

Summary
Animal testing is the current standard for drug and chemicals safety assessment, but hazards translation to human is uncertain. Human in vitro models can address the species translation but might not replicate in vivo complexity. Herein, we propose a network-based method addressing these translational multiscale problems that derives in vivo liver injury biomarkers applicable to in vitro human early safety screening. We applied weighted correlation network analysis (WGCNA) to a large rat liver transcriptomic dataset to obtain co-regulated gene clusters (modules). We identified modules statistically associated with liver pathologies, including a module enriched for ATF4-regulated genes as associated with the occurrence of hepatocellular single-cell necrosis, and as preserved in human liver in vitro models. Within the module, we identified TRIB3 and MTHFD2 as a novel candidate stress biomarkers, and developed and used BAC-eGFPHepG2 reporters in a compound screening, identifying compounds showing ATF4-dependent stress response and potential early safety signals.
Subject areas: Gene network, Bioinformatics, Transcriptomics
Graphical abstract
Highlights
-
•
A network-based method combining preclinical toxicogenomics and human disease data
-
•
An ATF4 network containing TRIB3 and MTHFD2 is associated with liver single cell necrosis
-
•
TRIB3 and MTHFD2 activation dynamics are translated across species
-
•
TRIB3 and MHTFD2 HepG2 reporter lines allow context-related screening for safety signals
Gene network; Bioinformatics; Transcriptomics
Introduction
Understanding when preclinical safety data is predictive of adverse outcome across species (e.g., rat to human) and scales of biological complexity (e.g., in vitro to in vivo) remains a vexing problem in drug discovery and development. The industry reports that unacceptable safety accounts for more than 50% of project closures, and that in particular failure to detect a safety signal in preclinical screens using in vitro, in vivo or in silico tools is responsible for ∼40% of safety attrition.1
Safety biomarkers are essential tools to monitor early signals for adverse events in clinical trials and ensure that effects are reversible with drug withdrawal. However, current clinical safety monitoring typically depends on traditional biomarkers that co-occur with toxicity and may not translate from preclinical studies to human clinical trials. An ideal translational safety biomarker would show a high association with pathology and be scalable (multiscale) from in vitro screens to in vivo outcomes, allowing hit and lead compound series to be rapidly triaged with confidence. In addition, it would translate across species so that results can be incorporated into a multiscale and translational decision framework designed to advance safer compound into clinical testing.
Liver is a common target organ for drug toxicity because of its role in absorption and metabolism, consequently drug induced liver injury (DILI) is a frequent and problematic safety concern.1,2 For these reasons considerable research focused on generating and leveraging animal-based liver datasets to investigate mechanisms involved in DILI and identify safety biomarkers.3,4,5 Although time-consuming and costly, animal (preclinical) studies remain the bulwark against advancing hepatotoxic compounds to the clinic, but the translational relevance of preclinical liver injury to human clinical trials is uncertain and there are no standard methods to determine the likelihood that a novel safety biomarker will translate across species.6 Thus, methods to identify early safety markers that have clinical relevance, i.e., preservation across species, as well as suitable platforms for cost-efficient early testing to avoid advancing problematic compounds, would be a significant advance in mechanism-based risk assessment practice.
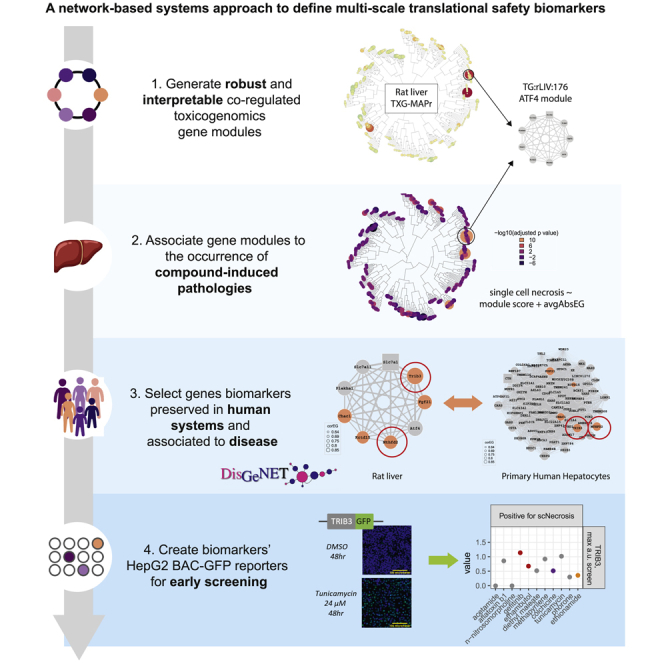
System biology and in particular network-based methods are uniquely suited to addressing the problem of translation across scales of complexity and species. System biology methods have been widely used across multiple domains, their success associated with the strength of analyzing networks of interaction rather than elements in isolation.7,8 Previous work demonstrates that co-regulated gene networks (modules), derived from in vivo transcriptomic data, with associated biologically relevant interpretation are statistically associated with the occurrence of liver toxicity phenotypes (pathology) in vivo5,9,10 Herein, we use weighted gene co-expression network analysis (WGCNA) to derive modules from the TG-GATEs (TG) dataset,11 and evaluate the robustness by comparing network preservation with co-expression modules derived from a separate rat liver dataset.5,12 We further leverage network preservation statistics13 to evaluate translation from preclinical to human relevant test systems using primary human hepatocytes and HepG2 transcriptomic data and identify novel candidate multiscale translational safety biomarkers. We anchor mechanistic interpretation based on network genes to databases of gene-disease associations and human expression14,15 to establish clinical relevance. Using this approach, we identified a novel ATF4/Atf4 regulated module robustly associated with hepatocellular single cell necrosis (apoptosis) in vivo. Gene co-expression is preserved across both rat liver datasets and as well as transcriptomic datasets from primary human hepatocytes (PHH) and HepG2.11 From this module, we identify TRIB3 and MTHFD2 as promising candidate translational biomarkers and developed HepG2 BAC-eGFP reporters useful for early drug screening in vitro. These reporter cell lines can be used to inform during early drug development about biological processes associated with in vivo adverse events and DILI risk and together with other safety signals make decisions about compound prioritization, as we demonstrated in a compound screen. In conclusion, we took advantage of large dataset of legacy in vivo data and created a pipeline approach to derive translationally conserved biomarkers that bridge between species and in vivo-to-in vitro gaps, as well as inform about the mechanisms underlying chemical-induced pathology.
Results
Rat liver gene co-expression modules consistently reflect pathogenesis across data sources
Previous work using co-expression modules derived from the Drug Matrix (DM) dataset demonstrated the utility in understand mechanisms of injury and linking network of genes to liver pathology.5,12 Before addressing translation of co-expression patterns from rat to human, we first tested the robustness and reproducibility of the co-expression patterns within the same test system, rat liver, by comparing the TG-GATES (TG) and DM datasets.11 We generated co-expression modules from the rat liver TG dataset, which contained a much larger set of treatments, some of which overlap between datasets, and treatments in both single- and repeated-dosing designs, and assessed preservation of co-expression and association with pathology. We obtained 316 co-expression modules (indicated further on as TG:rLIV) that include genes with similar correlation patterns across the 3528 experiments (time, compound, and dose combinations) (Table S1). To determine the robustness of the co-expression approach and to test the utility of preservation statistics to detect conservation of module topology, we calculated TG-modules preservation against the rat liver DM gene expression matrix and the previously derived set of modules based on DM data5 (Figure 1A and Table S1): 306 modules out of the 316 TG modules were preserved (Z summary ≥ 2) suggesting that co-expression is independent of the experimental system and that liver modules derived from the same species and target organ are showing moderate to high preservation. Most of the non-preserved modules showed very high eigengene scores (EGs) for cycloheximide, which is missing in the DM dataset (Table S1). Modules in DM (indicated further on as DM:rLIV) and TG builds whose gene sets significantly overlapped (Fisher test) had better EGs correlation (module’s EGs calculated with TG data for both builds) than modules that do not overlap (Figure 1B and Table S2). Only 16 TG-modules did not show a significant (pvalue <0.05) overlap with any DM-modules, among them modules TG:rLIV:122, 212 and 214, enriched for processes related to immune response (Table S1).
Figure 1.

Gene co-expression modules for TG-GATEs rat liver dataset
(A) Number of TG-based modules that show low (Z-summary <2), medium (2≤ Z-summary <10) and high (Z-summary ≥10) preservation toward DM rat liver dataset.
(B) Boxplot of Pearson correlation values between EGs of overlapping or non-overlapping modules pairs (color) between the 2 builds, shown by significance level of overlap (xaxis).
(C) −log10(pval) overlap between the gene sets consisting of the top 20, or bottom 20 associated modules in TG-GATEs to each toxicity phenotypes or 20 randomly selected modules in TG-GATEs and the gene sets consisting of the top 20, bottom 20 associated modules in DM to each toxicity phenotypes and 20 randomly selected modules in DM. The red solid line indicates-log10(0.0005). Toxicity phenotypes are abbreviated as specified in Table S3.
(D) Dendrogram of TG-modules, showing-log10 transformed adjusted p values for the association of each module with the occurrence of single cell necrosis (scNecrosis4). The size of each circle is proportional to the absolute log10(adjusted p value) and the color to the-log10(adjusted p value). Highlighted and zoomed out below the structure of module TG:rLIV:176. Each node corresponds to the genes included in the module, the square node corresponds to the hub gene and the edges weight indicates the correlation between each genes’ log2FC across TG-data.
(E) Count of experiments showing EGs for module TG:rLIV:176 higher than 2 or lower than −2 (points connected by lines) and showing positive score for the toxicity phenotype scNecrosis 4 (bar plots). Different colors indicated the three dosage levels.
We then focused on the concordance of module to pathology associations by comparing module-pathology association in TG and DM modules using the same classification scheme for toxicity phenotypes (pathology) and level described previously (lower levels have fewer co-occurring pathologies, see (5, Table S3 and Figure S2A). Considering that modules are dynamic structures, we collapsed the top 20 modules associated with each toxicity phenotypes for each module build into one gene set for each toxicity phenotype, as well as the bottom 20 associated and a random selection for background comparison. Top associated gene sets overlapped across module builds with highly significant p values (Fisher’s test) with the exception of vacuolation (Figure 1C, red solid line indicates p = 0.0005). Similar results are obtained when considering one-to-one module comparisons (data not shown). Overall, the data suggest that co-expression modules in the same species and organ are preserved and comparable information content and pathology associations are maintained.
Finally, we focused on the preserved TG-modules associated with early response and pathologies reasoning these were more likely to represent cell-autonomous responses associated with early toxicity signals. In particular, module TG:rLIV:176 scored first for association with the toxicity phenotype single cell necrosis, a descriptive morphological term indicating lesions comprising scattered dead cells of apoptotic or oncotic morphology,16 although in liver pathology it is generally considered synonymous with apoptosis17 (scNecrosis4, selectivity level 4, from now on labeled scNecrosis, Figure 1D). Higher scores for module TG:rLIV:176 and positive single cell necrosis scores are more frequently found in short exposure experiments (<1day), than repeated and late exposure experiments (Figure 1E). Module TG:rLIV:176 contained the Atf4 transcription factor and three high confidence Atf4-target genes (Fgf21, Trib3 and Chac1) and three lower confidence Atf4 target (Slc7a1, Slc7a11 and Mthfd2), TG:rLIV:176 was also enriched for terms related to apoptosis (GO:0070059, pvalue <0.001), response to unfolded protein (GO:0006986, pvalue <0.001) and PERK-mediated unfolded protein response (GO:0036499, pvalue = 0.0074), indicating this module could represent a more general integrated stress response (ISR) gene network (Table S1).
In summary, we constructed co-expression modules from the TG-GATEs dataset, found high within-species preservation with another rat perturbation dataset, and identified specific modules showing high preservation and overlapping properties. In particular, we selected TG:rLIV:176 as an exemplar candidate module with high within-species preservation to select biomarker genes for monitoring risk of early single cell necrosis in vivo, and to further investigate its potential for translational to human test systems.
Preservation in human in vitro systems and prioritization of candidate translational safety biomarkers
An ideal safety biomarker would translate across boundaries defined by scales of complexity, e.g., in vitro to in vivo, and species, e.g., rat to human. There is little well annotated transcriptomic data available from in vivo human liver samples with drug-induced toxicity, therefore, we evaluated the preservation of network topology of TG-based rat in vivo modules toward two separate human in vitro datasets, the public PHH TG-GATEs dataset and an in-house dataset of HepG2 cell line exposed to multiple DILI compounds (11,13, manuscript in preparation, Figure 2A right panel in logarithmic scale, Table S4). This approach allowed us to address translation across scales of complexity and species in a stepwise fashion by first comparing rat liver modules to a rat primary hepatocytes dataset, also available in TG-GATEs (RPH, Figure 2A left panel in logarithmic scale,11) and then preservation with PHH and HepG2. Of the 316 rat liver modules, 214 had a Z-summary score ≥2 and a Median Rank of <100 and were considered preserved in the intra-species test.13,18 For the more complex comparisons introducing differences in both scale and species (rat liver versus PHH) only 92 modules were preserved. Even fewer modules were preserved when considering a human in vitro model more distant from in vivo hepatocyte physiology (rat liver versus HepG2, 49 preserved modules). Stress responses to oxidative stress and DNA damage are often considered signals for adverse outcomes.19 Of interest, only early responding modules enriched for Nrf2 target genes (TG:rLIV:108, TG:rLIV:101) were preserved in the human sets, whereas late responding Nrf2 modules (TG:rLIV:262, TG:rLIV:49) and modules in the Tp53 DNA-damage response (TG:rLIV:288, TG:rLIV:19) were not among the preserved set.
Figure 2.

TG-modules preservation towardin vitro datasets
(A) Visual representation of the suggested comparisons, then on the left scatterplot of the natural logarithm transformed Z-summary of TG-modules toward PHH data (xaxis) and RPH data (yaxis). Each dot correspond to a module and the color indicates whether it is associated with single cell necrosis (top 10), any pathology (top 10), or none in the top 10. The shape indicates Median Rank statistic, the lower the more preserved. On the right, same visualization but for the natural logarithm transformed Z-summary of TG-modules toward PHH data (xaxis) and HepG2 data (yaxis).
(B) Modules’ dendrograms of TG rat liver (right) and PHH (left). Circles correspond to modules and color and size to EGs for the conditions of Tunicamycin 300 μg/kg 9 h (rat liver) and Tunicamycin 10 μg/mL 8 h (PHH). Highlighted and zoomed out in the center, overlapping modules TG:rLIV:176 and PHH:15. Each node corresponds to the genes included in the module, the square node corresponds to the hub gene and the edges weight indicates the correlation between each genes’ log2FC across TG-data. In salmon the overlapping genes between the two modules.
(C) Tables indicating the main enriched terms and TFs for the two modules shown in B.
By contrast with other stress networks, TG:rLIV:176, the Atf4 enriched module, and TG:rLIV:118 (both had Z summary ≥ 2 and Median Rank <100 for RPH, and PHH and HepG2 datasets) were among the preserved set of modules and ranked first and second for single cell necrosis, (Figure 2A in logarithmic scale, Table S5 and Figure S2B). Of interest, module TG:rLIV:118 included Ddit3, a well-known ER stress and ISR inducible protein under the direct transcriptional control of Atf4,20 already in the interest of our research and for which we previously developed the stress reporter cell line HepG2 CHOP-GFP21 Using previously generated PHH WGCNA modules,18 we looked for PHH modules with significant gene overlap (Table S6). Module TG:rLIV:176, overlaps with PHH:15 (Figure 2B, Fisher’s overlap test, p value 3.0E-09), and the comparison of modules pathway and TF enrichment strengthened the conclusion that the biological processes reflected in the module genes are preserved across systems (Figure 2C).
We next prioritized genes overlapping between TG:rLIV:176 and PHH:15 to identify candidate biomarker genes relevant for translation from non-clinical to human risk assessment and suitable for screening using higher throughput microscopy-based fluorescent human in vitro reporters.21 For each candidate gene, we took into account the correlation with the EG, or CorEG, to reflect how well the gene reflects the response of the entire network18 and the degree of perturbation in TG-GATEs, as well as human liver expression, from the human protein atlas (HPA) and information on associations to known human liver disease phenotypes (DisGeNet) (Figure 3A). FGF21 and TRIB3 had high corEG and were positively perturbed by a large number of conditions in TG-GATEs in both PHH and rat liver. With complementary methods (principal component analysis applied to the gene matrices), overlapping genes between species showed still high contributions to the obtained principal components (Tables S7 and S8). Protein levels were found to be elevated in the liver compared to the other tissues (enriched or enhanced, Human Protein Atlas) and both genes are associated with a number of hepatic disease phenotypes in DisGeNet including Liver Fibrosis, Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis (Figure 3A). Fibroblast growth factor 21 (FGF21) is a secreted growth factor (https://www.proteinatlas.org/ENSG00000105550-FGF21) and, therefore, less suitable as a target for in vitro screening methods based on microscopy detection of fluorescent reporters. Another module gene, Tribbles homolog 3 (TRIB3), is a nuclear located pseudokinase with regulatory function toward MAPK, directly regulated by ATF4, and part of the ISR involved in DDIT3/CHOP-dependent cell death following ER stress and ISR.22,23 methylenetetrahydrofolate dehydrogenase (MTHFD2), is a mitochondrial enzyme involved in folate metabolism, suspected to also serve in an antioxidative role but most widely known for its role in cancer progression and increase of proliferation.24 MTHFD2 was predicted as target of Atf4 (although with low confidence) but is associated with different hepatic disease phenotypes compared to TRIB3 suggesting distinct roles liver damage and recovery.
Figure 3.

TRIB3 and MTHFD2 are promising candidate translational biomarkers for in vivo single cell necrosis
(A) Properties of the overlapping genes between TG:rLIV:176 and PHH:15.
(B) Dose-response plots of modules and genes responses across systems for tunicamycin (TUN) and diethyl maleate (DEM). In the top row, module TG:rLIV:176 EG calculated with rat in vivo, or PHH, or HepG2 log2FC. In the middle and bottom row, log2FC of MTHFD2 and TRIB3 across rat in vivo (Low/Medium/High doses respectively: TUN: 30, 100, 300 μg/kg, DEM: 80, 240, 800 mg/kg), PHH (Low/Medium/High doses respectively: TUN: 0.4, 2, 10ug/mL, DEM: 60, 300, 1500 μM) and HepG2 (Low/Medium/High/vHigh doses respectively: TUN: 0.2, 1, 5, 10 μM, DEM: 3, 16, 80, 160 μM). The shape indicates whether the log2FC is associated with an adjusted p value lower than 0.1.
(C). Boxplot (and over imposed jittered points) of rLIV:176 maximum EGs over time and concentration (left panel), Trib3 (rat, middle panel) and TRIB3 (PHH, right panel) maximum log2FC over time and concentration, for all TG compounds, ranked as vMost-DILI-Concern (Positive) or vNo/vLes—DILI-Concern (Negative) (xaxis). The size of the circles is proportional to the average score of single cell necrosis in rat. The green dotted line represents EGs = 2 for the top panel, and log2FC = 0.5 for the mid and lower panel.∗ indicates p value <0.05 in a Student’s ttest.
In summary, we prioritized genes belonging to module TG:rLIV:176 to select biomarkers by comparing with in vitro human datasets, association to diseases and tissue expression in human. Based on these results, we selected MTHFD2 and TRIB3 as human liver disease relevant candidates for development of an in vitro reporter gene assay using a HepG2 platform.21
TRIB3 and MTHFD2 respond similarly across rat in vivo and human in vitro systems
Before creating the reporter cell lines, we assessed how well expression of TRIB3 and MTHFD2 transcripts correlated with module EGs in all three systems (rat liver, PHH and HepG2) using two compounds tested in all three experimental models in the considered datasets, i.e., tunicamycin (TUN) a known ER stressor and diethyl maleate (DEM) which activates the Nrf2-dependent oxidative stress response (Figure 3B). We calculated EGs for module TG:rLIV:176 using log2FC from the three test systems (as described in the section “preservation analysis and calculation of module scores for external datasets” of STAR Methods). In all three systems, TG:rLIV:176 responded with both stressors, peaking at 8 h or earlier for DEM in vivo. Both genes followed the same trend as the modules’ EGs, TRIB3 showing higher log2FC values. Of interest, the HepG2 cell line showed good sensitivity at lower concentrations at 24 h, particularly to tunicamycin, suggesting it could be a suitable timepoint for compound screening.
Module TG:rLIV:176 EG scores, as well as the individual Trib3 gene log2FC in rat in vivo and TRIB3 log2FC in PHH, were activated by multiple compounds, not just those inducing single cell necrosis in vivo (Figure 3C, size of the circles indicating the single cell necrosis average score, Figure S4B for MTHFD2). We then grouped compounds for the DILI concern score,25 to evaluate whether the gene activation levels could correlate with the DILI risk. Compounds with ranked as “vMost-DILI-Concern” (Positive in Figure 3C)(25) showed on average stronger activation of module rLIV:176 scores, Trib3 log2FC in rat liver and TRIB3 log2FC in PHH than compounds ranked as “vNo-DILI-Concern” or “vLess-DILI-Concern” (Negative in Figure 3C). Of interest, TRIB3 in PHH showed stronger differences between positive and negative DILI categories (p value in Student’s ttest 0.012, compared to Trib3 in rat, p value of 0.033), as well as less sensitivity to one false positive (Ethionamide, Figure 3C) indicating a possible broader human DILI relevance.
In summary, we confirmed similar induction patterns of the selected biomarkers in the three evaluated test systems. Thus, the selected candidate biomarker genes capture a common ATF4-mediated stress response to multiple upstream initiating events and are connected to a common adverse outcome suggesting their activation in both in vivo rodent and in vitro human models can be indicative of a general DILI risk in patients.
Transcriptional regulation of candidate safety translational biomarkers by ATF4
Having established that TG:rLIV:176 is conserved across systems, including HepG2, we next generated BAC-eGFP HepG2 cell lines for the two novel candidate translational safety biomarkers, TRIB3 and MTHFD2, to be used for compound screening (Figure 4A). TRIB3 and MTHFD2 genes were tagged with eGFP within a bacterial artificial chromosomes (BAC) construct containing at least 10 kb of the gene flanking regions ensuring physiological regulation of the reporter response. The recombinant BACs were transfected into HepG2 cells and clonal HepG2 MTHFD2-eGFP and HepG2 TRIB3-eGFP cell lines were established. The increase in expression and localization of the TRIB3-eGFP and MTHFD2-eGFP signal in the nucleus (Figure 4B) and cytoplasm (Figure 4F), respectively, after tunicamycin exposure confirmed that both fusion proteins were induced and located in the appropriate cellular locations (https://www.proteinatlas.org).
Figure 4.

HepG2 BAC TRIB3-eGFP and MTHFD2-eGFP reporters functionality results
(A) Schematic of the BAC recombineering process and establishment of HepG2 BAC eGFP reporter cell line.
(B and F) Microscopic images (scale bar is 100 μm) of siRNA knock down in HepG2 BAC TRIB3-eGFP cells/HepG2 BAC MTHFD2-eGFP cells. Solvent control (DMSO), tunicamycin C exposure at a concentration of 6 μM (Tun. 6 μM), transfectant control (mock). siRNA knockdown of TRIB3 (siTRIB3), MTHFD2 (siMTHFD2), DDIT3 (siDDIT3), ATF4 (siATF4). Top row pictures are the images in the Hoechst channel (Hoechst), and second row contains the eGFP channel images (eGFP) at 48h after exposure.
(C and G) Nuclear/cytoplasmic eGFP intensity of HepG2 BAC TRIB3-eGFP cells/HepG2 BAC MTHFD2-eGFP after siRNA knock down immediately after (0) or 48 h after exposure (48).
(D and H) mRNA fold change of HepG2 BAC TRIB3-eGFP cells/HepG2 BAC MTHFD2-eGFP cells after exposure to solvent control (DMSO) or to 24 μM of tunicamycin C (Tunicamycin) at 48 h after exposure.
(E and I) Western blot of HepG2 BACTRIB3-eGFP cells/HepG2 BAC MTHFD2-eGFP 48 h after exposure to medium control, solvent control or a dose range of tunicamycin C.
The physiological regulation of the HepG2 eGFP reporter cell lines were investigated by targeted knock down of the reporter genes as well as transcription factors ATF4 and DDIT3 because both are reported in the Dorothea database26 as “high confidence” regulators of TRIB3 transcription. Gene knock-down with siTRIB3, siDDIT3 and siATF4 in HepG2 TRIB3-eGFP cells reduced the eGFP signal in the nucleus as expected (Figure 4C). siMTHFD2 transfection in HepG2 MTHFD2-eGFP cells led to a decreased eGFP signal on tunicamycin exposure, but siDDIT3 knock down did not decrease the eGFP signal, and siATF4 transfection only slightly reduced it compared to mock transfection (Figure 4G). These results are consistent with the Dorothea database information (no regulation of MTHFD2 by DDIT3, and low confidence, D level, for ATF4/Atf4 direct regulation) and suggest a possible indirect regulation of MTHDF2 by the ATF4/Atf4 pathway that contributed to inclusion of MTHFD2 in modules TG:rLIV:176 and PHH:15. We further validated the linkage between the eGFP signal and differential expression of the target genes, the transcript levels and protein levels of TRIB3 and MTHFD2 by RT-qPCR and western blot (Figures 4D and 4E, 4G–4I, and S5–S10). In summary, we created HepG2 reporter cell lines for the selected biomarkers and confirmed and expanded the current knowledge of transcriptional regulation of TRIB3 and MTHFD2.
HepG2 in vitro reporters for TRIB3 and MTHFD2 generate general translational safety signals
Having defined the reporter functionalities and regulation, we investigated their utility in compound screening to generate translational safety signals as well as the correlation with the known pathology outcome in rat, scNecrosis, typically the first preclinical species tested for safety concerns. We selected 24 compounds belonging to four main categories: 12 compounds inducing single cell necrosis in vivo with an average histology score higher than 0.66 in at least one dose-time point combination (“Positive compounds for scNecrosis”), 3 compounds that did not induce any pathological outcome in TG-GATEs (“Negative for scNecrosis no known pathology”), 7 compounds annotated for liver relevant pathologies other than single cell necrosis, and 3 compounds not tested in TG-GATEs but are either prototypical ATF4 activators or have DILI concerns based on literature (“No data in TG-GATES”) (Table S9, see details regarding compound selection in the STAR Methods “compound screening”). We included the DILIRank status in the analysis, when available, to indicate human patient risk. We also compared the results to CHOP using a previously established HepG2 CHOP-eGFP cell line21; CHOP is integral to the ATF4-mediated ISR and is encoded by DDIT3 which is a member of TG:rLIV:118, the second ranked single cell necrosis module which is also preserved in PHH and HepG2.
Figure 5A shows the maximum TG:rLIV:176 EG scores found in the rat studies, and the maximum fraction of positive cells values obtained with the focused ISR reporter panel for each compound after screening 7 concentrations and 3 time points (24, 48 and 72 h, the complete heatmap is shown in Figure 5B and for of all readouts in Figure S8B). Thapsigargin and tunicamycin, prototypical ER stress inducers (indicated with arrows in Figure 5A), both showed strong activation of all three cell lines and clustered together when considering the whole time and concentration range for all three reporters (Figure 5B). Generally, MTHFD2 showed lower values and a downregulation at 24 h for most compounds potentially because of higher eGFP levels at baseline (Figures S8B and 4F) compared to CHOP and TRIB3. TRIB3 showed an improved sensitivity over CHOP, perhaps because of the higher dynamic range of the protein expression (Figure S14).
Figure 5.

HepG2 BAC-eGFP reporter screening results of CHOP, MTHFD2 and TRIB3
(A) Scatterplot of rLIV:176 maximum EGs over time and concentration in TG rat in vivodata, HepG2-TRIB3, HepG2-MTHFD2 and HepG2-CHOP reporter maximum fraction of positive cells over time and concentration in the screening, for all tested compounds (xaxis). The color indicated the DILI Rank status, when available. Panels indicate the rat pathology category of each compound.
(B) Heatmap of GFP positive fraction results of screened compounds annotated with rat pathologies category and the DILI Rank status. Hierarchical clustering is based on Ward clustering including Ward’s clustering criterion. On the right, the concentration range tested is shown (bar) with the Cmax value (salmon dot), when available (Table S9).
Generally, scNecrosis compounds (Figure 5, most left panel) that had high EG scores for TG:rLIV:176 in rat liver also had high maximum fraction of positive HepG2 reporter cells, especially for HepG2-TRIB3 (the intermediate activity cluster in Figure 5B). Two scNecrosis compounds (acetamide and N−nitrosomorpholine) that showed no activity in the reporter cell lines also had low EGs for the rat module, suggesting a non-ATF4 mode of action, and are present in the low activity cluster (Figure 5B) together with acetamidofluorene which had a pathology mechanistically distinct from hepatocellular scNecrosis (hyperplasia of bile duct epithelia, Table S9 and S7), and the three “no pathology in liver” compounds triamterene, famotidine and cimetidine. Aflatoxin B1 showed higher values in the reporter cell lines, likely because of early onset of cytotoxicity (Figure S14). The three compounds without data in TG-GATES showed high reporter responses (Figure 5A, and in the cluster of high activity in Figure 5B). Their risk is confirmed by the DILIRank (“vMost-DILI-Concern” for nefadozone) and the literature information on their ability to induce apoptosis (Thapsigargin and tert butyl hydroxyanisol in the Comparative Toxicogenomics Database, Table S11). Compounds that showed no scNecrosis in rat and no other pathologies, did not respond in our reporters (Figure 5). Some compounds inducing pathologies other than scNecrosis were inducing the reporter response and intermediate activation EGs for rLIV:176 (mid panels, Figure 5A). Of interest, compounds ranked as “vMost-DILI-Concern”, like diclofenac, or the “v-Less-DILI-Concern” omeprazole triggered early safety signals using the reporters, in particular the HepG2-TRIB3. Also, other “v-Less-DILI-Concern” compounds showed activation of the reporters, simvastatin and dexamethasone, but showed lower DILIRank severity (3, compared to 4 of omeprazole) and rLIV:176 EG compared to omeprazole and activity occurs at concentration exceeding 100∗Cmax (Figure 5B).
In summary, we challenged the reporter cell lines with a compound screen including TG-GATEs compounds inducing a variety of rat liver pathologies and belonging to several DILI categories. Altogether, our data demonstrate that TRIB3 activation in vitro correctly identifies compound with scNecrosis in vivo but also represents a more general safety signal in vitro, suggesting risk that should be considered in the context of dose and intended therapeutic use.
Discussion
Safety decisions are context dependent - tolerance for false positives (predicted unsafe but safe) versus false negatives (predicted safe but unsafe), and may differ depending on the stage of development and the therapeutic indication (e.g., cancer versus obesity). Unacceptable safety findings can arise at all stages of drug discovery and development from early screening to post-marketing withdrawals.27 High rates of attrition, particularly late in development, or post-marketing, contribute to low pipeline efficiency and high opportunity cost for testing clinical hypotheses; late failures put patients at risk.27,28 Underlying safety failure for leads, drug candidates and marketed drugs are three distinct translational safety problems – (1) Lack of translation across scales of complexity when lead compounds transition from in vitro safety testing to preclinical testing in vivo; (2) lack of translation across scales of evolution when a clinical candidate transitioning from preclinical species to clinical testing in human; and, (3) inability to scale safety data from controlled clinical trials to diverse populations subject to complex genetic and environmental influences.
In this investigation we addressed the first two translational safety problems using a network-based approach to determine when safety signals are more likely to translate from one complex system to another. We built a set of co-expression modules that capture information from gene networks responsive to drug toxicity in rat liver and used network preservation analysis to determine which networks are preserved from liver in vivo to hepatocytes in vitro within and across species boundaries. In the set of preserved modules, we identified a gene network (TG:LIV:176) associated with an adverse outcome (single cell necrosis) enriched for gene markers associated with the ISR. Human homologues for two of the highly connected genes, TRIB3 and MTHFD2, are also associated with human liver disease phenotypes. Using the human liver cell line HepG2, we identify ATF4, a critical regulator of the ISR23 as an upstream transcription factor. We then identified candidate safety biomarker genes from TG:rLIV:176 and developed a high throughput screen to generate safety signals before transition to non-clinical studies. Taken together, our results suggest a stepwise network-based approach to identify biomarker genes signals that translate across scales of complexity and can add confidence to early safety decisions in context. The third translational problem, not addressed in this research, could be further included by considering and comparing large human disease datasets to controlled (pre)-clinical data, focusing on biomarkers that show variations in a diverse human population disease model.
Disease processes, of which drug-toxicities are a subset, can be viewed as emergent properties of complex adaptive systems in response to shifts in the environment.29 Network-based approaches, like WGCNA, are ideally suited for modeling the behavior of complex adaptive systems. Unlike signature-based classification approaches to drug toxicity,30 WGCNA does not provide any a priori information on compound classification, rather it defines self-organizing co-expressed modules of genes within a high dimensional data matrix of genes responses to variation in dose, time and treatment. A unique feature of network-based methods is the ability to measure preservation of node-edge relationship across systems.13 We exploited this property to address the conservation of biological response networks from in vitro to in vivo and across species from rodent to human. By comparing gene networks from two large rat liver toxicity datasets, TG-GATEs and Drug Matrix, we show that modules in the two datasets are highly concordant, suggesting that the modular approach is overcoming inter-dataset reproducibility, increasing the confidence of transcriptomic data source for drug/chemical risk assessment.31 We then addressed the first translational problem, translation across scales of complexity, by comparing the rat-based liver networks to rat primary hepatocytes in vitro. We found a large number of modules (21⅔16) that show mild to high preservation. Conversely, comparing TG:rLIV modules with PHH showed lower preservation (37 modules were preserved in both PHH and HepG2). In general, preservation was higher for modules annotating for processes with a high degree of evolutionary conservation such as cell cycle, RNA processing, fatty acid metabolism, mitochondrion, as well as some early stress responsive modules, such as oxidative responsive, immune process, ER/Golgi and proteasome. These results confirmed previous findings concerning preservation of PHH co-expression modules toward rat-based systems.18 The lack of preservation according to our analysis can be interpreted in multiple ways: modules not preserved could include genes that have no unique ortholog in humans, that are lowly expressed in humans, or for which the dynamic of activation differ between the species, leading to a different network structure. Further studies are necessary to shed more light in these possible interpretations of the lack of preservation, however our results support the application of network preservation approaches to tackle translational components of complex adaptive systems.
To demonstrate the utility of the approach, we focused on preserved module linked to early pathologies, e.g., single cell necrosis (apoptosis), expecting that early cell autonomous events might be better captured in less complex in vitro systems (Figure S2C). Modules associated with single cell necrosis in vivo (TG:rLIV:176, TG:rLIV:118) were preserved in all in vitro systems and enriched for ATF4-governed response in each model. Module TG:rLIV:176 contained a number of candidate safety biomarkers applicable to a higher throughput format for compound screening. We confirmed ATF4 transcriptional regulatory role for two of the candidate markers present in both rat and PHH modules, TRIB3 and MTHFD2 by developing HepG2 BAC reporter cell lines and by performing specific siRNA knockdowns. Of interest, the two genes show different regulation (for example, by DDIT3), despite being part of the same module. Transcriptional responses are multi-factorial, e.g., involving more than one TF, thus assuring system robustness in their adaptive behavior. The fact that TG:rLIV:176 is highly preserved suggests that complex interactions across elements of multiple systems are preserved and captured by co-expressed gene sets in all systems. The two candidate markers, TRIB3 and MTHFD2, are expressed in liver (Human Protein Atlas), and have clear human disease connections (DisGenNet mining), suggesting the BAC-eGFP cell lines could be used to generate safety signals in a high-throughput screening scenario.
A variety of cell-based screening platforms can generate safety signals early in drug discovery and development.21,32 However, safety decisions are not binary safe versus unsafe calls – they are highly context dependent on when the signal is detected, e.g., early versus late in the pipeline, what is the multiple between the signal and the efficacy exposures (margin of safety) and species differences in responses, e.g., PPARalpha-induced peroxisome proliferation occurs in rodent but not human. The results of both the TG:rLIV:176 and the HepG2-eGFP screens suggest that predictions are not absolutely perfect but highly useful to detect safety signals (Figure 5A). For example, in rat liver 8 of the 12 compounds exceeding the TG:rLIV:176 EG threshold were positive for single cell necrosis while four, WY-14643, diclofenac, omeprazole and ticlopidine, were the exceptions (although with lower activity) and might be considered false positives – the latter two are strong activators of Nrf2 modules which could act as an adaptive buffer against ATF4 activation and cell death.33 Three positive compounds, acetamide, aflatoxin and N-nitrosomorpholine might be considered false negatives because they did not exceed the TG:rLIV:176 EG threshold. The latter two are genotoxic carcinogens and in the DILI-TXG-MAPr showed a strong TG:rLIV:288 linked p53 activation signal (data not shown) suggesting an alternate p53-dependent upstream signaling event distinct from ATF4.34 Screening in the human HepG2-eGFP reporters suggests that both TRIB3 and MTHFD2 lines detected all of the single cell necrosis positive compounds as well as the three putative false positives (WY-14463, omeprazole and ticlopidine) but also responded to compound not flagged by TG:rLIV:176 in rat liver but had varying levels of DILI concern, and for which rat pathologies other than single cell necrosis were noted. None of the negative compounds were flagged in any of the three systems (except famotidine in the MTHFD2 line). Thus, a key to making the right safety decision in the right context is combining of generation of safety signals that are likely to translate across scales of complexity, for which the HepG2-eGFP cell are well suited, and understanding the biological context for that safety signal using the mechanistic analysis capabilities of the DILI-TXG-MAPr tool. When we combined the signals from the HepG2-eGFP reporters in a cluster correlation analysis, there were three distinct clusters (top, middle and bottom, Figure 5B). In general, the top cluster was driven by the enhanced CHOP, TRIB3 and MTHFD2 signals while the bottom cluster contained compounds that activated TRIB3 only. The three negative controls (no pathology) were in the middle cluster along with acetamide and 2 genotoxic carcinogens that depend on activation to reactive species by cytochromes P450 (acetamidofluorene, N-nitrosomorpholine).
The predictions obtained with our method could be improved both from the computational as from the experimental side. Increasing the number of non-DILI compounds in the rat experiments could strengthen the distinction of safety signals. Also, it is well known that HepG2 cells have low metabolic activation capability35; however they are ideal for high throughput screening because they can be grown, plated reproducibly, and screened in higher density multi-well formats and can be grown in a more differentiated 3D spheroid format allowing repeated dose evaluation36 As noted above, exposure relative to the anticipated efficacy exposure is a critical determinant of safety –in the bottom cluster the Cmax values for 5 of the 7 compounds for which Cmax information was available was either in the lower end of the exposure range (ethambutol and methapyrilene) or well below (colchicine, simvastatin and dexamethasone) the concentration range in which signals were generated from the HepG2-eGFP reporter cells.
In conclusion, we believe network methods can capture the interconnected nature of disease processes, which are responses of complex adaptive systems with emergent properties to environmental changes, e.g. nutrition, inflammation, etc., not captured in a purely reductionist approach.7,29,37 Modeling and understanding the complex and dynamic adaptive responses that alter the emergent properties of complex biological systems is necessary to unravel these complex interaction disease and drug toxicity phenotypes. The methodology presented here takes such an approach to define a multiscale, network-based approach to identify safety biomarkers quantifiable as part of an early screening platform and with increased confidence to be relevant for human safety.
Limitations of the study
A limitation of the study is determined by the datasets size: the more variability is included in the dataset used to generate modules or to calculate preservation statistics, the more descriptive and comprehensive of the studied biology will be the analysis. TG-GATEs and Drug Matrix are extensive datasets, but including more drugs and triggered responses might be beneficial. A second limitation of the study regards the in vitro model applied for the screening: the HepG2 cell line has well known metabolic limitations as well as cancer-like alterations and it represents a simplified model of hepatocytes. Applying more elaborated protocols such as improved medium or 3D culturing could partly overcome these limitations, although for practical reasons these protocols might reduce the high-throughput and cost-convenience of the method.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MTHFD2 | ABCAM | 4G7-2G3; RRID: AB_2147537 |
| Rabbit monoclonal anti-TRIB3 | ABCAM | EPR3151Y; RRID: AB_1267365 |
| Mouse monoclonal anti-Tubulin A | Sigma | DM 1A; RRID: AB_261775 |
| Chemicals, peptides, and recombinant proteins | ||
| Acetamide | Sigma | CAT#A0500-1G |
| Acetamidofluorene | Sigma | CAT#A7015-5G |
| Aflatoxine B1 | Sigma | CAT#A6636 |
| Cimetidine | Sigma | CAT#C4522-5G |
| Colchicine | Sigma | CAT#C9754 |
| Dexamethasone | Sigma | CAT#D4902 |
| Diclofenac | Sigma | CAT#D6899 |
| Diethyl maleate | Sigma | CAT#D97703 |
| Ethambutol | Sigma | CAT#E4630-25G |
| Ethionamide | Sigma | CAT#E6005-5G |
| Famotidine | Merck | CAT#F6889-500MG |
| Gefitinib | Sigma | CAT#SML1657 |
| Methapyrilene | Supelco | CAT#442641 |
| Nefazodone | Sigma | CAT#N5536 |
| N-Nitrosomorpholine | Sigma | CAT#N7382-1ML |
| Omeprazole | Supelco | CAT#PHR1059 |
| Phorone | Sigma | CAT#MAN424067694-10G |
| Simvastatin | Supelco | CAT#PHR1438-1G |
| Tert-Butylhydroquinone | Sigma | CAT#112941 |
| Tharpsigargin | Sigma | CAT#T9033 |
| Ticlopidine | Sigma | CAT#T6654 |
| Triamterene | Merck | CAT#T4143-10G |
| Tunicamycin C | Sigma | CAT#T7765 |
| WY-14643 | Sigma | CAT#C7081 |
| DMSO | Sigma | CAT#D2650 |
| Critical commercial assays | ||
| TempO-Seq | BioClavis J. M. Yeakley et al.38 |
https://www.bioclavis.co.uk/overview |
| Rneasy kit | Qiagen | CAT#74004 |
| RevertAid H Minus First strand cDNA Synthesis Kit | Thermo Scientific | CAT#K1682 |
| PowerUp SYBR Green Master Mix | Sigma | CAT#KCQS02 |
| Experimental models: Cell lines | ||
| HepG2 cell line | ATCC | Clone HB8065 |
| Oligonucleotides | ||
| siRNA siGENOME MTHFD2 SMARTpool | GE Dharmacon | gene ID 10797 |
| siRNA siGENOME TRIB3 SMARTpool | GE Dharmacon | gene ID 57761 |
| siRNA siGENOME ATF4 SMARTpool | GE Dharmacon | gene ID 468 |
| siRNA siGENOME DDIT3 SMARTpool | GE Dharmacon | gene ID 1649 |
| Primer real time quantitative PCR are in suppl. Table SZ | This paper | N/A |
| Recombinant DNA | ||
| Bacterial Artificial Chromosomes MTHFD2 | BACPAC Genomics (Emeryville, California, USA) | RP11-101L15 |
| Bacterial Artificial Chromosomes TRIB3 | BACPAC Genomics (Emeryville, California, USA) | RP11-99E14 |
| Software and algorithms | ||
| affy R package version 1.52.0 | L. Gautier et al.38 | http://bioconductor.org/packages/release/bioc/html/affy.html |
| BrainArray chip description file (CDF) version 19 Rat2302 | MicroArray Lab | http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download.asp |
| BrainArray chip description file (CDF) version 19 PHH |
MicroArray Lab G. Callegaro et al.16 |
http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download.asp |
| limma R package version 3.30.13 | M. E. Ritchie et al.39 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| DESeq2 R package version 1.28.1 | M. I. Love et al.40 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| WGCNA R package version 1.51 | P. Langfelder et al.9 | https://www.rdocumentation.org/packages/WGCNA/versions/1.51 |
| OverRepresentation Analysis (ORA) via Consensus Pathway DB (cpdb version 34) | A. Kamburov et al.41 | http://cpdb.molgen.mpg.de/CPDB |
| topGO R package version 2.26.0 | A. Alexa et al.42 | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| stat R package (R version 3.5.1) | R Core Team | https://cran.r-project.org/bin/windows/base/old/3.5.1/ |
| DoRothEA | L. Garcia-Alonso et al.26 | https://bioconductor.org/packages/release/data/experiment/html/dorothea.html |
| TXG-MAPr | G. Callegaro et al.18 | https://txg-mapr.eu/WGCNA_liver/TGGATEs_LIVER/ |
| DisGeNET database v7.0 | J. Piñero et al.14 | https://www.disgenet.org/ |
| Human Protein Atlas | M. Uhlén et al.15 | https://www.proteinatlas.org/ |
| Other | ||
| Dulbecco’s modified Eagle’s medium | Gibco | 11504496 |
| Penicillin-Streptomycin | Gibco | 15070063 |
| PageRuler™ Plus | Thermo Scientific | 26620 |
| Deposited data | ||
| TG-GATEs dataset | Igarashi et al.11 | Open TG-GATEs data available at http://toxico.nibio.go.jp/english/index.html |
| Drug Matrix dataset, rat in vivo experiments | Ganter et al. 12 | GEO Series: GSE57815 |
| Rat Genome Database, orthology table | Smith et al.43 | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bob van de Water (b.water@lacdr.leidenuniv.nl).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cells
This study used HepG2 cell line (HB-8065, ATCC) and HepG2 reporter cell lines obtained with bacterial artificial chromosome recombineering as described below in the method details section. All cells were seeded it 384 well plates (CellCarrierTM Perkin Elmer, cat. 6057302) at a density of 8000 cells per well. Cells were grown, treated and imaged under standard cell culture conditions (37°C, 5% CO2, saturated humidity) in DMEM.
Method details
Gene expression data processing
Rat liver gene expression dataset was downloaded by TG-GATEs (TG) repository (https://dbarchive.biosciencedbc.jp/en/open-tggates/download.html), consisting of 3528 combinations of compound, exposure time (single and repeated exposure) and exposure dose, tested in three different animals.11 Microarrays were jointly normalized with Robust Multi-array Average (RMA) method within the affy R package version 1.52.0.38 Probe sets were mapped to Entrez ID with the BrainArray chip description file (CDF) version 19 (http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download.asp, Rat2302), resulting in 13,877 unique gene IDs, mapped one to one to the probe sets. TG-GATEs experiments were designed so that every compound-time point combination is matched to a specific control. Log2 fold-change values of treatment conditions versus respective controls were calculated by building a linear model including concentration levels in the design as covariate and computing the log-odds of differential expression by empirical Bayes moderation (limma R package version 3.30.1339). Significance of log2FC was determined by using Benjamini–Hochberg multiple testing correction. Drug Matrix rat liver dataset and Primary Human Hepatocyte (PHH) TG dataset were analyzed following the same steps, but with suitable CDF description as described above.18 The HepG2 gene expression dataset includes 40 compounds tested in 4 to 6 concentration levels and 2 to 3 time points (4, 8, 24 hrs). Gene expression was measured with TempOseq technology in combination with the whole transcriptome gene set.44 Raw counts were normalized with CPM normalization, log2 transformed and log2FC calculated with DESeq2 (R package version 1.28.1).40
WGCNA module generation
Co-expressed gene sets were identified with the WGCNA R package version 1.519 and applied to the matrix consisting of 3258 rows (rat liver experiments) and 13,877 columns (log2 fold change values for genes). We selected as optimal soft-power threshold 6 (unsigned method, average method for clustering, deep-split = 4, min cluster size = 5), by maximizing both the scale-free network topology using standard power-law plotting tool in WGCNA9 and the exclusion of genes having low gene intensity in the vehicle controls (determined via t-tests). For each experiment and module, we calculated the eigengene score (EGs, or module score), the z-scored 1st principal component of the PCA performed on the gene matrix of each module, which summarizes log2 fold change of their constituent genes, as described previously.18 After merging modules having correlation of their EGs ≥0.8, we obtained 317 modules containing 13,095 genes. As described previously,18 we then identified the hub gene in each module network and characterized each gene in a module by calculating the correlation between the log2 fold change versus the EGs of the parent module across the 3258 experiments (termed ‘corEG’). Modules are displayed in a folded hierarchical tree (dendrogram), based on Ward’s hierarchical clustering of pair-wise Pearson correlations for each module across all treatment conditions. Module locations on dendrogram branches were identified with a hierarchical anti-clockwise nomenclature system, and branches length adjusted to improve visualization (Figures S1A and S1B).
Module annotation
Modules’ gene sets were used to perform OverRepresentation Analysis (ORA) via Consensus Pathway DB (cpdb version 34), including the following databases: BioCarta, EHMN, HumanCyc, INOH, KEGG, NetPath, Reactome, Signalink, SMPDB, Wikipathways, UniProt, InterPro.41 Additionally, GO enrichment was obtained with the R package topGO version 2.26.0, algorithm = “classic”, statistic = “fisher”.42 For both resources, only terms enriched with a hypergeometric test p-value <0.01 were considered. Transcription Factors (TFs) were associated to each module by performing a hypergeometric test on gene members using the function phyper within the stat package in R (version 3.5.1). The gene set of TFs and their regulated genes (regulons) are derived from DoRothEA.26 DoRothEA database does not provide rat specific annotation, therefore the human targets and TFs were translated into rat gene names IDs with the Rat Genome Database, which provided one-to-one ortholog matches,43 assuming similar regulation in the two species. We used two sets of confidence levels: the “high confidence” level comprises categories A, B and C while the “high coverage” level comprises categories A, B, C and D. The enriched TFs with p-value less than 0.01 were included in the study.
Module to pathology association
Modules are associated with pathologies and previously described5 and summarized in brief in the following: 1) histology findings and chemistry results in TG were binarized (0 or 1) for each compound-time-dose combination, by averaging the severity score of each animal for each finding. A severity average grade above 0.67 was considered positive. 2) we defined ‘toxicity phenotypes’ consisting of an ‘anchoring phenotype’, i.e. a given histologically defined lesion (for example, single-cell necrosis) or clinical chemistry result (for example, increased total bilirubin), and defined multiple selectivity levels allowing co-occurrence of other findings (e.g. no other findings, allowing only specific other findings, any other findings). Findings occurring in isolation, can be considered non-adverse, while in combination there is stronger indication of organ damage. Following this strategy, we defined 26 toxicity phenotypes with at least 7 treatments showing a positive average score (>0.67). 3). For each module, the association with a toxicity phenotype was quantified using the effect size measurement Cohen’s d.45 The average absolute transcriptional activity (quantified through EGs, avgAbsEG) can have a large effect size alone for many toxicity phenotypes, therefore we performed logistic regression to determine the contribution of module scores after accounting for the avgAbsEG, and reported the significance of each module as the p-adj associated with the module score. More details can be found in5 and Table S3 and Figure S2B. Modules’ structure, annotation, pathology associations and more are available at the TXG-MAPr user interface (https://txg-mapr.eu/WGCNA_liver/TGGATEs_LIVER/), with a similar structure as the sister application for PHH (Figure S3,18).
Preservation analysis and calculation of module scores for external datasets
Preservation between TG-based modules and other datasets was performed with the WGCNA R package and thresholds for interpretation were adopted by relevant literature,13 and rat gene IDs have been converted to human gene IDs with the Rat Genome Database, which provided only one-to-one ortholog matches.43 Complete Z-summary scores and overlap Fisher t test p values are provided in Tables S1, S2, S5, and S6. TG-based modules’ scores were calculated as described previously18 and considering the same one-to-one ortholog matching as above. In brief, new EGs scores are calculated by summing the Z-scored gene log2FC values of the module genes, normalized by the standard deviation of the raw module score in the TG-GATEs dataset.
Biomarker prioritization
We used DisGeNET database v7.014 and the Human Protein Atlas (HPA,15) to annotate the biomarkers with information about human diseases and phenotypes and proteomic and transcriptomics information. We computed the number of hepatic phenotypes, and the number of annotations in DisGeNET for the gene, that includes all diseases, and phenotypes. According to the HPA, a gene can be classified as tissue-enriched for liver (meaning that the normalized RNA expression level (NX) in liver is at least four times any other tissue), as group-enriched (NX levels of a group (of 2–5 tissues including liver) are at least four times larger than any other tissue type), as tissue-enhanced (NX levels of a group of 1–5 tissues including liver are at least four times the mean of other tissue, or with low tissue specificity. Finally, we included information about the plasma proteome and an attribute to reflect if the protein is present in the cell membrane (HPA).
BAC HepG2 reporter generation and validation
Bacterial artificial chromosome recombineering
The Bacterial Artificial Chromosomes (BAC) were recombined as described by Poser et al.46 The HepG2 reporter cell lines were generated as described by Wink et al.47 MTHFD2 (clone RP11-101L15) and TRIB3 (clone RP11-99E14) containing BACs were purchased from BACPAC Genomics (Emeryville, California, USA).
siRNA knock down
siRNAs were obtained from GE Dharmacon (siGENOME) against MTHFD2 (gene ID 10797), TRIB3 (57761), ATF4 (468), DDIT3 (1649). For all genes a SMARTpool was used to ensure high knock down efficiency. Cells were reversely transfected with siRNA at a concentration of 50 nM and a cell density of 2.4∗10ˆ4 cells per well in a 96 well plate (CellCarrierTM-96 ultra, Perkin Elmer, cat. 6057300). Cells were exposed to tunicamycin C or solvent control (DMSO 0.1% v/v) 72h after seeding. Cells were grown, treated and imaged under standard cell culture conditions (37°C, 5% CO2, saturated humidity) in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, cat. 11504496) supplemented with 10% (v/v) FBS and 0.5%(v/v) Penicillin-Streptomycin (Gibco, 15070063). All results of the siRNA KD experiments are displayed in Figure S5 (TRIB3) and S8 (MTHFD2).
Western blotting WB
Cells were seeded in 24 well plates (CELLSTAR®, Greiner Bio-One, cat. 662160) at 2∗10ˆ5 cells per well. 48h after seeding cells were exposed to tunicamycin C (see supplemental table) or solvent controls. 48h After exposures, cells, were washed with ice-cold PBS and directly lysed with SPB buffer (10% v/v beta-mercaptoethanol). The antibodies used were MTHFD2 (ABCAM, 4G7-2G3), TRIB3 (ABCAM, EPR3151Y) and tubulin A (Sigma, DM 1A). The protein protein size was estimated in comparison to a protein ladder (PageRuler™ Plus, Thermo Scientific, 26620). Original blots are displayed in Figure S6 and S7 (TRIB3) and in S9, S10 (MTHFD2).
Real time quantitative PCR
Cells were seeded in 24 well plates (CELLSTAR®, Greiner Bio-One, cat. 662160) at 2∗10ˆ5 cells per well. 48h after seeding cells were exposed to tunicamycin C (see supplemental table) or solvent controls. 48h After exposures, cells, were washed with ice-cold PBS and directly lysed with lysis buffer provided in the Rneasy kit (Qiagen).
Total RNA was isolated using the Rneasy kit (Qiagen) and yield and purity was measured by spectrophotometry (DeNovix DS-11). 800 ng RNA was used to generate cDNA with the RevertAid H Minus First strand cDNA Synthesis Kit (Thermo Scientific). 5x diluted cDNA, 1 μM forward and reverse primers, and PowerUp SYBR Green Master Mix was mixed and used for RT-qPCR. Parameters for RT-qPCR were as followed: 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute, and final steps of 15 seconds at 95°C, 10 seconds at 60°C, 95°C for 30 seconds and 15 seconds at 60°C. mRNA expression was quantified using the delta-delta Ct (2-ΔΔCt) method and normalized to the housekeeping genes GAPDH and TBP. The primer sequences used are displayed in Table S12.
Compound screening
Details about the compounds can be found in Table S9. Compounds were freshly dissolved in DMSO.
We selected compounds for the proof of concept screening belonging to five categories: compounds inducing single cell necrosis in vivo, i.e. scoring average grade higher than 0.66 in at least one concentration-time point combination (“Positive for scNecrosis”), compounds not inducing single cell necrosis in vivo and no other pathologies (vacuolation, hypertrophy and bile duct hyperplasia, “Negative for scNecrosis and no other pathologies”), compounds not inducing single cell necrosis in vivo but inducing other pathologies considered non-adverse (“Negative for scNecrosis + non-adverse other path.”), compounds not inducing single cell necrosis in vivo but inducing other cell death pathologies (“Negative for scNecrosis + other cell death path.”), and compounds not tested in TG-GATEs (“No data in TG”). The complete compound table, including concentrations tested and concentration interval design is available as Table S9. We further selected in the screening compounds belonging to the category “Positive compounds for scNecrosis4” when showing a strong positive correlation between module’s TG:rLIV:176 EGs and single cell necrosis score (Pearson R > 0.5, Figures S11A and S11B, a total of 12 compounds). In the group “No data in TG” we included three compounds because of known molecular mechanisms that could trigger the Atf4 response or DILI annotations: nefazodone, Most-DILI-Concern according to DILI ranking25 and ER stress and mitochondrial dysfunction inducer,48,49,50 thapsigargin, ER stress inducer21 and tert-butylhydroquinone, Nrf2 activator.51 To select compounds for the group “Negative for scNecrosis no path”, we focused on compounds having Less-DILI-Concerns in the FDA DILI ranking.25
Image analysis
Images were analyzed as described in.47 For analysis, the threshold of a GP positive cell was set to the mean eGFP intensity of either the nucleus (CHOP, TRIB3) or the cytoplasm (MTHFD2) of all cells visualized in the DMSO control of each cell line. Any cell above this threshold was considered eGFP positive. PI positive cells were identified based on the overlap of an PI object with the cells’ nucleus.52 Images were analyzed as described in Wink et al.47 For analysis, the threshold of a GP positive cell was set to the mean eGFP intensity of either the nucleus (CHOP, TRIB3) or the cytoplasm (MTHFD2) of all cells visualized in the DMSO control of each cell line. Any cell above this threshold was considered eGFP positive. PI positive cells were identified based on the overlap of an PI object with the cells’ nucleus.52 Example images can be found in Figure S13, correlation analysis across features in Table S10.
Additional resources
WGCNA modules derived from TG-GATEs rat in vivo experiments are available at the TXG-MAPr user interface (https://txg-mapr.eu/WGCNA_liver/TGGATEs_LIVER/), with a similar structure as the sister application for PHH.
Acknowledgments
This work was supported by the EU-EFPIA Innovative Medicines Initiative 2 (IMI2) Joint Undertaking TransQST project (grant number 116030) and eTRANSAFE project (grant number 777365) (this Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA), the EC Horizon2020 EU-ToxRisk project (grant number 681002), the European Research & Innovation project funded under the European Commission’s Horizon 2020 program RISK-HUNT3R project (grant number 964537) and the Cosmetics Europe/CEFIC Liver Ontology project.
This publication was neither originated nor managed by AbbVie, and it does not communicate results of AbbVie-sponsored Scientific Research. Thus, it is not in scope of the AbbVie Publication Procedure (PUB-100).
Author contributions
Conceptualization: G.C., J.P.S., J.L.S., B.vd.W.
Methodology: G.C., J.P.S., L.vd.B.
Investigation: G.C., J.P.S., L.vd.B, J.P.G., L.F., K.B., L.W., S.J.K., J.J.S., J.M.
Visualization: G.C., J.P.S.
Funding acquisition: B.vd.W.
Supervision: B.vd.W, J.L.S.
Writing – original draft: G.C., J.P.S.
Writing – review and editing: J.L.S., B.vd.W., S.J.K., J.J.S.
Declaration of interests
L.I.F. and J.P.G. are employees and shareholders of MedBioInformatics Solutions SL.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106094.
Contributor Information
James L. Stevens, Email: j.l.stevens@lacdr.leidenuniv.nl.
Bob van de Water, Email: b.water@lacdr.leidenuniv.nl.
Supplemental information
The lower p value of possible overlaps is reported, and only if < 0.05, related to Figure 1.
The lower p value of possible overlaps is reported, and only if < 0.05, related to Figure 2.
Data and code availability
-
•
Data. This paper analyzes existing, publicly available data of TG-GATEs and DrugMatrix datasets. These accession numbers for the datasets are listed in the key resources table. The HepG2 dataset reported in this paper will be shared by the lead contact upon request and is currently under consideration for publication.
-
•
Code. This paper does not report original code, but uses of published CRAN packages detailed in the STAR Methods section.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Cook D., Brown D., Alexander R., March R., Morgan P., Satterthwaite G., Pangalos M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 2014;13:419–431. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 2.Watkins P.B. Drug safety sciences and the bottleneck in drug development. Clin. Pharmacol. Ther. 2011;89:788–790. doi: 10.1038/clpt.2011.63. [DOI] [PubMed] [Google Scholar]
- 3.Monroe J.J., Tanis K.Q., Podtelezhnikov A.A., Nguyen T., Machotka S.v., Lynch D., Evers R., Palamanda J., Miller R.R., Pippert T., et al. Application of a rat liver drug bioactivation transcriptional response assay early in drug development that informs chemically reactive metabolite formation and potential for drug-induced liver injury. Toxicol. Sci. 2020;177:281–299. doi: 10.1093/toxsci/kfaa088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podtelezhnikov A.A., Monroe J.J., Aslamkhan A.G., Pearson K., Qin C., Tamburino A.M., Loboda A.P., Glaab W.E., Sistare F.D., Tanis K.Q. Quantitative transcriptional biomarkers of xenobiotic receptor activation in rat liver for the early assessment of drug safety liabilities. Toxicol. Sci. 2020;175:98–112. doi: 10.1093/toxsci/kfaa026. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland J.J., Webster Y.W., Willy J.A., Searfoss G.H., Goldstein K.M., Irizarry A.R., Hall D.G., Stevens J.L. Toxicogenomic module associations with pathogenesis: a network-based approach to understanding drug toxicity. Pharmacogenomics J. 2018;18:377–390. doi: 10.1038/tpj.2017.17. [DOI] [PubMed] [Google Scholar]
- 6.Dirven H., Vist G.E., Bandhakavi S., Mehta J., Fitch S.E., Pound P., Ram R., Kincaid B., Leenaars C.H.C., Chen M., et al. Performance of preclinical models in predicting drug-induced liver injury in humans: a systematic review. Sci. Rep. 2021;11:6403. doi: 10.1038/s41598-021-85708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barabási A.L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ankley G.T., Gray L.E. Cross-species conservation of endocrine pathways: a critical analysis of tier 1 fish and rat screening assays with 12 model chemicals. Environ. Toxicol. Chem. 2013;32:1084–1087. doi: 10.1002/etc.2151. [DOI] [PubMed] [Google Scholar]
- 9.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B., Horvath S. A general framework for weighted gene Co- expression network analysis A general framework for weighted gene Co- expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi Y., Nakatsu N., Yamashita T., Ono A., Ohno Y., Urushidani T., Yamada H. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 2015;43:D921–D927. doi: 10.1093/nar/gku955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganter B., Tugendreich S., Pearson C.I., Ayanoglu E., Baumhueter S., Bostian K.A., Brady L., Browne L.J., Calvin J.T., Day G.-J., et al. Development of a large-scale chemogenomics database to improve drug candidate selection and to understand mechanisms of chemical toxicity and action. J. Biotechnol. 2005;119:219–244. doi: 10.1016/j.jbiotec.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Langfelder P., Luo R., Oldham M.C., Horvath S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011;7 doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piñero J., Ramírez-Anguita J.M., Saüch-Pitarch J., Ronzano F., Centeno E., Sanz F., Furlong L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48:D845–D855. doi: 10.1093/nar/gkz1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;347:347. doi: 10.1126/SCIENCE.1260419. [DOI] [PubMed] [Google Scholar]
- 16.Levin S., Bucci T.J., Cohen S.M., Fix A.S., Hardisty J.F., Legrand E.K., Maronpot R.R., Trump B.F. Focus on: nomenclature of cell death the nomenclature of cell death: recommendations of an ad hoc committee of the society of toxicologic pathologists∗. Toxicol. Pathol. 1999;27:484–490. doi: 10.1177/019262339902700419. [DOI] [PubMed] [Google Scholar]
- 17.Liver . Hepatocyte - Apoptosis - Nonneoplastic Lesion Atlas. 2017. https://ntp.niehs.nih.gov/nnl/hepatobiliary/liver/hapop/index.htm [Google Scholar]
- 18.Callegaro G., Kunnen S.J., Trairatphisan P., Grosdidier S., Niemeijer M., den Hollander W., Guney E., Piñero Gonzalez J., Furlong L., Webster Y.W., et al. The human hepatocyte TXG-MAPr: gene co-expression network modules to support mechanism-based risk assessment. Arch. Toxicol. 2021;95:3745–3775. doi: 10.1007/s00204-021-03141-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L., Yamazaki T., Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:731–745. doi: 10.1038/S41580-018-0068-0. [DOI] [PubMed] [Google Scholar]
- 20.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 21.Wink S., Hiemstra S.W., Huppelschoten S., Klip J.E., van de Water B. Dynamic imaging of adaptive stress response pathway activation for prediction of drug induced liver injury. Arch. Toxicol. 2018;92:1797–1814. doi: 10.1007/s00204-018-2178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin S., Buel G.R., Wolgamott L., Plas D.R., Asara J.M., Blenis J., Yoon S.O. ERK2 mediates metabolic stress response to regulate cell fate. Mol. Cell. 2015;59:382–398. doi: 10.1016/J.MOLCEL.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costa-Mattioli M., Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368:eaat5314. doi: 10.1126/SCIENCE.AAT5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Z., Leung G.K.K. More than a metabolic enzyme: MTHFD2 as a novel target for anticancer therapy? Front. Oncol. 2020;10:658. doi: 10.3389/fonc.2020.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen M., Suzuki A., Thakkar S., Yu K., Hu C., Tong W. DILIrank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today. 2016;21:648–653. doi: 10.1016/j.drudis.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Alonso L., Holland C.H., Ibrahim M.M., Turei D., Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29:1363–1375. doi: 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waring M.J., Arrowsmith J., Leach A.R., Leeson P.D., Mandrell S., Owen R.M., Pairaudeau G., Pennie W.D., Pickett S.D., Wang J., et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015;14:475–486. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- 28.Butler L.D., Guzzie-Peck P., Hartke J., Bogdanffy M.S., Will Y., Diaz D., Mortimer-Cassen E., Derzi M., Greene N., DeGeorge J.J. Current nonclinical testing paradigms in support of safe clinical trials: an IQ Consortium DruSafe perspective. Regul. Toxicol. Pharmacol. 2017;87:S1–S15. doi: 10.1016/j.yrtph.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Finzer P. How we become ill. EMBO Rep. 2017;18:515–518. doi: 10.15252/embr.201743948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander-Dann B., Pruteanu L.L., Oerton E., Sharma N., Berindan-Neagoe I., Módos D., Bender A. Developments in toxicogenomics: understanding and predicting compound-induced toxicity from gene expression data. Mol. Omics. 2018;14:218–236. doi: 10.1039/C8MO00042E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauer U.G., Deferme L., Gribaldo L., Hackermüller J., Tralau T., van Ravenzwaay B., Yauk C., Poole A., Tong W., Gant T.W. The challenge of the application of ’omics technologies in chemicals risk assessment: background and outlook. Regul. Toxicol. Pharmacol. 2017;91:S14–S26. doi: 10.1016/j.yrtph.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 32.Hatherell S., Baltazar M.T., Reynolds J., Carmichael P.L., Dent M., Li H., Ryder S., White A., Walker P., Middleton A.M. Identifying and characterizing stress pathways of concern for consumer safety in next-generation risk assessment. Toxicol. Sci. 2020;176:11–33. doi: 10.1093/TOXSCI/KFAA054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarcinelli C., Dragic H., Piecyk M., Barbet V., Duret C., Barthelaix A., Ferraro-Peyret C., Fauvre J., Renno T., Chaveroux C., Manié S.N. ATF4-Dependent NRF2 transcriptional regulation promotes antioxidant protection during endoplasmic reticulum stress. Cancers. 2020;12 doi: 10.3390/CANCERS12030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J., Thomas H.R., Li Z., Yeo N.C.F., Scott H.E., Dang N., Hossain M.I., Andrabi S.A., Parant J.M. Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis. 2021;12:659. doi: 10.1038/S41419-021-03902-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeCluyse E.L., Witek R.P., Andersen M.E., Powers M.J. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit. Rev. Toxicol. 2012;42:501–548. doi: 10.3109/10408444.2012.682115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiemstra S., Ramaiahgari S.C., Wink S., Callegaro G., Coonen M., Meerman J., Jennen D., van den Nieuwendijk K., Dankers A., Snoeys J., et al. High-throughput confocal imaging of differentiated 3D liver-like spheroid cellular stress response reporters for identification of drug-induced liver injury liability. Arch. Toxicol. 2019;93:2895–2911. doi: 10.1007/S00204-019-02552-0. [DOI] [PubMed] [Google Scholar]
- 37.Esmaili S., Langfelder P., Belgard T.G., Vitale D., Azardaryany M.K., Alipour Talesh G., Ramezani-Moghadam M., Ho V., Dvorkin D., Dervish S., et al. Core liver homeostatic co-expression networks are preserved but respond to perturbations in an organism- and disease-specific manner. Cell Syst. 2021;12:432–445.e7. doi: 10.1016/J.CELS.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Gautier L., Cope L., Bolstad B.M., Irizarry R.A. Affy - analysis of affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 39.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamburov A., Stelzl U., Lehrach H., Herwig R. The ConsensusPathDB interaction database: 2013 Update. Nucleic Acids Res. 2013;41:D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexa A., Rahnenführer J. Vol. 27. R package; 2007. (Gene Set Enrichment Analysis with topGO). [Google Scholar]
- 43.Smith J.R., Hayman G.T., Wang S.J., Laulederkind S.J.F., Hoffman M.J., Kaldunski M.L., Tutaj M., Thota J., Nalabolu H.S., Ellanki S.L.R., et al. The year of the rat: the rat Genome database at 20: a multi-species knowledgebase and analysis platform. Nucleic Acids Res. 2020;48:D731–D742. doi: 10.1093/nar/gkz1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeakley J.M., Shepard P.J., Goyena D.E., Vansteenhouse H.C., McComb J.D., Seligmann B.E. A Trichostatin a expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS One. 2017;12:e0178302. doi: 10.1371/journal.pone.0178302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen Y., Cohen J.Y. Wiley; 2008. Statistics and Data with R: An Applied Approach through Examples. [Google Scholar]
- 46.Poser I., Sarov M., Hutchins J.R.A., Hériché J.K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A.W., et al. BAC TransgeneOmics. Nat. Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wink S., Hiemstra S., Huppelschoten S., Danen E., Niemeijer M., Hendriks G., Vrieling H., Herpers B., van de Water B. Quantitative high content imaging of cellular adaptive stress response pathways in toxicity for chemical safety assessment. Chem. Res. Toxicol. 2014;27:338–355. doi: 10.1021/tx4004038. [DOI] [PubMed] [Google Scholar]
- 48.Silva A.M., Barbosa I.A., Seabra C., Beltrão N., Santos R., Vega-Naredo I., Oliveira P.J., Cunha-Oliveira T. Involvement of mitochondrial dysfunction in nefazodone-induced hepatotoxicity. Food Chem. Toxicol. 2016;94:148–158. doi: 10.1016/j.fct.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 49.Ren Z., Chen S., Zhang J., Doshi U., Li A.P., Guo L. Endoplasmic reticulum stress induction and ERK1/2 activation contribute to nefazodone-induced toxicity in hepatic cells. Toxicol. Sci. 2016;154:368–380. doi: 10.1093/toxsci/kfw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fredriksson L., Wink S., Herpers B., Benedetti G., Hadi M., de Bont H., Groothuis G., Luijten M., Danen E., de Graauw M., et al. Drug-induced endoplasmic reticulum and oxidative stress responses independently sensitize toward TNFα-mediated hepatotoxicity. Toxicol. Sci. 2014;140:144–159. doi: 10.1093/toxsci/kfu072. [DOI] [PubMed] [Google Scholar]
- 51.Bischoff L.J.M., Kuijper I.A., Schimming J.P., Wolters L., Braak B.T., Langenberg J.P., Noort D., Beltman J.B., van de Water B. A systematic analysis of Nrf2 pathway activation dynamics during repeated xenobiotic exposure. Arch. Toxicol. 2019;93:435–451. doi: 10.1007/s00204-018-2353-2. [DOI] [PubMed] [Google Scholar]
- 52.Schimming J.P., ter Braak B., Niemeijer M., Wink S., van de Water B. System microscopy of stress response pathways in cholestasis research. Methods Mol. Biol. 2019;1981:187–202. doi: 10.1007/978-1-4939-9420-5_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The lower p value of possible overlaps is reported, and only if < 0.05, related to Figure 1.
The lower p value of possible overlaps is reported, and only if < 0.05, related to Figure 2.
Data Availability Statement
-
•
Data. This paper analyzes existing, publicly available data of TG-GATEs and DrugMatrix datasets. These accession numbers for the datasets are listed in the key resources table. The HepG2 dataset reported in this paper will be shared by the lead contact upon request and is currently under consideration for publication.
-
•
Code. This paper does not report original code, but uses of published CRAN packages detailed in the STAR Methods section.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.