


Keywords: cardiac function, hypertrophy, ischemia-reperfusion injury, nitric oxide synthase, tetrahydrobiopterin
Abstract
The cofactor tetrahydrobiopterin (BH4) is a critical regulator of nitric oxide synthase (NOS) function and redox signaling, with reduced BH4 implicated in multiple cardiovascular disease states. In the myocardium, augmentation of BH4 levels can impact on cardiomyocyte function, preventing hypertrophy and heart failure. However, the specific role of endothelial cell BH4 biosynthesis in the coronary circulation and its role in cardiac function and the response to ischemia has yet to be elucidated. Endothelial cell-specific Gch1 knockout mice were generated by crossing Gch1fl/fl with Tie2cre mice, generating Gch1fl/flTie2cre mice and littermate controls. GTP cyclohydrolase protein and BH4 levels were reduced in heart tissues from Gch1fl/flTie2cre mice, localized to endothelial cells, with normal cardiomyocyte BH4. Deficiency in coronary endothelial cell BH4 led to NOS uncoupling, decreased NO bioactivity, and increased superoxide and hydrogen peroxide productions in the hearts of Gch1fl/flTie2cre mice. Under physiological conditions, loss of endothelial cell-specific BH4 led to mild cardiac hypertrophy in Gch1fl/flTie2cre hearts. Endothelial cell BH4 loss was also associated with increased neuronal NOS protein, loss of endothelial NOS protein, and increased phospholamban phosphorylation at serine-17 in cardiomyocytes. Loss of cardiac endothelial cell BH4 led to coronary vascular dysfunction, reduced functional recovery, and increased myocardial infarct size following ischemia-reperfusion injury. Taken together, these studies reveal a specific role for endothelial cell Gch1/BH4 biosynthesis in cardiac function and the response to cardiac ischemia-reperfusion injury. Targeting endothelial cell Gch1 and BH4 biosynthesis may provide a novel therapeutic target for the prevention and treatment of cardiac dysfunction and ischemia-reperfusion injury.
NEW & NOTEWORTHY We demonstrate a critical role for endothelial cell Gch1/BH4 biosynthesis in coronary vascular function and cardiac function. Loss of cardiac endothelial cell BH4 leads to coronary vascular dysfunction, reduced functional recovery, and increased myocardial infarct size following ischemia/reperfusion injury. Targeting endothelial cell Gch1 and BH4 biosynthesis may provide a novel therapeutic target for the prevention and treatment of cardiac dysfunction, ischemia injury, and heart failure.
INTRODUCTION
Cardiovascular diseases including coronary artery disease, myocardial infarction, and heart failure are leading causes of global mortality and disability (1). A hallmark of cardiovascular diseases is an early reduction in nitric oxide (NO) bioavailability and an increase in reactive oxygen species (ROS) production. Tetrahydrobiopterin (BH4) is a critical regulator of nitric oxide synthases (NOS) function and NOS-derived NO and ROS signaling in cardiovascular physiology (2, 3). Biosynthesis of BH4 is catalyzed by GTPCH (GTP cyclohydrolase 1, encoded by Gch1). We have previously shown that Gch1 expression is a key determinant of BH4 bioavailability, NOS regulation, and NO generation (4, 5). When BH4 bioavailability is limited, NOS is unable to generate NO from l-arginine and becomes “uncoupled,” resulting in generation of superoxide anion and other ROS, rather than NO, contributing to disturbed redox signaling (3, 6–8).
Clinically, genetic variants in GCH1 functionally associated with altered GCH1 expression appear to be associated with alterations in markers of cardiac function and cardiovascular risk (9, 10). Rare genetic variants causing loss of BH4 synthesis are also associated with alterations of NOS-mediated vascular function and cardiovascular physiology (9, 10). These observations from clinical studies have been supported by preclinical models. Reduced BH4 bioavailability and NOS uncoupling are associated with various heart diseases including cardiac hypertrophy (11) and ischemia-reperfusion injury (12–17). Oral supplementation with BH4 or the BH4 precursor sepiapterin has been shown to prevent or reduce cardiac hypertrophy and failure (11, 18–21). For example, pressure overload induced by transverse aortic constriction (TAC) in mice reduced cardiac BH4 levels and promoted eNOS uncoupling, leading to myocyte hypertrophy, cardiac dilation, interstitial fibrosis, and ventricular dysfunction (22). Treatment with oral BH4 prevented the NOS uncoupling and reduced the TAC-induced hypertrophy. Furthermore, exogenous BH4 was able to recouple eNOS and reverse preexisting cardiac hypertrophy and fibrosis caused by TAC-induced pressure overload (11).
However, these and other studies do not address the important question of which cell types in the heart may mediate the effects of BH4 on cardiac function. NO is generated from coronary endothelial cells by eNOS, whereas in cardiac myocytes, both eNOS and nNOS have been shown to contribute to myocardial function. In the endothelium, NO mediates coronary vascular function and flow, whereas in cardiac myocytes, NO regulates LV relaxation by effects on myofilament calcium sensitivity and calcium handling (23). Although prior studies have suggested a critical role of endothelial NO and BH4 levels on cardiac function and injury (17, 21, 24–26), no prior study has specifically addressed the requirement for endothelial cell BH4 in the regulation of cardiac function, particularly in myocardial ischemia-reperfusion (I/R) injury where roles for both eNOS and nNOS are implicated (27). Interpreting the specific roles of NO in the heart using knockouts of either eNOS or nNOS is limited by loss of all NOS-related functions, including ROS generation, subcellular localization, and protein-protein interactions, whereas the requirement for BH4 in the generation of NO by both eNOS and nNOS enables selective targeting of NOS-mediated NO generation, without primary alterations in either NOS protein levels or other NOS functions.
Accordingly, we sought to investigate how selective targeting of endothelial cell BH4 biosynthesis, without alteration of cardiac myocyte BH4, would alter cardiac function, focusing on ischemia-reperfusion injury, where changes in coronary endothelial function play an important pathophysiological role.
METHODS
Animals
All animal studies were conducted under project licenses PPL 30/3080 and P0C27F69A with ethical approval from the Local Ethical Review Committee and in accordance with the United Kingdom Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act), 1986, with procedures reviewed by the clinical medicine Animal Care and Ethical Review Body (AWERB). Animals were housed in individually ventilated cages (between 4 and 6 mice per cage of mixed genotypes) in specific pathogen-free conditions. All animals were provided with standard chow (Teklad global 16% protein diet, Harlan Laboratories) and water ad libitum and maintained on a 12-h:12-h light/dark cycle at controlled temperature (20–22°C) and humidity.
Gch1 Knockout Mice
We generated mice with a Gch1 conditional knockout (floxed) allele, as previously described (28–30). Gch1fl/fl animals were bred with Tie2cre transgenic mice to produce Gch1fl/flTie2cre mice where Gch1 is deleted in endothelial cells, generating a mouse model of endothelial cell-specific BH4 deficiency. Since the Tie2cre transgene is active in the female germline, only male animals are used to establish breeding pairs to maintain conditional endothelial cell expression. Mice were genotyped according to the published protocol (29, 31). Briefly, mice were genotyped by polymerase chain reactions using DNA prepared from ear biopsies. For Gch1fl/fl genotyping, PCR was performed using the following primers: Gch1fl/fl, forward 5′-GTC CTT GGT CTC AGT AAA CTT GCC AGG-3′; Gch1fl/fl, reverse 5′-GCC CAG CCA AGG ATA GAT GCA G-3′. The Gch1 floxed allele showed as 1,030 bp. For Tie2cre genotyping, PCR was performed using the following primers: Tie2cre, forward 5′-GCA TAA CCA GTG AAA CAG CAT TGC TG-3′; Tie2cre, reverse 5′-GGA CAT GTT CAG GGA TCG CCA GGC G-3′. The Tie2cre allele amplified as 280-bp fragment. Adult male Gch1fl/flTie2cre mice and their Gch1fl/fl littermates (hereafter referred to as wild type) on a pure (>10 generations) C57BL6/J background were bred in house and were used for all experiments at 20 to 24 wk.
Determination of Tissue Tetrahydrobiopterin Levels
BH4 and oxidized biopterins (BH2 and biopterin) were determined by high-performance liquid chromatography (HPLC) followed by electrochemical and fluorescence detection, respectively, following an established protocol (32). Briefly, frozen heart samples were homogenized in ice-cold resuspension buffer, consisting of (in mmol · L−1 ) 50 phosphate-buffered saline, 1 dithioerythriol, and 1 EDTA at pH 7.4. After centrifugation at 13,200 rpm for 10 min at 4°C, the supernatant was removed, and ice-cold acid precipitation buffer, consisting of (in mmol · L−1) 1 phosphoric acid, 2 trichloroacetic acid, and 1 dithioerythritol, was added. Samples were vigorously mixed and then centrifuged for 15 min at 13,000 rpm and 4°C. Samples were injected into an isocratic HPLC system and quantified using sequential electrochemical (Coulochem III, ESA, Inc.) and fluorescence (Jasco) detection. HPLC separation was performed using a 250-mm ACE C-18 column (Hichrom) and a mobile phase comprised of 50 mM sodium acetate, 5 mM citric acid, 48 µΜ EDTA, and 160 µΜ dithioerythritol (pH 5.2) (all ultrapure electrochemical HPLC grade) at a flow rate of 1.3 mL/min. Background currents of +500 μA and −50 μA were used for the detection of BH4 on electrochemical cells E1 and E2, respectively. 7,8-BH2 and biopterin were measured using a Jasco FP2020 fluorescence detector set at 510-nm excitation and 595-nm emission. Quantification of BH4, BH2, and B was done by comparison with authentic external standards and normalized to sample protein content.
Endothelial Cell Isolation
Primary heart endothelial cells were isolated using MACS beads (Miltenyi Biotec), as previously described (29). Briefly, mice were euthanized by an overdose of inhaled isoflurane. Hearts were harvested and digested in DMEM containing 0.18 U/mL Liberase (Roche) and 0.1 mg/mL DnaseI (Roche) for 1 h at 37°C. The digested tissue was filtered through 100- and 70-μm cell strainers. The cell suspension was then incubated with rat anti-CD31 antibody (BD PharMingen) for 15 min at 4°C and then with anti-rat secondary antibody coated immune magnetic beads for a further 15 min at 4°C. Bead-bound endothelial cells were selected using a magnetic column. Endothelial cells were collected and stored at −80°C for further analysis.
Cardiomyocyte Isolation
Cardiac myocytes were isolated using an enzymatic dispersion technique (23). Briefly, the heart was perfused with Ca2+-free isolation solution (37°C, oxygenated) for 3 min and then with 1 mg/mL collagenase type II solution (Worthington Biochemical) for a further 9 min. The myocytes were pelleted by centrifugation at a low speed (600 rpm). The supernatant was then spun down at 10,000 rpm; the pellet was considered as the nonmyocyte fraction.
Echocardiography
Left ventricular (LV) size and function were investigated in vivo using a high-resolution two-dimensional (2D) echocardiography system (Vevo 2100, VisualSonics, Canada) in isoflurane (1%–1.5%) anesthetized mice. LV wall thickness and chamber dimensions were determined in the parasternal short-axis view (M-mode), from which measures of LVEF and fractional shortening were derived. 2-D images of the heart were obtained from the four-chamber apical view to assess mitral blood inflow and tissue-Doppler velocities.
Quantification of Superoxide Production by Dihydroethidine (DHE)-HPLC
Superoxide production was quantified by measuring the production of 2-hydroxyethidium from dihydroethidium, using HPLC (29). Briefly, frozen heart homogenate was preincubated with serum-free DMEM with or without 100 μM NG-nitro-l-arginine methyl ester (l-NAME; Sigma). Samples were then incubated with 25 μM DHE (Invitrogen) for 20 min before being harvested for separation of 2-hydroxyethidium using a gradient HPLC system (Jasco, UK) with an ODS3 reverse phase column (250 mm, 4.5 mm, Hichrom UK) and quantified using a fluorescence detector set at 510 nm (excitation) and 595 nm (emission).
Langendorff Heart Preparation
Mice were heparinized (300 U) and anesthetized with ketamine (75 mg/kg) plus medetomidine hydrochloride (1 mg/kg), with the adequacy of anesthesia confirmed by the absence of a pedal reflex. Hearts were quickly excised and immersed in KH buffer. The aorta was then cannulated onto the Langendorff perfusion system for retrograde perfusion. The heart was perfused with 37°C KH buffer and gassed with 95% O2-5% CO2, at 2 mL/min, and cardiac function was assessed using a fluid-filled balloon inserted into the left ventricle, which connected to a pressure transducer and a PowerLab system (ADInstruments). Left ventricular developed pressure (LVDP), calculated by the difference between systolic and diastolic pressure, was recorded continuously via LabChart software v.7.0. After 25-min equilibration, hearts were subjected to 35 min global ischemia followed by 60 min of reperfusion. For triphenyltetrazolium chloride (TTC) staining, hearts were removed from the Langendorff following ex vivo I/R, briefly frozen, and then sliced into six 1-mm-thick transverse sections. To distinguish viable (stained) versus necrotic (pale, unstained) tissue, sections were incubated in 1% TTC for 30 min at 37°C. Sections were then scanned, and the area of infarction (TTC negative) was quantified as a percentage of the area at risk (entire area of the section) using ImageJ.
Quantification of Gene Expression by Real-Time RT-PCR
RNA was prepared using the RNeasy kit (Qiagen) and was reverse transcribed using Superscript II (Life Technologies) according to standard protocols. RNA equivalent cDNA (5 ng) was used to perform real-time PCR using predesigned tag-man gene expression assays (Life Technologies) using a BioRad CFX1000. Gene expression levels of mouse Gch1, Nos1, Nos2, and Nos3 were normalized to the housekeeping gene GAPDH using the ΔCt method.
Western Blot Analysis
Immunoblotting in LV homogenates was performed to evaluate protein levels of GTPCH (1:10,000 dilution; a gift from S.Gross, Cornell University; New York), iNOS (1: 1,100 dilution; Abcam), nNOS (1:1,000 dilution; Santa Cruz Biotechnology), eNOS (1:5,000 dilution; BD Bioscience), CD102 (1:1,000; R&D systems), SERCA2A (1:5,000 dilution; Santa Cruz Biotechnology), total phospholamban (1:2,000 dilution; PLB, Badrilla), phosphor-Thr17-PLB (1:2,000 dilution; Badrilla), phosphor-Ser16-PLB (1:2,000 dilution; Badrilla), NCX1 (1:1,000 dilution, Santa Cruz), phospho-extracellular signal-regulated protein kinases (1:500 dilution; ERK1/2), total ERK1/2 (1:500 dilution;), catalase (1:5,000 dilution; Calbiochem), MnSOD (1:5,000 dilution; Stressgen Bioreagents), EcSOD (1:750 dilution; Stressgen Bioreagents), Cu/ZnSOD (1:500 dilution; Stressgen Bioreagents), and β-tubulin (1:20,000; Abcam), followed by appropriate HRP-conjugated secondary antibody (1:10,000–20,000 dilution; Promega). Protein bands were visualized by enhanced chemiluminescence (Super West Pico Chemiluminescence, Thermo Scientific).
Blood Pressure Measurement by Tail-Cuff Plethysmography
Systolic blood pressure in conscious wild-type and Gch1fl/flTie2cre mice was determined using the VisitechR computerized tail-cuff plethysmography system (Visitech) following 5 days of training and 3 days baseline periods. Experiments were performed between the hours of 8:00 and 12:00 am. The animal tails were passed through a cylindrical latex tail-cuff and taped down to reduce movement. Twenty readings were taken per mouse of which the first five readings were discarded. The remaining 15 readings were used to calculate the mean systolic blood pressure in each mouse.
Statistical Analysis
All data are reported as means ± SE. The experimental unit (n) was defined as a single animal, animals of both genotypes were caged together, and animals of both genotypes were derived from more than one cage in all experiments. Statistical analyses were performed using GraphPad Prism v. 9.3.0. (San Diego, CA). Normality was tested using D’Agostino and Pearson omnibus normality test. Groups were compared using the Mann–Whitney U test for nonparametric data or an unpaired Student’s t test for parametric data. When comparing multiple groups, data were analyzed by analysis of variance (ANOVA) with Newman–Keuls posttest for parametric data or Kruskal–Wallis test with Dunn’s posttest for nonparametric data. When more than two independent variables were present, a two-way ANOVA with Tukey’s multiple comparisons test was used. When within-subject repeated measurements were present, a repeated-measures (RM) ANOVA was used. A value of P < 0.05 was considered statistically significant. Data were collected and analyzed with the operator blind of treatment allocation. Randomization was performed by cage.
RESULTS
Endothelial Cell-Targeted Gch1 Deletion in the Heart Causes Selective Endothelial Cell BH4 Deficiency
We generated matched litters of Gch1fl/flTie2cre and Gch1fl/fl mice (hereafter referred to as wild type) by crossing male Gch1fl/flTie2cre and female Gch1fl/fl mice. Body weights between the groups were similar (36 ± 1.5 g in wild type and 36 ± 1.1 g in Gch1fl/flTie2cre; n = 6 to 8 animals per group). Genomic polymerase chain reaction demonstrated efficient excision of the floxed Gch1 allele in isolated endothelial cells from Gch1fl/flTie2cre hearts (Fig. 1A). Endothelial cell-specific Gch1 deletion resulted in a significant reduction in Gch1 expression (Fig. 1, B and C), GTPCH protein in whole heart tissue, and barely detectable levels in endothelial cells isolated from the hearts. However, the GTPCH protein in isolated cardiomyocytes was similar between the groups (Fig. 1, D–I). Accordingly, BH4 levels were significantly decreased in hearts and barely detected in isolated endothelial cells from Gch1fl/flTie2cre hearts (Fig. 1, J, K, and M). Despite marked BH4 deficiency, absolute BH2 levels in heart tissue were comparable between wild-type and Gch1fl/flTie2cre mice, such that the BH4/BH2 and biopterin ratio was significantly reduced in Gch1fl/flTie2cre hearts (Fig. 1L). However, the BH4/BH2 and biopterin ratio in isolated endothelial cells was comparable between wild-type and Gch1fl/flTie2cre mice (Fig. 1N). In contrast to the observations in endothelial cells, BH4 levels in isolated cardiomyocytes were similar between wild-type and Gch1fl/flTie2cre mice, indicating that the reduction in overall heart tissue BH4 levels in Gch1fl/flTie2cre mice is due to specific deletion of endothelial cell Gch1. However, BH2 levels were significantly increased in cardiomyocytes isolated from Gch1fl/flTie2cre mice, such that the BH4/BH2 and biopterin ratio was significantly decreased in cardiomyocytes from Gch1fl/flTie2cre mice (Fig. 1, O and P), suggesting that selective endothelial cell BH4 deficiency in the heart leads to secondary effects on BH4 oxidation and/or recycling in cardiomyocytes, independent of changes in de novo BH4 biosynthesis. Importantly, plasma BH4 levels were similar between the groups, indicating that endothelial cell BH4 biosynthesis by GTPCH1 is not a major contributor to circulating BH4 levels (Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21732581.v1).
Figure 1.
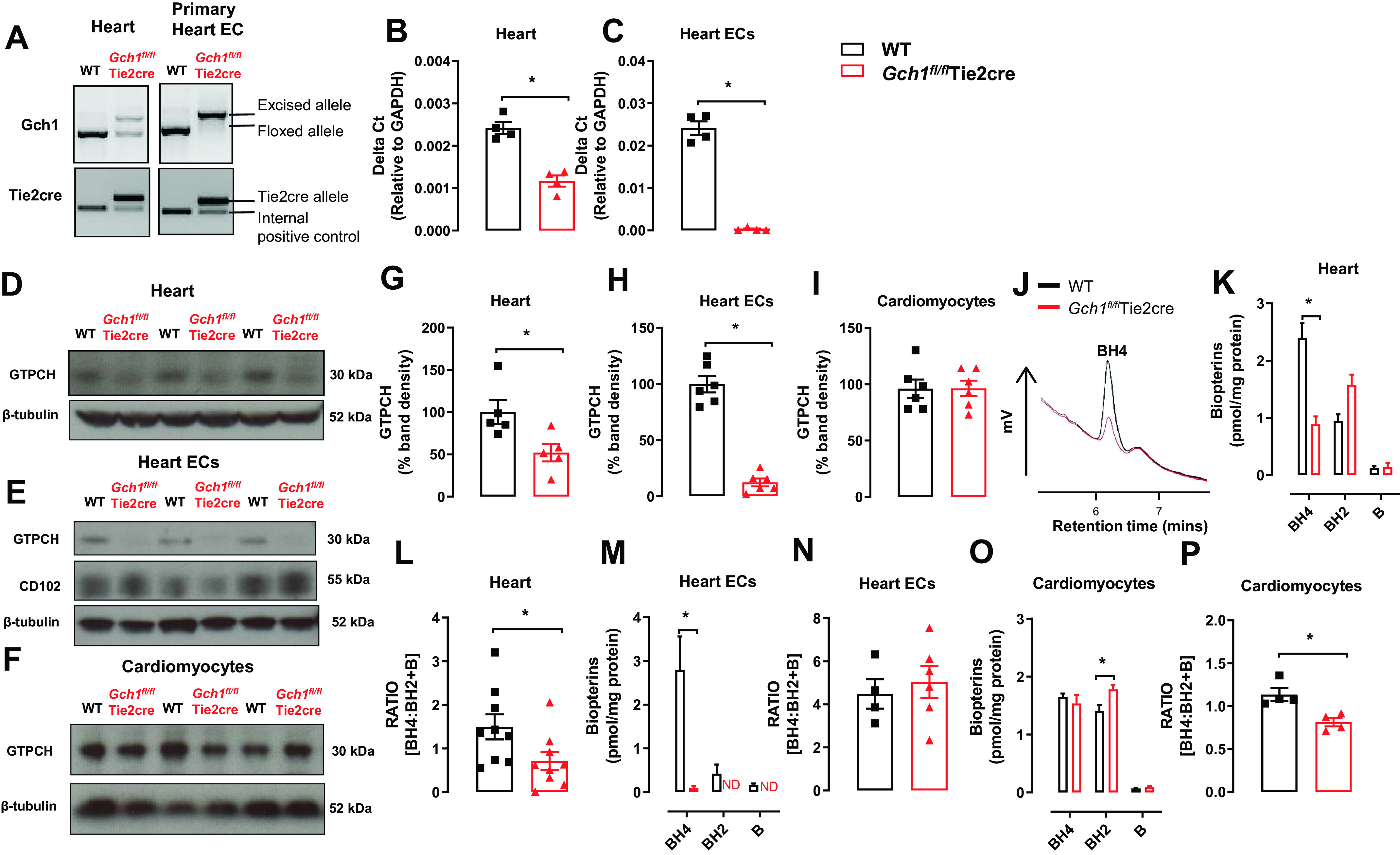
Myocardial endothelial cell targeted Gch1 deletion causes a tissue-specific decrease in Gch1 gene, GTPCH protein, and biopterin content. A: evaluation of Tie2cre-mediated excision of the loxP flanked DNA in heart tissues and primary heart endothelial cells derived from Gch1fl/flTie2cre and Gch1fl/fl [wild-type mice (WT)]. The predicted 1,030-bp product was detected in WT mice. In the presence of Tie2cre transgene a 1,392-bp knockout allele was detected, with efficient excision in primary endothelial cells from hearts. B and C: quantitative real-time PCR was used to quantify Gch1 gene expression in hearts and primary endothelial cells from hearts (*P < 0.05; n = 4 per group). D–F: representative immunoblot of GTPCH proteins in hearts, isolated primary endothelial cells, and isolated cardiomyocytes from WT and Gch1fl/flTie2cre hearts, respectively, with quantitative data, measured as percent band density in G–I: CD102 and β-tubulin were used as endothelial cell marker and loading control respectively. J: representative chromatograms of BH4 traces in hearts from WT and Gch1fl/flTie2cre mice. K and L: BH4 levels and BH4/BH2 + B ratio were reduced in hearts from Gch1fl/flTie2cre mice compared with wild-type littermates (*P < 0.05; n = 8 and 9 per group). M and N: BH4 levels were barely detectable in primary ECs from Gch1fl/flTie2cre compared with WT mice (*P < 0.05; n = 4–6 per group). O and P: BH4 levels were comparable between primary cardiomyocytes from Gch1fl/flTie2cre mice and wild-type littermates. Absolute BH2 levels in cardiomyocytes were significantly increased in Gch1fl/flTie2cre mice compared with wild-type mice, such that the BH4/BH2 and biopterin ratio was significantly reduced in cardiomyocytes in Gch1fl/flTie2cre mice (*P < 0.05; n = 4 per group). Each data point represents an individual adult male mouse.
Endothelial Cell BH4 Deficiency Leads to Cardiac NOS Uncoupling with Increased Superoxide Production and Loss of Cardiac NO Generation
We next determined the effects of altering cardiac endothelial cell BH4 availability on NOS function. We first measured basal superoxide productions in whole heart homogenate by quantification of 2-hydroxyethidium (2-HE) production from dihydroethidine, using high-performance liquid chromatography (HPLC). Basal superoxide production was significantly elevated in hearts from Gch1fl/flTie2cre mice compared with wild-type controls (P < 0.05, Fig. 2, A and B). In the presence of the nonselective nitric oxide synthase inhibitor l-NAME (100 µM), the levels of NOS-derived superoxide production in wild-type hearts were significantly increased compared with untreated hearts, suggesting a tonic scavenging effect of cardiac NO on superoxide. Furthermore, there was significant inhibition of superoxide production in Gch1fl/flTie2cre hearts by the NOS inhibitor l-NAME, (P < 0.05, Fig. 2, A–D), suggesting that NOS is a source of superoxide production in Gch1fl/flTie2cre endothelial cells.
Figure 2.
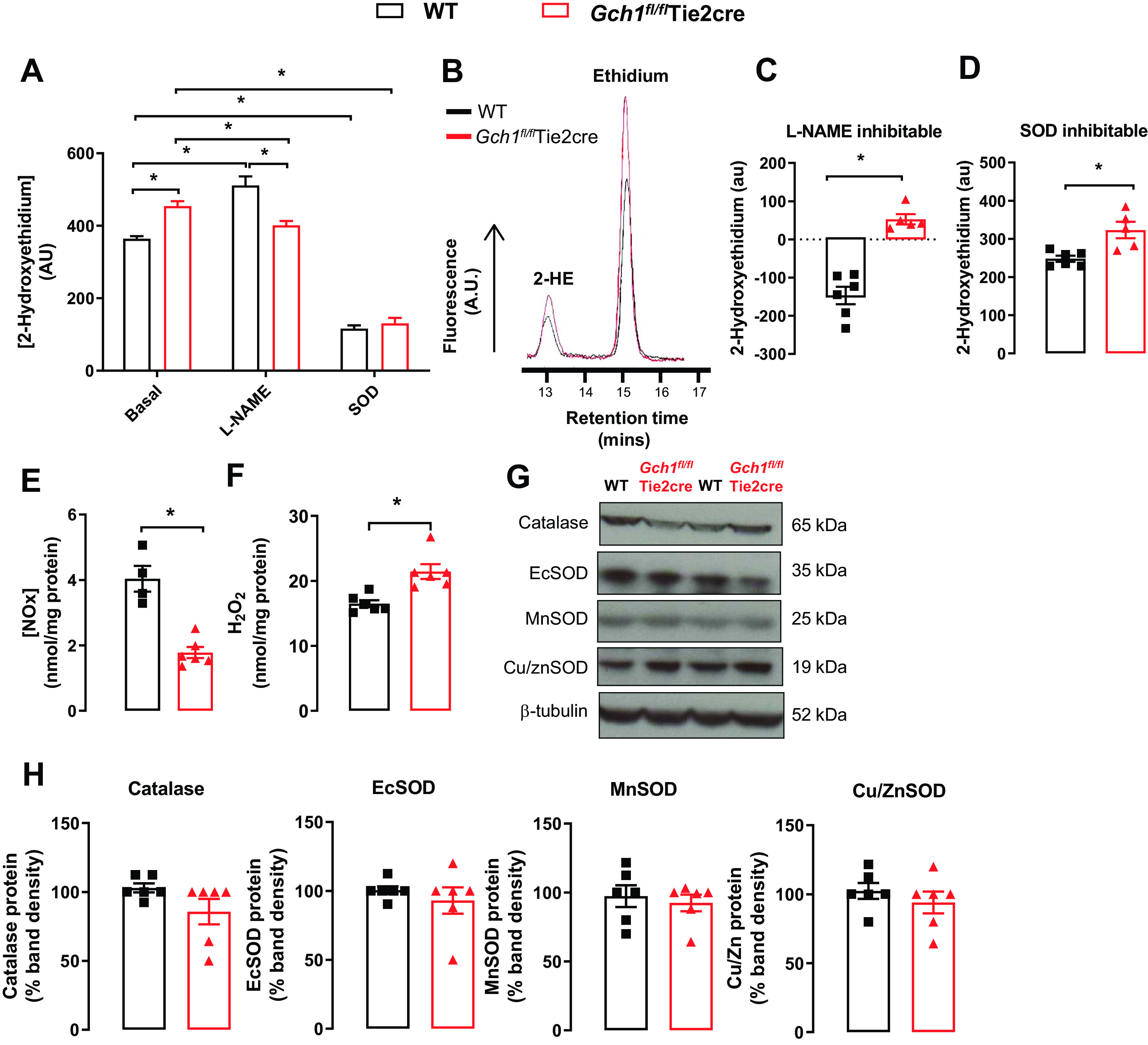
Superoxide production is increased, and nitric oxide bioavailability is reduced in cardiac endothelial cell BH4-deficient mice. Quantification of superoxide production, as measured by 2-hydroxyethidium (2-HE), in whole heart homogenate from Gch1fl/flTie2cre and wild-type (WT) mice using dihydroethidine (DHE) high-performance liquid chromatograph (HPLC). A: superoxide production was markedly increased in hearts from Gch1fl/flTie2cre mice compared with wild-type controls (*P < 0.05, n = 5–6 per group). B: representative trances of 2-HE and ethidium peaks in hearts from WT and Gch1fl/flTie2cre mice detected by DHE HPLC. C and D: nonselective nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 100 µM)-inhibitable fraction and polyethylene glycol superoxide dismutase, PEG-SOD (100 U/ml)-inhibitable fraction were greatly increased in Gch1fl/flTie2cre hearts compared with wild-type controls (*P < 0.05, n = 5–6 per group), respectively. E: nitrite/nitrate production in whole heart homogenate. Nitrite/nitrate production in heart homogenate from Gch1fl/flTie2cre mice was significantly decreased when compared with that from WT controls (*P < 0.05; n = 4–6 animals per group). F: levels of hydrogen peroxide from wild-type and Gch1fl/flTie2cre hearts were determined using an amperometric hydrogen peroxide microsensor electrode. Level of hydrogen peroxide production was significantly increased in endothelial cell BH4-deficient hearts (Gch1fl/flTie2cre) compared with wild-type controls (*P < 0.05; n = 6 per group). G: representative immunoblots for antioxidant proteins: catalase, Es-SOD, Mn-SOD, and Cu/Zn-SOD in Gch1fl/flTie2cre and wild-type hearts, with quantitative data, measured as percent band density in H (n = 6 per group). Each data point represents an individual adult male mouse.
To determine the effects of endothelial cell BH4 deficiency on NO bioactivity, we measured nitrate and nitrite in heart homogenates using ozone chemiluminescence. Nitrite and nitrate production were significantly reduced in Gch1fl/flTie2cre hearts compared with wild-type controls (P < 0.05, Fig. 2E). Furthermore, hydrogen peroxide production was significantly elevated in endothelial cell BH4-deficient hearts compared with wild-type controls (P < 0.05, Fig. 2F). To investigate whether increased productions of superoxide and hydrogen peroxide and reduced NO bioactivity in Gch1fl/flTie2cre heart altered antioxidant defenses, we measured protein levels of antioxidant enzymes by Western blot. There was no change in protein levels of catalase, extracellular superoxide dismutase (ecSOD), manganese superoxide dismutase (MnSOD), or Cu/ZnSOD between Gch1fl/flTie2cre and wild-type hearts (Fig. 2, G and H). Taken together, these data demonstrate that deficiency in coronary endothelial cell BH4 leads to eNOS uncoupling, increased superoxide and hydrogen peroxide productions, and decreased NO bioactivity in myocardium from Gch1fl/flTie2cre mice.
Specific Loss of Endothelial Cell BH4 Leads to Cardiac Dysfunction and Hypertrophy
We next investigated the effect of endothelial cell Gch1 and BH4 deficiency on cardiac function, using M-mode echocardiography (Fig. 3A). There was no difference in either fractional shortening or ejection fraction or peak systolic velocity in wild-type and Gch1fl/flTie2cre mice (Fig. 3, B–D). However, left ventricular (LV) diastolic volume and LV end dimensions were significantly reduced in Gch1fl/flTie2cre mice compared with wild-type littermates (Fig. 3E). LV diastolic and systolic volume were also significantly reduced in Gch1fl/flTie2cre mice compared with wild-type littermates (Fig. 3, F and G). LV end-diastolic thickness was significantly increased in Gch1fl/flTie2cre mice compared with wild-type littermates (Fig. 3H). Cardiac output was significantly depressed in Gch1fl/flTie2cre mice compared with wild-type controls (Fig. 3I). We next measured blood pressure in Gch1fl/flTie2cre and wild-type littermate controls using tail-cuff plethysmography. We observed that Gch1fl/flTie2cre mice have a mild increased (∼5–7 mmHg) systolic blood pressure compared with wild-type littermate controls (102 ± 3 mmHg WT vs. 109 ± 2 mmHg in Gch1fl/flTie2cre mice; Fig. 3J).
Figure 3.
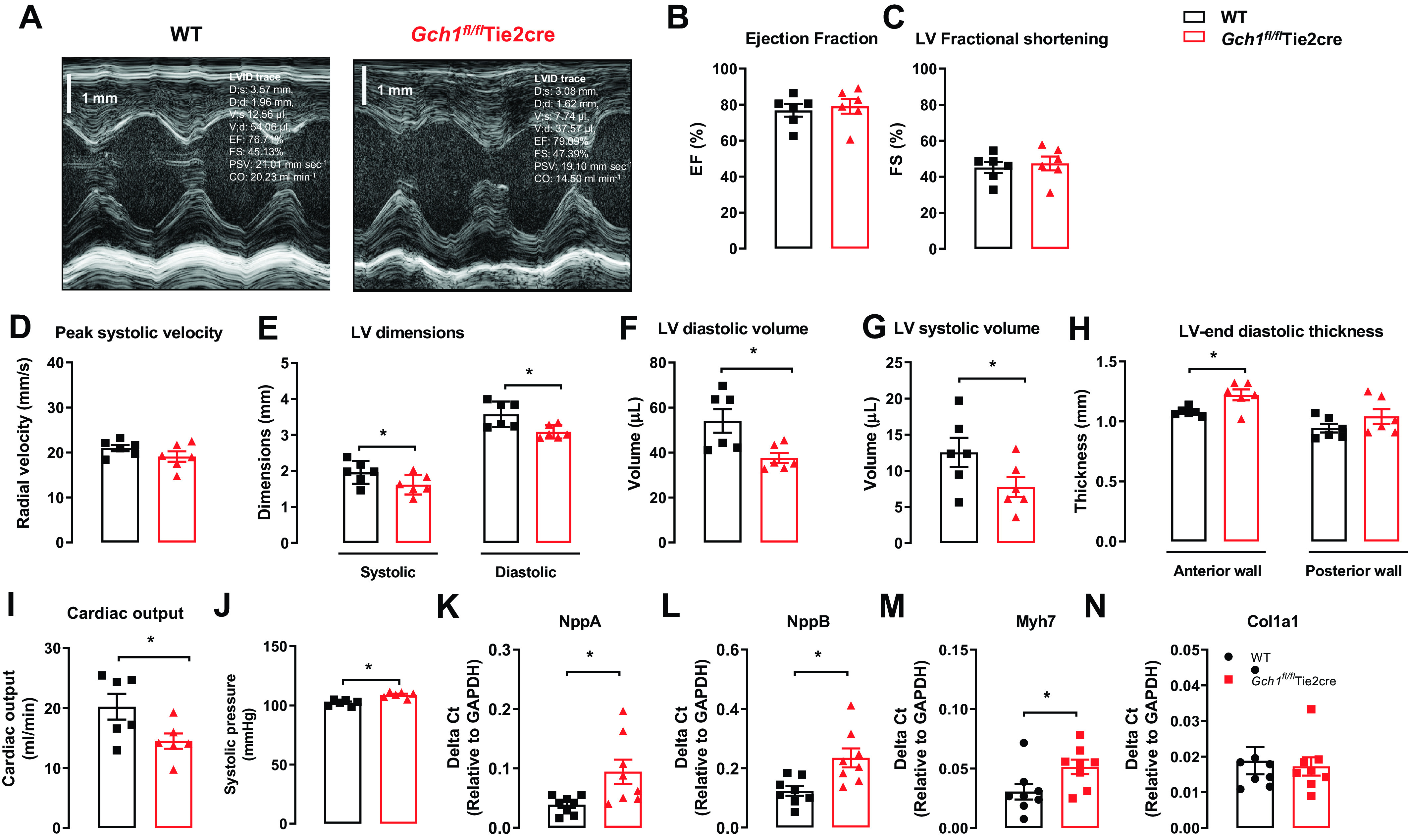
Loss of cardiac endothelial cell BH4 leads to cardiac dysfunction and hypertrophy. A: example of M-mode echocardiograms from Gch1fl/flTie2cre and wild-type littermates controls. B: ejection fraction. C: LV fractional shortening. D: peak systolic velocity, cardiac output (mL/min). E: systolic and diastolic LV end dimensions (mm). F and G: LV diastolic and systolic volume (μL). H: anterior and posterior LV end-diastolic thickness (mm). I: cardiac output (mL/min) in WT and Gch1fl/flTie2cre mice (*P < 0.05, n = 6 per group). J: systolic blood pressure in Gch1fl/flTie2cre and wild-type littermates were determined using tail-cuff plethysmography. K–M: gene expression of hypertrophic markers and fibrosis marker (N) in hearts from Gch1fl/flTie2cre and WT littermates (*P < 0.05; n = 7–8 per group). Each data point represents an individual adult male mouse.
In addition, we have undertaken further studies to examine the effect of endothelial cell BH4 deficiency on blood glucose levels and lipid profiles in Gch1fl/flTie2cre mice. First, we observed that blood glucose levels after 6 h of fasting were also comparable between wild-type and Gch1fl/flTie2cre mice (Supplemental Fig. S1). Lipid profiles including total cholesterol, triglycerides, LDL, and HDL were also comparable between the groups (Supplemental Fig. S2). These findings indicate that endothelial cell BH4 deficiency does not affect the metabolic or lipid profile in Gch1fl/flTie2cre mice.
To investigate the downstream effect of endothelial cell BH4 deficiency and NOS uncoupling on myocardial remodeling and hypertrophy, we measured mRNA expression of fetal genes in heart homogenates from Gch1fl/flTie2cre mice and their littermates. The mRNA expression of hypertrophic markers including natriuretic factor type A (Nppa), type B (Nppb), and β-myosin heavy chain (Myh7) were significantly increased in Gch1fl/flTie2cre hearts compared with littermate WT controls (Fig. 3, K–M). In contrast, there was no difference in Col1a1 (a fibrosis marker) expression between the genotypes (Fig. 3N). Taken together, these findings suggest that endothelial cell BH4 deficiency leads to LV dysfunction and hypertrophy.
Endothelial Cell BH4 Deficiency Leads to Activation of Cardiomyocytes, Increased nNOS, and Decreased eNOS Protein in Gch1fl/flTie2cre Hearts
To investigate the mechanism by which cardiac endothelial cell-specific BH4 deficiency impairs of cardiac function and increases hypertrophy, we isolated primary endothelial cells and primary cardiomyocytes from Gch1fl/flTie2cre and wild-type mice and tested whether NOS uncoupling from endothelial cells could mediate changes in cardiomyocytes. First, we observed that Erk1/2 phosphorylation was significantly increased in the whole hearts from Gch1fl/flTie2cre mice compared with wild-type littermate controls (Fig. 4, A and B). Furthermore, we found that Erk1/2 phosphorylation was significantly increased in primary cardiomyocytes from Gch1fl/flTie2cre compared with wild-type controls (Fig. 4, A and B). In contrast, we did not observe an increase in Erk1/2 phosphorylation in endothelial cells isolated from Gch1fl/flTie2cre hearts compared with littermate controls (Fig. 4, A and B). These findings suggest that loss of BH4 leads to NOS uncoupling in cardiac endothelial cells, which in turn causes changes in cardiomyocyte signaling pathways.
Figure 4.

Increased phosphorylated extracellular signal-regulated kinases 1/2 in hearts and cardiomyocytes from Gch1fl/flTie2cre mice. A: representative immunoblots for phosphorylated and total proteins for extracellular signal-regulated kinases 1/2 (ERK1/2) in hearts, isolated endothelial cells (ECs), and cardiomyocytes from Gch1fl/flTie2cre mice and wild-type littermate controls. B: summary data (*P < 0.05, n = 5–6 per group). Each data point represents an individual adult male mouse.
We next determined whether cardiac dysfunction and hypertrophy in Gch1fl/flTie2cre hearts are associated with changes in myocardial NOS isoforms. Quantitative real-time PCR and Western blot analysis demonstrated a significant increase in nNOS mRNA and protein in Gch1fl/flTie2cre hearts, accompanied by a reduction in eNOS mRNA and protein (Fig. 5, A–C). There was no significant difference in iNOS mRNA and protein expression between Gch1fl/flTie2cre hearts and wild-type hearts (Fig. 5, A–C). To investigate which cell type is responsible for the upregulation of nNOS and downregulation of eNOS protein, NOS isoforms were determined in isolated cardiomyocytes from Gch1fl/flTie2cre mice, revealing increased nNOS protein and decreased eNOS protein in isolated cardiomyocytes from Gch1fl/flTie2cre hearts (Fig. 5, D and E).
Figure 5.
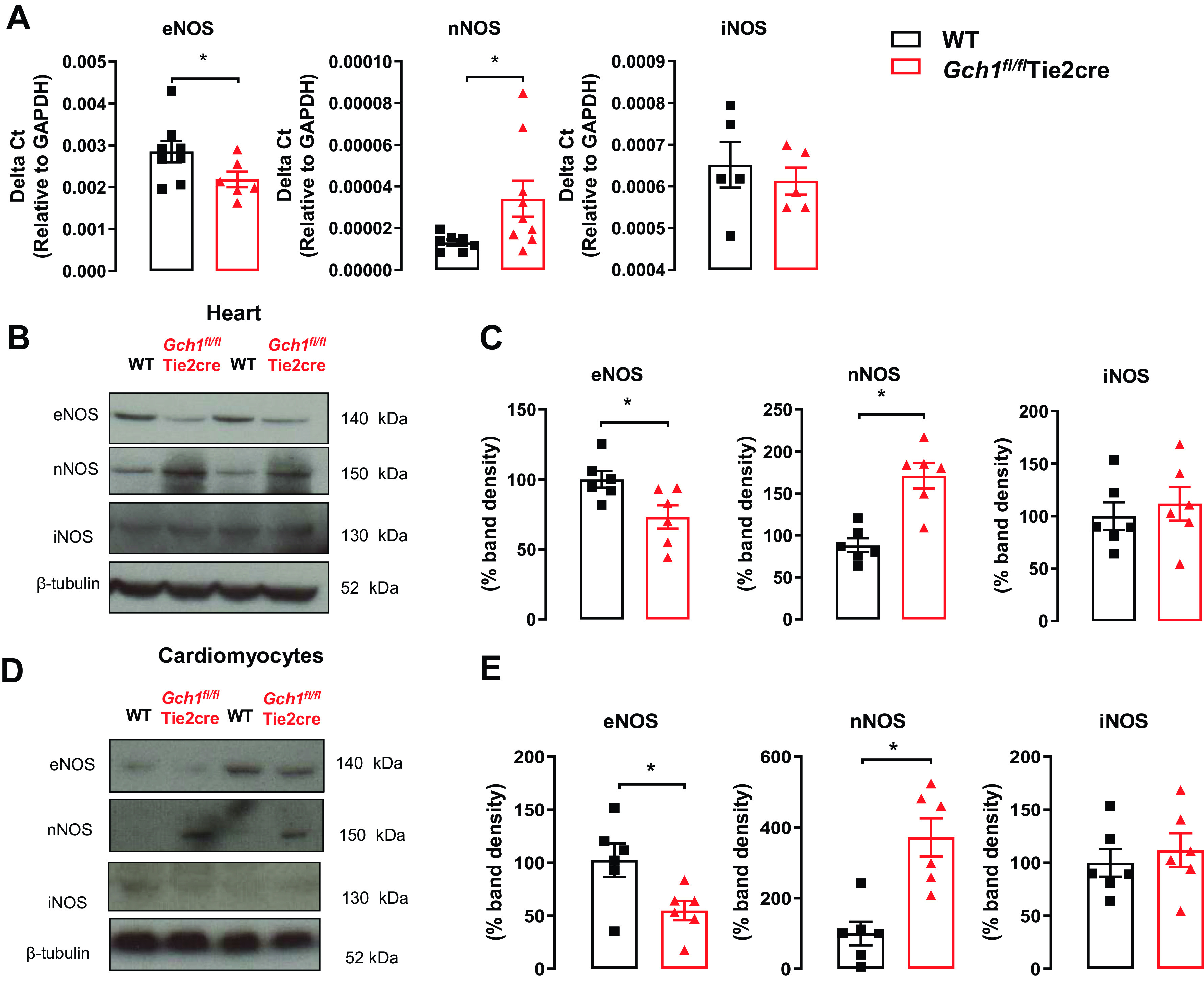
Deficiency in endothelial cell BH4 causes an increased nNOS expression and reduced eNOS expression in the hearts specifically in cardiomyocytes from Gch1fl/flTie2cre mice. A: quantitative real-time PCR was used to quantify endothelial cell nitric oxide synthase (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS) gene expression in hearts from Gch1fl/flTie2cre mice and wild-type controls (*P < 0.05; n = 5–9 per group). B: representative immunoblots of eNOS, nNOS, and iNOS isoforms in hearts from Gch1fl/flTie2cre mice and wild-type controls. C: summary data (*P < 0.05; n = 6 per group). D: representative immunoblots of eNOS, nNOS, and iNOS isoforms in cardiomyocytes from Gch1fl/flTie2cre mice and wild-type controls. E: summary data (*P < 0.05; n = 6 per group). Each data point represents an individual adult male mouse.
We further investigated whether loss of Gch1/BH4 in endothelial cells alters calcium-handling proteins in cardiomyocytes. We found that phospholamban phosphorylation at the calmodulin-dependent kinase II (CaMKII)-specific site (PLB-Thr17) was increased in myocardium from endothelial cell BH4-deficient mice, but not at the protein kinase A-specific site (PLB-Ser16) (Fig. 6, A and B). There was no change in overall protein levels of phospholamban, NCX, or SERCA2A in cardiomyocytes between Gch1fl/flTie2cre and wild-type mice (Fig. 6, C and D). Taken together, these data demonstrate that endothelial cell BH4 deficiency leads to activation of cardiomyocytes, increased nNOS, and decreased eNOS protein in Gch1fl/flTie2cre hearts, associated with alteration in cardiomyocyte calcium handling.
Figure 6.
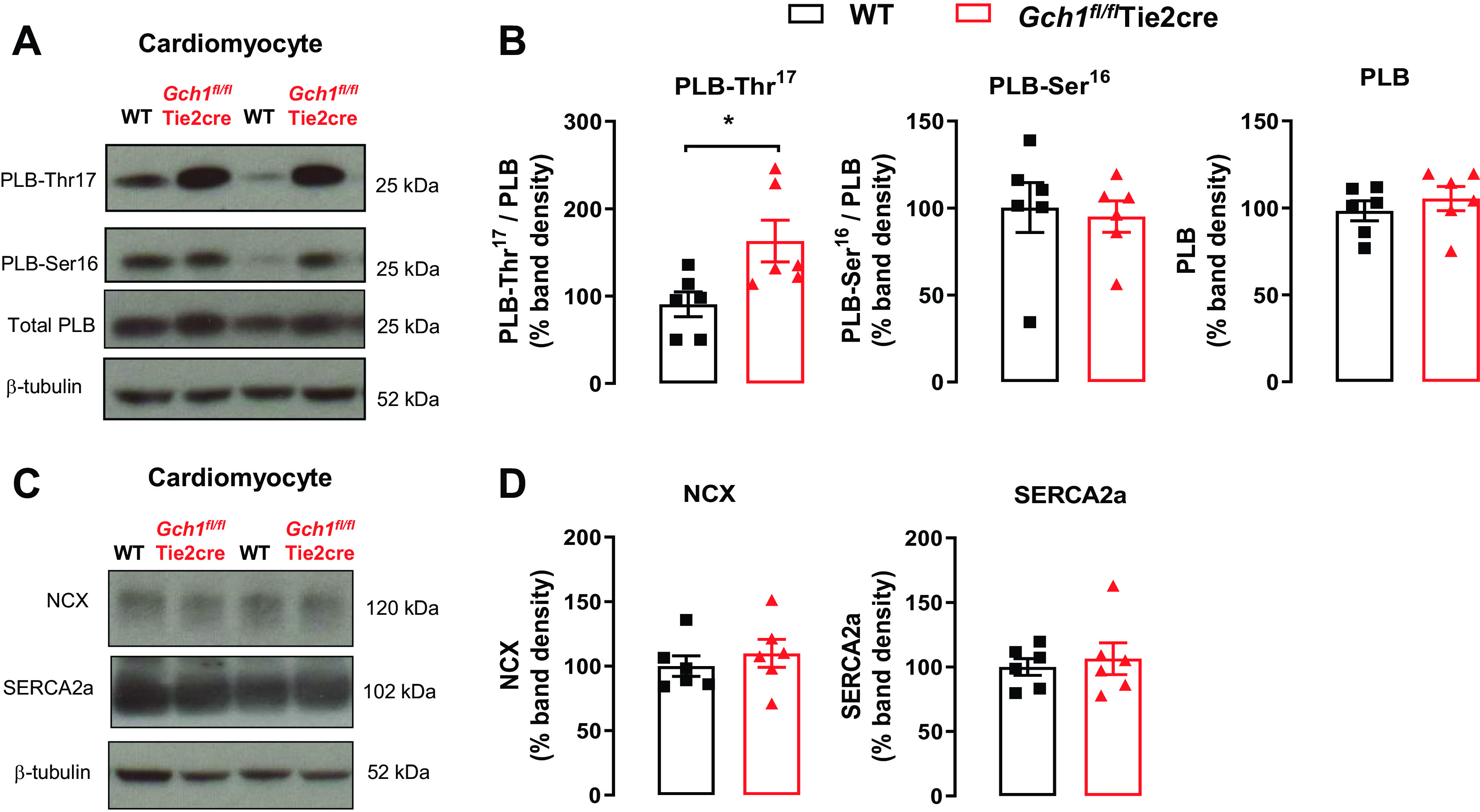
Calcium-handling proteins in cardiomyocytes from wild-type and Gch1fl/flTie2cre mice. A and B: phosphorylation of phospholamban (PLB) at Thr (17) was significantly increased in cardiomyocytes from Gch1fl/flTie2cre mice (*P < 0.05; n = 6 per group), whereas the phosphorylation of phospholamban at Ser (16) was unchanged (n = 6 per group). C: representative immunoblots for sodium-calcium exchanger (NCX) and sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) in cardiomyocytes from Gch1fl/flTie2cre mice and wild-type littermate controls. D: summary data (n = 6 per group). Each data point represents an individual adult male mouse.
Deficient Endothelial Cell Gch1/BH4 Biosynthesis Leads to Coronary Vascular Dysfunction and Injury Following Cardiac Ischemia-Reperfusion
To investigate the specific role of endothelial cell BH4 in postischemic myocardial function and injury following ischemia-reperfusion, Langendorff perfused wild-type and Gch1fl/flTie2cre hearts were subjected to 35 min global ischemia followed by 60-min reperfusion (Fig. 7A). Myocardial function was measured using a ventricular balloon to determine LV developed pressure, LV end-diastolic pressure, and rate pressure product. LV functional recovery after 35 min of global ischemia was significantly impaired in Gch1fl/flTie2cre hearts compared with wild-type controls. This impairment in recovery was manifest as decreased LV-developed pressure (Fig. 7B) and increased diastolic pressure (Fig. 7C). The maximal rates of contraction (LV dP/dtmax) and relaxation (LV dP/dtmin) were also significantly lower in the Gch1fl/flTie2cre hearts than in the wild-type group (P < 0.05) (Fig. 7, D and E). The rate pressure product (RPP), an index of workload, was significantly decreased in Gch1fl/flTie2cre hearts compared with wild-type controls (Fig. 7F). The coronary perfusion pressure (CPP), a direct measure of coronary vascular resistance, was greatly increased in Gch1fl/flTie2cre hearts compared with wild-type controls (Fig. 7G). Importantly, the coronary flow was significantly reduced after ischemia in Gch1fl/flTie2cre hearts compared with wild-type hearts (Fig. 7I). In addition, in Gch1fl/flTie2cre hearts, infarct size, assessed by TTC staining, was significantly larger than in wild-type mice (Gch1fl/flTie2cre, 62 ± 6% of the risk region; wild type, 38 ± 7%, P < 0.05; Fig. 7, J and K). Collectively, these data demonstrate a critical role for endothelial cell Gch1/BH4 biosynthesis on coronary vascular function, cardiac function, and myocardial injury following ischemia-reperfusion injury.
Figure 7.
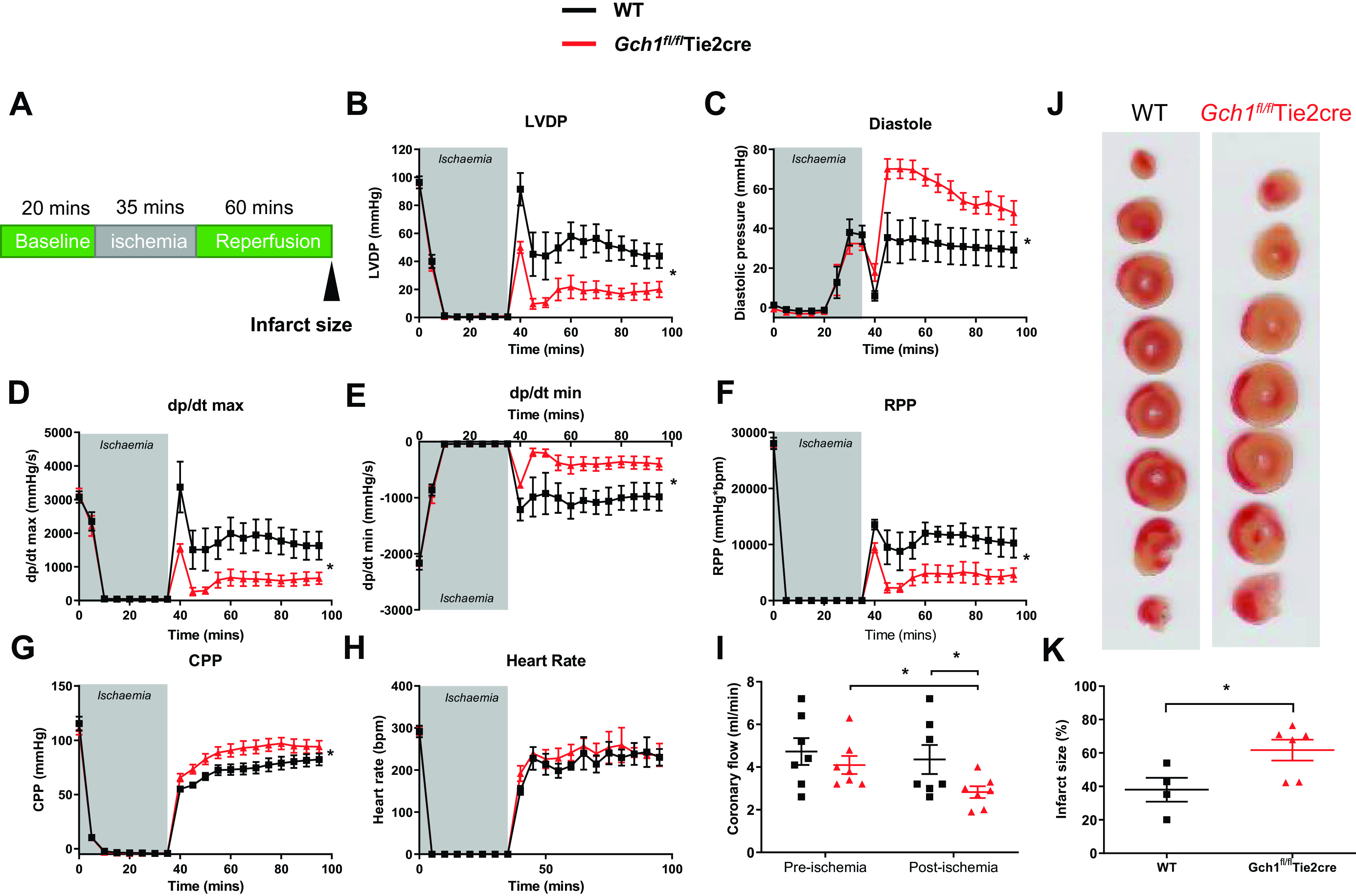
Loss of endothelial cell Gch1/BH4 biosynthesis leads to cardiac dysfunction and injury following cardiac ischemia-reperfusion. Cardiac function and infarct size were measured in isolated wild-type (WT) and Gch1fl/flTie2cre hearts perfused using Langendorff model. A: after baseline stabilization, wild-type and Gch1fl/flTie2cre hearts were subjected to 35 min of global ischemia and 60 min of reperfusion. B: LV developed pressure (LVDP). C: LV diastolic pressure. D: LV dP/dtmax. E: LV dP/dtmin. F: rate pressure product (RPP; mmHg). G: coronary perfusion pressure (CPP; mmHg). H: heart rate (beats/min). I: coronary flow was measured in hearts before ischemia and after periods of ischemia followed by 60 min of reperfusion (n = 7 per group). J and K: infarct size, defined by triphenyltetrazolium chloride staining and measurement as percentage of risk region, was significantly increased in hearts from Gch1fl/flTie2cre mice compared with wild-type littermate controls. Values are the means ± SE (*P < 0.05; n = 4–6 animals per group). Each data point represents an individual adult male mouse.
DISCUSSION
In this study, we used a mouse model of endothelial cell-targeted Gch1 deletion to test the specific requirement for endothelial cell BH4 in the regulation of cardiac function under both physiological and pathophysiological conditions. The major findings of this study are as follows: 1) endothelial cell-targeted Gch1 deletion leads to selective loss of endothelial cell BH4 in the myocardium; 2) loss of endothelial cell BH4 leads to NOS uncoupling, increased superoxide and hydrogen peroxide productions, and decreased NO bioactivity, resulting in cardiac dysfunction and mild myocardial hypertrophy; 3) selective loss of endothelial cell BH4 is associated with changes in cardiomyocytes, including increased nNOS protein, loss of eNOS protein, and increased in phospholamban phosphorylation at Ser17; and 4) specific loss of cardiac endothelial cell BH4 leads to coronary vascular dysfunction, cardiac dysfunction, and increased myocardial infarct size following ischemia-reperfusion injury. Collectively, these studies reveal specific effects of endothelial cell Gch1/BH4 biosynthesis on cardiomyocytes and on overall cardiac function under both physiological and pathophysiological conditions.
An important observation underpinning the interpretation of selective and specific roles for endothelial cell BH4 is that endothelial cell-targeted Gch1 deletion abolishes GTPCH protein expression and de novo BH4 biosynthesis in endothelial cells, and this loss of endothelial cell BH4 synthesis is not rescued by normal levels of BH4 in plasma or adjacent cells. This indicates that BH4 in endothelial cells is compartmentalized and is not amenable to uptake or recycling, at least under conditions of normal BH4 levels. High-level supplementation of sepiapterin (20, 21, 33) or BH4 (34, 35) has been shown to prevent NOS uncoupling and improved left ventricular function and I/R injury. However, it is not clear whether these effects of supraphysiological BH4 supplementation are mediated by known BH4 functions, and if so via which cell type and which mechanisms. Previous observations suggest that exogenous BH4 is not sufficient to rescue or augment BH4 levels in endothelial cells (30), whereas cardiomyocytes are amenable to exogenous BH4 supplementation (18, 23, 36).
Our study emphasizes the importance of the cross talk between the coronary endothelium and cardiomyocytes in cardiac function (37). We found that selective loss of endothelial cell BH4 led to changes in cardiomyocyte redox signaling, gene expression, and function. Increased superoxide and hydrogen peroxide generation in endothelial cell BH4-deficient hearts is consistent with the induction of fetal gene expression (nppa, nppb, and myh7) contributing to cardiac hypertrophy. Indeed, emerging evidence suggests that H2O2 can mediate and cause cardiac dysfunction, hypertrophy, and heart failure (38–40). In eNOS knockout mice, cardiac structure, LV function, and heart rate were similar to that of wild-type mice (41). These findings suggest that the combination of increased NOS-derived H2O2 and reduced NOS-derived NO production are likely to be important contributors to the development of cardiac dysfunction in Gch1fl/flTie2cre mice and contrasts with eNOS knockout mice where all functions of eNOS (i.e., both NO and ROS generation) are deleted.
Neuronal NOS (nNOS) has also been implicated in the regulation of basal and β-adrenergic inotropy in normal and chronically infarcted hearts. We confirmed that nNOS was upregulated and eNOS downregulated in cardiomyocytes in endothelial cell BH4-deficient mice. Consistent with these findings, increased nNOS and decreased eNOS expression have been observed in the failing rat and human hearts (42–44). It is possible that upregulation of eNOS-derived H2O2 generation from coronary endothelial cells from Gch1fl/flTie2cre mice affects eNOS protein and activity in the cardiomyocytes. Consistent with this idea, several reports have shown that H2O2 decreases eNOS protein expression and activity, at least in part by an inhibition of c-Jun activity and thus leading to a reduction in AP-1 transcription factor binding to the eNOS promoter (45–47). Thus, eNOS expression and activity have been found to be suppressed in the failing hearts, suggesting that the eNOS-mediated regulation of cardiac function and β-adrenergic response may be reduced in myocardial disease.
We have shown that Gch1fl/flTie2cre mice have a slightly increased (∼5–7 mmHg) in systolic blood pressure compared with wild-type littermate controls. Thus, it is possible that increased systolic blood pressure in Gch1fl/flTie2cre mice may contribute to cardiac dysfunction and hypertrophy in this study. However, this degree of blood pressure elevation is very modest and would not usually be considered sufficient to constitute a model of “hypertension.” Indeed, the eNOS knockout mouse has systemic hypertension with a 20–30-mmHg increase in systolic blood pressure, but at baseline, cardiac contractility was reported to be normal in eNOS knockout mice (48, 49). Therefore, it is unlikely that a mild increase in systolic blood pressure in Gch1fl/flTie2cre mice was responsible for the cardiac dysfunction and hypertrophy in this study.
Increased superoxide and hydrogen peroxide productions in the myocardium have been shown to impair calcium handling and induce pathological cardiac changes such as fibrosis, apoptosis, and hypertrophy (50–52). We found that endothelial cell BH4-deficient mice had increased phospholamban phosphorylation at the calmodulin-dependent kinase II (CaMKII)-specific site (PLB-Thr17) but not at the protein kinase A-specific site (PLB-Ser16). nNOS modulates cardiac relaxation via effects on phospholamban phosphorylation (53). PLB has inhibitory effect on SERCA activity and reuptake of Ca2+. Therefore, increased phospholamban phosphorylation at Ser17 abrogates the inhibitory effect of PLB on SERCA, thereby increasing SR Ca2+ reuptake and ultimately increased myocyte contractility and relaxation. This finding suggests that increased nNOS gene expression and protein in Gch1fl/flTie2cre hearts and cardiomyocytes may contribute to an increase in contraction and accelerated SR Ca2+ reuptake in cardiomyocytes, possibly by increased basal PLB phosphorylation.
Myocardial ischemia-reperfusion is associated with markedly elevated levels of ROS production (14, 54), and these ROS are central mediators of postischemic injury. In the postischemic heart, changes in coronary endothelial vascular function occur because of a reduction in eNOS-derived NO production which in turn impairs coronary flow (24). Evidence from isolated rat hearts suggests that ischemia-induced oxidative stress leads to enhanced BH4 oxidation (11, 24), contributing to postischemic eNOS uncoupling with a resultant loss of coronary endothelium-dependent vasodilation (24). Our observation of greatly increased coronary perfusion pressure (CPP), a direct measure of coronary vascular resistance, and reduced coronary flow in Gch1fl/flTie2cre hearts now demonstrates a specific role of endothelial cell BH4 in determining the response of the coronary microcirculation to ischemia-reperfusion. Importantly, we found that loss of endothelial cell BH4 caused a greater myocardial infarct size compared with wild-type hearts following I/R.
Collectively, these data demonstrate a critical role for endothelial cell Gch1/BH4 biosynthesis in coronary vascular function, cardiac function, and the response to ischemia-reperfusion injury. Thus, targeting endothelial cell Gch1 and BH4 biosynthesis may provide a novel therapeutic target for the prevention and treatment of cardiac dysfunction, ischemia injury, and heart failure.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1 and S2: https://doi.org/10.6084/m9.figshare.21732581.v1.
GRANTS
This study was supported by British Heart Foundation (BHF) Programme Grants RG/12/5/29576 and RG/17/10/32859, BHF Chair Award CH/16/1/32013, Wellcome Trust Grant 090532/Z/09/Z, BHF Centre of Research Excellence (Oxford) Grants RE/13/1/30181 and RE/18/3/34214, and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.C. and K.M.C. conceived and designed research; S.C., S.M.C., R.C., M.K., J.K.B., J.N.S., G.D., and M.J.C. performed experiments; S.C., S.M.C., R.C., M.K., J.K.B., J.N.S., G.D., and M.J.C. analyzed data; S.C., B.C., and K.M.C. interpreted results of experiments; S.C. prepared figures; S.C. and K.M.C. drafted manuscript; S.C. and K.M.C. edited and revised manuscript; S.C., S.M.C., R.C., M.K., J.K.B., J.N.S., G.D., M.J.C., B.C., and K.M.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful for the excellent support by the university facility staff of the Wellcome Trust Centre for Human Genetics (University of Oxford) for animal well-being.
REFERENCES
- 1. Kim H, Yun J, Kwon SM. Therapeutic strategies for oxidative stress-related cardiovascular diseases: removal of excess reactive oxygen species in adult stem cells. Oxid Med Cell Longev 2016: 2483163, 2016. doi: 10.1155/2016/2483163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal 20: 3040–3077, 2014. doi: 10.1089/ars.2013.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase - A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 4. Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J 362: 733–739, 2002. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, Channon KM, Alp NJ. Quantitative regulation of intracellular endothelial nitric oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: Insights from cells with tet-regulated GTP cyclohydrolase I expression. J Biol Chem 284: 1136–1144, 2009. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- 6. Channon KM. Tetrahydrobiopterin - regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med 14: 323–327, 2004. doi: 10.1016/j.tcm.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 7. Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BSS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225, 1998. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chuaiphichai S, Crabtree MJ, Mcneill E, Hale AB, Trelfa L, Channon KM, Douglas G. A key role for tetrahydrobiopterin- dependent endothelial NOS regulation in resistance arteries: studies in endothelial cell tetrahydrobiopterin-deficient mice. Br J Pharmacol 174: 657–671, 2017. doi: 10.1111/bph.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang L, Rao F, Zhang K, Khandrika S, Das M, Vaingankar SM, Bao X, Rana BK, Smith DW, Wessel J, Salem RM, Rodriguez-Flores JL, Mahata SK, Schork NJ, Ziegler MG, O'Connor DT. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest 117: 2658–2671, 2007. doi: 10.1172/JCI31093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mayahi L, Mason L, Bleasdale-Barr K, Donald A, Trender-Gerhard I, Sweeney MG, Davis MB, Wood N, Mathias CJ, Watson L, Pellerin D, Heales S, Deanfield JE, Bhatia K, Murray-Rust J, Hingorani AD. Endothelial, sympathetic, and cardiac function in inherited (6R)-L-erythro-5,6,7,8-tetrahydro-L-biopterin deficiency. Circ Cardiovasc Genet 3: 513–522, 2010. doi: 10.1161/CIRCGENETICS.110.957605. [DOI] [PubMed] [Google Scholar]
- 11. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin - efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 117: 2626–2636, 2008. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cubberley RR, Alderton WK, Boyhan A, Charles IG, Lowe PN, Old RW. Cysteine-200 of human inducible nitric oxide synthase is essential for dimerization of haem domains and for binding of haem, nitroarginine and tetrahydrobiopterin. Biochem J 323: 141–146, 1997. doi: 10.1042/bj3230141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimizu S, Ishii M, Kawakami Y, Momose K, Yamamoto T. Protective effects of tetrahydrobiopterin against nitric oxide-induced endothelial cell death. Life Sci 63: 1585–1592, 1998. doi: 10.1016/s0024-3205(98)00427-5. [DOI] [PubMed] [Google Scholar]
- 14. Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA 86: 4695–4699, 1989. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamashiro S, Noguchi K, Kuniyoshi Y, Koja K, Sakanashi M. Role of tetrahydrobiopterin on ischemia-reperfusion injury in isolated perfused rat hearts. J Cardiovasc Surg (Torino) 44: 37–49, 2003. [PubMed] [Google Scholar]
- 16. Settergren M, Bohm F, Malmstrom RE, Channon KM, Pernow J. L-Arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis 204: 73–78, 2009. doi: 10.1016/j.atherosclerosis.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 17. Perkins KAA, Pershad S, Chen Q, McGraw S, Adams JS, Zambrano C, Krass S, Emrich J, Bell B, Iyamu M, Prince C, Kay H, Teng JCW, Young LH. The effects of modulating eNOS activity and coupling in ischemia/reperfusion (I/R). Naunyn Schmiedebergs Arch Pharmacol 385: 27–38, 2012. doi: 10.1007/s00210-011-0693-z. [DOI] [PubMed] [Google Scholar]
- 18. Takimoto E, Champion HC, Li MX, Ren SX, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang YB, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115: 1221–1231, 2005. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silberman GA, Fan THM, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121: 519–528, 2010. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tiefenbacher CP, Lee CH, Kapitza J, Dietz V, Niroomand F. Sepiapterin reduces postischemic injury in the rat heart. Pflugers Arch 447: 1–7, 2003. doi: 10.1007/s00424-003-1131-y. [DOI] [PubMed] [Google Scholar]
- 21. Baumgardt SL, Paterson M, Leucker TM, Fang J, Zhang DX, Bosnjak ZJ, Warltier DC, Kersten JR, Ge ZD. Chronic co-administration of sepiapterin and L-citrulline ameliorates diabetic cardiomyopathy and myocardial ischemia/reperfusion injury in obese type 2 diabetic mice. Circ Heart Fail 9: e002424, 2016. doi: 10.1161/CIRCHEARTFAILURE.115.002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49: 241–248, 2007. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 23. Carnicer R, Hale AB, Suffredini S, Liu X, Reilly S, Zhang MH, Surdo NC, Bendall JK, Crabtree MJ, Lim GB, Alp NJ, Channon KM, Casadei B. Cardiomyocyte GTP cyclohydrolase 1 and tetrahydrobiopterin increase NOS1 activity and accelerate myocardial relaxation. Circ Res 111: 718–727, 2012. doi: 10.1161/CIRCRESAHA.112.274464. [DOI] [PubMed] [Google Scholar]
- 24. Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci USA 104: 15081–15086, 2007. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giraldez RR, Panda A, Zweier JL. Endothelial dysfunction does not require loss of endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 278: H2020–H2027, 2000. doi: 10.1152/ajpheart.2000.278.6.H2020. [DOI] [PubMed] [Google Scholar]
- 26. Tiefenbacher CP, Chilian WM, Mitchell M, DeFily DV. Restoration of endothelium-dependent vasodilation after reperfusion injury by tetrahydrobiopterin. Circulation 94: 1423–1429, 1996. doi: 10.1161/01.cir.94.6.1423. [DOI] [PubMed] [Google Scholar]
- 27. Aragón JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB, Gundewar S, Jha S, Calvert JW, Barouch LA, Lavu M, Wright HM, Lefer DJ. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol 58: 2683–2691, 2011. doi: 10.1016/j.jacc.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chuaiphichai S, Rashbrook VS, Hale AB, Trelfa L, Patel J, McNeill E, Lygate CA, Channon KM, Douglas G. Endothelial cell tetrahydrobiopterin modulates sensitivity to Ang (Angiotensin) II-induced vascular remodeling. Hypertension 72: 128–138, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chuaiphichai S, McNeill E, Douglas G, Crabtree MJ, Bendall JK, Hale AB, Alp NJ, Channon KM. Cell-autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension 64: 530–540, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chuaiphichai S, Yu GZ, Tan CMJ, Whiteman C, Douglas G, Dickinson Y, Drydale EN, Appari M, Zhang W, Crabtree MJ, McNeill E, Hale AB, Lewandowski AJ, Alp NJ, Vatish M, Leeson P, Channon KM. Endothelial GTPCH (GTP cyclohydrolase 1) and tetrahydrobiopterin regulate gestational blood pressure, uteroplacental remodeling, and fetal growth. Hypertension 78: 1871–1884, 2021. doi: 10.1161/HYPERTENSIONAHA.120.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chuaiphichai S, Starr A, Nandi M, Channon KM, McNeill E. Endothelial cell tetrahydrobiopterin deficiency attenuates LPS-induced vascular dysfunction and hypotension. Vascul Pharmacol 77: 69–79, 2016. doi: 10.1016/j.vph.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem 284: 28128–28136, 2009. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jo H, Otani H, Jo F, Shimazu T, Okazaki T, Yoshioka K, Fujita M, Kosaki A, Iwasaka T. Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice. Clin Exp Pharmacol Physiol 38: 485–493, 2011. doi: 10.1111/j.1440-1681.2011.05535.x. [DOI] [PubMed] [Google Scholar]
- 34. Xie L, Talukder MAH, Sun J, Varadharaj S, Zweier JL. Liposomal tetrahydrobiopterin preserves eNOS coupling in the post-ischemic heart conferring in vivo cardioprotection. J Mol Cell Cardiol 86: 14–22, 2015. doi: 10.1016/j.yjmcc.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Katano T, Ito S, Iwasaka T. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J Mol Cell Cardiol 50: 534–544, 2011. doi: 10.1016/j.yjmcc.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 36. Hashimoto T, Sivakumaran V, Carnicer R, Zhu GS, Hahn VS, Bedja D, Recalde A, Duglan D, Channon KM, Casadei B, Kass DA. Tetrahydrobiopterin protects against hypertrophic heart disease independent of myocardial nitric oxide synthase coupling. J Am Heart Assoc 5: e003208, 2016. doi: 10.1161/JAHA.116.003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang M, Shah AM. ROS signalling between endothelial cells and cardiac cells. Cardiovas Res 102: 249–257, 2014. doi: 10.1093/cvr/cvu050. [DOI] [PubMed] [Google Scholar]
- 38. Raut GK, Manchineela S, Chakrabarti M, Bhukya CK, Naini R, Venkateshwari A, Reddy VD, Mendonza JJ, Suresh Y, Nallari P, Bhadra MP. Imine stilbene analog ameliorate isoproterenol-induced cardiac hypertrophy and hydrogen peroxide-induced apoptosis. Free Radic Biol Med 153: 80–88, 2020. doi: 10.1016/j.freeradbiomed.2020.04.014. [DOI] [PubMed] [Google Scholar]
- 39. Song R, Zhang J, Zhang LJ, Wang GH, Wo D, Feng J, Li XC, Li J. H2O2 induces myocardial hypertrophy in H9c2 cells: a potential role of Ube3a. Cardiovasc Toxicol 15: 23–28, 2015. doi: 10.1007/s12012-014-9264-0. [DOI] [PubMed] [Google Scholar]
- 40. Steinhorn B, Sorrentino A, Badole S, Bogdanova Y, Belousov V, Michel T. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nature communications 9: 4044, 2018. doi: 10.1038/s41467-018-06533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 104: 448–454, 2001. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 42. Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, Ebrahimian T, Marotte F, Samuel JL, Heymes C. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J 17: 1–22, 2003. doi: 10.1096/fj.02-1208fje. [DOI] [PubMed] [Google Scholar]
- 43. Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 363: 1365–1367, 2004. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 44. Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation 110: 2368–2375, 2004. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 45. Kumar S, Sun X, Wiseman DA, Tian J, Umapathy NS, Verin AD, Black SM. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of Sp1 activity. DNA Cell Biol 28: 119–129, 2009. doi: 10.1089/dna.2008.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumar S, Sun XT, Wedgwood S, Black SM. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of AP-1 activity. Am J Physiol Lung Cell Mol Physiol 295: L370–L377, 2008. doi: 10.1152/ajplung.90205.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wedgwood S, Steinhorn RH, Bunderson M, Wilham J, Lakshminrusimha S, Brennan LA, Black SM. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 289: L660–L666, 2005. doi: 10.1152/ajplung.00369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang PL, Huang ZH, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric-oxide synthase. Nature 377: 239–242, 1995. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 49. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93: 13176–13181, 1996. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, Hirsch E, Lederer WJ, Shah AM. Contractile function during angiotensin-ii activation increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J Am Coll Cardiol 66: 261–272, 2015. doi: 10.1016/j.jacc.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joseph LC, Avula UMR, Wan EY, Reyes MV, Lakkadi KR, Subramanyam P, Nakanishi K, Homma S, Muchir A, Pajvani UB, Thorp EB, Reiken SR, Marks AR, Colecraft HM, Morrow JP. Dietary saturated fat promotes arrhythmia by activating NOX2 (NADPH oxidase 2). Circ Arrhythm Electrophysiol 12: e007573, 2019. doi: 10.1161/CIRCEP.119.007573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Donoso P, Finkelstein JP, Montecinos L, Said M, Sanchez G, Vittone L, Bull R. Stimulation of NOX2 in isolated hearts reversibly sensitizes RyR2 channels to activation by cytoplasmic calcium. J Mol Cell Cardiol 68: 38–46, 2014. doi: 10.1016/j.yjmcc.2013.12.028. [DOI] [PubMed] [Google Scholar]
- 53. Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res 102: 242–249, 2008. doi: 10.1161/CIRCRESAHA.107.164798. [DOI] [PubMed] [Google Scholar]
- 54. Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci 84: 1404–1407, 1987. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1 and S2: https://doi.org/10.6084/m9.figshare.21732581.v1.
Data Availability Statement
Data will be made available upon reasonable request.