

Abstract
Muscle atrophy is debilitating and can be induced by several stressors. Unfortunately, there are no effective pharmacological treatment until now. MicroRNA (miR)-29b is an important target that we identified to be commonly involved in multiple types of muscle atrophy. Although sequence-specific inhibition of miR-29b has been developed, in this study, we report a novel small-molecule miR-29b inhibitor that targets miR-29b hairpin precursor (pre-miR-29b) (Targapremir-29b-066 [TGP-29b-066]) considering both its three-dimensional structure and the thermodynamics of interaction between pre-miR-29b and the small molecule. This novel small-molecule inhibitor has been demonstrated to attenuate muscle atrophy induced by angiotensin II (Ang II), dexamethasone (Dex), and tumor necrosis factor α (TNF-α) in C2C12 myotubes, as evidenced by increase in the diameter of myotube and decrease in the expression of Atrogin-1 and MuRF-1. Moreover, it can also attenuate Ang II-induced muscle atrophy in mice, as evidenced by a similar increase in the diameter of myotube, reduced Atrogin-1 and MuRF-1 expression, AKT-FOXO3A-mTOR signaling activation, and decreased apoptosis and autophagy. In summary, we experimentally identified and demonstrated a novel small-molecule inhibitor of miR-29b that could act as a potential therapeutic agent for muscle atrophy.
Keywords: MT: Non-coding RNAs, miR-29b, muscle atrophy, Ang II, Dex, TNF-α, C2C12 myotubes, small-molecule inhibitor
Graphical abstract
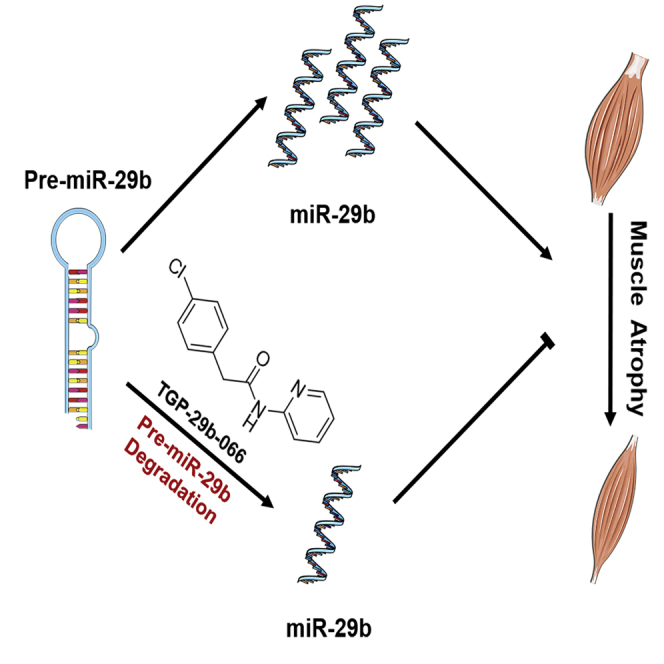
MicroRNA (miR)-29b contributes to muscle atrophy. Xiao and Xu et al. select and identify a novel small-molecule miR-29b inhibitor that specifically binds to pre-miR-29b and leads to the pre-miR-29b degradation. This inhibitor may be used to treat muscle atrophy.
Introduction
Muscle atrophy is an important source of disability and mobility that is caused by a reduction of protein synthesis along with increased protein degradation.1,2,3 It accompanies several diseases and conditions including inactivity, denervation, cancer, and heart failure. In spite of this, apart from exercise, muscle atrophy lacks any effective clinical intervention.4,5,6 Thus, developing novel therapeutics for muscle atrophy is the need of the hour.7,8
MicroRNA (miR)-29b has been identified to be commonly increased in muscle atrophy induced in the following conditions including dexamethasone (Dex), tumor necrosis factor α (TNF-α), and H2O2 in vitro as well as muscle atrophy induced by fasting, cancer cachexia, aging, denervation, immobilization, and angiotensin II (Ang II) in vivo.9,10 Also, a long non-coding RNA, long non-coding muscle-atrophy-associated transcript (lncMAAT), has been found to be a common regulator of muscle atrophy by negatively regulating the transcription of miR-29b through SOX6.11 Moreover, we have also established CRISPR-Cas9-based miR-29b editing as a therapy for several types of muscle atrophy in mice.12 Thus, inhibition of miR-29b could act as an attractive therapeutic strategy for muscle atrophy.9,12,13
Currently, methods using sequence-specific inhibition of miRNA (or miR) including antisense oligonucleotides, locked nucleic acids, antagomirs, and miRNA sponge are actively used by pharmaceutical industry for miRNA-based drug development.9,14,15,16,17,18 Here, we have identified a novel small-molecule miR-29b inhibitor that targets miR-29b hairpin precursor (pre-miR-29b) (Targapremir-29b-066 [TGP-29b-066]) by taking into account the three-dimensional structure as well as the thermodynamics of the interaction between pre-miR-29b and the small molecule. We found that this novel small-molecule inhibitor of miR-29b could attenuate muscle atrophy both in vitro and in vivo. Taken together, our results show that TGP-29b-066, a novel small-molecule inhibitor of miR-29b, could act as a promising therapy agent for muscle atrophy.
Results
Virtual and experimental screening for miR-29b inhibitor
miR-29b is an important therapeutic target for muscle atrophy.12,13 To identify potential compounds that can effectively inhibit miR-29b, we chose the compound that selectively and potently targets pre-miR-29b (TGP-29b) for miR-29b inhibition. Dozens of candidates with a variety of skeletons were screened virtually by checking the binding interaction between the candidate and the well-documented three-dimensional structure of pre-miR-29b. The candidates for screening mostly are N-containing heterocycles or their derivatives, including but not limited to pyridines, isoxazolines, quinolones, and azaindoles. Among these, 10 compounds were chosen as privileged compounds, according to the results of primary virtual screening (Figure 1A). The results are based on the scores obtained from two docking programs, GOLD and CDOCKER (Discovery Studio), respectively. Scores of the 10 best compounds are shown in Figures 1B and 1C.
Figure 1.

Compounds after first virtual screening
(A) Compounds that scored high according to virtual screening using GOLD and CDOCKER. (B) –CDOCKER_Energy of selected compounds. (C) Score of top-ranked 10 compounds according to GOLD.
Since the extent of miR-29b reduction was the intended endpoint, TGP-29b-066 was identified from various other compounds to effectively decrease miR-29b expression levels in C2C12 myoblast (Figure 2). The biological tests illustrated that aminopyridine might be an appropriate chemical functional moiety for further investigation. Inspired by this result, a second virtual screening was conducted with aminopyridine-derived compounds and other N-containing heterocycles, resulting in another 18 top-ranked compounds for in vitro biological tests (Figure 3A). Applying the method described before, scores of 18 chosen compounds were illustrated from GOLD and CDOCKER docking programs (Figures 3B and 3C). Among them, three compounds including TGP-29b-066, TGP-29b-054, and TGP-29b-281 were found to decrease miR-29b expression in C2C12 myoblast (Figure 4A). The effects of these three compounds were further verified in C2C12 myotubes, and we observed that TGP-29b-066 and TGP-29b-281 could decrease miR-29b expression without affecting miR-29a, miR-29c, and miR-30d (an unrelated miRNA) (Figures 4B and 4C). These results demonstrate that TGP-29b-066 and TGP-29b-281 can specifically inhibit miR-29b in C2C12 myotubes. To evaluate the efficacy of TGP-29b-066 and TGP-29b-281 treatment, miR-29b expression was tested by using qRT-PCR. TGP-29b-066 and TGP-29b-281 inhibited mature miR-29b generation in a dose-independent manner after 24 h of treatment (Figures S1A and S1B). Furthermore, TGP-29b-066 and TGP-29b-281 (30 μM) caused an approximate 50% inhibition after 24 h in C2C12 myotubes (Figures S1C and S1D). These data indicate that TGP-29b-066 and TGP-29b-281 could inhibit mature miR-29b generation.
Figure 2.

Screening for miR-29b inhibitor
qRT-PCR analysis of the miR-29b expression in C2C12 myoblasts treated with 10 compounds (n = 3). Data are presented as mean ± SD. Statistical significance was determined by Student t test. ∗∗p < 0.01.
Figure 3.

Compounds after second virtual screening
(A) Compounds that scored high according to virtual screening using GOLD and CDOCKER. (B) –CDOCKER_Energy of selected compounds. (C) Score of top-ranked 18 compounds according to GOLD.
Figure 4.

TGP-29b-066 and TGP-29b-281 decrease miR-29b specifically
(A) qRT-PCR analysis of the miR-29b expression in C2C12 myoblasts treated with 19 compounds (n = 4). (B) qRT-PCR analysis of the miR-29b expression in C2C12 myotubes treated with 3 selected compounds (n = 6). (C) qRT-PCR analysis of the expression of miR-29a, miR-29c, and miR-30d (an unrelated miRNAs) in C2C12 myotubes treated with 3 selected compounds (n = 6). Data are presented as mean ± SD. Statistical significance was determined by Student t test. ∗p < 0.05; ∗∗p < 0.01.
Compound synthesis and docking models
Compounds TGP-29b-066 and TGP-29b-281 could be synthesized according to previously published methods (Figure S2A).19 From the docking model, both of the two compounds have interactions with pre-miR-29b, including conventional hydrogen bond interactions (Figure S2B). These interactions are strong enough to show biological activity. In the case of TGP-29b-066, there are additional π-alkyl interactions (in pink) and attractive charge interactions (in brown). There is a π-π T-shaped interaction (in pink) between TGP-29b-281 and pre-miR-29b, which comes from two perpendicular aromatic rings attached to the bicyclic skeleton.
TGP-29b-066 attenuates muscle atrophy in vitro
Differentiated C2C12 myotubes were used to analyze the in vitro function of these two compounds in muscle atrophy. We assessed the influence of TGP-29b-066 and TGP-29b-281 in C2C12 myoblast differentiation firstly. By immunofluorescence staining assay, the number of MyHC-positive cells was not affected when C2C12 myoblasts were treated with TGP-29b-066 and TGP-29b-281 (Figure S3). Thus, TGP-29b-066 and TGP-29b-281 did not affect C2C12 differentiation. Then, in C2C12 myotubes treated with Ang II, TGP-29b-066 was used to ameliorate muscle atrophy. Ang II treatment decreased the diameter of myotube and elevated Atrogin-1 and MuRF-1 in C2C12 myotubes. TGP-29b-066 significantly attenuated muscle atrophy induced by Ang II (Figures 5A and 5B). Consistently, in C2C12 myotubes treated with Dex and TNF-α, TGP-29b-066 was observed to also increase the diameter of myotube and decrease Atrogin-1 and MuRF-1 expression levels (Figures 5C–5F). Collectively, in all three in vitro muscle atrophy models, TGP-29b-066 consistently attenuates muscle atrophy.
Figure 5.

TGP-29b-066 attenuates muscle atrophy in vitro
(A) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-066 in angiotensin II (Ang II)-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (B) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-066 in Ang II-induced muscle atrophy model (n = 6 per group). (C) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-066 in dexamethasone (Dex)-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (D) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-066 in Dex-induced muscle atrophy model (n = 6 per group). (E) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-066 in tumor necrosis factor α (TNF-α)-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (F) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-066 in TNF-α-induced muscle atrophy model (n = 6 per group). Green: MF-20; blue: DAPI. Data are presented as mean ± SD. Statistical significance was determined by two-way ANOVA with post hoc Tukey. ∗∗p < 0.01; ∗∗∗p < 0.001.
TGP-29b-281 attenuates muscle atrophy in vitro
Similarly, TGP-29b-281 could significantly attenuate muscle atrophy induced by Ang II in C2C12 myotubes, which was evidenced by increased diameter of myotube and decreased Atrogin-1 and MuRF-1 expression (Figures 6A and 6B). Besides, in C2C12 myotubes treated with Dex and TNF-α, TGP-29b-281 could also increase diameter of myotube and attenuate Atrogin-1 and MuRF-1 expression (Figures 6C–6F). Thus, in all three in vitro muscle atrophy models, TGP-29b-281 consistently attenuates muscle atrophy.
Figure 6.

TGP-29b-281 attenuates muscle atrophy in vitro
(A) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-281 in Ang II-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (B) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-281 in Ang II-induced muscle atrophy model (n = 6 per group). (C) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-281 in Dex-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (D) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-281 in Dex-induced muscle atrophy model (n = 6 per group). (E) Immunofluorescent staining and quantification of diameters of C2C12 myotubes treated with TGP-29b-281 in TNF-α-induced muscle atrophy model (n = 6 per group; scale bar: 100 μm). (F) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in C2C12 myotubes treated with TGP-29b-281 in TNF-α-induced muscle atrophy model (n = 6 per group). Green: MF-20; blue: DAPI. Data are presented as mean ± SD. Statistical significance was determined by two-way ANOVA with post hoc Tukey. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
After clarifying that TGP-29b-066 and TGP-29b-281 could attenuate muscle atrophy in vitro, we further explored whether TGP-29b-066 and TGP-29b-281 could treat established muscle atrophy in vitro. After being incubated with Ang II for 24 h, differentiated C2C12 myotubes were treated with TGP-29b-066 and TGP-29b-281 for another 48 h. We found that TGP-29b-066 and TGP-29b-281 treatment increased the diameter of myotube and decreased Atrogin-1 and MuRF-1 expression in Ang II-treated differentiated C2C12 myotubes (Figure S4). These data suggest that TGP-29b-066 and TGP-29b-281 can treat established muscle atrophy in vitro.
TGP-29b-066 ameliorates muscle atrophy in vivo
Based on the protective effects of TGP-29b-066 and TGP-29b-281 in muscle atrophy in vitro, we further investigated their effects in vivo. In the Ang II-induced muscle atrophy mice model, we treated mice with TGP-29b-066 or TGP-29b-281 by intraperitoneal injection (gastrocnemius muscles). We found that Ang II could significantly decrease grip strength and gastrocnemius weight, while TGP-29b-066 could increase grip strength and gastrocnemius weight, whereas TGP-29b-281 failed to have the same effects (Figures 7A–7C). In addition, TGP-29b-066 reduced the elevation of Atrogin-1 and MuRF-1 (Figures 7D and 7E). Furthermore, some other new “atroprotein” markers were also introduced to assess muscle atrophy.20 We found that Ndufb2 was downregulated and Clusterin was upregulated in Ang II-induced muscle atrophy, while TGP-29b-066 could rescue the changes (Figure 7F). Moreover, TGP-29b-066 attenuated the reduction of the cross-sectional area of myofibers in Ang II-treated mice (Figures 7G and S5A). Moreover, the downregulation of phosphorylation of AKT at serine 473 (Ser473), FOXO3A at Ser253, mTOR, P70S6K, and EIF-4EBP1 was reversed by TGP-29b-066 in Ang II-treated mice (Figure 7H). Additionally, TGP-29b-066 reduced cell apoptosis and autophagy in Ang II-treated mice (Figures S5B–S5D). As IGF-1 and PI3K(p85α) have been identified as target genes of miR-29b,9 we determined their expressions in TGP-29b-066-treated mice. We found that IGF-1 and PI3K(p85α) were downregulated in Ang II-treated mice, while TGP-29b-066 increased their expressions (Figure S5E).
Figure 7.

TGP-29b-066 attenuates muscle atrophy in vivo
(A) Protocol of the experiment. (B) The grip strength of right hindlimb of Ang II-treated mice injected with TGP-29b-066 and TGP-29b-281 (n = 7–8). (C) Gastrocnemius muscle morphology, gastrocnemius weight, and gastrocnemius weight/tibial length (G/TL) of Ang II-treated mice injected with TGP-29b-066 and TGP-29b-281 (n = 7–8). (D) qRT-PCR analysis for the expression of Atrogin-1 and MuRF-1 in Ang II-treated mice injected with TGP-29b-066 (n = 7–8). (E) Western blot analysis of the protein expression of Atrogin-1 and MuRF-1 in Ang II-treated mice injected with TGP-29b-066 (n = 6). (F) Western blot analysis of the protein expression of Ndufb2 and Clusterin in Ang II-treated mice injected with TGP-29b-066 (n = 6). (G) WGA staining showed the cross-sectional area of myofibers of Ang II-treated mice injected with TGP-29b-066 (n = 6, scale bar: 50 μm), Red, WGA; blue, DAPI. (H) Western blot analysis for the protein expression of phosphorylation of AKT at Ser473, FOXO3A at Ser253, mTOR, P70S6K, and EIF-4EBP1 in Ang II-treated mice injected with TGP-29b-066 (n = 6). Data are presented as mean ± SD. Statistical significance was determined by two-way ANOVA with post hoc Tukey. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
TGP-29b-066 represses miR-29b generation via interfering with the stability of pre-miR-29b
To confirm how TGP-29b-066 represses the abundance of miR-29b, we firstly detected whether TGP-29b-066 represses the abundance of direct targets pre-miR-29b. qRT-PCRs showed that TGP-29b-066 significantly repressed the expression of pre-miR-29b-1/2 (Figure 8A). Then, we performed differential scanning fluorimetry (DSF) to identify the direct interaction between TGP-29b-066 and pre-miR-29b-1/2. Results from DSF revealed that TGP-29b-066 could change the DSF patterns in that the addition of TGP-29b-066 lowered the melting temperature (Tm) of pre-miR-29b-1/2 (Figure 8B). Our data also showed that the direct binding between TGP-29b-066 and pre-miR-29b-1/2 led to inhibition. Besides, we further determined whether TGP-29b-066 affected the stability of pre-miR-29b-1/2. TGP-29b-066-treated C2C12 myotubes were detected at different time points upon transcriptional inhibition with actinomycin D. qRT-PCRs showed that TGP-29b-066 addition led to shortened RNA lifetimes of pre-miR-29b-1/2 and miR-29b (Figure 8C). These data demonstrated that TGP-29b-066 could directly bind to pre-miR-29b-1/2, which in turn reduced pre-miR-29b-1/2 stability. In addition, we compared the inhibition efficiency of TGP-29b-066 and antisense oligodeoxynucleotides of miR-29b (ASO-miR-29b) and found that TGP-29b-066 had a longer inhibitory effect than ASO-miR-29b (Figure 8D).
Figure 8.

TGP-29b-066 decreases RNA stability of pre-miR-29b-1/2
(A) qRT-PCR analysis of the pre-miR-29b-1/1 expression in C2C12 myotubes treated with TGP-29b-066 for 24 h at a dose of 30 μM (n = 6). (B) Differential scanning fluorimetry (DSF) analysis of the fluorescence intensity change after pre-miR-29b-1/2 incubated with TGP-29b-066 for 10 min (n = 4). (C) qRT-PCR analysis for the expression of pre-miR-29b-1/2 and miR-29b in TGP-29b-066 incubated C2C12 myotubes when treated with actinomycin D (Act D) for 0, 4, 8, and 12 h (n = 3 per group). (D) qRT-PCR of analysis the miR-29b expression in C2C12 myotubes treated with TGP-29b-066 and ASO-miR-29b at different time points (n = 6). ∗∗ and ∗∗∗, vs. with TGP-29b-066 treated for 0 h; #, vs. with ASO-miR-29b treated for 0 h; &&&, vs. TGP-29b-066 treated with ASO-miR-29b treated for 120 h. Data are presented as mean ± SD. Statistical significance was determined by Student t test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Discussion
Aging and many chronic diseases including heart failure, cancer cachexia, etc., show a common complication called muscle atrophy.21,22 Due to its complex etiology, exploring the common regulator of many types of muscle atrophy is necessary. Using transcript profiling, some atrogenes such as Fbxo32 (atrogin-1/MAFbx), Trim63 (MuRF1), and Fbxo30 (MUSA) have been found to be upregulated in muscle atrophy induced by diverse stimuli.23,24 Recently, some new atroproteins were also identified by transcriptome-proteome disconnect in distinct atrophic stimuli (corticosteroids, cancer cachexia, and aging). Protein levels of Cyr61, clusterin, Apod, Clu, Fhad1, and Pla2g7 were increased in these muscle atrophy models, whereas protein levels of Ndufb2 were reduced.20 These atrogenes and atroproteins may be used as markers or antiatrophy interventions for muscle atrophy induced by diverse stimuli. In addition, miRNAs are powerful regulators of gene expression and play essential roles in many biological processes including cell proliferation, differentiation, and apoptosis.25,26 miRNAs play a critical role in maintaining tissue homeostasis such as in skeletal muscles and participate in many diseases including heart failure and muscle atrophy.13,25,27 Some miRNAs have been reported in a single type of muscle atrophy: e.g., miR-21, miR-181a, and miR-432 in aging-induced muscle atrophy; miR-450a-5p, miR-450b-5p, miR-424-5p, miR-424-3p, miR-199a, miR-140, miR-147-3p, miR-511-3p, miR-223-3p, and miR-205-5p in cancer cachexia-induced muscle atrophy; and miR-1, miR-499, miR-133, miR-206, miR-27a, and miR-675 in chronic obstructive pulmonary disease (COPD)-induced muscle atrophy.13 Interestingly, we report miR-29b as a common regulator of many types of muscle atrophy including denervation, Dex, fasting, cancer cachexia, aging, and Ang II.9,10 In this study, we provide a novel inhibitor of this miRNA that can specifically downregulate miR-29b and ameliorate Ang II-induced muscle atrophy in mice.
Currently, there are three types of commercially available miR-29b inhibitors, namely antisense oligonucleotides, locked nucleic acids, and antagomirs.15,16 However, all of them are specially modified oligonucleotides that are derived from complicated structures and are hard to be further modified.15 Thus, it is of great value to design and synthesize small organic molecules as alternatives to miR-29b inhibitors. The core skeleton could be easily synthesized through a previously developed method, leading to diverse synthesis of molecules that compose the compound library. Both of the screenings were performed through docking optimization by GOLD and CDOCKER (Discovery Studio), followed by final scoring by calculating binding energies, which concluded interactions between aminopyridine- or isoxazoline-derived compounds and miR-29b. Here, we identified a novel small molecule TGP-29b-066 as a miR-29b inhibitor that could be easily synthesized via condensation of commercially available amine and carboxylic acid in high yield.
miR-29b is a promising therapeutic target of many types of muscle atrophy.9 In addition, we found that miR-29b was a key mediator for the protective effects of exercise in Ang II-induced muscle atrophy.5 Besides, we identified a lncRNA (lncMAAT) as a regulator of many types of muscle atrophy that negatively regulates the transcription of miR-29b through SOX6 by a trans-regulatory module.11 Moreover, we developed a novel CRISPR-based genome editing in miR-29b that can target miR-29b specifically and can effectively prevent several types of muscle atrophy including Ang II-induced muscle atrophy.12 Although no clinical trials have been conducted in muscle atrophy based on miRNAs, several clinical trials are ongoing in other diseases by targeting miRNAs, e.g., anti-miR-92a and anti-miR-132.28 The novel small molecule TGP-29b-066 reported in the present study considered both the three-dimensional structure of miR-29b and the thermodynamics of interactions between miR-29b and the small molecule. As a pilot compound, TGP-29b-066 will pave a way for developing more effective small molecules for miR-29b inhibition and, therefore, be used as a potential therapy for muscle atrophy.
Intraperitoneal injection is one of the most commonly used injection methods in animal experiments. To evaluate the pro-atrophy of compounds in mice, we also choose intraperitoneal injection in our animal experiments. As miR-29b has been reported to be involved in many specific functions in different organs, systemic suppression of miR-29b via intraperitoneal injection may not originally be the best choice for muscle atrophy therapy. Intramuscular injection might be an optimized administration route of TGP-29b-066 in muscle atrophy therapy. This injection method is widely used for precise intervention of muscle diseases, such as Duchenne.29,30,31,32
In conclusion, our study identified a novel small-molecule inhibitor of miR-29b that attenuates muscle atrophy, which provided a pilot compound to combat muscle atrophy.
Materials and methods
Molecular modeling
Pre-miRNA sequence of miR-29b was obtained from miRBase database. The hairpin loop of miR-29b was chosen to predict the three-dimensional structure using the MC-Fold/MC-Sym pipeline, which was widely used for the prediction of RNA secondary and tertiary structures. After the RNA sequence was uploaded to MC-Fold, the resulting secondary structures were directly used as input for MC-Sym to afford the corresponding tertiary structures.
Docking studies
The predicted three-dimensional structure of miR-29b was then used for docking studies. To prepare the receptor structure, polar hydrogen atoms were added, while the energy was minimized using the CHARMm force field. The compound library was built by our group (Prof. Bin Xu) and contains more than 3,000 chemical structures. All the compounds were also prepared before docking. After filtering compounds by the Lipinski’s Rule of Five, two different docking programs, GOLD and CDOCKER (Discovery Studio), were employed successively to conduct docking-based virtual screening. Protein-ligand interactions were analyzed and visualized using Gold.Goldscore.Fitness and -CDOCKER_ENERGY.
Procedure of virtual screening
The structure-based virtual screening was carried out using GOLD and CDOCKER (Discovery Studio) after preparing the three-dimensional structure of miR-29b. The binding site was formed from receptor cavities. In the first selection step, the GOLD program was utilized to calculate the binding mode of miR-29b and ligands. The GA parameter was set as automatic or default, while other parameters were set to their default values, and Gold.Goldscore.Fitness was employed for ranking molecules. Subsequently, the CDOCKER program was used as the secondary filter to prioritize the compounds. Docking was performed with all the parameters set to their default values. The top-ranked docking conformations were determined using -CDOCKER_ENERGY.
Compounds
All reagents and compounds were purchased commercially without further purification unless noted. Compounds TGP-29b-230, TGP-29b-282, TGP-29b-286, TGP-29b-291, TGP-29b-293, TGP-29b-318, TGP-29b-345, TGP-29b-347, TGP-29b-348, TGP-29b-349, TGP-29b-705,19 TGP-29b-259, TGP-29b-266, TGP-29b-316, TGP-29b-558,25 TGP-29b-553, TGP-29b-555, TGP-29b-666, TGP-29b-273, TGP-29b-402,33 TGP-29b-056,34 and TGP-29b-06835 were synthesized according to reported or modified methods.
Synthetic procedure access to TGP-29b-066 and TGP-29b-281

2-(4-chlorophenyl)-N-(pyridin-2-yl)acetamide (TGP-29b-066)
To a solution of the 2-(4-chlorophenyl)acetic acid (853 mg, 5 mmol) and dicyclohexylcarbodiimide (DCC) (1.24 g, 6 mmol) in CH2Cl2 (6 mL) was added pyridin-2-amine (564.7 mg, 6 mmol) at room temperature. Upon the completion of reaction monitored by thin layer chromatography (TLC), the mixture was filtered through a thin pad of Celite, and the filtrate was concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate = 8:1) to give product as a pale-yellow solid (1.14 g, 93%). 1H nuclear magnetic resonance (NMR) (500 MHz, CDCl3): δ 8.23 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H); 8.20 (d, J = 8.3 Hz, 1H); 7.98 (s, 1H); 7.73–7.67 (m, 1H); 7.39–7.32 (m, 2H); 7.30–7.25 (m, 2H); 7.03 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H); and 3.72 (s, 2H). The data are consistent with a previous report.36

Butyl 6-hydroxy-4-oxo-5-phenyl-3a,5,6,6a-tetrahydro-4H-pyrrolo[3,4-d]isoxazole -3-carboxylate (TGP-29b-281)
To a test tube, butyl acrylate (43 μL, 0.3 mmol), 1-phenyl-1H-pyrrole-2,5-dione (207.6 mg, 1.2 mmol), Cu(NO3)2·3H2O (290 mg, 1.2 mmol), KI (49.8 mg, 0.3 mmol), B(OH)3 (37.1 mg, 0.6 mmol), and CH3CN/PhCN (2:1, v/v, 3.0 mL) were added. The reaction was stirred at 80°C under air. Upon completion as monitored by TLC, the reaction mixture was cooled down to room temperature, quenched with ammonium hydroxide, and extracted with ethyl acetate (3 × 10 mL). The combined organic phase was washed with water and brine and dried over anhydrous Na2SO4. After filtration through a thin pad of Celite, the filtrate was evaporated under reduced pressure to give the crude product, which was further purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate = 10:1, v/v) to give a pale-yellow solid (55.4 mg, 58%). 1H NMR (CDCl3, 500 MHz): δ 7.49–7.34 (m, 3H); 7.20 (d, J = 7.0 Hz, 2H); 5.68 (d, J = 10.0 Hz, 1H); 4.87 (d, J = 10.0 Hz, 1H); 4.39–4.21 (m, 2H); 1.78–1.64 (m, 2H); 1.48–1.33 (m, 2H); and 0.93 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz): δ 170, 168.7, 158.6, 147.8, 130.7, 129.4, 129.3, 126.3, 82.4, 66.9, 53.9, 30.3, 19, and 13.6.
To a flask, isoxazoline (63.2 mg, 0.2 mmol) and dichloromethane/EtOH (4:1, v/v, 5 mL) were added. Then, to the mixture, sodium borohydride (9.1 mg, 0.24 mmol) was added and further dissolved in EtOH (1 mL) dropwise at −80°C during 0.5 h. The mixture was strictly controlled between −80°C and −70°C for 1.5 h. Upon completion, the reaction mixture was warmed to room temperature, quenched with saturated NH4Cl aqueous solution (5 mL), and extracted with dichloromethane (3 × 10 mL). The combined organic phase was washed with brine and dried over Na2SO4. After filtration through a thin pad of Celite, the filtrate was evaporated under reduced pressure to give the crude product, which was further purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 3:1, v/v) to give the product as a pale-yellow solid (61.2 mg, 96%). 1H NMR (CDCl3, 500 MHz): δ 7.44–7.22 (m, 5H); 5.59–5.44 (m, 1H); 5.19 (d, J = 9.0 Hz, 1H); 5.06 (d, J = 9.5 Hz, 1H); 4.74 (d, J = 9.0 Hz, 1H); 4.30 (t, J = 7.0 Hz, 2H); 1.78–1.63 (m, 2H); 1.49–1.35 (m, 2H); and 0.94 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz): δ 167.6, 159.3, 149.2, 135.6, 129.3, 127.6, 124.8, 88.7, 88.6, 66.6, 54.3, 30.4, 19, and 13.7. The data obtained are consistent with a previous report.19
Cell culture and treatment
C2C12 myoblasts were maintained in Dulbecco’s modified Eagle’s medium mixture with 10% fetal bovine serum and 1% penicillin-streptomycin, and cells were cultured in a 37°C incubator with 5% CO2. To obtain myotubes, C2C12 myoblasts cells were cultured in differentiation medium (Dulbecco’s modified Eagle’s medium [DMEM] containing 2% horse serum and 1% penicillin and streptomycin), and the full differentiation myotubes were obtained after differentiation for 4 days.
To induce muscle atrophy in vitro, Dex (50 mM for 24 h), Ang II (500 nM for 48 h), and TNF-α (100 ng/mL for 48 h) were used to incubate with C2C12 myotubes as described previously.9,10 To evaluate the function of compounds (TGP-29b-066 and TGP-29b-281) in muscle atrophy in vitro, the compounds were added into the C2C12 myotube at a dose of 30 μΜ, and then muscle atrophy was induced. To evaluate the therapeutic function of compounds (TGP-29b-066 and TGP-29b-281), Ang II (500 nM) was used to induce muscle atrophy model for 24 h, and these compounds were added to the model at the dose of 30 μΜ for another 48 h.
Immunofluorescence staining
C2C12 myotubes were fixed in 4% paraformaldehyde (PFA) and permeabilized with 0.5% Triton, then blocked with 5% BSA at room temperature. After that, cells were incubated with primary antibody MF-20 (DSHB, 1:100) and secondary antibody FITC-AffiniPure rabbit antimouse immunoglobulin G (IgG) (heavy chain + light chain [H + L]) (Jackson ImmunoResearch, 1:200), continuously. Images were acquired by fluorescence microscope (20× magnification) (Leica). The diameter of the myotube was measured using ImageJ software in randomly selected fields of view.
Animal studies
All animal experiments were conducted under the guidelines approved by the committee on the Ethics of Animal Experiments of Shanghai University. Male C57BL/6J mice at 8–10 weeks old were purchased from Charles River (Beijing, China). All mice were maintained in a specific-pathogen-free (SPF) laboratory animal facility of Shanghai University (Shanghai, China).
All procedures conform to the regulations detailed in the National Institutes of Health Guide for the Care and Use of Laboratory Animals (no. 85-23, revised 1996). Animal groups received either PBS or inhibitor intraperitoneally at the dose of 20 mg/kg every day. Then, Ang II-induced muscle atrophy model was achieved through implanting a pump (Alzet 2001) containing 2 μg/μL Ang II into the back of mice. The same was achieved for the control mice by implanting a pump containing PBS, and they received the same amount of food as the experimented mice.
All mice were sacrificed 1 week later, and muscles were dissected and rapidly frozen in liquid nitrogen for subsequent analyses.
RT and quantitative real-time PCRs
Total RNA from cells and skeletal muscle were isolated and purified with Qiagen RNeasy Mini Kit (Qiagen). 300 ng RNA was used to reverse transcribe into cDNA by SuperScript First-Strand Synthesis System (Thermo Fisher Scientific). Quantitative real-time PCR was performed with a LightCycler 480 instrument (Roche) using TB Green Premix Ex Taq II (Takara). The BulgeLoop miRNA qPCR Primer Set (RiboBio) was used to determine the expression levels of miRNAs. The expression of Atrogin-1 and MuRF-1 were normalized to 18S rRNA, while the expression of miR-29b, miR-29a, miR-29c, miR-30d, and pre-miR-29b-1/2 were normalized to 5S rRNA. 10 ng cDNA was applied in a PCR reaction. The relative expression level of miRNA and gene was calculated using the 2−ΔΔct method. The primer sequences used are as follows: mmu-Atrogin-1 forward: 5′-CAGCTTCGTGAGCGACCTC-3′; mmu-Atrogin-1 reverse: 5′-GGCAGTCGAGAAGTCCAGTC-3′; mmu-MuRF-1 forward: 5′-GTGTGAGGTGCCTACTTGCTC-3′; mmu-MuRF-1 reverse: 5′-GCTCAGTCTTCTGTCCTTGGA-3′; mmu-18S forward: 5′-TCAAGAACGAAAGTCGGAGG-3′; mmu-18S reverse: 5′-GGACATCTAAGGGCATCAC-3′.
Western blot
Total protein was extracted from skeletal muscle by the protein extraction Kit (KeyGEN). Protein concentration was determined with BCA Protein Assay Kit (Takara), and an equal amount of protein was separated on SDS-polyacrylamide gel electrophoresis. After transferring to a polyvinylidene fluoride (PVDF) membrane (Millipore), the primary antibody and the corresponding secondary antibody were used to detect protein of interest. The antibody used in this study were as follows: Atrogin-1 (Abclonal); MuRF-1 (Abclonal); LC3 (Abclonal); P62 (Abclonal); Ndufb2 (Abclonal); Clusterin (Abclonal); IGF1 (Abclonal); PI3K(p85α) (Abclonal); P-mTOR (Abclonal); mTOR (Abclonal); P-P70S6K (Abclonal); P70S6K (Abclonal); P-FOXO3A (Abclonal); FOXO3A (Abclonal); P-EIF4EBP1 (Abclonal); EIF4EBP1 (Abclonal); P-AKT (Cell Signaling Technology); AKT (Proteintech); and GAPDH (Bioworld Technology). The protein was visualized using High-sig ECL Western Blotting Substrate (Tanon) and imaged using the Tanon-5200S Chemiluminescent Imaging System (Tanon).
Staining
Hematoxylin and eosin (H&E)-stained and wheat germ agglutinin (WGA) staining were used to determine the cross-sectional area of myofibers. 10 μm thickness paraffin sections were subjected to H&E staining using an H&E staining kit (KeyGEN) according to published protocols. 10 μm cryosections were subjected to WGA staining using standard protocols. WGA (1:100; Sigma) and Hoechst 33342 (1:2000, KeyGEN) were used to label the cell membrane and nucleus, separately. The images of the sections were taken with microscope Leica (Wetzlar, Germany, DM3000). Cross-sectional areas of myofibers were measured by using ImageJ software.
TdT mediated dUTP nick end labeling (TUNEL) staining was used to detect and quantitate apoptotic myofibers. 10 μm thick frozen sections were subjected to DeadEnd Fluorometric TUNEL System (Promega) according to standard protocols. Images were taken by Confocal Imaging Microscope System (20× magnification) (Olympus). TUNEL-positive cells (FITC) and all cells (DAPI) were counted in a slide.
DSF
The DSF was performed in a total reaction volume of 6 μL containing 1 mM pre-miR-29b-1 (or pre-miR-29b-2), 1×SYBR Green Dye (Takara), 2% DMSO, and buffer (0.25 mM Tris-HCl and 0.25 mM NaCl) in a 384-well white polypropylene reaction plate (Roche) and treated with 10 μM TGP-29b-066. DSF analysis was performed by Roche LightCycler 480 Instrument II by using the following parameters: manual mode with integration time set at 0.75 s, continuous acquisition mode up to 99°C, ramp rate of 0.11°C/s, and 5 acquisitions per °C. Raw data were imported into GraphPad Prism 8.0 for analysis and high-resolution figures.
RNA stability assay
After incubation with TGP-29b-066 for 48 h, C2C12 myotubes were treated with 10 μg/mL actinomycin D (Sigma). Then, the cells were harvested after 0, 4, 8, and 12h. The total RNA was extracted and quantified, and the expressions of pre-miR-29b-1/2 and miR-29b were analyzed by qRT-PCR.
Quantification and statistical analysis
All data were analyzed by SPSS 20.0 and presented as mean ± SD using GraphPad Prism 8.0. An unpaired, two-tailed Student’s t test was used for comparisons between the two groups. Two-way ANOVA with Tukey test was performed to compare multiple groups. Differences were considered significant with p <0.05.
Acknowledgments
This work was supported by the National Key R&D Program of China (2020YFA0803800 to J.L.); National Natural Science Foundation of China (82020108002 and 82225005 to J.X., 21871174 to B.X., and 81900359 and 82271623 to J.L.); Innovation Program of Shanghai Municipal Education Commission (2019-01-07-00-09-E00008 to B.X.); grants from Science and Technology Commission of Shanghai Municipality (20DZ2255400 and 21XD1421300 to J.X. and 22010500200 to J.L.); the “Dawn” Program of Shanghai Education Commission (19SG34 to J.X.); the Shanghai Sailing Program (19YF1416400 to J.L.); the “Chen Guang” project supported by the Shanghai Municipal Education Commission; and and Shanghai Education Development Foundation (19CG45 to J.L.).
Author contributions
Q.L., W.Y., and Y.Y. conducted experiments. B.J., M.Y., T.L., M.G., and J.L. acquired and analyzed the data. P.G., G.V., and G.L. contributed to the helpful discussion. J.X. and B.X. directed the project and designed the research studies and wrote and edited the manuscript. All authors contributed to the editing of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.02.003.
Contributor Information
Bin Xu, Email: xubin@shu.edu.cn.
Junjie Xiao, Email: junjiexiao@shu.edu.cn.
Supplemental information
Data availability
All data generated or analyzed during this study are included in this published article (and its supplemental information files).
References
- 1.Bamba R., Okamura T., Hashimoto Y., Majima S., Senmaru T., Ushigome E., Nakanishi N., Asano M., Yamazaki M., Takakuwa H., et al. Extracellular lipidome change by an SGLT2 inhibitor, luseogliflozin, contributes to prevent skeletal muscle atrophy in db/db mice. J. Cachexia Sarcopenia Muscle. 2022;13:574–588. doi: 10.1002/jcsm.12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song Y.H., Li Y., Du J., Mitch W.E., Rosenthal N., Delafontaine P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Invest. 2005;115:451–458. doi: 10.1172/JCI22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X.H., Mitch W.E., Price S.R. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat. Rev. Nephrol. 2022;18:138–152. doi: 10.1038/s41581-021-00498-0. [DOI] [PubMed] [Google Scholar]
- 4.Feng L., Li B., Xi Y., Cai M., Tian Z. Aerobic exercise and resistance exercise alleviate skeletal muscle atrophy through IGF-1/IGF-1R-PI3K/Akt pathway in mice with myocardial infarction. Am. J. Physiol. Cell Physiol. 2022;322:C164–C176. doi: 10.1152/ajpcell.00344.2021. [DOI] [PubMed] [Google Scholar]
- 5.Liu Q., Chen L., Liang X., Cao Y., Zhu X., Wang S., Li J., Gao J., Xiao J. Exercise attenuates angiotensin-induced muscle atrophy by targeting PPARgamma/miR-29b. J. Sport Health Sci. 2022;11:696–707. doi: 10.1016/j.jshs.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C., Wang P., Du J., Chen J., Liu W., Ye K. LncRNA RAD51-AS1/miR-29b/c-3p/NDRG2 crosstalk repressed proliferation, invasion and glycolysis of colorectal cancer. IUBMB Life. 2021;73:286–298. doi: 10.1002/iub.2427. [DOI] [PubMed] [Google Scholar]
- 7.Rong S., Wang L., Peng Z., Liao Y., Li D., Yang X., Nuessler A.K., Liu L., Bao W., Yang W. The mechanisms and treatments for sarcopenia: could exosomes be a perspective research strategy in the future? J. Cachexia Sarcopenia Muscle. 2020;11:348–365. doi: 10.1002/jcsm.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen C., Zhou J., Wang X., Yu X.Y., Liang C., Liu B., Pan X., Zhao Q., Song J.L., Wang J., et al. Angiotensin-II-induced muscle wasting is mediated by 25-hydroxycholesterol via GSK3beta signaling pathway. EBioMedicine. 2017;16:238–250. doi: 10.1016/j.ebiom.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J., Chan M.C., Yu Y., Bei Y., Chen P., Zhou Q., Cheng L., Chen L., Ziegler O., Rowe G.C., et al. miR-29b contributes to multiple types of muscle atrophy. Nat. Commun. 2017;8:15201. doi: 10.1038/ncomms15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J., Yang T., Sha Z., Tang H., Hua X., Wang L., Wang Z., Gao Z., Sluijter J.P.G., Rowe G.C., et al. Angiotensin II-induced muscle atrophy via PPARgamma suppression is mediated by miR-29b. Mol. Ther. Nucleic Acids. 2021;23:743–756. doi: 10.1016/j.omtn.2020.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Yang T., Tang H., Sha Z., Chen R., Chen L., Yu Y., Rowe G.C., Das S., Xiao J. Inhibition of lncRNA MAAT controls multiple types of muscle atrophy by cis- and trans-regulatory actions. Mol. Ther. 2021;29:1102–1119. doi: 10.1016/j.ymthe.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia H., Li Z., Chang Y., Fang B., Zhou Y., Ma H. Downregulation of long noncoding RNA TUG1 attenuates MTDH-mediated inflammatory damage via targeting miR-29b-1-5p after spinal cord ischemia reperfusion. J. Neuropathol. Exp. Neurol. 2021;80:254–264. doi: 10.1093/jnen/nlaa138. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q., Deng J., Qiu Y., Gao J., Li J., Guan L., Lee H., Zhou Q., Xiao J. Non-coding RNA basis of muscle atrophy. Mol. Ther. Nucleic Acids. 2021;26:1066–1078. doi: 10.1016/j.omtn.2021.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herkt M., Thum T. Pharmacokinetics and proceedings in clinical application of nucleic acid therapeutics. Mol. Ther. 2021;29:521–539. doi: 10.1016/j.ymthe.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C.K., Kafert-Kasting S., Thum T. Preclinical and clinical development of noncoding RNA therapeutics for cardiovascular disease. Circ. Res. 2020;126:663–678. doi: 10.1161/CIRCRESAHA.119.315856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Z., Zhang J., Qian X., Han L., Zhang K., Chen L., Liu J., Ren Y., Yang M., Zhang A., et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013;73:5519–5531. doi: 10.1158/0008-5472.CAN-13-0280. [DOI] [PubMed] [Google Scholar]
- 17.Singh A., Dashynam M., Chim B., Escobar T.M., Liu X., Hu X., Patnaik S., Xu X., Southall N., Marugan J., et al. Identification of small molecule inhibitors of a Mir155 transcriptional reporter in Th17 cells. Sci. Rep. 2021;11:11498. doi: 10.1038/s41598-021-90944-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Chen R., Zheng Y., Yuan W., Yang T., Zhu X., Yan Y., Jin B., Xu W., Zhang Z., et al. Engineered circular RNA CircmiR-29b attenuates muscle atrophy by sponging MiR-29b. Adv. Ther. 2022;5:2200029. doi: 10.1002/adtp.202200029. [DOI] [Google Scholar]
- 19.Gao M., Gan Y., Xu B. From alkenes to isoxazolines via copper-mediated alkene cleavage and dipolar cycloaddition. Org. Lett. 2019;21:7435–7439. doi: 10.1021/acs.orglett.9b02748. [DOI] [PubMed] [Google Scholar]
- 20.Hunt L.C., Graca F.A., Pagala V., Wang Y.D., Li Y., Yuan Z.F., Fan Y., Labelle M., Peng J., Demontis F. Integrated genomic and proteomic analyses identify stimulus-dependent molecular changes associated with distinct modes of skeletal muscle atrophy. Cell Rep. 2021;37:109971. doi: 10.1016/j.celrep.2021.109971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Pan X., Sun Y., Geng Y.J., Yu X.Y., Li Y. The molecular mechanisms and prevention principles of muscle atrophy in aging. Adv. Exp. Med. Biol. 2018;1088:347–368. doi: 10.1007/978-981-13-1435-3_16. [DOI] [PubMed] [Google Scholar]
- 22.Zhou J., Liu B., Liang C., Li Y., Song Y.H. Cytokine signaling in skeletal muscle wasting. Trends Endocrinol. Metab. 2016;27:335–347. doi: 10.1016/j.tem.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K., et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 24.Sartori R., Schirwis E., Blaauw B., Bortolanza S., Zhao J., Enzo E., Stantzou A., Mouisel E., Toniolo L., Ferry A., et al. BMP signaling controls muscle mass. Nat. Genet. 2013;45:1309–1318. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- 25.Bär C., Chatterjee S., Falcão Pires I., Rodrigues P., Sluijter J.P.G., Boon R.A., Nevado R.M., Andrés V., Sansonetti M., de Windt L., et al. Non-coding RNAs: update on mechanisms and therapeutic targets from the ESC working groups of myocardial function and cellular biology of the heart. Cardiovasc. Res. 2020;116:1805–1819. doi: 10.1093/cvr/cvaa195. [DOI] [PubMed] [Google Scholar]
- 26.Jaé N., Dimmeler S. Noncoding RNAs in vascular diseases. Circ. Res. 2020;126:1127–1145. doi: 10.1161/CIRCRESAHA.119.315938. [DOI] [PubMed] [Google Scholar]
- 27.Bei Y., Xiao J. MicroRNAs in muscle wasting and cachexia induced by heart failure. Nat. Rev. Cardiol. 2017;14:566. doi: 10.1038/nrcardio.2017.122. [DOI] [PubMed] [Google Scholar]
- 28.Das S., Shah R., Dimmeler S., Freedman J.E., Holley C., Lee J.M., Moore K., Musunuru K., Wang D.Z., Xiao J., et al. Noncoding RNAs in cardiovascular disease: current knowledge, tools and technologies for investigation, and future directions: a scientific statement from the American heart association. Circ. Genom. Precis. Med. 2020;13:e000062. doi: 10.1161/HCG.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 29.Malerba A., Thorogood F.C., Dickson G., Graham I.R. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum. Gene Ther. 2009;20:955–965. doi: 10.1089/hum.2008.157. [DOI] [PubMed] [Google Scholar]
- 30.Schaakxs D., Wiberg M., Kingham P.J., Kalbermatten D.F. Intramuscular stem cell injection in combination with bioengineered nerve repair or nerve grafting reduces muscle atrophy. Plast. Reconstr. Surg. 2022;149:905e–913e. doi: 10.1097/PRS.0000000000009031. [DOI] [PubMed] [Google Scholar]
- 31.Mann C.J., Honeyman K., Cheng A.J., Ly T., Lloyd F., Fletcher S., Morgan J.E., Partridge T.A., Wilton S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA. 2001;98:42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vulin A., Barthélémy I., Goyenvalle A., Thibaud J.L., Beley C., Griffith G., Benchaouir R., le Hir M., Unterfinger Y., Lorain S., et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. 2012;20:2120–2133. doi: 10.1038/mt.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goswami S., Ghosh K., Dasgupta S. Troger's base molecular scaffolds in dicarboxylic acid recognition. J. Org. Chem. 2000;65:1907–1914. doi: 10.1021/jo9909204. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein J., Stearns B., Dexter M., Lott W.A. Derivatives of aminopyridines. J. Am. Chem. Soc. 1947;69:1147–1150. doi: 10.1021/ja01197a047. [DOI] [PubMed] [Google Scholar]
- 35.Berger R., Duff K., Leighton J.L. Enantioselective allylation of ketone-derived benzoylhydrazones: practical synthesis of tertiary carbinamines. J. Am. Chem. Soc. 2004;126:5686–5687. doi: 10.1021/ja0486418. [DOI] [PubMed] [Google Scholar]
- 36.Basilio-Lopes A., de Aquino T.M., Mongeot A., Bourguignon J.-J., Schmitt M. Toward versatile methods leading to highly functionalized imidazo[1,2-a]pyridines. Tetrahedron Lett. 2012;53:2583–2587. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplemental information files).