibition of cell adhesion demonstrated by our lead antibodies provides an additional advantage to impair tumor growth.41,62
In addition, several therapeutic antibodies have been redeveloped as ADCs to deliver cytotoxic drugs to antigen-positive tumor cells.63 Here, selected antibodies were evaluated with a secondary ADC killing assay against MDA-MB-231 cells, and lead antibodies (3159, 8163, and 11857) showed the potential use of uPAR-targeting ADCs. Further confocal microscopy analysis revealed that the antibody-uPAR complex was internalized and delivered to the lysosome for the proper release of MMAE in the low pH and protease-rich environment. According to previous studies, uPAR can be internalized by tumor cells via clathrin-mediated endocytosis or via a clathrin-independent mechanism mediated by LRP-1, both of which are responsible for trafficking uPAR to the lysosome for degradation and recycling.64,65 This could provide an additional advantage because all known internalization mechanisms of uPAR cause it to dissociate from its co-receptors, which include matrix-engaged integrins and other bonafide ligands, and therefore abrogate the downstream signaling.66
Finally, we propose a binding model for three lead antibodies (3159, 8163, and 11857) to uPAR. Human and cyno uPAR share 96% sequence identity, where most of the sequence variation between the homologs lies at the uPA-binding site (Figure 9(a)), resulting in the species-specific interaction between uPA and uPAR.67,68 On the other hand, the VN-binding site is located on the opposite side and is more conserved between uPAR homologs (Figure 9(a)). Based on the BLI epitope binning results, the binding epitopes of lead antibodies (3159, 8163, and 11857) are close or on the VN-binding site, and they are distinct from the previously reported uPAR-binders, 2G10 and 3C6. The binding epitopes of 3159 and 8163 on uPAR are independent, and the 11857 binding epitope overlaps considerably with their binding sites, but is not identical (Figure 9(b)). All of them exhibited an inhibitory effect on cell adhesion, and 3159 binds to an epitope on the VN-binding site and therefore showed the strongest inhibitory effect. 11857 binds to a spot resulting in a synergistic effect on ADCC, uPAR internalization, and blocking cell adhesion, showing the advantage of having antibodies with ADCC and direct biological effects for impairing tumor growth.
Figure 9.
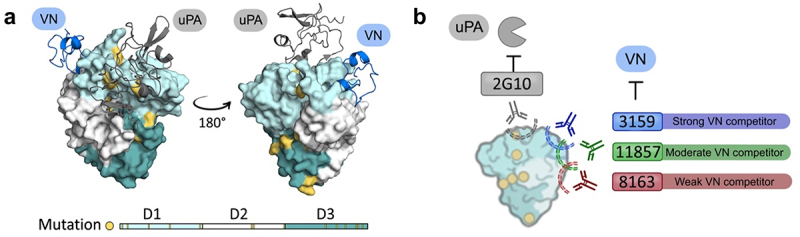
(a) Molecular surface representation of human uPAR-ATF-SMB complex. The uPA N-terminal fragment (ATF) is shown as a ribbon diagram in gray, and the somatomedin-B like domain of VN is shown as a ribbon diagram in blue. (PDB ID: 3BT1).69 Mutation variants between human and cyno uPAR are highlighted in yellow. (b) Proposed binding model for lead antibodies to uPAR, highlighting their inhibitory effects for vitronectin binding and distinct binding epitopes compared to 2G10. 3159 and 8163 recognize distinct epitopes on uPAR, and 11857 has a partial overlapping epitope with 3159 and 8163.
In conclusion, we used the Beacon platform to screen single B cells from uPAR-immunized mice to identify novel cross-reactive antibodies against human and cyno uPAR. Selected antibody candidates were characterized in vitro and shown to recognize uPAR on human breast cancer cells and induce tumor cell killing via ADCC. In addition, lead antibodies showed potent ADC cytotoxicity against breast cancer cells in the presence of secondary ADCs by inducing receptor internalization to lysosomes. The lead antibodies were also capable of inhibiting cell adhesion by blocking VN binding to uPAR, thus resulting in the functional effect on cancer cell adhesion. Moreover, an in vivo study demonstrated three lead antibodies (3159, 8163, and 11857) were able to inhibit tumor growth in an orthotopic mouse model of human breast cancer. Finally, a binding model for our lead antibodies was proposed, providing a better understanding of their interactions to uPAR.
Materials and methods
Antigen production
A HEK293 cell line, stably expressed human suPAR, was generously provided by the Chapman Lab at the University of California, San Francisco. For suPAR production, 3.7 × 107 cells were seeded in a 5-stack Corning® CellSTACK® Culture Chamber and maintained in the DMEM complete medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/L streptomycin (Gibco) at 5% CO2 and 37°C. The protein was harvested at day 5 and purified using a Ni-NTA column followed by gel-filtration using a Hiload 16/600 Superdex 200 prep-grade column. The production of suPAR was confirmed by immunoblot analysis using a mouse anti-human uPAR monoclonal antibody clone R-3 (Invitrogen, #MON R-3-02) and a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) antibody (Biorad, #1706516). Purified suPAR samples were also characterized by LC-MS/MS and further subject to endotoxin removal using Pierce™ High-Capacity Endotoxin Removal Spin Columns. After endotoxin removal, samples were characterized for their total endotoxin level and only those not exceeding 0.5 U/mL were approved for the preparation of immunogen injections.
Animal immunization strategy
The immunization strategy consisted of a primary intraperitoneal injection with 50 μg of antigen prepared as an emulsion in Freund’s Complete Adjuvant (FCA). Eight SJL mice (6–8 weeks old) received a boost injection containing 25 μg of antigen every other week for three boosts, with bleeds being intercalated to each boost injection for 60 days. A pre-bleed was performed for each mouse prior to the start of the immunization campaign in order to control for non-anticipated exposure to the antigen. All blood samples were allowed to clot and 100–200 μL of serum was stored at −80°C for antibody titer determination. All animal studies were conducted under an animal use protocol (IACUC ID: AN179937) approved by the University of California, San Francisco animal care and use committee approved on February 7, 2022.
Harvest and enrichment of mouse antibody-secreting cells
Animals were euthanized in accordance with approved IACUC protocols. Spleens and bone marrows were harvested and processed into single-cell suspension in RPMI. Total B cells were isolated by magnetic negative selection using EasySep Mouse Pan-B Cell Kit (StemCell) to deplete non-B cells from the single-cell suspension. ASCs were enriched from single B cell suspensions by magnetic positive selection using EasySep Mouse CD138+ Kit (StemCell) and a FACS-based positive selection through the gating of CD45R(B220)-/CD138high cells, which are traditionally used to broadly define the population of plasma cells.70
Nanofluidic opto-electropositioning for screening of single B cells
Direct screening of secreted antibodies from ASCs was achieved with the Beacon platform. Enriched ASCs were injected into OptoSelectTM 3500 and 14k chips. The platform utilizes OEP technology to effectively isolate single ASCs into nanopens. ASCs were individually cultured for 1 hr within the chip and screened for both IgG secretion and antigen specificity using an in-channel multiplex bead-based fluorescent assay. Briefly, beads coated with rabbit anti-mouse IgG (H+L) (Biorad, #5180–2104) were imported to the chip where active accumulation of secreted antibodies was identified by the binding of a FITC-labeled goat anti-mouse secondary antibody (Biorad, # STAR70) to the beads. Antigen specificity was evaluated by importing fluorescently labeled human (Alexa Fluor 488), cynomolgus monkey (R&D Systems, #10949-UK), or mouse (Sino Biologicals, #50160-M08H) (Alexa Fluor 647) uPAR into the chip. The binding of ASC-derived IgGs to the antigen was monitored by the time-dependent increase of uPAR-derived fluorescence on the beads at the mouth of the nanopen. HEK293T/17 cells expressing 2G10 were used as a positive control for both IgG secretion and the production of anti-uPAR antibodies.
Sequencing, recombinant cloning, expression, and purification of recombinant antibodies
Selected single B cells were exported to 96-well plates containing lysis buffer and mineral oil. RNA was captured onto Agencourt RNAclean XP Beads (Beckman Coulter), and cDNA was synthesized using an MMLV-RT (Thermo). Identical priming sites were added to the cDNA as part of the oligo-T and TSO primers and used to amplify total cDNA. cDNA was purified using Agencourt AMPure XP Beads (Beckman Coulter), and size distribution was assessed using capillary electrophoresis (Labchip, Perkin Elmer). Selected human and cyno uPAR cross-reactive binders were then amplified using a commercial Ig Primer set (MilliporeSigma). Clones showed amplicons within 500–700 bp and were sequenced using NGS method. The NGS library preparation was done using cDNA samples of the selected clones by indexing universal forward and mouse reverse constant primers (MilliporeSigma) for VH and VL separately. Then, the samples were run on MiSeq (Illumina), resulting in VH/VL sequences. The CDR sequences were aligned using Clustal Omega,71 and a phylogenetic tree was constructed using MEGAX with the neighbor-joining method.72
Selected mouse VH and VL sequences were amplified and cloned into pcDNA3.4-hCg1 and pcDNA3.4-hCk mammalian expression vectors, which contain the trastuzumab CH and CL sequences, respectively. The transfection and expression of recombinant IgG were performed based on the manufacturer’s protocol. Briefly, HEK293F cells were seeded in FreeStyleTM media and incubated at 130 rpm and 37°C with 8% CO2. Transfection was carried out using polyethylenimine (PEI) keeping a 1:2 ratio of DNA/PEI. A 5% solution of peptone was added at 0.1 equivalent volumes of the original cell suspension to improve recombinant protein synthesis. On day 6–7 post-transfection, the IgG-enriched media were collected, and IgGs were purified using protein A column (GE MabSelect™ SuRe™) and dialyzed overnight against phosphate-buffered saline (PBS) (pH 7.4) at 4°C.
Biolayer interferometry analysis
The binding affinity of anti-uPAR antibodies was measured using an Octet RED384 System at 25°C, and samples were prepared in assay buffer (PBS with 1% bovine serum albumin (BSA)). Octet Streptavidin (SA) biosensor was used to immobilize biotinylated human or cyno uPAR at 2 µg/mL. After a short baseline, the biosensor was dipped into each well containing anti-uPAR antibodies and followed by a dissociation step.
The epitope binning experiments were carried out in two ways. First, biotinylated uPAR was immobilized on anti-penta-His (HIS1K) biosensors (Sartorius), followed by two association steps with the antibody candidate and vitronectin. Second, the first biotinylated antibody was immobilized on the SA biosensors. After a wash step, uPAR was bound to reach saturation. The biosensors were then moved to the next well for the association of the second antibody. The data analysis was performed using Octet Data Analysis software, and figures were made with Matlab.
Cross-reactivity of antibodies by ELISA
Nunc MaxiSorp™ flat-bottom 96-well plates were coated with human or cyno uPAR (3.19 μg/mL) at 4°C overnight, and plates were washed with wash buffer and blocked with 5% nonfat dry milk. A standard log serial dilution of each antibody was added to the uPAR-coated plates and incubated at 4°C overnight. Plates were washed three times and incubated with 50 μL of HRP-conjugated goat anti-human (H+L) antibody (Biorad, #1721050). After a two-hour incubation, plates were washed, and 100 μL of 1-Step™ Turbo TMB-ELISA Substrate Solution (Thermo Scientific) was added to each well. The reaction was quenched with 2 M H2SO4 for 5 min at room temperature, and the optical density of each well was measured at 450 nm using a SpectraMax190 microplate reader. The resulting dose-response curves were used to determine the minimum dose of antibodies required to achieve 50% of the saturation signal and enable a quantitative comparison of binding affinity.
Antibody recognition of cell-surface uPAR by FACS
MDA-MB-231 cells were harvested with TrypLE (Gibco), resuspended in FACS buffer (PBS+1% BSA) to 2 × 106 cells/mL, and aliquoted to a 96-well plate (100 μL, 2 × 105 cells/well). A serial dilution of antibody was added to a maximum concentration of 600 nM, followed by a 50 min incubation at 4°C. Cells were then washed three times with PBS and incubated with an Alexa Fluor 488-conjugated goat anti-human IgG for 50 min at 4°C in the dark. Finally, cells were washed twice with FACS buffer and resuspended in 80 μL for FACS analysis in a Bio-rad S3e Cell Sorter.
Inhibition of cell adhesion to vitronectin
MDA-MB-231 cells were cultured in the complete medium in a humidified atmosphere of 5% CO2 at 37°C. MaxiSorp 96-wells plates were coated with VN (corning, #354238) at 4°C overnight. The wells were washed with PBS and blocked for 1 hr with 1% BSA in PBS. A total of 50,000 MDA-MB-231 cells were seeded in each well and a serial dilution of antibody or RGDS peptide was added, and the plate was incubated at 5% CO2 and 37°C overnight. All wells were washed with PBS, and ice-cold methanol was added to fix cells for 10 min at room temperature. After fixation, a 5% crystal violet solution was used to stain cells. Wells were washed three times with PBS, and cells were lysed with 2% SDS lysis buffer. Each lysate was transferred to a transparent 96-well plate, and the absorption at 590 nm was recorded using a Synergy Neo2 Multi-Mode Microplate Reader (BioTek Instruments, Inc.) to quantify the adherent cells.
ADCC in vitro using NK cells
ADCC on MDA-MB-231 by NK cells was detected by DELFIA® EuTDA Cytotoxicity Reagents (Perkinelmer). Briefly, MDA-MB-231 cells were harvested and labeled by incubation with 2 μL/mL of the fluorescence-enhancing ligand (ParkinElmerDELFIA® BATDA Labeling Reagent) for 20 min at 37°C. After the diffusion of BATDA into the cells, it was hydrolyzed and converted to 2,2‘:6’,2“-terpyridine-6,6”-dicarboxylic acid (TDA) by cytosolic acetyl esterase. Since TDA is a non-cell permeable hydrophobic ligand, it can be trapped inside the live target cells. The solution was centrifuged, and cells were washed three times with PBS. The labeled cells were reconstituted in RPMI 1640 media without phenol red and then seeded to a 96-well U-bottom sterile microplate (100 μL, 1 × 104 cells/well). Next, 50 μL of a serial dilution of each antibody was added to the assay plate and incubated at 37°C for 5–10 min. Separately, NK-92 CD16a 176 V effector cells were harvested and concentrated to approximately 1.2 × 106 cells/mL before the addition of 50 μL to the assay plate resulting in an effector to target cell ratio of 6:1 in each well. The plate containing the antibodies, target, and effector cells was then incubated for 4 hr at 37°C and 5% CO2. After the incubation, the plate was centrifuged for 5 min at 400 RCF, and 25 μL of the supernatant was transferred to a flat-bottom detection plate. Finally, 200 μL of Europium solution (PerkinElmer, DELFIA® Eu-Solution) was added to each well, and the plate was incubated for 15 min at room temperature to allow the formation of a highly fluorescent stable-chelate (Eu-TDA). The resulting fluorescent signal was obtained in a time-resolved fluorimeter within 5 hr. The background death control was determined by diluting target cells with media, and the maximum death control was determined by incubating cells with 10 µL of lysis buffer (1% Triton X-100) for 30 min prior to centrifuging the plate.
ADCC in vitro using human PBMCs isolated from healthy donors
Frozen PBMC cells were obtained commercially from AllCells (FPB004F-C). The cells were isolated from human blood by the Leuko Pak-Density gradient method, then stored in liquid nitrogen. Cells were thawed at 37°C, suspended in RPMI1640 + 10% FBS, and incubated at 37°C overnight. MDA-MB-231 target cells are labeled with DELFIA BATDA in accordance with the manufacturer’s instructions. Then, the effector cells PBMC cells from each donor were plated with target cells into a 96-well plate at a ratio of 50:1. The induction of ADCC was triggered upon the addition of each antibody to the mix, which was incubated for 4 hr at 37°C. Finally, the supernatant was collected and mixed with Europium solution. Time-resolved fluorescent (TRF) signal intensity was used to determine the degree of cytotoxicity. Control groups are set for data normalization, including target spontaneous group (Target cells), target maximum group (Target cells lysised by Triton) and background group (Supernatant of target cell). ADCC effect is determined by the formula: Calculated ADCC was defined by the formula: % ADCC=(Sample Cytotoxicity – Target cell and Effector cell mixture spontaneous cytotoxicity)/(Target cell Maximum cytotoxicity (Triton X-100 treatment) - Target cell and Effector cell mixture spontaneous cytotoxicity)*100%. Dose-response effect was analyzed with Graphpad Prism.
Antibody-drug conjugates cytotoxicity screening
MDA-MB-231 cells were seeded at 2,500 cells/well in 96-well plates (Corning) at 37°C and 5% CO2 overnight, and cells were grown for five days in the presence of serial dilutions of antibodies ranging from 0.0032 nM to 10 nM in triplicates, combined with a Fab fragment of an anti-human IgG Fc-specific antibody conjugated to MMAE (Fab-HFc-CL-MMAE, Moradec) in a final concentration of 20 nM. The number of live cells was quantified by the CellTiter-Glo luminescent cell viability assay (Promega) based on luminescent detection of ATP, which is directly proportional to the number of cells present in each well. After the incubation, the luminescence was recorded using a Synergy Neo2 Multi-Mode Microplate Reader (BioTek Instruments, Inc.)
Antibody internalization and Abs-lysosomes colocalization
Antibodies were conjugated with AF568 NHS ester (ThermoFisher Scientific), and MDA-MB-231 cells were seeded in an ibidi8 well µ-slide at a density of 3 × 104 cells/well and incubated at 37°C with 5% CO2 overnight. After 2 times of wash with PBS, the cells were incubated with 0.035 mg/mL of AF568-conjugated antibody and 50 nM of Lysotracker Green DND-26 (Cell Signaling Technology) in DMEM at 37°C with 5% CO2 for 2 hr before live-cell imaging to allow antibody internalization and lysosomal staining. The cells were washed with PBS and treated with acidic glycine buffer (50 mM glycine, 150 mM NaCl, pH 3.0) for 1 min at room temperature to strip any membrane-bound antibodies. After the final wash with PBS, DMEM without phenol red was supplemented to avoid a background fluorescence, and fluorescence images were acquired on a Yokagawa CSU22 spinning disk confocal (488 nm laser with chroma ET525/50 m filter, and 561 nm laser with chroma ET645/75 m filter in addition to brightfield imaging). Image processing was done using Fiji with the JACoP colocalization analysis plug-in.
Small interfering RNA knockdown and flow cytometry
Knockdown of human uPAR expression in MDA-MB-231 cells was carried out by transfection of siRNA (Thermo Fisher s10614, s10615, and 111193). MDA-MB-231 cells were seeded in T75 flasks at a density of 1.25 × 106 cells/flask in the DMEM complete medium and incubated at 37°C with 5% CO2 overnight. The next day, MDA-MB-231 cells were transfected with the siRNA pool using DharmaFECT 4 transfection reagent (Dharmacon) following the DharmaFECT transfection protocol. After 48 hr of incubation, the MDA-MB-231 and uPAR-knockdown MDA-MB-231 cells were dissociated using TrypLE select (Gibco) and resuspended at a density of 106 cells/mL in the cold flow medium (PBS, 2% FSB, and 2 mM EDTA). Collected cells were incubated with lead antibodies and 2G10 (2 µg/mL) at 4°C for an hour. Non-relevant isotype controls (P2B2, human IgG1;40 PE-conjugated mouse IgG1, Thermo Fisher, #12-4714-42) were used as negative controls. Cells were washed twice by centrifugation at 90 g for 5 min and resuspended with ice-cold flow buffer, followed by incubation with PE-conjugated anti-human Fc secondary antibody (1 µg/mL) at 4°C for an hour. After twice of wash with the flow buffer, the cells were analyzed on flow cytometry (BD Cytoflex), and data analysis was performed using FlowJo software.
Therapeutic efficacy in an orthotopic animal model of human breast cancer
A group of 16 female Foxn1nu mice were orthotopically implanted with 1 × 106 MDA-MB-231 cells and monitored for several days until tumor volume reached 75–100 mm3. Once tumor volumes were reached animals were considered eligible for therapeutic intervention starting three days after such tumor volumes were achieved. Therapeutic intervention commenced with each experimental arm receiving antibodies administered intravenously at a concentration of 30 mg/kg. A 30-day treatment regimen was conducted, with animals receiving weekly antibody treatment over a period of 30 days (Days 3, 10, 17, and 24). Animal welfare, body weight, and tumor volume were monitored continuously throughout the study. Upon the completion of the therapeutic intervention regimen tumors were harvested and prepared for histological analysis. All animal studies were conducted under an animal use protocol (IACUC ID: AN179937) approved by the University of California, San Francisco animal care and use committee approved on February 7, 2022.
Statistical analysis
All statistical analysis was performed in GraphPad Prism version 8.0 (GraphPad Software, Inc., San Diego, CA). Dose-response curves were driven from non-linear fitting to raw values run in a minimal of three experimental replicates. Statistical analysis for all data acquisition was performed as two-way ANOVA with a post-hoc multiple-comparison Tukey test. Differences between groups were considered significant at a P value≤0.05.
Supplementary Material
Acknowledgments
The authors thank Dr. Cheng-I Wang for helpful comments and discussion during the preparation of this article. We also acknowledge the Nikon Imaging Center at UCSF for providing access to the spinning disk confocal microscope.
Funding Statement
This work was funded by the Target Validation Initiative grant from the UCSF Helen Diller Family Comprehensive Cancer Center; by the National Cancer Institute, grant number P41 CA196276, Shang Pharma-UCSF partnership A133575, and National Science Foundation-Science Technology Center DBI1548297.
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- ACSs
antibody-secreting cells
- ADCs
antibody-drug conjugates
- BLI
biolayer interferometry
- CDR
complementarity-determining region
- Cyno
cynomolgus monkeys
- FACS
fluorescence-activated cell sorting
- FcγR
Fc gamma receptor
- GPI
glycosylphosphatidylinositol
- HER2
human epidermal growth factor receptor-2
- NGS
next-generation sequencing
- NK cells
natural killer cells
- OEP
opto-electropositioning
- PBMCs
peripheral blood mononuclear cells
- rAbs
recombinant antibodies
- suPAR
soluble urokinase-type plasminogen activator receptor
- Tras
trastuzumab
- TNBC
triple-negative breast cancer
- uPAR
urokinase-type plasminogen activator receptor
- uPA
urokinase-type plasminogen activator
- VN
vitronectin
Disclosure statement
Charles S. Craik is a co-founder of Dandelion Biosciences. Robert Drakas is the president of ShangPharma Innovation.
Data availability
The data that support the findings of this study are available from the corresponding author, CSC, upon reasonable request.
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2184197
References
- 1.Smith HW, Marshall CJ.. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–15. [DOI] [PubMed] [Google Scholar]
- 2.Lund IK, Illemann M, Thurison T, Christensen IB, Høyer-Hansen G.. uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr Drug Targets. 2011;12:1744–60. [DOI] [PubMed] [Google Scholar]
- 3.Mazar AP, Ahn RW, O’halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr Pharm Des. 2011;17:1970–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stroomberg HV, Kristensen G, Drimer Berg K, Lippert S, Brasso K, Røder MA. The association between plasma levels of intact and cleaved uPAR levels and the risk of biochemical recurrence after radical prostatectomy for prostate cancer. Diagnostics. 2020;10:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu KL, Fan JH, Wu J. Prognostic role of circulating soluble uPAR in various cancers: a systematic review and meta-analysis. Clin Lab. 2017;63:871–80. [DOI] [PubMed] [Google Scholar]
- 6.de Bock CE, Wang Y. Clinical significance of urokinase-type plasminogen activator receptor (uPAR) expression in cancer. Med Res Rev. 2004;24:13–39. [DOI] [PubMed] [Google Scholar]
- 7.Yuan C, Guo Z, Yu S, Jiang L, Huang M. Development of inhibitors for uPAR: blocking the interaction of uPAR with its partners. Drug Discov Today. 2021;26:1076–85. [DOI] [PubMed] [Google Scholar]
- 8.Meijer-van Gelder ME, Look MP, Peters HA, Schmitt M, Brünner N, Harbeck N, Klijn JGM, Foekens JA. Urokinase-type plasminogen activator system in breast cancer: association with tamoxifen therapy in recurrent disease. Cancer Res. 2004;64:4563–68. [DOI] [PubMed] [Google Scholar]
- 9.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konecny G, Untch M, Arboleda J, Wilson C, Kahlert S, Boettcher B, Felber M, Beryt M, Lude S, Hepp H, et al. Her-2/neu and urokinase-type plasminogen activator and its inhibitor in breast cancer. Clin Cancer Res. 2001;7(8):2448–57. [PubMed] [Google Scholar]
- 11.Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem. 2009;284:22825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandran VI, Eppenberger-Castori S, Venkatesh T, Vine KL, Ranson M. HER2 and uPAR cooperativity contribute to metastatic phenotype of HER2-positive breast cancer. Oncoscience. 2015;2:207–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhr J. uPAR and HER2 genes are usually co-amplified in individual breast cancer cells from blood and tissues. Breast Care. 2008;3:16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Cao S, Liu Z, Ye X, Chen L, Meng S. Rnai-mediated downregulation of uPAR synergizes with targeting of HER2 through the ERK pathway in breast cancer cells. Int J Cancer. 2010;127:1507–16. [DOI] [PubMed] [Google Scholar]
- 15.Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11:263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LeBeau AM, Duriseti S, Murphy ST, Pepin F, Hann B, Gray JW, VanBrocklin HF, Craik CS. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013;73:2070–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao B, Gandhi S, Yuan C, Luo Z, Li R, Gårdsvoll H, de Lorenzi V, Sidenius N, Huang M, Ploug M. Stabilizing a flexible interdomain hinge region harboring the SMB binding site drives uPAR into its closed conformation. J Mol Biol. 2015;427:1389–403. [DOI] [PubMed] [Google Scholar]
- 19.Rabbani SA, Ateeq B, Arakelian A, Valentino ML, Shaw DE, Dauffenbach LM, Kerfoot CA, Mazar AP. An anti-urokinase plasminogen activator receptor antibody (ATN-658) blocks prostate cancer invasion, migration, growth, and experimental skeletal metastasis in vitro and in vivo. Neoplasia. 2010;12:778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rullo AF, Fitzgerald KJ, Muthusamy V, Liu M, Yuan C, Huang M, Kim M, Cho AE, Spiegel DA. Re-engineering the immune response to metastatic cancer: antibody-recruiting small molecules targeting the urokinase receptor. Angew Chem. 2016;128:3706–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani T, Wang F, Knabe WE, Sinn AL, Khanna M, Jo I, Sandusky GE, Sledge GW, Jones DR, Khanna R, et al. Small-molecule inhibition of the uPar·upa interaction: synthesis, biochemical, cellular, in vivo pharmacokinetics and efficacy studies in breast cancer metastasis. Bioorg Med Chem. 2013;21(7):2145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minopoli M, Polo A, Ragone C, Ingangi V, Ciliberto G, Pessi A, Sarno S, Budillon A, Costantini S, Carriero MV. Structure-function relationship of an urokinase receptor-derived peptide which inhibits the formyl peptide receptor type 1 activity. Sci Rep. 2019;9:12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag A, Im HK, Jagadeeswaran S, Shaw DE, Mazar AP, Lengyel E. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. 2011;17:459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harel ET, Drake PM, Barfield RM, Lui I, Farr-Jones S, Van’t Veer L, Gartner ZJ, Green EM, Lourenço AL, Cheng Y, et al. Antibody-drug conjugates targeting the urokinase receptor (uPAR) as a possible treatment of aggressive breast cancer. Antibodies. 2019;8(4):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kriegbaum MC, Persson M, Haldager L, Alpízar-Alpízar W, Jacobsen B, Gårdsvoll H, Kjær A, Ploug M. Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes. Curr Drug Targets. 2011;12:1711–28. [DOI] [PubMed] [Google Scholar]
- 26.Baart VM, Houvast RD, de Geus-Oei LF, Quax PHA, Kuppen PJK, Vahrmeijer AL, Sier CFM. Molecular imaging of the urokinase plasminogen activator receptor: opportunities beyond cancer. EJNMMI Res. 2020;10:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duriseti S, Goetz DH, Hostetter DR, LeBeau AM, Wei Y, Craik CS. Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signaling and migration. J Biol Chem. 2010;285:26878–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwasaki K, Uno Y, Utoh M, Yamazaki H. Importance of cynomolgus monkeys in development of monoclonal antibody drugs. Drug Metab Pharmacokinet. 2019;34:55–63. [DOI] [PubMed] [Google Scholar]
- 29.Winters A, McFadden K, Bergen J, Landas J, Berry KA, Gonzalez A, Salimi-Moosavi H, Murawsky CM, Tagari P, King CT. Rapid single B cell antibody discovery using nanopens and structured light. mAbs. 2019;11:1025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedrioli A, Oxenius A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021;42:1143–58. [DOI] [PubMed] [Google Scholar]
- 31.Yeku O, Frohman MA. Rapid amplification of cDNA ends (RACE). Methods Mol Biol. 2011;703:107–22. [DOI] [PubMed] [Google Scholar]
- 32.Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, Cabaret V, Fermeaux V, Bertheau P, Garnier J, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. 2019;51:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petricevic B, Laengle J, Singer J, Sachet M, Fazekas J, Steger G, Bartsch R, Jensen-Jarolim E, Bergmann M. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med. 2013;11:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forsström B, Bisławska Axnäs B, Rockberg J, Danielsson H, Bohlin A, Uhlen M, Mantis NJ. Dissecting antibodies with regards to linear and conformational epitopes. PLoS ONE. 2015;10:e0121673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKeage K, Perry CM. Trastuzumab: a review of its use in the treatment of metastatic breast cancer overexpressing HER2. Drugs. 2002;62:209–43. [DOI] [PubMed] [Google Scholar]
- 37.Mazzotta M, Krasniqi E, Barchiesi G, Pizzuti L, Tomao F, Barba M, Vici P. Long-term safety and real-world effectiveness of trastuzumab in breast cancer. J Clin Med. 2019;8:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J. Antibody-dependent cellular cytotoxicity (ADCC) activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3:1148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dean AQ, Luo S, Twomey JD, Zhang B. Targeting cancer with antibody-drug conjugates: promises and challenges. mAbs. 2021;13:1951427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Rohweder PJ, Ongpipattanakul C, Basu K, Bohn M-F, Dugan EJ, Steri V, Hann B, Shokat KM, Craik CS. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40:1060–9.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferraris GMS, Schulte C, Buttiglione V, De Lorenzi V, Piontini A, Galluzzi M, Podestà A, Madsen CD, Sidenius N. The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins. Embo J. 2014;33:2458–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LeBeau AM, Sevillano N, King ML, Duriseti S, Murphy ST, Craik CS, Murphy LL, VanBrocklin HF. Imaging the urokinase plasminongen activator receptor in preclinical breast cancer models of acquired drug resistance. Theranostics. 2014;4:267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gårdsvoll H, Ploug M. Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J Biol Chem. 2007;282:13561–72. [DOI] [PubMed] [Google Scholar]
- 44.Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): diagnostic, prognostic, and therapeutic applications. Front Oncol. 2018;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahmood N, Arakelian A, Khan HA, Tanvir I, Mazar AP, Rabbani SA. uPAR antibody (huATN-658) and zometa reduce breast cancer growth and skeletal lesions. Bone Res. 2020;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han C, Gunn GR, Marini JC, Shankar G, Han Hsu H, Davis HM. Pharmacokinetics and immunogenicity investigation of a human anti-interleukin-17 monoclonal antibody in non-naïve cynomolgus monkeys. Drug Metab Dispos. 2015;43:762–70. [DOI] [PubMed] [Google Scholar]
- 47.Derebe MG, Nanjunda RK, Gilliland GL, Lacy ER, Chiu ML. Human IgG subclass cross-species reactivity to mouse and cynomolgus monkey Fcγ receptors. Immunol Lett. 2018;197:1–8. [DOI] [PubMed] [Google Scholar]
- 48.Zost SJ, Gilchuk P, Chen RE, Case JB, Reidy JX, Trivette A, Nargi RS, Sutton RE, Suryadevara N, Chen EC, et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat Med. 2020;26(9):1422–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and Fc receptors to enhance ADCC by human NK cells. Cancers. 2021;13:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.St-Pierre F, Bhatia S, Chandra S. Harnessing natural killer cells in cancer immunotherapy: a review of mechanisms and novel therapies. Cancers. 2021;13:1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yogo R, Yamaguchi Y, Watanabe H, Yagi H, Satoh T, Nakanishi M, Onitsuka M, Omasa T, Shimada M, Maruno T, et al. The fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci Rep. 2019;9(1):11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang W, Chen Q. Antigen improves binding of IgGs to FcγRs in SPR analysis. Anal Biochem. 2022;640:114411. [DOI] [PubMed] [Google Scholar]
- 53.Sun Y, Izadi S, Callahan M, Deperalta G, Wecksler AT. Antibody–receptor interactions mediate antibody-dependent cellular cytotoxicity. J Biol Chem. 2021;297:100826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Acharya P, Tolbert WD, Gohain N, Wu X, Yu L, Liu T, Huang W, Huang CC, Kwon YD, Louder RK, et al. Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J Virol. 2014;88(21):12895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tolbert WD, Sherburn RT, Van V, Pazgier M. Structural basis for epitopes in the gp120 cluster a region that invokes potent effector cell activity. Viruses. 2019;11:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mielke D, Bandawe G, Pollara J, Abrahams MR, Nyanhete T, Moore PL, Thebus R, Yates NL, Kappes JC, Ochsenbauer C, et al. Antibody-dependent cellular cytotoxicity (ADCC)-mediating antibodies constrain neutralizing antibody escape pathway. Front Immunol. 2019;10:2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4:511–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pirazzoli V, Ferraris GMS, Sidenius N. Direct evidence of the importance of vitronectin and its interaction with the urokinase receptor in tumor growth. Blood. 2013;121:2316–23. [DOI] [PubMed] [Google Scholar]
- 59.Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269:32380–88. [PubMed] [Google Scholar]
- 60.Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ. Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J Cell Biol. 1996;134:1563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng G, Curriden SA, Hu G, Czekay RP, Loskutoff DJ. Plasminogen activator inhibitor-1 regulates cell adhesion by binding to the somatomedin B domain of vitronectin. J Cell Physiol. 2001;189:23–33. [DOI] [PubMed] [Google Scholar]
- 62.Madsen CD, Ferraris GMS, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. J Cell Biol. 2007;177:927–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferraro E, Drago JZ, Modi S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: state of the art and future directions. Breast Cancer Res. 2021;23:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vilhardt F, Nielsen M, Sandvig K, van Deurs B, Pfeffer SR. Urokinase-type plasminogen activator receptor is internalized by different mechanisms in polarized and nonpolarized madin–darby canine kidney epithelial cells. Mol Biol Cell. 1999;10:179–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cortese K, Sahores M, Madsen CD, Tacchetti C, Blasi F, Blagosklonny MV. Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS ONE. 2008;3:e3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noh H, Hong S, Huang S. Role of urokinase receptor in tumor progression and development. Theranostics. 2013;3:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem. 1987;262:4437–40. [PubMed] [Google Scholar]
- 68.Estreicher A, Wohlwend A, Belin D, Schleuning WD, Vassalli JD. Characterization of the cellular binding site for the urokinase-type plasminogen activator. J Biol Chem. 1989;264:1180–89. [PubMed] [Google Scholar]
- 69.Huai Q, Zhou A, Lin L, Mazar A P, Parry G C, Callahan J, Shaw D E, Furie B, Furie B C and Huang M. (2008). Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat Struct Mol Biol, 15(4), 422–423. 10.1038/nsmb.1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilmore JR, Jones DD, Allman D. Improved resolution of plasma cell subpopulations by flow cytometry. Eur J Immunol. 2017;47:1386–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, Madhusoodanan N, Kolesnikov A, Lopez R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022;50:W276–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar S, Stecher G, Li M, Knyaz C, Tamura K, Mega X, Battistuzzi FU. Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, CSC, upon reasonable request.